
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and properties of hypervalent electron-rich pentacoordinate nitrogen compounds†
Chenting
Yan
 a,
Masato
Takeshita
a,
Jun-ya
Nakatsuji
a,
Akihiro
Kurosaki
a,
Kaoko
Sato
a,
Rong
Shang
*a,
Masaaki
Nakamoto
a,
Yohsuke
Yamamoto
*a,
Yohei
Adachi
b,
Ko
Furukawa
c,
Ryohei
Kishi
d and
Masayoshi
Nakano
de
a,
Masato
Takeshita
a,
Jun-ya
Nakatsuji
a,
Akihiro
Kurosaki
a,
Kaoko
Sato
a,
Rong
Shang
*a,
Masaaki
Nakamoto
a,
Yohsuke
Yamamoto
*a,
Yohei
Adachi
b,
Ko
Furukawa
c,
Ryohei
Kishi
d and
Masayoshi
Nakano
de
aDepartment of Chemistry, Graduate School of Science, Hiroshima University, 1-3-1 Kagamiyama, Higashi-Hiroshima 739-8526, Japan. E-mail: yohsuke@hiroshima-u.ac.jp; rshang@hiroshima-u.ac.jp
bDepartment of Applied Chemistry, Graduate School of Engineering, Hiroshima University, 1-3-1 Kagamiyama, Higashi-Hiroshima 739-8526, Japan
cCenter for Coordination Research Facilities, Institute for Research Promotion, Niigata University, 8050 Ikarashi 2-no-cho, Nishi-ku, Niigata 950-2181, Japan
dDepartment of Materials Engineering Science, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan
eDepartment of Materials Engineering Science, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan
First published on 22nd April 2020
Abstract
Isolation and structural characterization of hypervalent electron-rich pentacoordinate nitrogen species have not been achieved despite continuous attempts for over a century. Herein we report the first synthesis and isolation of air stable hypervalent electron-rich pentacoordinate nitrogen cationic radical (11-N-5) species from oxidation of their corresponding neutral (12-N-5) species. In the cationic radical species, the nitrogen centers adopt a trigonal bipyramidal geometry featuring a 3-center-5-electron hypervalent attractive interaction. The combination of single crystal X-ray diffraction analysis and computational studies revealed weak N–O interactions between the central nitrogen cation and oxygen atoms. This successful design strategy and isolation of air-stable pentacoordinate hypervalent nitrogen species allow further investigations on reactivity and properties resulting from these unusually weakly coordinating interactions in nitrogen compounds.
1 Introduction
Hypervalent compounds1,2 have been defined as main group element compounds which contain a number of formally assignable electrons of more than the octet in a valence shell directly associated with the central atom.1,3,4 More recently, Parkin5 clearly distinguished main group element compounds that feature three-center four-electron (3c-4e) interactions from those that feature 3c-2e interactions. The former, in which one of the atoms appears to have an expanded octet, are termed “electron-rich” hypervalent molecules. The latter are invoked for so-called “electron deficient” hypercoordinate molecules.6–14The N-X-L nomenclature15 has been used to classify hypervalent compounds, where N is the number of valence electrons formally present on the central element according to the Lewis diagram, X is the identity of the central element and L is the number of ligand atoms bonded to the central atom. Parkin proposed MLlXxZzHh classifications,5 which are more detailed descriptions of the N-X-L nomenclature.
The debate on the fundamental bonding descriptions of this class of compounds to understand why they do not comply to the Lewis “octet rule” has lasted for decades on the theoretical front.4,7–10,16–27 In a recent review, Crabtree28 pointed out the similarity among hypervalency and other types of weak bonding and the valence shell of the central atom is essentially an octet and the electrons beyond 8e are predominantly located on ligands, not on the central atom. The hypervalent bonding nature and geometry have significant implications in several current fast-growing areas of synthetic organic chemistry, such as hypervalent iodine reagents,29–31 sterically constrained T-shaped phosphorus(III) compounds in small molecule activation and catalysis,32–39 and application of new heavier group 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 40 and 1541–47 Lewis acids in frustrated Lewis pair chemistry. While hypervalent compounds of heavy main group elements are common,2,47–53 those of the light, second row elements still remain a synthetic challenge, with only a handful of isolated and structurally confirmed examples of penta-/hexa-coordinate carbon54–57 and boron58–61 compounds reported (Chart 1B and C).
40 and 1541–47 Lewis acids in frustrated Lewis pair chemistry. While hypervalent compounds of heavy main group elements are common,2,47–53 those of the light, second row elements still remain a synthetic challenge, with only a handful of isolated and structurally confirmed examples of penta-/hexa-coordinate carbon54–57 and boron58–61 compounds reported (Chart 1B and C).
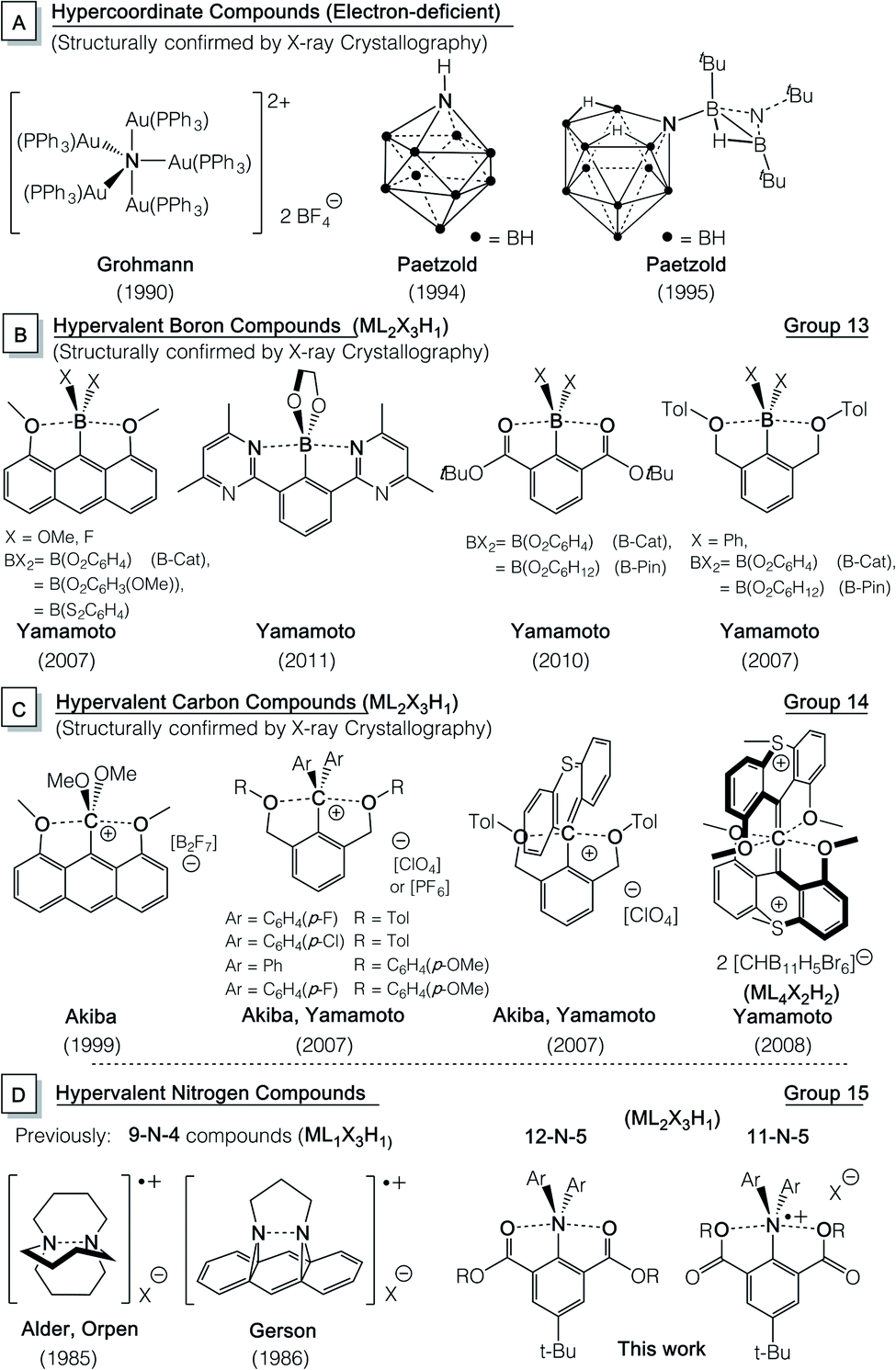 | ||
| Chart 1 Structurally confirmed hypercoordinate (A) nitrogen and hypervalent (B) boron, (C) carbon and (D) nitrogen compounds. | ||
Hypervalent pentacoordinate nitrogen species (10-N-5) are some of the most fundamental hypervalent compounds, with their synthetic attempts traced back to 1916.62–70 Despite theoretical calculations predicting their existence and structural stability,71–76 thermally stable hypervalent nitrogen compounds (N–N–L; N > 10, L > 4) have not been reported to date.68,70,77,78 Some success has been achieved by detection,79–83 isolation and structural characterization of transient, as well as stable84–86 hypervalent tetracoordinate nitrogen radical species (9-N-4, Chart 1D).
Here we report the first hypervalent “electron-rich” pentacoordinate nitrogen cationic radical (11-N-5 or ML2X3H1) species with 3c-5e interactions and their corresponding neutral (12-N-5) compounds although “electron-deficient” pentacoordinate nitrogen compounds6–9,87,88 have been reported as shown in Chart 1A.
2 Results and discussion
To stabilize the trigonal-bipyramidal (TBP) geometry of a 10-N-5 structure, which is typically a transition state resembling an SN2 reaction,1,3 we targeted the tridentate 2,6-di(alkoxycarbonyl)phenyl framework60 in consideration of two crucial factors: (i) electronegative elements (oxygen) are positioned at apical positions to localize the electron density at the 3-center-4-electron (3c-4e) hypervalent bond and (ii) the apical 3c-4e attractive interaction should be weak enough (O⋯N attractive interactions) to avoid a “bell-clapper” type equilibrium.89,90 The alkoxycarbonyl groups provide some degree of steric rigidity, which plays an important role in balancing the two factors and thus stabilizing the hypervalent compounds. The tert-butyl group at the para position increases the solubility.We synthesized neutral triarylamine precursors 2Mea-b and 2iPra-b from the Ullmann coupling reaction of methoxyl- and isopropoxyl-carbonyl substituted bromobenzenes (1Me and 1iPr) with chloro- and trifluoromethyl-substituted diarylamines (Scheme 1). Compounds 2Mea-b and 2iPra-b were isolated as white to pale yellow solids and found fluorescent in solution. Their absorption spectra obtained in CH2Cl2 showed two absorption bands at approximately 310 nm and 350 nm, which are assignable to HOMO–LUMO+2 and HOMO–LUMO transitions, respectively (Fig. S7†). The similar absorption wavelengths and oscillator strengths between methyl- and isopropyl-substituted analogues suggest very similar electronic structures of 2Mea-b and 2iPra-b in their ground state. In contrast to the similarities of absorption, the fluorescence emission of 2Mea-b and 2iPra-b measured in CH2Cl2 at 25 °C showed substituent-dependence on the aryl groups (X = Cl or CF3, see Fig. S1 and Table S1†). The ester groups (R = Me or i-Pr) do not exert clear influence. The quantum yields of 2iPra-b (9.43%, 13.95% respectively) with R = i-Pr are obviously higher than those of 2Mea-b (3.22%, 3.50% respectively) with R = Me, likely due to the more restricted rotation around the N-Ph bonds with the sterically larger i-Pr groups.
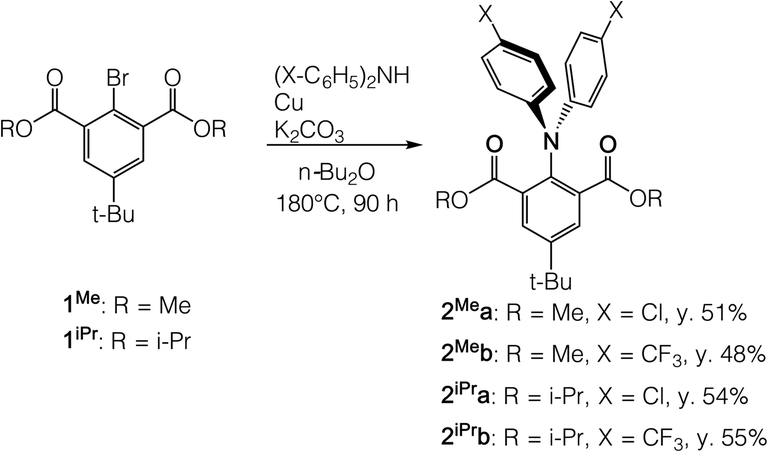 | ||
| Scheme 1 Preparation of neutral triarylamine precursors 2Mea-b and 2iPra-b. | ||
Cyclic voltammetry (CV) of 2Mea-b and 2iPra-b in CH2Cl2 at room temperature with TBAPF6 as a supporting electrolyte revealed a reversible one electron oxidation wave at formal potentials of +1.196 V, +1.485 V, +1.177 V and +1.477 V versus Fc/Fc+ respectively, indicating stability of the corresponding monocationic species (Fig. S3†). The σ withdrawing effect of trifluoromethyl groups is clearly shown in the higher oxidation potentials observed in 2Meb and 2iPrb. The σ donating effect between the methyl and isopropyl substituents is much less prominent. A 2nd electron oxidation wave was not observed under this condition between −2.0 and 2.0 volts, which is the limit of our instrument.
The one electron oxidation reaction of 2Mea-b and 2iPra-b (Scheme 2) was easily achieved by using 1 equiv. (2,4-Br2C6H3)3NSbCl6 as an oxidant in CH2Cl2. Dark blue-green solids 3Mea-b and 3iPra-b were obtained in moderate to high yields. The absorption spectra of 3Mea-b and 3iPra-b showed bands at 600–800 nm, significantly red-shifted from those of the neutral compounds in the visible region (ca. 310 nm, Fig. 1). This is due to the smaller HOMO–SOMO energy gaps of radical species in comparison to the HOMO–LUMO energy gaps in neutral species. The chloro-analogues 3Mea (745 nm) and 3iPra (739 nm) showed absorption maxima at noticeably longer wavelengths compared to their CF3-substituted 3Meb (688 nm) and 3iPrb (681 nm), revealing that electron-donating substituents decrease HOMO–SOMO energy gaps. The chloro-substituted cationic radical species 3Mea and 3iPra are stable in air and moisture for several days at room temperature without detectable decomposition. In contrast, the trifluoromethyl derivatives 3Meb and 3iPrb decompose readily when taken out of an inert atmosphere. All of them appear to be light-sensitive.
 | ||
| Scheme 2 One electron oxidation of 2Mea-b and 2iPra-b. | ||
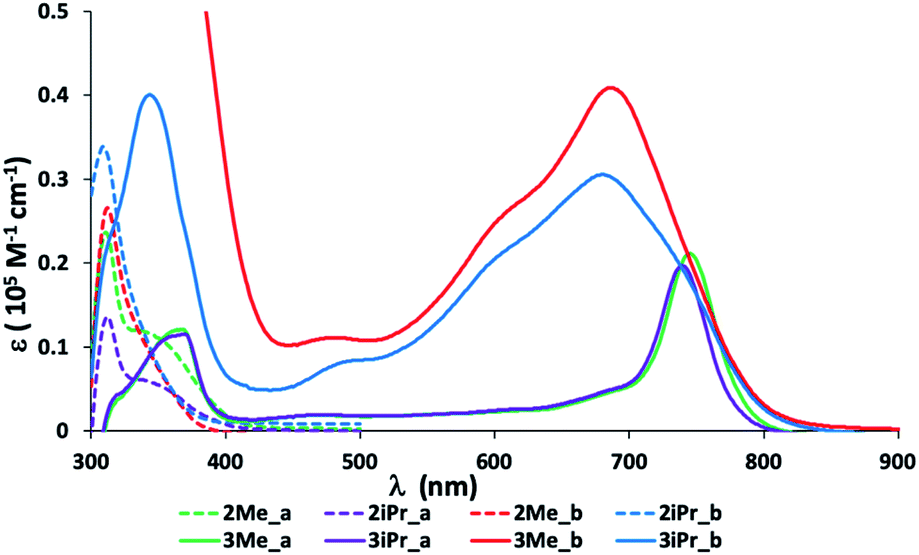 | ||
| Fig. 1 Absorption spectra of 10−5 M 2Mea-b and 2iPra-b (dashed line) and 3Mea-b and 3iPra-b (solid line) in CH2Cl2 at 25 °C. | ||
Single crystals suitable for X-ray crystallographic studies were obtained by recrystallization from CH2Cl2/hexane solution mixtures of 2Mea-b, 2iPra-b, 3Mea and 3iPra at room temperature. Their solid-state molecular structures and selected bonding parameters are shown in Fig. 2 and Table 1. Due to proneness to decomposition, publishable data of the trifluoromethyl-substituted derivatives 3Meb and 3iPrb could not be obtained. In all structures, the central nitrogen atoms are essentially planar, with the sum of the angles around nitrogen (∑Nα) being larger than 359°. The ester groups seem to rotate along the carbon–carbon single bond, resulting in conformational differences between the solid-state structures of neutral (2Mea and 2iPra) and oxidized compounds (3Mea and 3iPra). The sp3 alkoxyl oxygen (OR) atoms align almost coplanar with the central nitrogen atom in 2Mea and 3iPra, while in the case of 2Meb, 2iPra-b and 3Mea, the sp2 carbonyl (CO) oxygen atoms and the nitrogen atom are aligned. Calculations show that the energy differences resulting from the two coordination modes are trivial (<3 kcal mol−1, see Table S12 and Fig. S15†). Therefore, conformational preferences observed in the crystals may be influenced significantly by crystal packing energy. This lack of consistency of coordination modes makes the structural comparison difficult to elucidate.
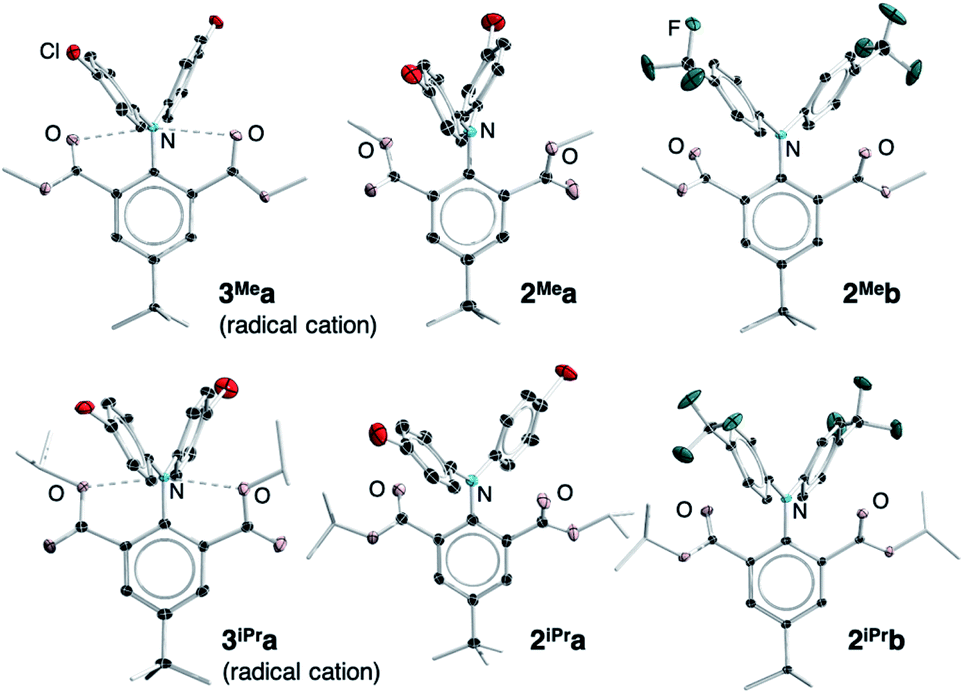 | ||
| Fig. 2 Solid-state molecular structures of 2Mea-b, 2iPra-b, 3Mea and 3iPra. Thermal ellipsoids are set at 30% probability. Ellipsoids of periphery atoms, hydrogen atoms, and counter ions are omitted for clarity. A side-view is included in the ESI.† CCDC numbers: 1945530–1945536.91 | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Parameters | Ionic radical | Neutral | Ionic radical | Neutral | ||
| 3Mea | 2Mea | 2Meb | 3iPra | 2iPra | 2iPrb | |
| Coord. | (CO) | (OMe) | (CO) | (OiPr) | (CO) | (CO) |
| N–O1 | 2.721(3) | 2.764(18) | 2.945(15) | 2.662(5) | 2.891(3) | 2.843(12) |
| N–O2 | 2.790(3) | 2.823(18) | 3.015(16) | 2.676(5) | 2.952(3) | 2.868(13) |
| N–OAve | 2.755(3) | 2.793(18) | 2.980(16) | 2.669(5) | 2.921(3) | 2.855(13) |
| N–C1 | 1.440(3) | 1.423(2) | 1.422(18) | 1.454(6) | 1.409(5) | 1.428(12) |
| N–C2 | 1.376(3) | 1.408(2) | 1.411(17) | 1.383(5) | 1.408(5) | 1.405(13) |
| N–C3 | 1.409(3) | 1.414(2) | 1.415(17) | 1.397(6) | 1.420(5) | 1.404(14) |
| ΣNα | 360.0(6) | 359.1(39) | 359.8(31) | 359.9(12) | 359.5(9) | 360.0(24) |
| Φ OCCO | 47.7 | 77.8 | 90.7 | 49.0 | 81.9 | 77.3 |
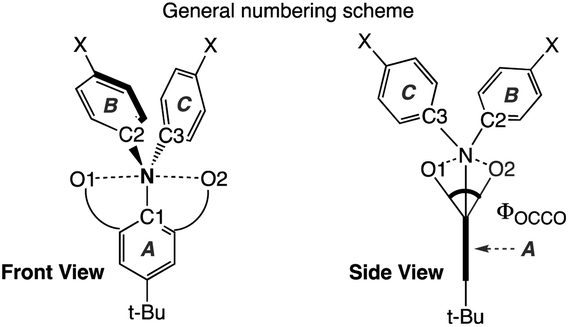
In all the structures, the ester groups twist out of the plane of their attached phenyl ring (Fig. S5† for a side view). Although the N–O bond lengths in all neutral and cationic species are longer than the sum of the covalent bond radii of N and O atoms (1.34 Å),92 they are shorter than the sum of van der Waals radii of N and O atoms (2.79–3.16 Å),93–99 falling in the upper range of previously reported hypervalent bonds.56 This suggests possible weak interactions between the central nitrogen atom and both oxygen atoms in both cationic and neutral structures. The torsion angles (Φ) between CO1 and CO2 (Table 1) are the smallest in the formally 11-N-5 cationic species 3Mea and 3iPra (47.7° and 49.0° respectively). They correspond to the shortest N–O distances, 2.721(3) Å and 2.790(3) Å (3Mea) and 2.662(5) Å, 2.676(5) Å (3iPra) among all structures, indicating the strongest N–O interactions.
Among the neutral compounds, the trend of torsion angles (Φocco) correlate with that of N–O distances, increasing from 2Mea (2.793(18) Å, 77.8°) and 2iPrb (2.855(13) Å, 77.3°) to 2iPra (2.921(3) Å, 81.9°) and lastly, 2Meb (2.980(16) Å, 90.7°). These N–O distances are slightly shorter than those in Shimizu and co-worker's parent 2-aminoisophthalic acid diester, Ph2N[C6H3(COOMe)] (3.093 Å and 2.984 Å), which also displayed a larger Φocco (96.6°).100
As an additional comparison, we investigated the molecular structure of the bromo-substituted starting material 1Me, which may not be expected to form the same type of pentacoordinate hypervalent interaction. (Fig. S4†). The smallest Φocco of 120.4° in 1Me is much larger than those found in our neutral compounds (2Mea-b and 2iPra-b) formed from the methoxy group on each side of the bromo-substituent. This shows that there is no preferred alignment of the two ester groups of 1Me through Cphenyl-Cester rotation for a small Φocco in the absence of the 3c-5e attractive interaction.
Overall, in the solid state, sp3 alkoxyl (OR) coordination forms shorter apical N–O interactions and thus stronger attractive interaction than those from the sp2 carbonyl (CO) coordination. In addition, the N–O interactions are stronger in cationic radical species than those in neutral compounds.
To confirm the interactions between the central nitrogen atom and the two oxygens in both neutral and cationic radical species, DFT calculations were carried out at the RCAM-B3LYP-D3/def2-SVP level for 2 and at the UCAM-B3LYP-D3/def2-SVP level for 3 using the Gaussian 09 program (Table S6†). The optimized structures corroborate well with the crystal structures except 2Meb, in which the N–O distances are underestimated.101 The unsystematic inconsistency between calculated and solid-state structures may be due to the solvent effect during the recrystallization process and packing effect of the crystal. Therefore, Atoms in Molecules (AIM) analysis of 2Mea-b, 2iPra-b, 3Mea and 3iPra was carried out based on single point calculations of the X-ray geometries (Fig. 3 and Table S10†).102
 | ||
| Fig. 3 Atom in molecules (AIM) analysis results based on X-ray geometries of 2Mea-b, 2iPra-b, 3Mea and 3iPra showing bond paths between the central nitrogen and the carbonyl oxygen, calculated at the RCAM- and UCAM-B3LYP-D3/def2-SVP levels in 2 and 3, respectively. | ||
The results of the cation radical species (3Mea and 3iPra) showed (3, −1) critical points between the central nitrogen atom and the ester oxygen atoms with small electron densities (ρ(r) 0.014–0.017 e/ao3) and small positive Laplacian values (∇2ρ(r) 0.051–0.064 e/ao5). This indicates that the N–O interactions in 3Mea and 3iPra are weak and polarized, similar to those of the reported second row hypervalent compounds (ρ(r), 0.014 − 0.022 e/ao3 and ∇2ρ(r), 0.051–0.078 e/ao5).54–57
According to the review paper by A. C. Tsipis,103 the following classifications are presented based on the character of the bond/interaction:
(1) ∇2ρ < 0 and Hcp < 0 indicate weakly polar and nonpolar covalent bonds.
(2) ∇2ρ > 0 and Hcp < 0 correspond to intermediate interactions including strong hydrogen bonds and most of the coordination bonds.
(3) ∇2ρ > 0, ρ(r) < 0.2 and Gcp/ρcp > 1 indicate closed-shell interactions such as weak hydrogen bonds and van der Waals interactions.
(4) ∇2ρ > 0 and |Vcp|/Gcp < 1 characterize ionic bonding.
The AIM results of 3Mea and 3iPra (Table S10†) match the criteria of the class 3 and 4 mostly, which revealed an ionic (non-covalent), weak secondary bonding nature of the N–O interactions. This provides further support for Crabtree's generalization of hypervalent, hydrogen and secondary bonding interactions.28
Among neutral species, although no hypervalent bonding was expected due to the repulsion between the nitrogen and oxygen lone pairs, the AIM analysis showed (3 −1) critical points between N and O atoms in 2Mea and 2iPrb. Their electron density (0.013–0.015 e/ao3) and Laplacian (0.048–0.052 e/ao5) values are slightly smaller than those found in cationic species (ρ(r) 0.014–0.017 e/ao3, ∇2ρ(r) 0.051–0.064 e/ao5). In contrast, no N–O (3, −1) critical points were found in 2Meb and 2iPra (Fig. 3). These AIM results corroborates well with the N–O distances: cation radicals show the shortest N–O distances (2.662–2.790 Å) and (3, −1) critical points with the highest ρ(r) and ∇2ρ(r) values, followed by 2Mea and 2iPrb with intermediate N–O distances (2.764–2.868 Å) and weaker (3, −1) critical points. The N–O distances in 2Meb and 2iPra are the longest (2.891–3.015 Å) and no (3, −1) critical points were found along the N–O paths.
Combined X-ray bonding analysis and theoretical AIM studies suggest that the N–O interactions in the cation radical cases are stronger than in the neutral cases. 3Mea and 3iPra should be regarded as hypervalent electron-rich pentacoordinate nitrogen species with a 3c-5e attractive interaction (ML2X3H1). The neutral compounds may or may not exert weak N–O attractive interactions. Any N–O interactions exhibited in the neutral case may originate from the effective delocalization of the nitrogen electron lone pair in the triarylamine structure (evident by the planar amine geometry and slightly shortened N–Cipso bond distances, Table 1). The slightly positive nitrogen atom then can form electrostatic interactions with the adjacent oxygen electron pairs, forming an 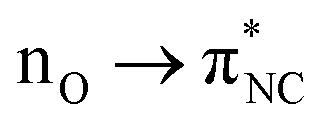 interaction (Fig. 4).
interaction (Fig. 4).
 | ||
| Fig. 4 The delocalization structures of neutral compounds (2Mea and 2iPrb) with electron-withdrawing groups (-Cl and CF3). | ||
The cationic radical compounds 3Mea-b and 3iPra-b were also characterized by Electron Paramagnetic Resonance (EPR) spectroscopy in CH2Cl2 (Fig. 5). In all cases, the spectrum showed a nitrogen centered radical with hyperfine coupling to hydrogen nuclei of the singly substituted aryl groups (rings B and C, Table 1), indicating that the radical delocalizes over the aryl groups B and C in all structures (Table S3 and Fig. S9–S12†). Larger hyperfine coupling constants (AH1 and AH2) in rings B and C and additional coupling contribution from the heteroatoms (Cl and F) were observed. This suggests that stronger electron-withdrawing CF3-substituents promote delocalization of the radical in the structures. The delocalization to the phenyl rings B and C is more effective than A, reflected by the significantly longer N–C1 distances in comparison to N–C2/C3 distances in the solid-state structures of 3Mea and 3iPra (Table 1).
 | ||
| Fig. 5 Observed (bottom) and simulated (top) EPR spectra of 3Mea-b and 3iPra-b in CH2Cl2 solution at 25 °C. Spin Hamiltonian parameters estimated from simulation are labelled in MHz (Table S3†). | ||
3 Conclusions
In conclusion, we successfully synthesized, isolated, and structurally characterized the first air stable hypervalent “electron-rich” pentacoordinate nitrogen compounds. The N–O bond distance in cationic radical species is shorter than the sum of N–O van der Waals radii, indicating attractive interactions between N and O atoms. AIM calculations showed the presence of (3, −1) critical points along the N–O bond paths in cationic radical species, confirming the formation of 11-N-5 hypervalent 3c-5e bonding. In addition, the shorter N–O bond distances and the presence of (3, −1) critical points in neutral compounds 2Mea and 2iPrb suggest possible electrostatic 12-N-5 interactions, likely due to an stabilization effect.
stabilization effect.
The isolation of these new nitrogen compounds and confirmation of their weak attractive interaction will allow us to explore the effect of weak electronic perturbation on the properties and/or reactivity of nitrogen containing compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Joji Ohshita and Prof. Kenji Komaguchi for the use of their instruments and discussion. This work was supported by the JSPS KAKENHI Grant (24109002) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. The authors also thank Research Center for Computational Science (Okazaki, Japan) for the supercomputer time for theoretical calculations.Notes and references
- J. C. Martin, Science, 1983, 221, 509–514 CrossRef CAS PubMed.
- K.-Y. Akiba, Heteroat. Chem., 2011, 22, 207–274 CrossRef CAS.
- J. I. Musher, Angew. Chem., Int. Ed., 1969, 8, 54–68 CrossRef CAS.
- W. B. Jensen, J. Chem. Educ., 2006, 83, 1751 CrossRef CAS.
- M. L. Green and G. Parkin, Dalton.Trans., 2016, 45, 18784–18795 RSC.
- A. Grohmann, J. Riede and H. Schmidbaur, Nature, 1990, 345, 140–142 CrossRef CAS.
- G. A. Olah, A. M. White and D. H. O'Brien, Chem. Rev., 1970, 70, 561–591 CrossRef CAS.
- G. A. Olah and G. Rasul, Acc. Chem. Res., 1997, 30, 245–250 CrossRef CAS.
- P. v. R. Schleyer, E. U. Wuerthwein, E. Kaufmann, T. Clark and J. A. Pople, J. Am. Chem. Soc., 1983, 105, 5930–5932 CrossRef CAS.
- H. Kudo, Nature, 1992, 355, 432–434 CrossRef CAS.
- P. v. R. Schleyer, B. Tidor, E. D. Jemmis, J. Chandrasekhar, E. U. Wuerthwein, A. J. Kos, B. T. Luke and J. A. Pople, J. Am. Chem. Soc., 1983, 105, 484–488 CrossRef CAS.
- Z. Varga, Struct. Chem., 2017, 28, 297–301 CrossRef CAS.
- W. Zhizhong, Z. Xiange and T. Auchin, J. Mol. Struc.Theochem., 1998, 453, 225–231 CrossRef.
- R. D. Harcourt, J. Organomet. Chem., 1994, 478, 131–140 CrossRef CAS.
- C. W. Perkins, J. C. Martin, A. J. Arduengo, W. Lau, A. Alegria and J. K. Kochi, J. Am. Chem. Soc., 1980, 102, 7753–7759 CrossRef CAS.
- A. E. Reed and P. v. R. Schleyer, J. Am. Chem. Soc., 1990, 112, 1434–1445 CrossRef CAS.
- J. A. Dobado, H. Martínez-García, J. M. Molina and M. R. Sundberg, J. Am. Chem. Soc., 1998, 120, 8461–8471 CrossRef CAS.
- R. J. Gillespie and B. Silvi, Coord. Chem. Rev., 2002, 233–234, 53–62 CrossRef CAS.
- X. Sun, Chem. Educat., 2002, 7, 261–264 CrossRef CAS.
- S. Noury, B. Silvi and R. J. Gillespie, Inorg. Chem., 2002, 41, 2164–2172 CrossRef CAS.
- S. C. A. H. Pierrefixe, C. Fonseca Guerra and F. M. Bickelhaupt, Chem. - Eur. J., 2008, 14, 819–828 CrossRef PubMed.
- P. G. Nelson, Chem. Educ. Res. Prac., 2001, 2, 67–72 RSC.
- A. Kalemos, J. Phys. Chem. A, 2018, 122, 2178–2183 CrossRef CAS PubMed.
- M. C. Durrant, Chem. Sci., 2015, 6, 6614–6623 RSC.
- R. D. Harcourt and T. M. Klapötke, Chem. Sci., 2016, 7, 3443–3447 RSC.
- M. C. Durrant, Chem.Sci., 2016, 7, 3448–3449 RSC.
- S. C. A. H. Pierrefixe, S. J. M. van Stralen, J. N. P. van Stralen, C. Fonseca Guerra and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 2009, 48, 6469–6471 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Soc. Rev., 2017, 46, 1720–1729 RSC.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- A. Yoshimura, M. S. Yusubov and V. V. Zhdankin, Org. Biomol. Chem., 2016, 14, 4771–4781 RSC.
- L. Catalano, G. Cavallo, P. Metrangolo, G. Resnati and G. Terraneo, Top. Curr. Chem., 2016, 373, 289–310 CrossRef CAS PubMed.
- E. E. Coyle and C. J. O'Brien, Nat. Chem., 2012, 4, 779 CrossRef CAS PubMed.
- M. Driess, N. Muresan, K. Merz and M. Päch, Angew. Chem., Int. Ed., 2005, 44, 6734–6737 CrossRef CAS PubMed.
- S. A. Culley and A. J. Arduengo, J. Am. Chem. Soc., 1984, 106, 1164–1165 CrossRef CAS.
- A. J. Arduengo, C. A. Stewart, F. Davidson, D. A. Dixon, J. Y. Becker, S. A. Culley and M. B. Mizen, J. Am. Chem. Soc., 1987, 109, 627–647 CrossRef CAS.
- A. J. Arduengo and C. A. Stewart, Chem. Rev., 1994, 94, 1215–1237 CrossRef CAS.
- N. L. Dunn, M. Ha and A. T. Radosevich, J. Am. Chem. Soc., 2012, 134, 11330–11333 CrossRef CAS PubMed.
- T. P. Robinson, D. M. De Rosa, S. Aldridge and J. M. Goicoechea, Angew. Chem., Int. Ed., 2015, 54, 13758–13763 CrossRef CAS PubMed.
- W. Zhao, S. M. McCarthy, T. Y. Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 17634–17644 CrossRef CAS PubMed.
- H. Kameo and H. Nakazawa, Chem. Rec., 2017, 17, 268–286 CrossRef CAS PubMed.
- J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765–774 RSC.
- D. W. Stephan, Angew. Chem., Int. Ed., 2017, 56, 5984–5992 CrossRef CAS PubMed.
- D. You and F. P. Gabbai, J. Am. Chem. Soc., 2017, 139, 6843–6846 CrossRef CAS PubMed.
- J. S. Jones, C. R. Wade, M. Yang and F. P. Gabbai, Dalton Trans., 2017, 46, 5598–5604 RSC.
- J. S. Jones and F. P. Gabbai, Chem. - Eur. J., 2017, 23, 1136–1144 CrossRef CAS PubMed.
- J. S. Jones and F. P. Gabbai, Acc. Chem. Res., 2016, 49, 857–867 CrossRef CAS PubMed.
- N. Tan, Y. Chen, S. Yin, R. Qiu, Y. Zhou and C. T. Au, Curr. Org. Chem., 2012, 16, 2462–2481 CrossRef CAS.
- G. He, O. Shynkaruk, M. W. Lui and E. Rivard, Chem. Rev., 2014, 114, 7815–7880 CrossRef CAS PubMed.
- C. I. Rat, C. Silvestru and H. J. Breunig, Coord. Chem. Rev., 2013, 257, 818–879 CrossRef CAS.
- R. M. Minyaev, T. N. Gribanova and V. I. Minkin, in Hyperbonding and Hypercoordination in Main-Group Chemistry, ed. J. Reedijk and K. Poeppelmeier, Comprehensive Inorganic Chemistry (II), Elsevier, Oxford, 2013, pp. 109–132 Search PubMed.
- G. Sean McGrady and J. W. Steed, Hypervalent Compounds Based in part on the article Indium: Inorganic Chemistry by Dennis G. Tuck which appeared in the Encyclopedia of Inorganic Chemistry, in Encyclopedia of Inorganic Chemistry, ed. R. B. King, R. H. Crabtree, C. M. Lukehart, D. A. Atwood and R. A. Scott, 1st edn, 2006, DOI:10.1002/0470862106.ia094.
- S. Sato, O. Takahashi and N. Furukawa, Coord. Chem. Rev., 1998, 176, 483–514 CrossRef CAS.
- R. Pajkert and G.-V. Roeschenthaler, Organophosphorus Chem., 2013, 42, 197–215 CAS.
- K.-y. Akiba, M. Yamashita, Y. Yamamoto and S. Nagase, J. Am. Chem. Soc., 1999, 121, 10644–10645 CrossRef CAS.
- M. Yamashita, Y. Yamamoto, K. Akiba, D. Hashizume, F. Iwasaki, N. Takagi and S. Nagase, J. Am. Chem. Soc., 2005, 127, 4354–4371 CrossRef CAS PubMed.
- K.-y. Akiba, Y. Moriyama, M. Mizozoe, H. Inohara, T. Nishii, Y. Yamamoto, M. Minoura, D. Hashizume, F. Iwasaki, N. Takagi, K. Ishimura and S. Nagase, J. Am. Chem. Soc., 2005, 127, 5893–5901 CrossRef CAS PubMed.
- T. Yamaguchi, Y. Yamamoto, D. Kinoshita, K.-y. Akiba, Y. Zhang, C. A. Reed, D. Hashizume and F. Iwasaki, J. Am. Chem. Soc., 2008, 130, 6894–6895 CrossRef CAS PubMed.
- M. Yamashita, Y. Yamamoto, K.-y. Akiba and S. Nagase, Angew. Chem., Int. Ed., 2000, 39, 4055–4058 CrossRef CAS PubMed.
- J.-Y. Nakatsuji, Y. Moriyama, S. Matsukawa, Y. Yamamoto and K.-Y. Akiba, Main Group Chem., 2007, 5, 277–285 CrossRef.
- N. Jun-ya and Y. Yohsuke, Bull. Chem. Soc. Jpn., 2010, 83, 767–776 CrossRef.
- Y. Hirano, S. Kojima and Y. Yamamoto, J. Org. Chem., 2011, 76, 2123–2131 CrossRef CAS PubMed.
- H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 619–635 CrossRef CAS.
- H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 608–611 CrossRef CAS.
- W. Schlenk and J. Holtz, Ber. Dtsch. Chem. Ges. B, 1917, 50, 274–275 CrossRef CAS.
- W. Schlenk and J. Holtz, Ber. Dtsch. Chem. Ges. B, 1916, 49, 603–608 CrossRef CAS.
- D. Hellwinkel and H. Seifert, Justus Liebigs Ann. Chem., 1972, 762, 29–54 CrossRef CAS.
- G. Wittig and M.-H. Wetterling, Justus Liebigs Ann. Chem., 1947, 557, 193–201 CrossRef CAS.
- K. O. Christe, W. W. Wilson, G. J. Schrobilgen, R. V. Chirakal and G. A. Olah, Inorg. Chem., 1988, 27, 789–790 CrossRef CAS.
- R. W. Johnson and E. R. Holm, J. Am. Chem. Soc., 1977, 99, 8077–8078 CrossRef CAS.
- G. A. Olah, D. J. Donovan, J. Shen and G. Klopman, J. Am. Chem. Soc., 1975, 97, 3559–3561 CrossRef CAS.
- H. F. Bettinger, P. v. R. Schleyer and H. F. Schaefer, J. Am. Chem. Soc., 1998, 120, 11439–11448 CrossRef CAS.
- C. S. Ewig and J. R. Van Wazer, J. Am. Chem. Soc., 1990, 112, 109–114 CrossRef CAS.
- C. S. Ewig and J. R. Van Wazer, J. Am. Chem. Soc., 1989, 111, 4172–4178 CrossRef CAS.
- H. H. Michels and J. A. M. Jr., J. Chem. Phys., 1990, 93, 1805–1813 CrossRef CAS.
- I. V. Getmanskii and R. M. Minyaev, J. Struct. Chem., 2008, 49, 973–978 CrossRef CAS.
- D. Kurzydłowski and P. Zaleski-Ejgierd, Sci. Rep., 2016, 6, 36049 CrossRef PubMed.
- A. R. Miller, R. R. Tsukimura and R. Velten, Science, 1967, 155, 688 CrossRef CAS PubMed.
- I. J. Solomon, J. N. Keith and A. Snelson, J. Fluorine Chem., 1972, 2, 129–136 CrossRef CAS.
- A. Hasegawa, R. L. Hudson, O. Kikuchi, K. Nishikida and F. Williams, J. Am. Chem. Soc., 1981, 103, 3436–3440 CrossRef CAS.
- K. Nishikida and F. Williams, J. Am. Chem. Soc., 1975, 97, 7166–7168 CrossRef CAS.
- S. A. Shaffer, M. Sadílek and F. Tureček, J. Org. Chem., 1996, 61, 5234–5245 CrossRef CAS.
- F. Kiyokazu and T. Ryozo, Bull. Chem. Soc. Jpn., 1995, 68, 3309–3318 CrossRef.
- V. Q. Nguyen, M. Sadilek, J. Ferrier, A. J. Frank and F. Tureček, J. Phys. Chem. A, 1997, 101, 3789–3799 CrossRef CAS.
- R. W. Alder, A. G. Orpen and J. M. White, J. Chem. Soc., Chem. Commum., 1985, 949–951 RSC.
- F. Gerson, G. Gescheidt, U. Buser, E. Vogel, J. Lex, M. Zehnder and A. Riesen, Angew. Chem., Int. Ed., 1989, 28, 902–904 CrossRef.
- F. Gerson, J. Knoebel, U. Buser, E. Vogel and M. Zehnder, J. Am. Chem. Soc., 1986, 108, 3781–3783 CrossRef CAS.
- L. Schneider, U. Englert and P. Paetzold, Z. Anorg. Allg. Chem., 1994, 620, 1191–1193 CrossRef CAS.
- M. Mueller, U. Englert and P. Paetzold, Inorg.Chem., 1995, 34, 5925–5926 CrossRef CAS.
- P. D. Livant, D. J. D. Northcott, Y. Shen and T. R. Webb, J. Org. Chem., 2004, 69, 6564–6571 CrossRef CAS PubMed.
- M. Yamashita, K. Kamura, Y. Yamamoto and K.-Y. Akiba, Chem. - Eur. J., 2002, 8, 2976–2979 CrossRef CAS.
- The solid-state molecular structure of 1Me is shown in ESI Fig. S5.† All cell parameters are listed in Table S5.†.
- P. Pyykkö and M. Atsumi, Chem. - Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
- J. K. Badenhoop and F. Weinhold, J. Chem. Phys., 1997, 107, 5422–5432 CrossRef CAS.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- The sum of N–O vdW values calculated based on crystallographic data ranges between 2.79 and 3.16 Å and that from pure theoretical methods ranges between 2.79 and 3.5 Å.
- M. Shimizu and M. Nakatani, Eur. J. Org. Chem., 2017, 2017, 4695–4702 CrossRef CAS.
- Similar deviation was observed from calculations carried out using B3PW91/6-31G(d), UB3PW91/6-31G(d), and B3LYP/6-31G(d), UB3LYP/6-31G(d), and B3LYP/cc-pVDZ, UB3LYP/cc-pVDZ (see Fig. S7–S9†).
- We also performed AIM analysis based on fully optimized geometries of 2 and 3. Please see the ESI, Fig. S14 and Table S11.†.
- A. C. Tsipis, Coord. Chem. Rev., 2017, 345, 229–262 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and computational details, X-ray crystallographic data and NMR spectra. CCDC 1945530–1945536, 1988910. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00002g |
| This journal is © The Royal Society of Chemistry 2020 |