
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFc-binding antibody-recruiting molecules exploit endogenous antibodies for anti-tumor immune responses†
Koichi
Sasaki
 a,
Minori
Harada
b,
Yoshiki
Miyashita
b,
Hiroshi
Tagawa
b,
Akihiro
Kishimura
abc,
Takeshi
Mori
*ab and
Yoshiki
Katayama
*abcd
a,
Minori
Harada
b,
Yoshiki
Miyashita
b,
Hiroshi
Tagawa
b,
Akihiro
Kishimura
abc,
Takeshi
Mori
*ab and
Yoshiki
Katayama
*abcd
aDepartment of Applied Chemistry, Graduate School of Engineering, Kyushu University, Fukuoka, 819-0395, Japan. E-mail: mori.takeshi.880@m.kyushu-u.ac.jp; ykatatcm@mail.cstm.kyushu-u.ac.jp
bGraduate School of Systems Life Sciences, Kyushu University, Fukuoka, 819-0395, Japan
cInternational Research Center for Molecular Systems, Kyushu University, Fukuoka, 819-0395, Japan
dDepartment of Biomedical Engineering, Chung Yuan Christian University, Taoyuan, Taiwan
First published on 25th February 2020
Abstract
Redirecting endogenous antibodies in the bloodstream to tumor cells using synthetic molecules is a promising approach to trigger anti-tumor immune responses. However, current molecular designs only enable the use of a small fraction of endogenous antibodies, limiting the therapeutic potential. Here, we report Fc-binding antibody-recruiting molecules (Fc-ARMs) as the first example addressing this issue. Fc-ARMs are composed of an Fc-binding peptide and a targeting ligand, enabling the exploitation of endogenous antibodies through constant affinity to the Fc region of antibodies, whose sequence is conserved in contrast to the Fab region. We show that Fc-ARM targeting folate receptor-α (FR-α) redirects a clinically used antibody mixture to FR-α+ cancer cells, resulting in cancer cell lysis by natural killer cells in vitro. Fc-ARMs successfully interacted with antibodies in vivo and accumulated in tumors. Furthermore, Fc-ARMs recruited antibodies to suppress tumor growth in a mouse model. Thus, Fc-ARMs have the potential to be a novel class of cancer immunotherapeutic agents.
Introduction
Immune cells, such as natural killer (NK) cells, recognize antibodies bound to target proteins and lyse target cells, and this process is termed antibody-dependent cell-mediated cytotoxicity (ADCC). Based on immune-mediated mechanisms of action including ADCC, therapeutic IgG antibodies have revolutionized the clinical outcomes for multiple types of cancers.1 However, the potential immunogenicity of monoclonal antibodies (mAbs) is one of their limitations, causing side effects such as IgE-mediated anaphylaxis and the production of anti-drug antibodies in the clinic.2,3As a potential solution to this issue, antibody-recruiting small molecules (ARMs) have been developed to induce antibody-mediated immune responses while circumventing antibody administration.4 ARMs are composed of an antibody-binding terminus (ABT) and a target-binding terminus (TBT) (Fig. 1A), and redirect endogenous antibodies in the bloodstream to target cells such as tumor cells. Subsequently, recruited antibodies trigger immune responses to eliminate the target (Fig. 1B, left panel). Notably, ARMs are thought to be less immunogenic due to their small molecular size.5,6 To date, various types of TBTs have been developed.7–22 By contrast, only a few types of ABTs have shown potential utility: galactose-α-1,3-galactose (α-Gal),12,13 rhamnose,21 nitroarenes,14–16,20,22 and phosphorylcholine.23 This is because ABTs have so far been limited to antigens against endogenous antibodies present in a wide range of population.
 | ||
| Fig. 1 Fc-binding antibody-recruiting molecule (Fc-ARM) concept in comparison with conventional ARMs. (A) Antibody-recruiting small molecule (ARM). ABT = antibody-binding terminus; TBT = target-binding terminus. (B) Conventional ARM-mediated (left) and Fc-ARM-mediated (right) induction of ADCC. (C) Molecular design of Fc-ARM1 and Fc-ARM2. | ||
However, these types of ABTs suffer from some shortcomings that limit their therapeutic application. For example, endogenous antibodies against ABTs are generally in short supply (e.g. anti-α-Gal accounts for ∼1% of endogenous IgG in human serum24), limiting the probability of encountering ARMs and antibodies in the human body. In addition, the efficacy of ARM-based therapy may be influenced by the polyclonal affinity of endogenous antibodies to the ABT.4 Although immunization of patients with ABTs may help to improve the characteristics of anti-ABT endogenous antibodies,4,9,17,20,22 it increases the complexity of treatment procedures, and may also induce additional side effects. Thus, new approaches that enable the exploitation of a larger proportion of pre-existing endogenous antibodies by constant affinity would be a breakthrough for the clinical translation of ARMs.
The Fc region of an antibody is characterized by its conserved molecular structure, as well as a variety of biological functions. It is the Fc module that is responsible for ADCC in the development of biopharmaceuticals.25,26 A recent study revealed that the Fc region of an antibody binds to virus-infected cells and NK cells simultaneously, resulting in ADCC against the infected cells.27 This report indicated that ADCC could be induced independently of antigen–Fab interactions.
Previously, we reported a new class of ARMs, namely Fc-ARMs, in which the Fc-binding cyclic peptide (Fc-III)28 is used as an ABT (Fig. 1B, right panel).29 Use of the Fc-binding peptide enables Fc-ARMs to at least access IgG1 and IgG2 with constant affinity.30 These two IgG subclasses account for more than 80% of the total IgG in humans.31,32 Thus, Fc-ARMs can theoretically exploit the majority of endogenous antibodies through their constant affinity to IgG-Fc. We previously demonstrated that Fc-ARMs can recruit human IgG on the surface of cancer cells in a selective manner.29 However, recruited antibodies did not mediate ADCC under the previous experimental conditions. Given that ADCC is regulated by the overall affinity of the antibody to its antigen33 and CD16a,34 we hypothesized that the replacement of the Fc-III peptide with a recently reported peptide, Fc-III4C, which has higher affinity for IgG-Fc,35 would strengthen the recruited antibody ability to activate effector cells such as NK cells for target cell destruction. We conducted quantitative evaluations of antibody recruitment and determined the anti-tumor effects of Fc-ARM both in vitro and in vivo to prove the concept of the Fc-ARM strategy.
Results and discussion
Fc affinity and antibody recruitment
We used an Fc-III peptide or Fc-III4C peptide35 as an ABT and folic acid (FA) as a TBT (Fig. 1C). FA has sub-nanomolar affinity to folate receptor-a (FR-α)36 and is therefore used for targeted drug delivery and imaging of cancers overexpressing FR-α.37 We connected the ABT and TBT using a hexaethylene glycol linker and synthesized two types of Fc-ARMs, namely Fc-ARM1 and Fc-ARM2. The Fc-ARMs were characterized by reverse phase high performance liquid chromatography (RP-HPLC, Fig. S1†) and matrix assisted laser desorption/ionization-time-of-flight mass spectrometry (MALDI-TOF MS, Fig. S2†).First, we used surface plasmon resonance (SPR) to measure the affinities between Fc-ARMs and the Fc region of the IgG1 antibody. We functionalized SPR chips with anti-HER2 humanized IgG1 mAb (Trastuzumab) and flowed Fc-ARMs over the chip surface. The results showed that Fc-ARM2 had higher affinity to IgG1 compared with Fc-ARM1 (Fig. 2A–C). The Kd values were 25.0 nM for Fc-ARM1 and 6.1 nM for Fc-ARM2. Next, we verified whether Fc-ARM2 could recruit IgG on FR-α+ cancer cells. Fluorescence microscopy revealed that Fc-ARM2 successfully recruited FITC-labeled human IgG (IgG-FITC) to IGROV-1 cells (Fig. S3†). Excess FA diminished IgG-derived fluorescence. Furthermore, Fc-ARM2 recruited the papain-digested Fc fragment of IgG1 labeled with fluorescein (Fc-fluo) on IGROV-1 cells (Fig. 2D and S4†). These results demonstrated that the bispecific affinities of Fc-ARM2 to FR-α and the Fc region of the antibody are crucial for antibody recruitment.
 | ||
| Fig. 2 Fc-ARMs bind to both the Fc region of the antibody and FR-α for antibody recruitment to cancer cells. (A–C) Dissociation constants of (A) Fc-ARM1 and (B) Fc-ARM2 to Trastuzumab were determined by SPR measurements. (C) kon, koff, and Kd values for Fc-ARMs are summarized. (D) IGROV-1 cells were seeded and incubated overnight. Fc-fluorescein (Fc-fluo, 500 nM), Fc-ARM2 (100 nM), and FA (100 μM) were used. Fc-fluo, Fc-fluo + Fc-ARM2, or Fc-fluo + Fc-ARM2 + FA were added to the cells. After washing, the cells were stained with Hoechst 33342. Scale bar = 20 μm. Representative pictures are shown. (E) IGROV-1 cells were treated with 10 nM of Fc-ARMs and increasing concentrations of IgG-FITC from serum. The mean fluorescence intensity (MFI) derived from the recruited antibodies was quantified by flow cytometry (n = 3, mean ± SEM). Two experimental repeats were performed. Statistical analyses were carried out using a two-tailed Welch's t test. *p < 0.05; **p < 0.01. | ||
Next, we conducted a quantitative evaluation of antibody recruitment using flow cytometry. Considering that 10 nM of Fc-ARM1 saturated the amount of recruited IgG (Fig. S5†), 10 nM of Fc-ARM (Fc-ARM1 or Fc-ARM2) and increasing concentrations of IgG-FITC were added to IGROV-1 cells. As a result, Fc-ARM2 recruited a higher amount of IgG-FITC compared with Fc-ARM1 (Fig. 2E). Notably, 1 μM of IgG-FITC reduced the mean fluorescence intensity (MFI), which was evident in three component systems (in our case, the ternary complex of FR-α, Fc-ARM, and IgG).38 These data demonstrated that Fc-ARM2 possesses improved affinity for IgG-Fc, enabling more efficient recruitment of antibodies on FR-α+ cancer cells.
Tumor cell killing by mAb recruitment
Next, we evaluated whether antibodies redirected to tumor cells by Fc-ARMs can activate NK cells and induce ADCC. Importantly, we previously reported that the binding sites of an Fc-binding peptide and CD16a to the Fc region of the antibody do not overlap,29 supporting the feasibility of the Fc-ARM strategy. Ofatumumab (anti-CD20 IgG1 mAb) was used as a source of antibodies since its ADCC capability is certified,39 and it does not bind to IGROV-1 cells by itself (IGROV-1 cells do not express CD20). IGROV-1 cells were co-cultured with human NK cells (KHYG-1/CD16a-158V)40 for 16 h in the presence of an Fc-ARM and anti-CD20, and then lactose dehydrogenase (LDH) released from lysed cells was quantified. Fc-ARM2 + anti-CD20 showed clear target cell killing in an effector/target (E/T) ratio-dependent manner, whereas anti-CD20 or Fc-ARM2 alone did not (Fig. 3A). Cytotoxicity was eliminated by excess FA. Upon recruitment of anti-CD20, NK cells showed secretion of interferon-γ (IFN-γ) (Fig. 3B) and surface mobilization of CD107a (an activation marker of NK cells,41Fig. 3C and S6†). ADCC was not induced against A549 lung cancer cells (FR-α−) (Fig. S7†). These results clearly demonstrated that Fc-ARM-mediated antibody recruitment activates NK cells and induces ADCC.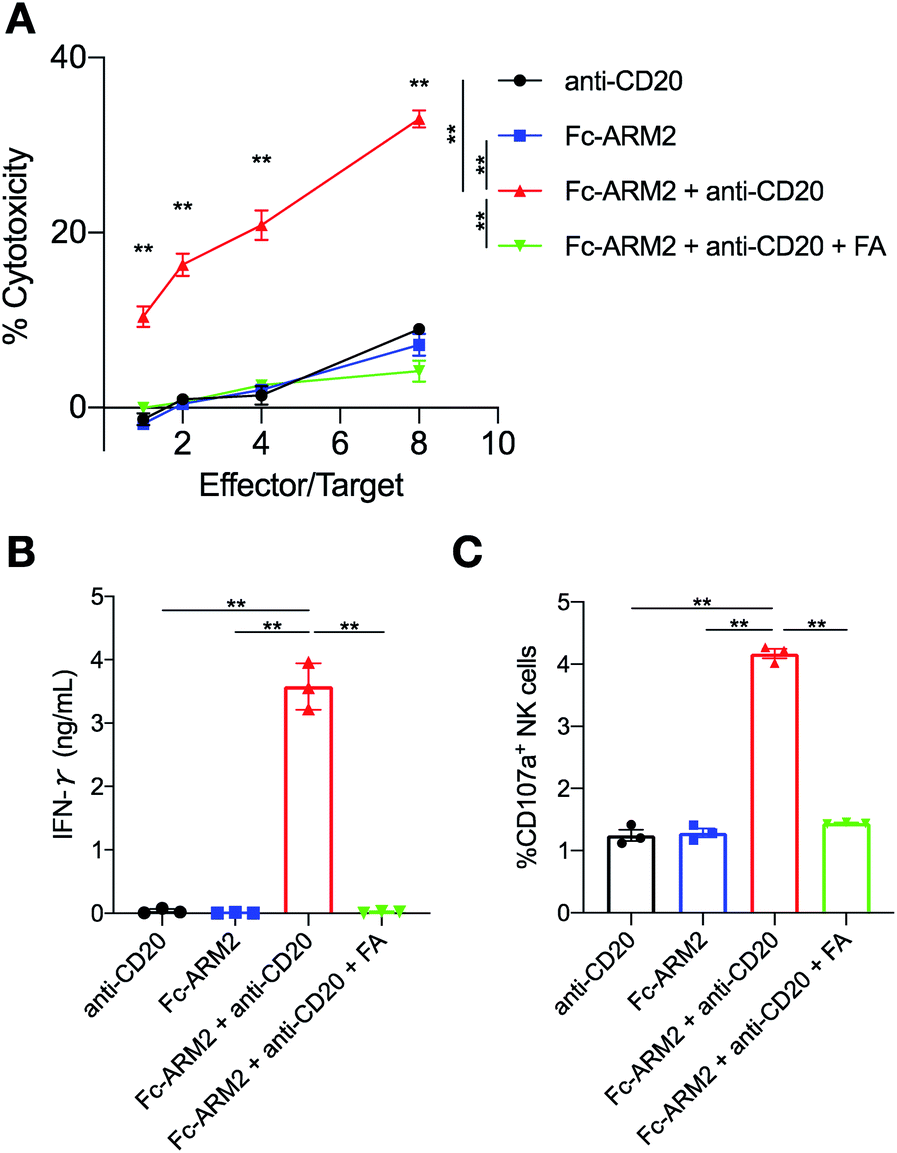 | ||
Fig. 3 Fc-ARM recruits anti-CD20 mAb for NK cell activation to eliminate cancer cells. (A) 5000 cells per well of IGROV-1 cells were treated with anti-CD20 (100 nM), Fc-ARM (10 nM), or Fc-ARM2 + anti-CD20, or Fc-ARM2 + anti-CD20 + FA (1 μM) and co-cultured with 5000–40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well of KHYG-1/CD16a-158V for 16 h. ADCC activity was quantified by an LDH assay (n = 3, mean ± SEM). Statistically significant differences between “Fc-ARM2 + anti-CD20” and all of the other groups were observed at all Effector/Target (E/T) ratios. (B) After co-culturing of IGROV-1 cells and KHYG-1/CD16a-158V, culture supernatants were collected for human IFN-γ detection by ELISA (E/T = 8, mean ± SEM). (C) Surface mobilization of CD107a on KHYG-1/CD16a-158V cells was evaluated after 6 h of co-culture with IGROV-1 cells in the presence of the indicated reagents by flow cytometry (E/T = 1, mean ± SEM). Two experimental repeats were performed. Statistical analyses were carried out using one-way ANOVA with Tukey's multiple comparison test. **p < 0.01. 000 cells per well of KHYG-1/CD16a-158V for 16 h. ADCC activity was quantified by an LDH assay (n = 3, mean ± SEM). Statistically significant differences between “Fc-ARM2 + anti-CD20” and all of the other groups were observed at all Effector/Target (E/T) ratios. (B) After co-culturing of IGROV-1 cells and KHYG-1/CD16a-158V, culture supernatants were collected for human IFN-γ detection by ELISA (E/T = 8, mean ± SEM). (C) Surface mobilization of CD107a on KHYG-1/CD16a-158V cells was evaluated after 6 h of co-culture with IGROV-1 cells in the presence of the indicated reagents by flow cytometry (E/T = 1, mean ± SEM). Two experimental repeats were performed. Statistical analyses were carried out using one-way ANOVA with Tukey's multiple comparison test. **p < 0.01. | ||
Fc affinity and NK cell activation
We next verified whether or not the Fc affinity of Fc-ARMs determines the immune-stimulatory activity of recruited antibodies. Specifically, we conducted NK cell lysis assays over different co-culture time periods as well as with different antibody concentrations. After 4 h of co-culture, Fc-ARM1 + anti-CD20 did not show detectable cytotoxicity (Fig. 4A), in line with our previous report.29 Fc-ARM2 + anti-CD20 showed higher cytotoxicity compared with Fc-ARM1 + anti-CD20 after 6, 8, and 16 h of co-culture (Fig. 4B), and it took more incubation time for Fc-ARM1 to reach comparable % cytotoxicity with Fc-ARM2. This result is in accordance with the literature showing that antibodies with enhanced affinity to their antigen42 or CD16a43 accelerate the kinetics of ADCC. Thus, we conclude that the reason why we could not detect ADCC using Fc-ARM1 in the previous report29 is insufficient incubation time. When compared with Fc-ARM1, Fc-ARM2 induced ADCC more effectively, along with increasing concentration of anti-CD20 (Fig. 4C). Similarly, Fc-ARM2 + anti-CD20 induced a higher amount of IFN-γ secretion from NK cells than Fc-ARM1 + anti-CD20 after 16 h of co-culture (Fig. S8†). Notably, the Fc-fragment of anti-CD20 induced cytotoxicity upon recruitment by Fc-ARM2 (Fig. 4D). Taken together, these data demonstrated that Fc affinity is a critical factor controlling both the efficacy and kinetics of ADCC in the Fc-ARM strategy, and that the Fab region of the antibody is not necessarily required for ADCC induction. | ||
| Fig. 4 Fc affinity controls both the efficacy and kinetics of ADCC in the Fc-ARM strategy. (A) 5000 cells per well of IGROV-1 cells were co-cultured with 5000–40000 cells per well of KHYG-1/CD16a-158V for 4 h. ADCC activity was quantified by an LDH assay (n = 3, mean ± SEM). (B) 5000 cells per well of IGROV-1 cells were co-cultured with 20000 cells per well of KHYG-1/CD16a-158V (E/T = 4) for 6 to 16 h. ADCC activity was then quantified (mean ± SEM). (C) 5000 cells per well of IGROV-1 cells were co-cultured with 40000 cells per well of KHYG-1/CD16a-158V in the presence of Fc-ARM (10 nM) and various concentrations of anti-CD20 IgG1 mAb for 16 h (n = 3, mean ± SEM). (D) An ADCC assay was performed using the Fc fragment of anti-CD20 mAb (n = 3, mean ± SEM). Statistically significant differences between “Fc-ARM2 + Fc fragment” and all of the other groups were observed at all E/T ratios. (A, B) One experimental replicate was performed. (C, D) Two experimental repeats were performed. Statistical analyses were carried out using (B, D) one-way ANOVA with Tukey's multiple comparison test or (C) a two-tailed Welch's t test. *p < 0.05; **p < 0.01. | ||
Partial inhibition of anti-EGFR by the recruited antibodies
Previously, we reported that Fc-ARM1 inhibited the ADCC activity of an anti-epidermal growth factor receptor (EGFR) mAb against IGROV-1 cells (EGFR/FR-α double positive).29 To investigate the mechanism of action, we first compared the amount of EGFR/anti-EGFR binary complex with that of the ternary complex formed on IGROV-1 cells using flow cytometry. IGROV-1 cells were incubated with fluorescein-labeled anti-EGFR mAb (Cetuximab). The results showed that 5 nM of anti-EGFR mAb was sufficient to saturate binary complex formation (Fig. S9A†). Furthermore, 10 nM of Fc-ARM1 induced about a six-fold increase in MFI, showing that a substantially higher amount of the ternary complex could be formed on IGROV-1 cells compared with the binary complex. Next, we evaluated the ADCC activity of both complexes with a co-culture time of 16 h (Fig. S9B†). The binary complex of EGFR/anti-EGFR mAb showed stronger ADCC activity compared with the ternary complex (Fc-ARM2 + anti-CD20), which is reasonable considering the overall affinity of those two forms of antibodies for IGROV-1 cells (Cetuximab reportedly possesses a Kd value of 0.49 nM against EGFR).44 Interestingly, the co-existence of the binary complex and the ternary complex (Fc-ARM2 + anti-EGFR) resulted in similar ADCC activity compared with the ternary complex (Fc-ARM2 + anti-CD20). These results indicated that the ternary complex not only activates NK cells, but also works as a competitive inhibitor of CD16a against stronger agonists (in this case, the binary complex of EGFR/anti-EGFR mAb). We do not discuss the potential contribution of the quaternary complex of FR-α, Fc-ARM2, anti-EGFR, and EGFR in this manuscript because we are currently incapable of isolating the quaternary complex.Pooled IgG from donor sera for NK cell activation
Motivated by the observation that IgG1 mAb can be used for tumor cell killing in the Fc-ARM strategy, we sought to test whether antibody mixtures could also be used. A clinically used IgG mixture from donor sera, known as intravenous immunoglobulin (IVIG), as well as an affinity-purified IgG mixture were used. Upon recruitment by Fc-ARM2, both IgG products clearly induced ADCC against IGROV-1, whereas IgG products alone did not (Fig. 5A). Notably, the levels of ADCC induced by these two conditions were similar to that induced by Fc-ARM2 + anti-CD20 (IgG1 mAb). It would be because IgG1 has the highest blood concentration among all IgG subclasses, given that IgG1 is the most potent IgG subclass for ADCC induction.45 Taken together, the results suggested that endogenous antibodies circulating in the human body could be an effective resource for ADCC induction in the Fc-ARM strategy. | ||
| Fig. 5 Fc-ARM effectively recruits IVIG for its enhanced blood retention time and tumor accumulation in vivo. (A) Two different products of IgG from human sera were compared with anti-CD20 mAb. IgG (100 nM) and Fc-ARM2 (10 nM) were added to 5000 cells per well of IGROV-1 cells. Then, 10000–80000 cells per well of KHYG-1/CD16a-158V cells were added and incubated for 16 h. LDH released from lysed cells was quantified (n = 3, mean ± SEM). Statistically significant differences between the “Fc-ARM2 + IgG” and “IgG only” groups were observed at all E/T ratios. (B) The molecular structure of Fc-ARM-Cy7. (C–E) 1 × 106 of IGROV-1 cells were inoculated into the left side of the back of each BALB/c nu/nu mouse. When the tumor volume reached about 300 mm3, the mice received 20 mg of IVIG intraperitoneally. After 2 h, the mice received 5 nmol of Fc-ARM-Cy7 intraperitoneally. (C) In vivo fluorescence imaging of Fc-ARM-Cy7 at different time points was performed using an IVIS Lumina II. White arrows indicate tumors. Representative images from four mice in each group are shown. (D) Time-course quantification of the fluorescence signals from tumors (n = 4, mean ± SEM). (E) 24 h after Fc-ARM-Cy7 injection, organs were harvested and imaged ex vivo. Then, fluorescence signals were quantified (mean ± SEM). Two experimental repeats were performed. Statistical analyses were carried out using (A) one-way ANOVA with Tukey's multiple comparison test or (D, E) a two-tailed Welch's t test. *p < 0.05; **p < 0.01; N.S. = not significant. | ||
In vivo interactions between Fc-ARMs and human antibodies
Next, we tested whether Fc-ARMs could interact with antibodies in vivo. Considering the ease of synthesis compared with Fc-ARM2, we prepared a derivative of Fc-ARM1 labeled with sulfo-cyanine7 (Fc-ARM-Cy7, Fig. 5B and S10†). Because Fc-ARM2 did not work with mouse IgG (mIgG) (Fig. S11†), we pre-injected tumor-bearing mice with IVIG. Injected IVIG remains in the blood circulation for an extended period of time as a macromolecule.46 Thus, we hypothesized that pre-injection of IVIG into mice would enhance the blood circulation time and the tumor accumulation of the Fc-ARM. Mice received 20 mg of IVIG to mimic the blood concentration of IgG in humans.47 Then, 2 h after intraperitoneal injection of IVIG, IGROV-1 tumor-bearing BALB/c nu/nu mice were injected intraperitoneally with 5 nmol of Fc-ARM-Cy7. In vivo time-course imaging showed that IVIG significantly improved the blood retention time and tumor-targeted delivery of Fc-ARM-Cy7 (Fig. 5C and D). Ex vivo imaging 24 h after Fc-ARM-Cy7 injection showed that significantly higher amounts of Fc-ARM-Cy7 remained in a range of dissected organs in the mice that received prior injection of IVIG (Fig. 5E and S12†). Considering that small molecules are readily removed from the systemic circulation through renal filtration, these data indicated that Fc-ARMs can hitchhike on human IgG antibodies and harness their pharmacokinetics in vivo, resulting in an enhanced blood circulation time and increased accumulation in tumors.Anti-tumor effect of the Fc-ARM strategy in vivo
Finally, we tested the anti-tumor efficacy of the Fc-ARM strategy in vivo. Because there is crosstalk between human antibodies and mouse effector cells including mouse NK cells,48 we hypothesized that the human IgG redirected to tumors can activate mouse NK cells and inhibit tumor growth. A summary of the treatment schedule is shown in Fig. 6A. Given that human IgG1 shows a blood half-life of approximately 10 days in mice,49 IVIG (1 g kg−1) was injected intraperitoneally into IGROV-1-bearing BALB/c nu/nu mice 0 and 9 days after tumor inoculation. Fc-ARM2 (4 mg kg−1) was intraperitoneally administered to the mice 0, 3, 6, 9, 12, and 15 days after tumor inoculation. Fc-ARM2 + IVIG suppressed tumor growth compared with all of the other groups (Fig. 6B and C). Although the therapeutic efficacy was not that dramatic, this is presumably due to the lower capacity of human antibodies to activate murine NK cells compared with their murine counterparts.48 | ||
| Fig. 6 Fc-ARM2 recruits IVIG to suppress IGROV-1 tumor growth. (A) The treatment schedule is shown. After inoculation of IGROV-1 cells, BALB/c nu/nu mice were injected intraperitoneally with 1 g kg−1 of IVIG (0 and 9 days after tumor inoculation) and 4 mg kg−1 of Fc-ARM2 (0, 3, 6, 9, 12, and 15 days after tumor inoculation). (B) The graph depicts the tumor volume until day 18 (n = 6 for untreated and IVIG and n = 5 for Fc-ARM2 and Fc-ARM2 + IVIG, mean ± SEM). (C) Individual growth curves for the IGROV-1 tumors shown in (B). (D) Body weight changes of mice during the treatment (mean ± SEM). Two experimental repeats were performed. Statistical analyses were carried out using one-way ANOVA with Tukey's multiple comparison test (on day 12). *p < 0.05; **p < 0.01. | ||
Usage of immunologically humanized mice may enable us to evaluate the therapeutic potential of the Fc-ARM strategy more accurately. In addition, a stronger Fc-binder or a targeting ligand would also enable the Fc-ARM to induce anti-tumor responses more strongly. In terms of the side effects, no abnormal weight loss of mice was observed during the treatment (Fig. 6D). Taken together, these data indicated that the Fc-ARM strategy can induce anti-tumor immune responses in vivo.
Conclusions
We have reported a novel class of antibody-recruiting small molecules (ARMs) that theoretically enables the recruitment of the majority of endogenous antibodies in the bloodstream for anti-tumor immune responses for the first time. Fc-ARMs recruited antibodies obtained from human sera to the FR-α+ cancer cell surface selectively and induced ADCC depending on the strength of their Fc affinity. Fc-ARMs successfully interacted with human antibodies in vivo, resulting in enhanced blood circulation and tumor accumulation. Furthermore, the Fc-ARM strategy suppressed the growth of IGROV-1 human ovarian adenocarcinoma in a mouse xenograft model. Fc affinity would provide the ARM strategy with efficient and robust opportunities to redirect endogenous antibodies to malignant cells. Therefore, our approach may lead to the re-emergence of ARMs as one of the promising modalities of cancer immunotherapy.Author contributions
K. S., Y. M., T. M., and Y. K. conceived and designed the project. K. S., M. H., Y. M., and H. T. performed the experiments. K. S., T. M., and Y. K. analyzed the data. K. S., A. K., T. M., and Y. K. wrote the manuscript.Ethical statement
All experiments using mice received approval from the Ethics Committee for Animal Experiments of Kyushu University and were in accordance with the guidelines of the Animal Care and Use Committee of Kyushu University.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was in part supported by Challenging Research (Exploratory) (Grant number: 18K19148) of MEXT, Japan. K. S. was supported by the Research Fellowship for Young Scientists (JSPS, 17J05032) and Advanced Graduate Course on Molecular Systems for Devices (Kyushu University). We thank Dr Y. Yonemitsu and Dr Y. Harada (Kyushu University) for fruitful discussions. We thank the Center for Advanced Instrumental and Educational Supports (Faculty of Agriculture, Kyushu University), and Dr M. Goto and Dr N. Kamiya (Kyushu University) for flow cytometer. We thank Dr K. Muguruma, (Tokyo University of Pharmacy and Life Sciences), Dr S. Kishimoto (Kagoshima University), and Dr Y. Ito (Kagoshima University) for assistance with SPR experiments. We thank K. Fox, DPhil, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.References
- A. M. Scott, J. D. Wolchok and L. J. Old, Nat. Rev. Cancer, 2012, 12, 278–287 CrossRef CAS PubMed
.
- T. T. Hansel, H. Kropshofer, T. Singer, J. A. Mitchell and A. J. George, Nat. Rev. Drug Discovery, 2010, 9, 325–338 CrossRef CAS PubMed
- G. M. Bartelds, C. A. Wijbrandts, M. T. Nurmohamed, S. Stapel, W. F. Lems, L. Aarden, B. A. Dijkmans, P. P. Tak and G. J. Wolbink, Ann. Rheum. Dis., 2007, 66, 921–926 CrossRef CAS PubMed
- P. J. McEnaney, C. G. Parker, A. X. Zhang and D. A. Spiegel, ACS Chem. Biol., 2012, 7, 1139–1151 CrossRef CAS PubMed
- K. Imai and A. Takaoka, Nat. Rev. Cancer, 2006, 6, 714–727 CrossRef CAS
- C. Rader, Nature, 2015, 518, 38–39 CrossRef CAS PubMed
- C. R. Bertozzi and M. D. Bednarski, J. Am. Chem. Soc., 1992, 114, 5543–5546 CrossRef CAS
- C. Rader, S. C. Sinha, M. Popkov, R. A. Lerner and C. F. Barbas III, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5396–5400 CrossRef CAS PubMed
- Y. Lu, E. Sega and P. S. Low, Int. J. Cancer, 2005, 116, 710–719 CrossRef CAS PubMed
- V. M. Krishnamurthy, L. J. Quinton, L. A. Estroff, S. J. Metallo, J. M. Isaacs, J. P. Mizgerd and G. M. Whitesides, Biomaterials, 2006, 27, 3663–3674 CAS
- V. R. Doppalapudi, N. Tryder, L. Li, T. Aja, D. Griffith, F.-F. Liao, G. Roxas, M. P. Ramprasad, C. Bradshaw and C. F. Barbas III, Bioorg. Med. Chem. Lett., 2007, 17, 501–506 CrossRef CAS PubMed
- C. B. Carlson, P. Mowery, R. M. Owen, E. C. Dykhuizen and L. L. Kiessling, ACS Chem. Biol., 2007, 2, 119–127 CrossRef CAS PubMed
- M. F. Perdomo, M. Levi, M. Sällberg and A. Vahlne, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12515–12520 CrossRef CAS
- M. K. O'Reilly, B. E. Collins, S. Han, L. Liao, C. Rillahan, P. I. Kitov, D. R. Bundle and J. C. Paulson, J. Am. Chem. Soc., 2008, 130, 7736–7745 CrossRef
- C. G. Parker, R. A. Domaoal, K. S. Anderson and D. A. Spiegel, J. Am. Chem. Soc., 2009, 131, 16392–16394 CrossRef CAS
- R. P. Murelli, A. X. Zhang, J. Michel, W. L. Jorgensen and D. A. Spiegel, J. Am. Chem. Soc., 2009, 131, 17090–17092 CrossRef CAS PubMed
- M. Popkov, B. Gonzalez, S. C. Sinha and C. F. Barbas, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4378–4383 CrossRef CAS PubMed
- J. Gavrilyuk, H. Uehara, N. Otsubo, A. Hessell, D. R. Burton and C. F. Barbas III, ChemBioChem, 2010, 11, 2113–2118 CrossRef CAS PubMed
- U. Wuellner, J. I. Gavrilyuk and C. F. Barbas III, Angew. Chem., Int. Ed., 2010, 49, 5934–5937 CrossRef CAS PubMed
- A. Dubrovska, C. Kim, J. Elliott, W. Shen, T.-H. Kuo, D.-I. Koo, C. Li, T. Tuntland, J. Chang, T. Groessl, X. Wu, V. Gorney, T. Ramirez-Montagut, D. A. Spiegel, C. Y. Cho and P. G. Schultz, ACS Chem. Biol., 2011, 6, 1223–1231 CrossRef CAS PubMed
- R. T. Sheridan, J. Hudon, J. A. Hank, P. M. Sondel and L. L. Kiessling, ChemBioChem, 2014, 15, 1393–1398 CrossRef CAS
- A. F. Rullo, K. J. Fitzgerald, V. Muthusamy, M. Liu, C. Yuan, M. Huang, M. Kim, A. E. Cho and D. A. Spiegel, Angew. Chem., Int. Ed., 2016, 55, 3642–3646 CrossRef CAS
- C. E. Jakobsche, C. G. Parker, R. N. Tao, M. D. Kolesnikova, E. F. Douglass Jr and D. A. Spiegel, ACS Chem. Biol., 2013, 8, 2404–2411 CrossRef CAS
- U. Galili, B. A. Macher, J. Buehler and S. B. Shohet, J. Exp. Med., 1985, 162, 573–582 CrossRef CAS PubMed
- H. Mitoma, T. Horiuchi, H. Tsukamoto, Y. Tamimoto, Y. Kimoto, A. Uchino, K. To, S. I. Harashima, N. Hatta and M. Harada, Arthritis Rheuma, 2008, 58, 1248–1257 CrossRef CAS
- R. Vazquez-Lombardi, C. Loetsch, D. Zinkl, J. Jackson, P. Schofield, E. K. Deenick, C. King, T. G. Phan, K. E. Webster, J. Sprent and D. Christ, Nat. Commun., 2017, 8, 15373 CrossRef CAS
- H.-S. Dai, N. Griffin, C. Bolyard, H. C. Mao, J. Zhang, T. P. Cripe, T. Suenaga, H. Arase, I. Nakano, E. Chiocca, B. Kaur, J. Yu and M. A. Caligiuri, Immunity, 2017, 47, 159–170 CrossRef CAS PubMed
- W. L. DeLano, M. H. Ultsch and J. A. Wells, Science, 2000, 287, 1279–1283 CrossRef CAS PubMed
- K. Sasaki, Y. Miyashita, D. Asai, D. Funamoto, K. Sato, Y. Yamaguchi, Y. Mishima, T. Iino, S. Takaishi, J. Nagano, A. Kishimura, T. Mori and Y. Katayama, MedChemComm, 2018, 9, 783–788 RSC
- Y. Jung, H. J. Kang, J. M. Lee, S. O. Jung, W. S. Yun, S. J. Chung and B. H. Chung, Anal. Biochem., 2008, 374, 99–105 CrossRef CAS PubMed
- M. French and G. Harrison, Clin. Exp. Immunol., 1984, 56, 473–475 CAS
- M. Klouche, A. R. Bradwell, D. Wilhelm and H. Kirchner, Clin. Chem., 1995, 41, 1475–1479 CrossRef CAS
- B. Li, L. Zhao, H. Guo, C. Wang, X. Zhang, L. Wu, L. Chen, Q. Tong, W. Qian, H. Wang and Y. Guo, Blood, 2009, 114, 5007–5015 CrossRef CAS PubMed
- G. Cartron, L. Dacheux, G. Salles, P. Solal-Celigny, P. Bardos, P. Colombat and H. Watier, Blood, 2002, 99, 754–758 CrossRef CAS PubMed
- Y. Gong, L. Zhang, J. Li, S. Feng and H. Deng, Bioconjugate Chem., 2016, 27, 1569–1573 CrossRef CAS PubMed
- C. Chen, J. Ke, X. E. Zhou, W. Yi, J. S. Brunzelle, J. Li, E.-L. Yong, H. E. Xu and K. Melcher, Nature, 2013, 500, 486–489 CrossRef CAS PubMed
- E. I. Sega and P. S. Low, Cancer Metastasis Rev., 2008, 27, 655–664 CrossRef CAS PubMed
- E. F. Douglass Jr, C. J. Miller, G. Sparer, H. Shapiro and D. A. Spiegel, J. Am. Chem. Soc., 2013, 135, 6092–6099 CrossRef PubMed
- M. J. Barth, C. Mavis, M. S. Czuczman and F. J. Hernandez-Ilizaliturri, Clin. Cancer Res., 2015, 21, 4391–4397 CrossRef CAS PubMed
- Y. Mishima, Y. Terui, Y. Mishima, R. Kuniyoshi, S. Matsusaka, M. Mikuniya, K. Kojima and K. Hatake, Int. Immunol., 2012, 24, 477–483 CrossRef CAS PubMed
- G. Alter, J. M. Malenfant and M. Altfeld, J. Immunol. Methods, 2004, 294, 15–22 CrossRef CAS PubMed
- M. P. Velders, C. M. van Rhijn, E. Oskam, G. J. Fleuren, S. O. Warnaar and S. V. Litvinov, Br. J. Cancer, 1998, 78, 478–483 CrossRef CAS PubMed
- G. Romain, V. Senyukov, N. Rey-Villamizar, A. Merouane, W. Kelton, I. Liadi, A. Mahendra, W. Charab, G. Georgiou, B. Roysam, D. A. Lee and N. Varadarajan, Blood, 2014, 124, 3241–3249 CrossRef CAS PubMed
- S. M. Lippow, K. D. Wittrup and B. Tidor, Nat. Biotechnol., 2007, 25, 1171–1176 CrossRef CAS PubMed
- M. Brüggemann, G. T. Williams, C. I. Bindon, M. R. Clark, M. R. Walker, R. Jefferis, H. Waldmann and M. S. Neuberger, J. Exp. Med., 1987, 166, 1351–1361 CrossRef PubMed
- J. T. Andersen, M. B. Daba, G. Berntzen, T. E. Michaelsen and I. Sandlie, J. Biol. Chem., 2010, 285, 4826–4836 CrossRef
- A. Gonzalez-Quintela, R. Alende, F. Gude, J. Campos, J. Rey, L. Meijide, C. Fernandez-Merino and C. Vidal, Clin. Exp. Immunol., 2008, 151, 42–50 CrossRef CAS
- M. B. Overdijk, S. Verploegen, A. Ortiz Buijsse, T. Vink, J. H. Leusen, W. K. Bleeker and P. W. Parren, J. Immunol., 2012, 189, 3430–3438 CrossRef CAS PubMed
- S. B. Petkova, S. Akilesh, T. J. Sproule, G. J. Christianson, H. Al Khabbaz, A. C. Brown, L. G. Presta, Y. G. Meng and D. C. Roopenian, Int. Immunol., 2006, 18, 1759–1769 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00017e |
| This journal is © The Royal Society of Chemistry 2020 |