
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect and quantitative monitoring of catalytic organic reactions under heterogeneous conditions using direct analysis in real time mass spectrometry†
Koichiro
Masuda
 and
Shū
Kobayashi
*
and
Shū
Kobayashi
*
Department of Chemistry, School of Science, The University of Tokyo. Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: shu_kobayashi@chem.s.u-tokyo.ac.jp
First published on 14th April 2020
Abstract
This new method overcomes problems of conventional analytical methodologies such as light scattering and sampling reproducibility issues. We used this method for mechanistic studies of catalytic reactions under heterogeneous conditions. Direct-type hydroxymethylation reactions and Mukaiyama-type hydroxymethylation reactions both catalyzed by a scandium–bipyridine ligand complex under micellar conditions were employed as examples of heterogeneous reactions. For direct-type hydroxymethylation reactions, initial reaction rate assays revealed first-order dependency on both substrate and catalyst. On the other hand, Mukaiyama-type hydroxymethylation reactions showed first-order rate dependency on substrate, zero-order on catalyst and saturation kinetics on formaldehyde.
Introduction
Among various types of organic reactions, heterogeneous reactions, have become more important in recent years. Heterogeneous reactions using solid catalysts are essential, especially in industrial fields.1,2 Indeed, currently, 80% of industrial catalytic processes are conducted under heterogeneous conditions.3 Among various kinds of heterogeneous reactions, the mechanism of surface reactions on a solid material has been well studied.4–6 However, heterogeneous reactions in other particular systems have not been investigated in detail from a macroscopic viewpoint because of technical difficulties. For example, liquid–liquid heterogeneous samples introduce difficulties with respect to phase separation, and optical spectroscopic analysis does not work well because of strong light scattering induced by heterogeneity. Instead, an aliquot sample analysis is often employed to elucidate the progress of a reaction;7,8 however, large deviations of material ratios in a heterogeneous sample mean that a large number (or amount) of samples is required to treat the data in a statistically valid manner. In our laboratory, a number of catalytic reactions have been developed in water, most of which form liquid–liquid heterogeneous mixtures. Our previous mechanistic study of the catalytic Mukaiyama aldol reaction employed a water–organic cosolvent system for homogenization, and the reaction was analyzed by HPLC with a quenching procedure for each datapoint.9 In the present article, we describe a direct and quantitative method to monitor reactions using direct analysis in real time mass spectrometry (DART-MS)10 to overcome the heterogeneity problem.Results and discussion
The introduction of an isotopic indicator as an internal standard would enable high levels of quantitation to be achieved by mass spectrometric analysis. Quantitative analysis with an isotopic indicator is a widely employed technique for static systems; however, to our knowledge, it has not been applied to a dynamic system such as organic reactions. We expected that although the mass distribution of materials would be totally different in a heterogeneous system, the isotopic distribution would be identical between the phases if diffusion is sufficiently rapid. Therefore, we envisaged that it would be possible to monitor a heterogeneous reaction by employing an isotope-labelled reaction product as an internal standard. The amount of an “artificial” isotope-labelled internal standard would remain constant during the reaction, whereas the amount of “natural” nonlabelled reaction product would increase. Thus, time-course analysis of the isotopic ratio of the reaction product would reflect the progress of the reaction (Fig. 1).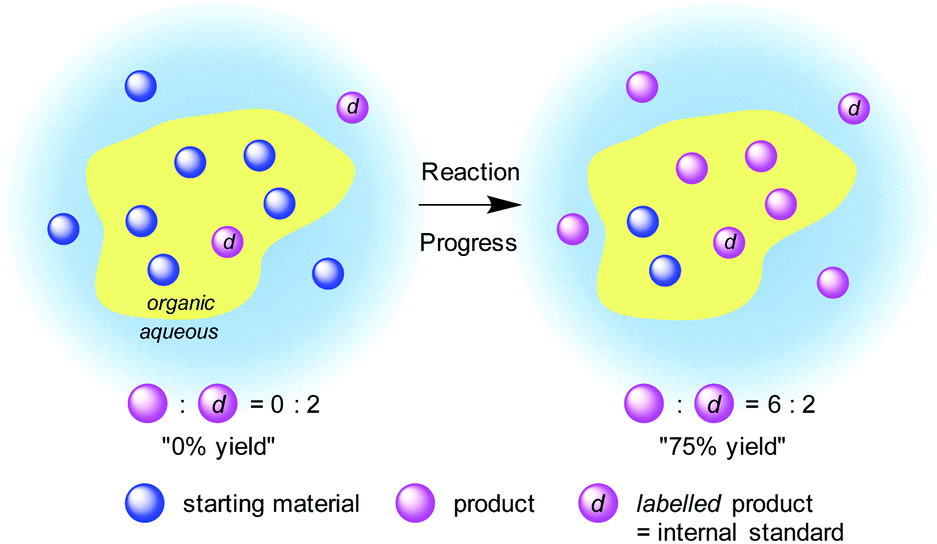 | ||
| Fig. 1 Concept art for our analysis design. | ||
While a variety of ambient ionization techniques11,12 have been employed for the on-line monitoring of homogeneous samples,13 direct analysis of heterogeneous mixture requires robust off-line sampling methods. To analyse heterogeneous reactions quantitatively, the reaction itself requires (1) no reverse reaction nor decomposition of product, and (2) fast matter exchange between phases; analysis requires (3) no progress of a target reaction during the ionization process and (4) fast processing without sample preparation (see 3E in the ESI† for details). Based on these requirements, DART-MS was chosen as the most appropriate ionization method because of its soft and robust ionization process. The ionization area of DART-MS is open to the air, which allows very high flexibility in positioning and introduction of a sample. Accurate control over sample treatment is crucial for stable detection of a peak with adequate intensity and reproducibility by using DART-MS techniques. Mechanically controlled sampling-ionization sequences allow samples to be analysed with good reproducibility. A semiquantitative sampling/ionization process that uses a robot arm autosampler with a glass capillary has been demonstrated for samples such as drugs in biological matrixes,14 allium chemistry,15 and additives in food packaging.16 An inexpensive autoionization system has been demonstrated that used cotton buds attached to an N-scale model railroad system.17–21 Surface contaminants,18 airborne-dispersed chemicals,19 or batch slurry reaction mixture21 were absorbed into a cotton bud head on a model train and was horizontally passed through the ionization area of a DART-MS instrument at a steady speed.
In our laboratory, a new autosampler system was designed to monitor the progress of reactions in organic synthesis. A reaction vessel with precise condition control and a robot arm autosampler were combined. A simplified image of the autosampler system is shown in Fig. 2 and photos of the setup are shown in Fig. 3. The reaction is conducted in a glass vessel in up to 15 mL scale under a positive flow of nitrogen. The reaction temperature and stirring rate can be controlled accurately (−20 to +80 °C, 0–1500 rpm). The autosampler takes a small aliquot sample from the vessel with the robot arm and a sample capillary and transfers this to the ionization area of the DART-MS within a few second.
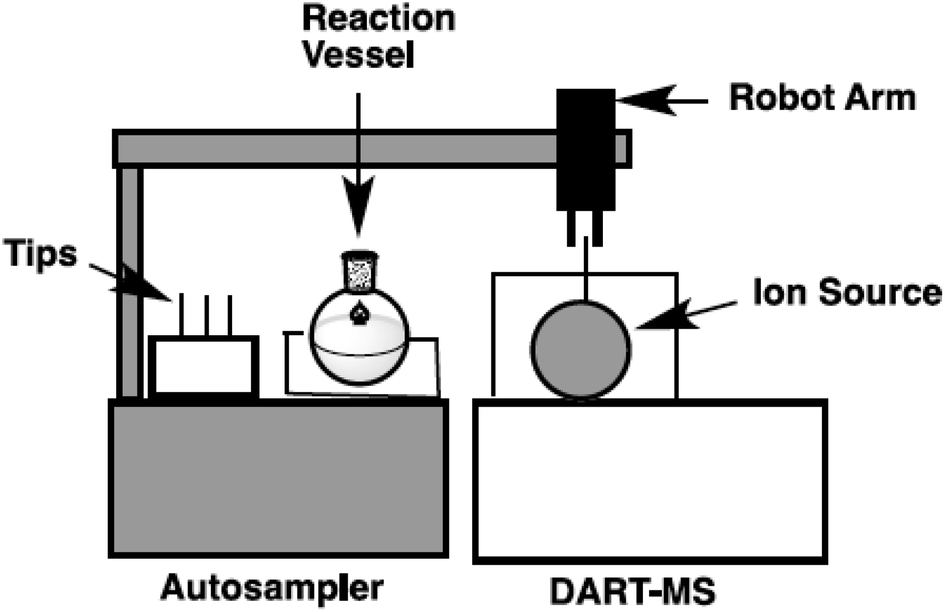 | ||
| Fig. 2 A simplified image of an autosampler for a DART-MS reaction monitoring system. | ||
 | ||
| Fig. 3 Autosampler system (left) and ionization with a DART-MS with autosampler (right). | ||
Hydroxymethylation reactions catalysed by Lewis acid–surfactant combined catalyst
Organic materials are often immiscible with water, and organic reactions in water are generally heterogeneous. In the presence of a surfactant, organic materials form colloidal dispersions (micelles) that extend the boundary between organic materials and water significantly, thereby enhancing surficial reaction rates. Our laboratory has discovered that the combination of a water-compatible Lewis acid and a surfactant moiety can work as an efficient catalyst in water; this system has been termed a Lewis acid-surfactant-combined catalyst (LASC).22–24 LASCs were found to be very efficient catalysts that form hydrophobic environments tightly surrounded by Lewis acid moieties.25 LASCs worked effectively in water to promote Mannich-type reactions,26 Mukaiyama aldol reactions,22 Diels–Alder reactions22 and Michael addition reactions.27 Enantioselective Mukaiyama aldol reactions in water were achieved by a combination of LASC with chiral ligands in 2008.28 In 2010, it was found that the combination of LASCs with chiral ligands could catalyse enantioselective direct-type aldol reactions in water.29 As a model study of our principle, we chose these aldol reactions with formaldehyde, namely hydroxymethylation reactions, catalysed by LASCs in water. After several investigations, a combination of Sc(DS)3 (DS = dodecylsulfate) with 2,2′-bipyridine ligand30,31 was found to be a good and simple catalytic system for both direct-type reaction and Mukaiyama-type reaction, furnishing the same product (Scheme 1). Due to the similarity of the catalytic systems, a valid comparison of the results is expected. Under these conditions, 1.5 equivalents of a surfactant assisted the formation of small particles of organic materials (see Fig. S10 in the ESI†). Given the formation of a good dispersion, high reproducibility of the reaction rate was expected.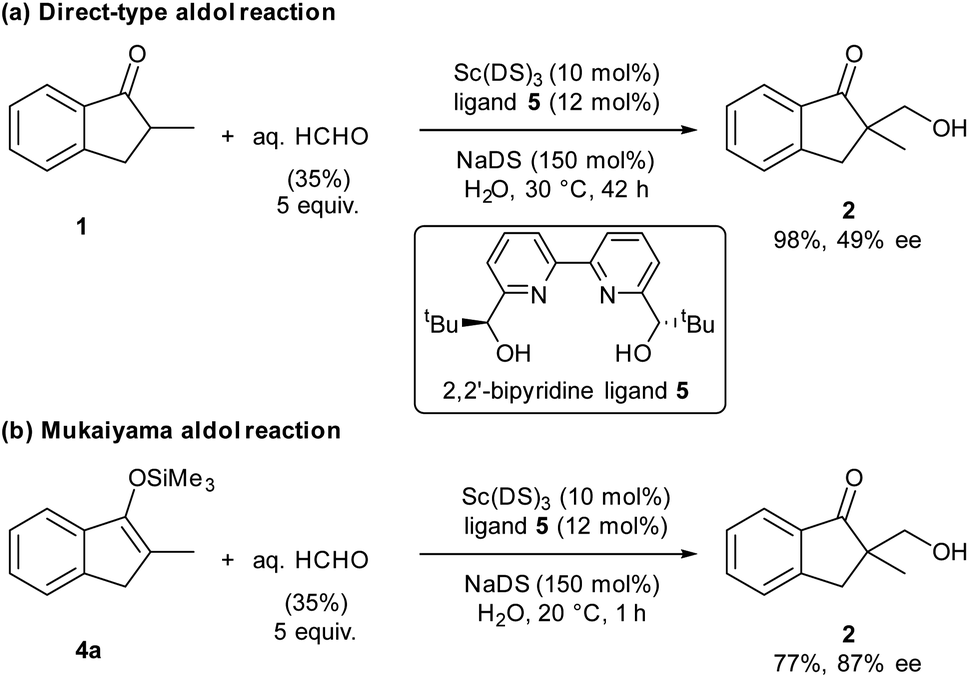 | ||
| Scheme 1 Modified reaction conditions for (a) direct- and (b)Mukaiyama-type reactions. | ||
Direct-type catalytic asymmetric hydroxymethylation reactions in water: monitoring trial and optimization of conditions
We assessed the reaction monitoring of a direct-type hydroxymethylation reaction that was conducted in the presence of isotope labelled compound 2-d4. The reaction was carried out under the conditions indicated in Scheme 1(a) using a 15 mL glass vessel in the autosampler. Small amounts of sample were taken from the reaction mixture with the autosampler with the closed end of a φ 1.8 mm tube and moved to the ionization area of the DART-MS. As initial conditions for ionization, helium gas at 200 °C was selected as the carrier gas. However, no peak was observed with sufficient intensity at m/z 177 or 181 (protonated cations of 2 and 2-d4, respectively), whereas strong peaks were observed at m/z 147 (substrate 1) and 329 (ligand 5). After optimization studies, nitrogen gas was found to be a good carrier gas; this allowed compounds 2 and 2-d4 to be ionized and produce peaks with good intensity at around 200–250 °C. Reaction monitoring was then conducted under the optimized conditions given in Scheme 1 and with modified DART-MS conditions (N2, 200 °C). As expected, peaks were observed at m/z 177 and 181 with a high signal-to-noise (S/N) ratio. The corresponding MS yield calculated from the ratio of these peak areas showed good linear reaction profiles (Fig. 4). Several control studies confirmed that there was no decomposition of internal standard under reaction conditions, which supports the reliability of this method (see 3G and 3I in the ESI†). In order to conduct kinetic analysis of reactions, calibration experiments were conducted. Aqueous solutions of compounds 2 and 2-d4 in several different ratios were analysed by DART-MS six times, and averages were taken for the calibration curve (see 3H in the ESI† for more details).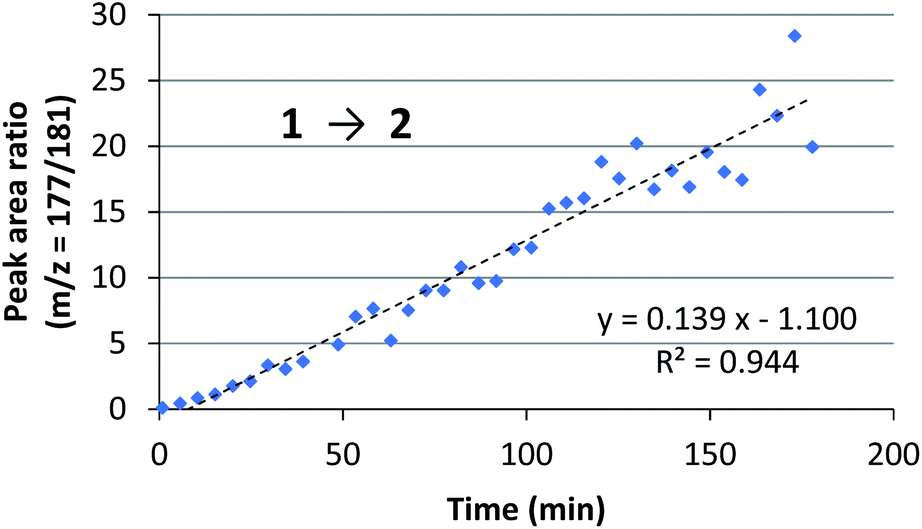 | ||
| Fig. 4 Initial trial of reaction monitoring. Reaction and monitoring conditions: Sc(DS)3 (10 mol%), ligand 5 (12 mol%), NaDS (150 mol%), 1 (0.8 mmol) and aqueous formaldehyde (35%, 5 equiv.) were mixed with an internal standard 2-d4 (0.5 mol%) in water (1.6 mL) at 30 °C. An aliquot was taken periodically with an autosampler and glass capillary, and analysed by DART-MS (positive mode, N2, 200 °C). | ||
Reaction kinetics for the direct-type hydroxymethylation reaction
Kinetic analysis of the enantioselective hydroxymethylation reaction was then conducted based on the established method using DART-MS with internal standard. By following the standard procedure, initial reaction rate assays were conducted by changing the concentration of each component (Scheme 2).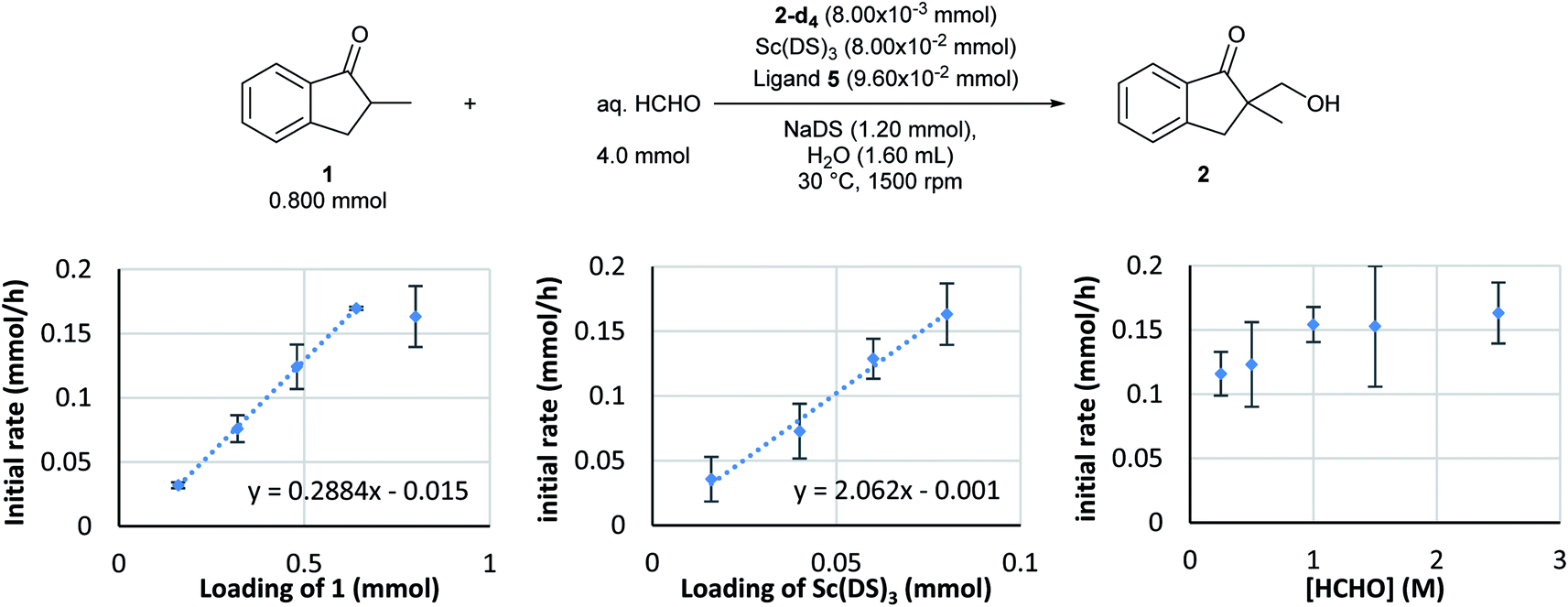 | ||
| Scheme 2 Kinetic analysis of the direct-type catalytic asymmetric hydroxymethylation reaction in water. Conditions: Sc(DS)3, ligand 5, and NaDS were mixed in H2O (0.9 mL) for 1 h at 30 °C. After catalyst preparation, ketone 1 and internal standard 2-d4 (solution in H2O) were added and the mixture was stirred for 10 min. The reaction was started by adding aqueous formaldehyde solution and was monitored by using the DART-MS autosampler system (positive, N2, 200 °C). The measurements were conducted three times for each data point and an average was taken for kinetic profiles. Initially, dependency of the rate of formation of product 2 on substrate 1 was investigated. The loading of 1 was changed from 0.160 to 0.80 mmol while those of the catalyst complex and formaldehyde were kept constant. | ||
As a result, first-order dependency was observed on the substrate 1 loading (Scheme 2, left). The dependency on the chiral Sc catalyst complex was then investigated. The loadings of Sc(DS)3 and 2,2′-bipyridine ligand 4 were varied from 0.0160 to 0.080 mmol while those of the substrate and formaldehyde were kept constant. The initial rate showed first-order dependency on the catalyst complex (Scheme 2, middle). In contrast, an investigation of formaldehyde kinetics revealed a different tendency. Concentration of formaldehyde was varied from 0.250 to 2.5 mol L−1 while those of the substrate and the catalyst were kept constant; as a result, each experiment showed a similar initial reaction rate. Judging from the kinetic study of formaldehyde for Mukaiyama-type hydroxymethylation reaction under dilute conditions (see below), this kinetic profile, showing essentially constant reaction rates for relatively concentrated conditions, indicates saturation kinetics on formaldehyde (Scheme 2, right).
Mechanistic considerations of direct-type hydroxymethylation reactions
The revealed first-order rate dependency of indanone 1 and the chiral Sc catalyst complex with a saturation kinetics on formaldehyde represents an overall experimental rate-law of(k1, k2 and k3 are arbitrary rate constants).
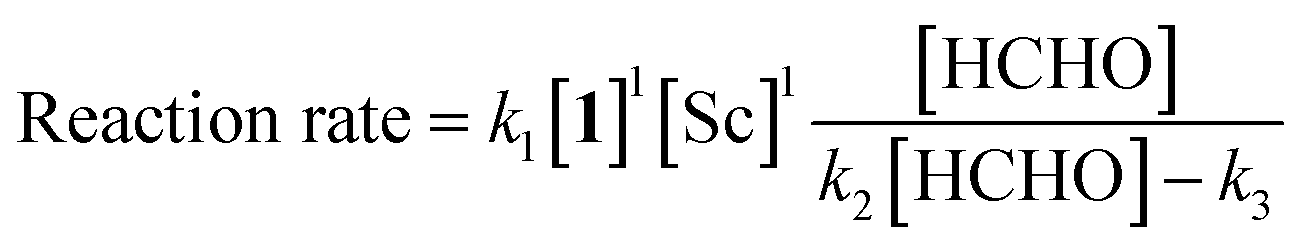
During the course of the reaction, the scandium catalyst and the two carbonyl species (indanone 1 and formaldehyde) form several intermediate complexes. Judging from the saturation kinetics of formaldehyde, formation of the formaldehyde–scandium complex is much faster than that of the indanone–scandium complex. A possible rate-determining step is (a) carbon–carbon bond formation or (b) the enolate formation step with deprotonation (Scheme 3). In a slightly acidic reaction mixture in protic media, substrate 1 become its enol form on tautomerization. In the first proposed pathway, coordination of 1 occurs in an enol form and the following intramolecular C–C bond formation is a plausible rate-determining step of the reaction. After C–C bond formation, intramolecular (or intermolecular) proton transfer leads to release of the product 2. In the second proposed pathway, deprotonation of coordinated enol (or ketone) would limit the overall reaction rate because the basicity of the proton scavenger (water or a dodecylsulfate anion) is very low. The subsequent C–C bond formation is also a potential rate-limiting step; however, we assume that enolate is much more nucleophilic than enol and the reaction would occur essentially instantaneously after the formation of the enolate complex (Scheme 4).
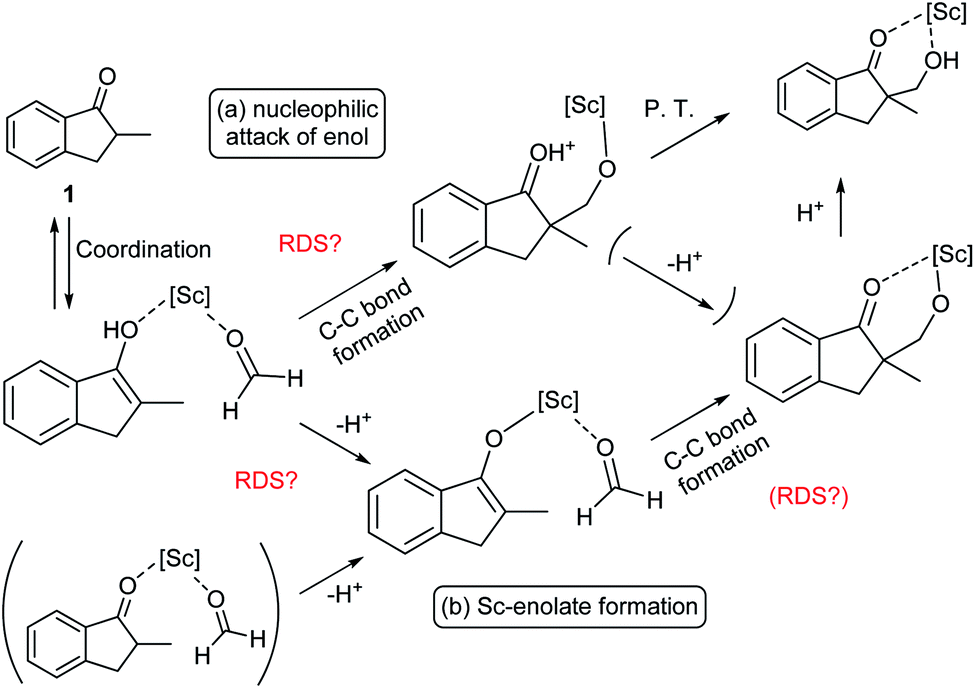 | ||
| Scheme 3 Possible rate-determining steps. | ||
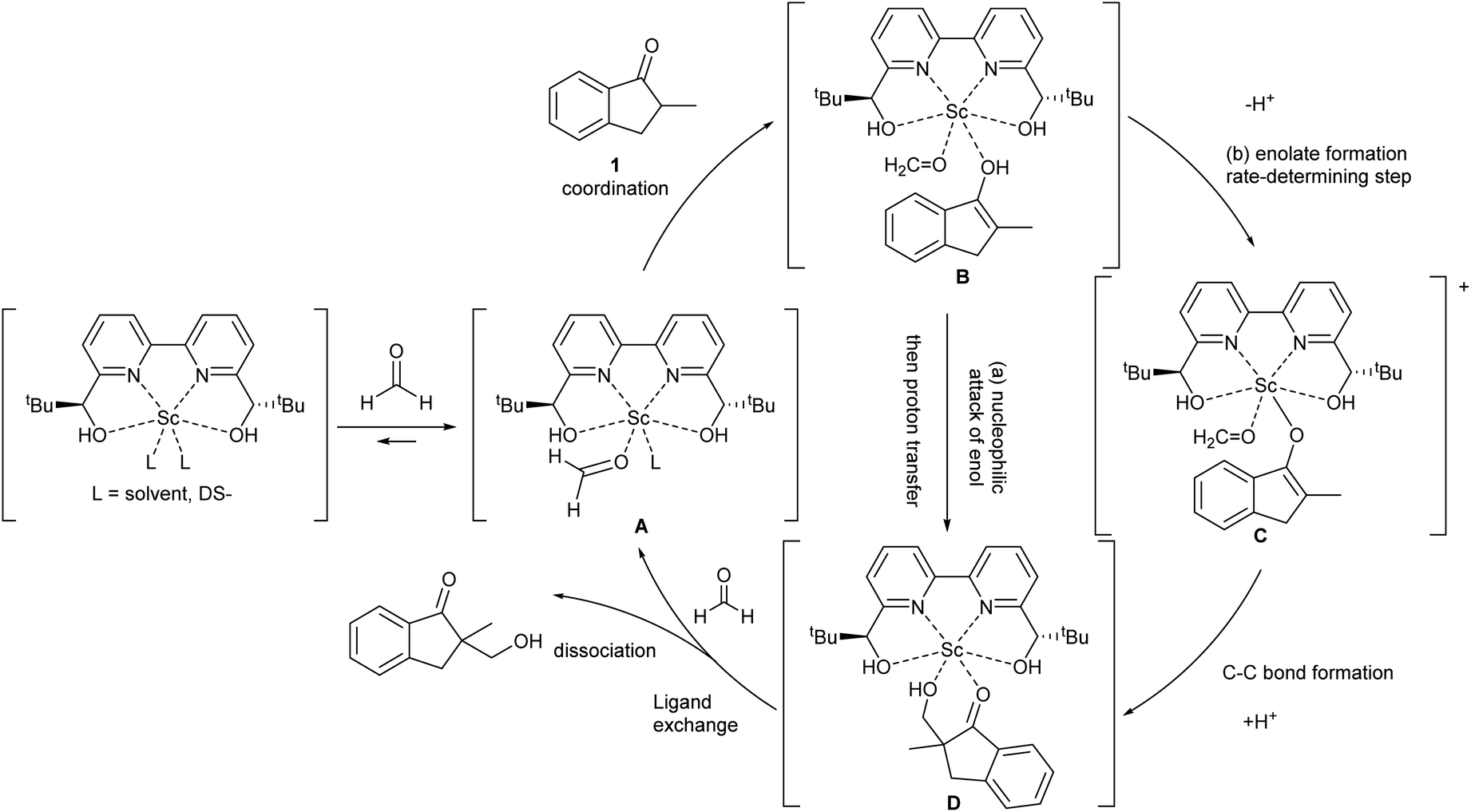 | ||
| Scheme 4 Assumed catalytic cycle of direct-type catalytic asymmetric hydroxymethylation reactions in water. | ||
Mukaiyama-type catalytic asymmetric hydroxymethylation reactions in water
Monitoring trial: To confirm whether the monitoring principle would work for the Mukaiyama-type hydroxymethylation reaction, a detection check experiment was conducted for a reaction under standard conditions with a slight modification (see Scheme 1(b)). An analytical mixture was prepared by following the standard reaction procedure in the absence of formaldehyde to avoid the formation of 2, and a deuterium-labelled reaction product 2-d4 was added (Scheme 5). The mixture was analysed under slightly modified DART-MS conditions of the direct-type hydroxymethylation reaction (N2 as carrier gas, 250 °C). It was found that the expected peak of an internal standard 2-d4 at m/z 181 was detected and no peak was observed at m/z 177, corresponding to nondeuterated reaction product 2.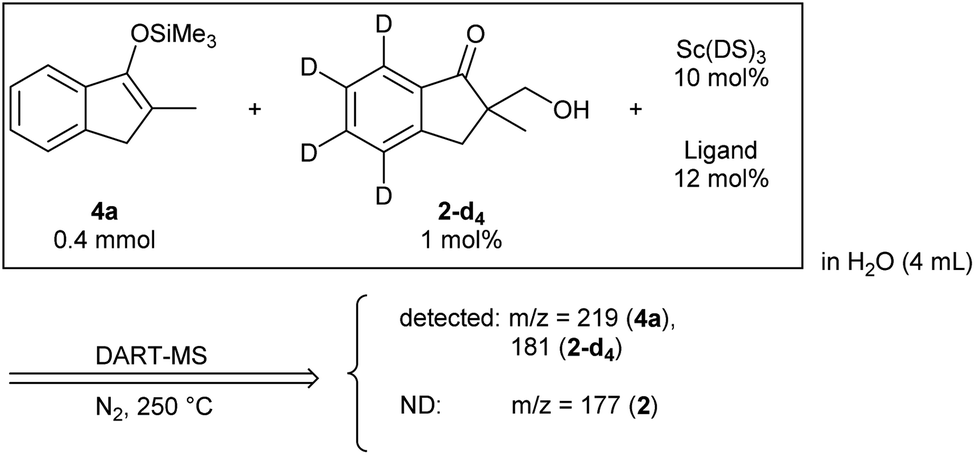 | ||
| Scheme 5 Initial detection verification. | ||
With the success of the detection verification experiment in hand, a monitoring trial was conducted for the Mukaiyama-type hydroxymethylation reaction (Fig. 5). In the presence of internal standard 2-d4, the reaction was conducted and monitored with the autosampler DART-MS system using a glass capillary as a sampling device. Gratifyingly, the 2/2-d4 isotope ratio showed a good reaction profile with a small spread of data points. The reaction profile (calibrated in Fig. S6 in ESI†) showed that the reaction was stopped at around 40 min with approximately 60–70% yield, which corresponded to 65% isolated yield of the product 2 after the monitoring study.
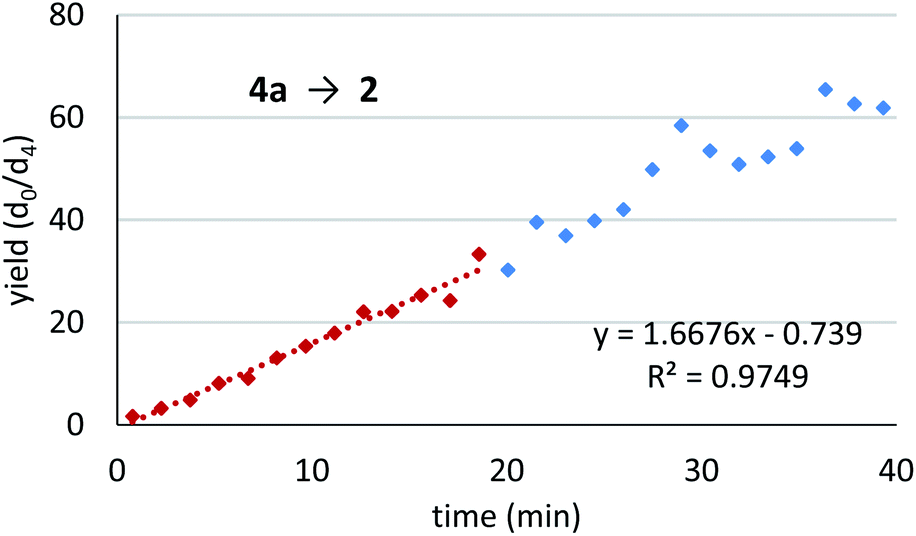 | ||
| Fig. 5 Reaction monitoring trial of Mukaiyama-type hydroxymethylation reactions. Conditions: 4a (0.4 mmol), Sc(DS)3 (5 mol%), ligand 5 (6 mol%), NaDS (150 mol%), and aqueous formaldehyde (35%, 5 equiv.) were mixed with an internal standard 2-d4 (1 mol%) in water (4.0 mL) at 20 °C. An aliquot was taken periodically with an autosampler and glass capillary, and analysed by DART-MS (positive mode, N2, 200 °C). After 1.5 h the reaction was quenched and 65% of compound 2 was isolated. Data points until 20 min (approximately below 30%, red) has been employed to determine initial rate. | ||
Reaction kinetics for Mukaiyama-type hydroxymethylation reactions
Based on the established monitoring conditions, kinetics experiments were performed with the Mukaiyama-type hydroxymethylation reaction of 4a with formaldehyde in water. By following the standard procedure, initial reaction rate assays were conducted by changing the concentration of each component.First, the dependency of the rate of formation of product 2 on the chiral scandium catalyst was investigated. The loading of Sc(DS)3 was changed from 0.0020 to 0.040 mmol and that of 2,2′-bipirydine ligand 5 was changed from 0.0024 to 0.048 while those of substrate 4a and formaldehyde were kept constant. Interestingly, an initial reaction rate assay revealed the zero-order dependency of this reaction on the catalyst, even though this is a catalytic reaction (Scheme 6, left).
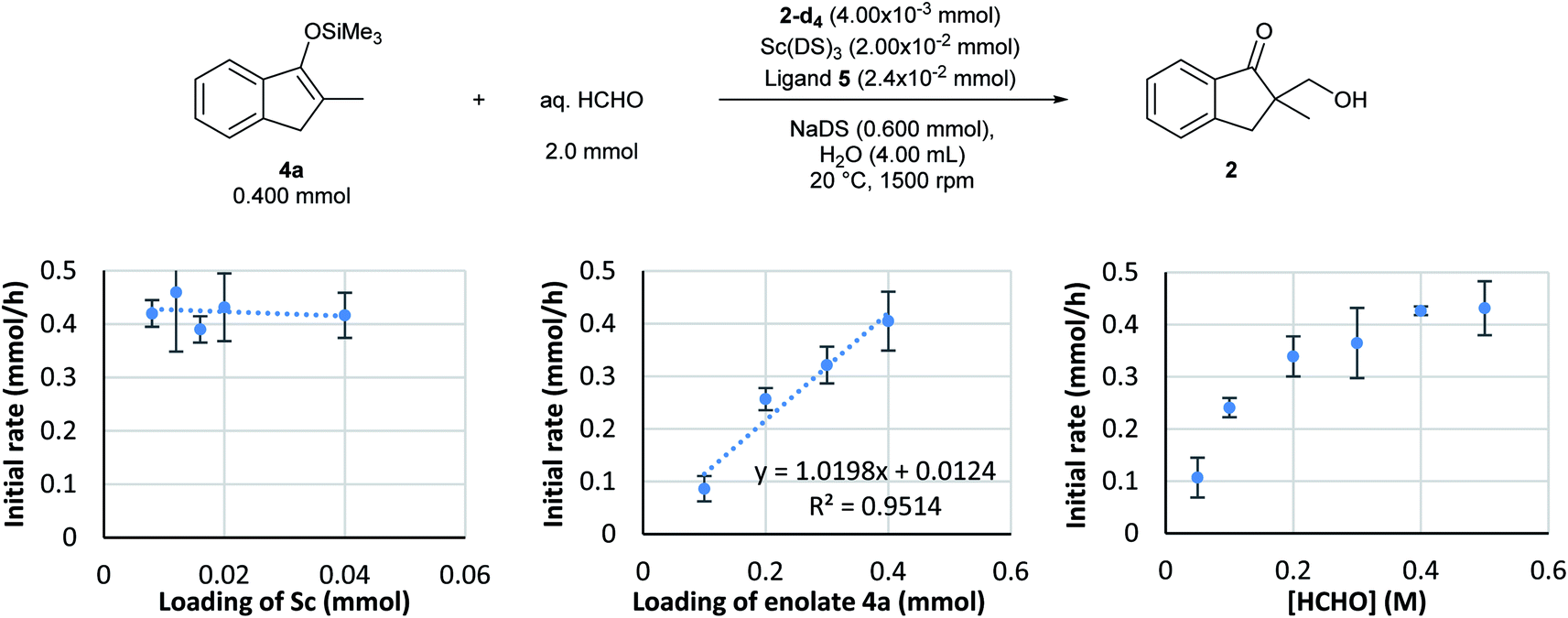 | ||
| Scheme 6 Kinetic analysis of Mukaiyama-type catalytic asymmetric hydroxymethylation reactions in water. Reaction and monitoring conditions: Sc(DS)3, ligand 5, and NaDS were mixed in water (3.7 mL) for 1 h at 20 °C. After catalyst preparation, silyl enol ether 4a and internal standard 2-d4 (solution in water) were added and the mixture was stirred for 10 min. The reaction was started by adding aqueous formaldehyde solution and was monitored with the DART-MS autosampler system (positive, N2, 200 °C). The monitoring was conducted three times for each data point and an average was taken for kinetic profiles. | ||
An initial reaction rate assay on loading of substrate 4a was then conducted. Although the data distribution was relatively wide, a positive correlation was identified between the reaction rate and the loading of substrate 4a (Scheme 6, middle). With an assumption of Gaussian distribution, the slope of the log–log plot was determined as 0.9 ± 0.26 in the 95% confidence interval.
On the other hand, the reaction showed interesting kinetics with respect to the concentration of formaldehyde. An initial reaction rate assay on formaldehyde was conducted by changing the concentration of formaldehyde from 0.050 to 0.50 M. The plot was not linear; rather, it showed a curved profile with saturation (Scheme 6, right). Given that this kinetics plot had some similarity to enzymatic reaction kinetics, the obtained data were plotted again as the double reciprocal. As a result, good linear relationships were observed between the inverse of formaldehyde concentration and the inverse of the reaction rate, except for the data point corresponding to the lowest formaldehyde concentration (Fig. 6). Different to enzymes, the coordination of formaldehyde to scandium is considered to be rapid and is under equilibrium; therefore, it is not limiting on the overall reaction rate.
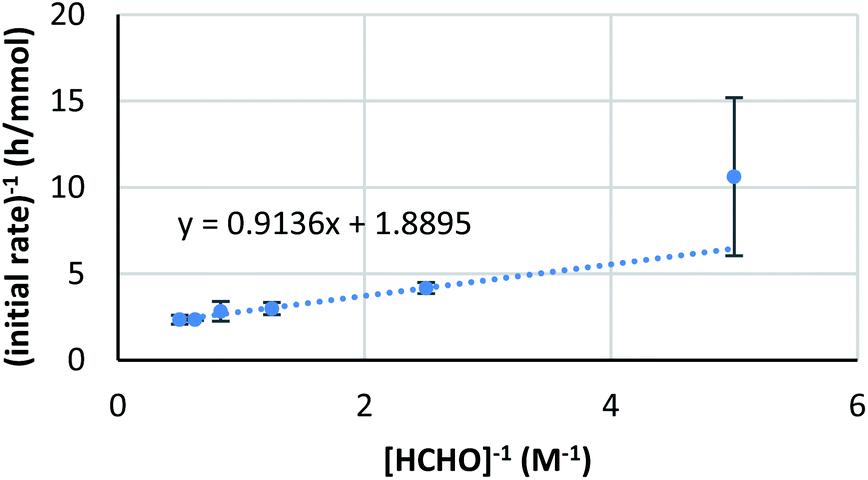 | ||
| Fig. 6 Double reciprocal plot of the kinetics on formaldehyde. | ||
Mechanistic considerations of Mukaiyama-type hydroxymethylation reactions
The obtained data indicated the following results; zero-order on catalyst, first-order on enolate, and saturation kinetics on formaldehyde. An overall experimental rate-law is described as(k4, k5 and k6 are arbitrary rate constants).
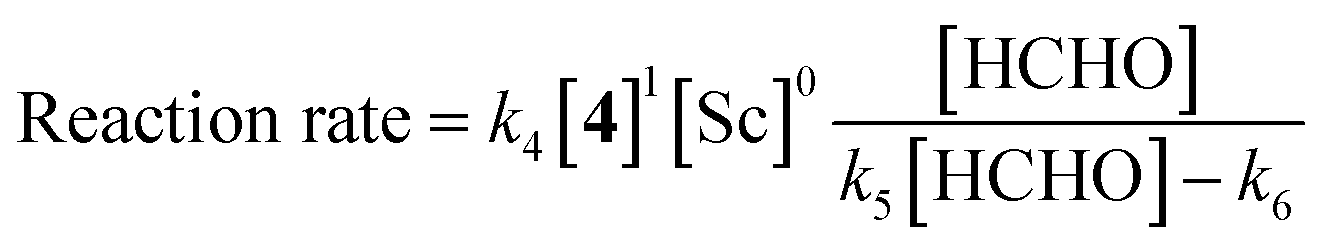
Notably, a zero-order rate law on a catalyst concentration is unique for a catalytic reaction. Indeed, zero-order dependence on a catalyst suggests that the step involving the catalyst is not the rate-determining step in the overall reaction mechanism. Different reaction kinetics in previously reported Mukaiyama-type hydroxymethylation reactions under homogeneous conditions in aqueous solution9,32 and direct-type hydroxymethylation reaction (see above) indicate the effect of heterogeneity in this reaction. The overall reaction system including heterogeneous issues is summarized in Scheme 7. The reaction may take place on the surface of a micelle, because silyl enol ether 4a exists in the micelle and the population of formaldehyde is considered to be greater in the aqueous phase. Therefore, (a) diffusion of formaldehyde or silyl enol ether, or (b) saturation of the Sc(DS)3–ligand complex on the micelle is considered to be a rate-determining factor under these conditions.
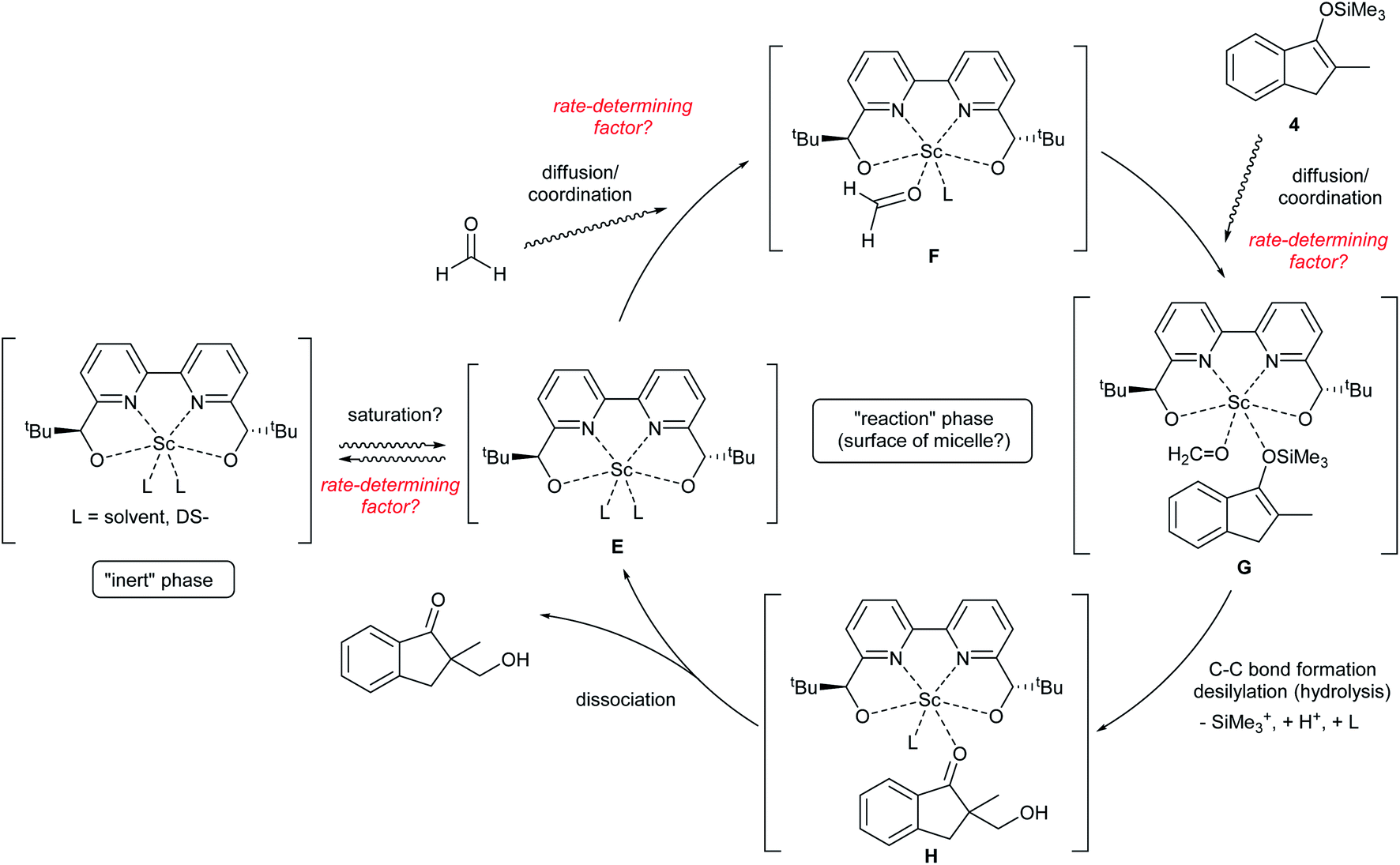 | ||
| Scheme 7 Assumed reaction pathway including heterogeneity on a micelle. | ||
Permeation of formaldehyde or silyl enol ether through a micelle
Diffusion rates of molecules on a micelle are dependent on their size and hydro- and lipophilicity. We assumed that the diffusion rate of formaldehyde is fast enough to be ignored, while that of a silyl enol ether is considerably slower. To confirm the effect of enolate diffusion, ethyldimethylsilyl (EtMe2Si) enol ether 4b was submitted to a kinetic study. The molecular weight and polarity of silyl enol ether 4b are similar to those of Me3Si enol ether 4a, so the diffusion rate of 4b would be expected to be close to that of 4a. On the other hand, a catalytic reaction, especially one involving a C–C bond-forming step with silyl cleavage, is expected to be slower for 4b because of the increased steric bulk on the silyl moiety.As a result, the zero-order dependency was again observed for the catalyst, and the initial reaction rate of enol ether 4b was almost three times slower than that of the Me3Si enol ether 4a (Fig. 7). This significant rate change indicates a C–C bond-forming step would be more plausible as a rate-determining step rather than diffusion of the silyl enol ether.
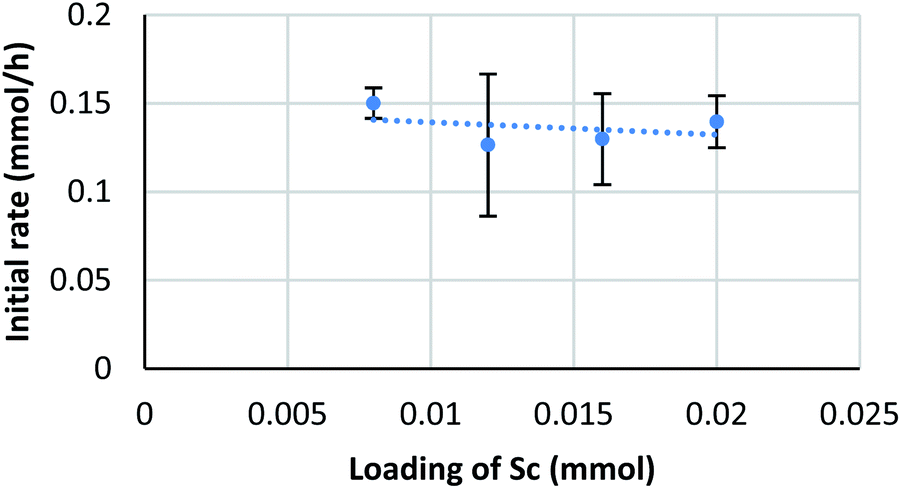 | ||
| Fig. 7 Kinetic analysis on EtMe2Si enol ether. Conditions: 4b (0.4 mmol), Sc(DS)3 (2–5 mol%), ligand 5 (6 mol%), NaDS (150 mol%), and aqueous formaldehyde (35%, 5 equiv.) were mixed with an internal standard 2-d4 (1 mol%) in water (4.0 mL) at 20 °C. Kinetic analysis was conducted in a same manner to Scheme 6. | ||
Saturation of scandium complex on a micelle
It is known that the composition of a mixed micelle is different from the loading of each surfactant. It is possible that the amount of actual “active” scandium species E is not proportional to the corresponding catalyst loading. In other words, the active species becomes “saturated” and a limited amount of scandium may work as a catalyst. Therefore, we hypothesize that the amount of “active species” could be proportional to the total surface area of micelles, which, in turn, is determined by the total amount of dodecylsulfate anions (DS−). Thus, an initial reaction rate assay was conducted on the loading of DS− to test this concept (Fig. 8).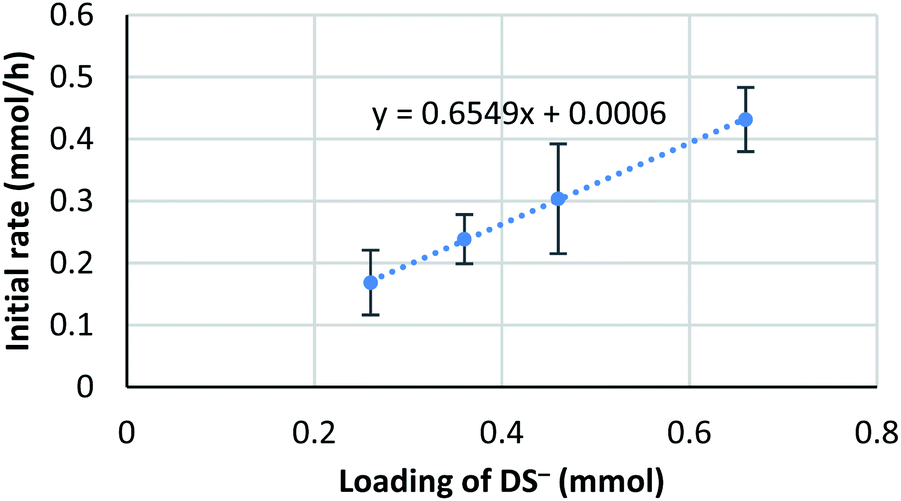 | ||
| Fig. 8 Kinetic analysis on the loading of NaDS. Conditions: 4a (0.4 mmol), Sc(DS)3 (5 mol%), ligand 5 (6 mol%), NaDS (50–150 mol%), and aqueous formaldehyde (35%, 5 equiv.) were mixed with an internal standard 2-d4 (1 mol%) in water (4.0 mL) at 20 °C. Loading of DS− indicates the sum amount of dodecylsulfate anion of Sc(DS)3 and NaDS. Kinetic analysis was conducted in a same manner to Scheme 6. | ||
The result showed a linear dependency of the initial reaction rate on the loading of NaDS. This relationship indicates that the amount of scandium cation on the micellar surface is constant while the ratio of surfactant loading is changed. From this experiment, saturation of the Sc(DS)3–ligand complex on the micelle is well supported. As already described, the catalytic cycle of the reaction is considered to be identical to the previous report9 under homogeneous conditions in aqueous media. Therefore, the zero-order dependency on the catalyst observed from the initial reaction rate assay is reasoned to stem from the saturation of the scandium species on the surface of the micelle. The first-order dependency observed in the direct-type reactions could be explained by the higher polarity of substrate 1, which enhanced the solubility of scandium complex in the organic phase, thereby avoiding the rate limitation.
Conclusions
Direct monitoring of heterogeneous reactions under micellar conditions was achieved by using DART-MS with an isotope-labelled internal standard. An internal standard 2-d4 was successfully designed to monitor the direct-type hydroxymethylation reaction of ketone 1 with formaldehyde catalysed by scandium tris(dodecylsulfate)–2,2′-bipyridine complex in water. Compound 2-d4 could also be used to monitor the Mukaiyama-type hydroxymethylation reaction in water catalysed by the same scandium complex. Further exploration revealed that the method is applicable to much more complex systems, including 1,4-addition reactions in water33 (liquid–liquid heterogeneous system without surfactant; see 3J in the ESI†) and catalytic allylation reactions in water34 (liquid–liquid heterogeneous system with solid catalyst; see 3J in the ESI†). As an extensive application of the monitoring methodology, kinetic analyses were conducted and overall experimental rate laws are described;for direct-type hydroxymethylation reactions
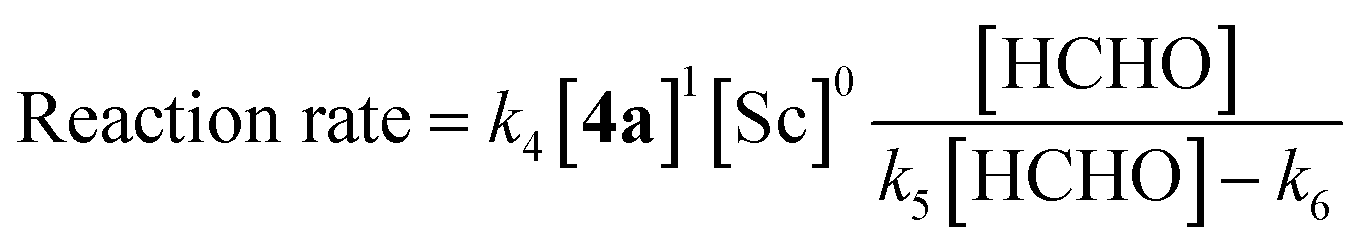
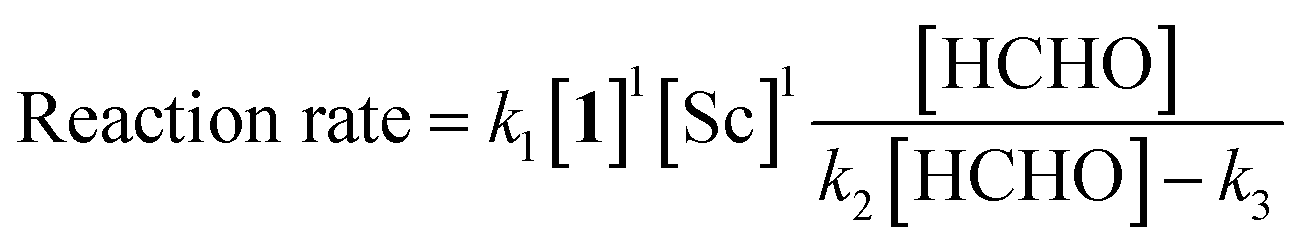
The completely different rate laws from homogeneous systems9 suggest distinct roles for the catalyst complex in the mechanism of the Mukaiyama-type hydroxymethylation reaction under heterogeneous conditions. For the zero-order dependency on the scandium complex in the Mukaiyama-type hydroxymethylation reaction, the saturation of the scandium species on micellar surface was suggested from several experiments.
This method is expected to be applicable to a wide variety of heterogeneous organic reactions, and it should be an efficient analytical tool for studying reactions in other complex media. Our previously developed heterogeneous reactions in water are of particular interest, and several reactions are under study in our group.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS (Research Fellowship for Young Scientists to KM). Dr M. Yamaguchi is acknowledged for her contributions on establishing the auto-sampler system and her early studies of homogeneous reactions.Notes and references
- H. Knözinger and K. Kochloefl, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA., Weinheim, Germany, 2000 Search PubMed.
- Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008 Search PubMed.
- J. R. H. Ross, Heterogeneous Catalysis, Elsevier, Amsterdam, 2012 Search PubMed.
- M. Boudart, Kinetics of heterogeneous catalytic reactions, Princeton University Press, Princeton, NJ, 1984 Search PubMed.
- J. A. Dumesic, The Microkinetics of heterogeneous catalysis, American Chemical Society, Washington, DC, 1993 Search PubMed.
- S. H. Lin and H. Eyring, Proc. Natl. Acad. Sci. U. S. A., 1970, 65, 47–50 CrossRef CAS PubMed.
- J. H. Ramsden, R. S. Drago and R. Riley, J. Am. Chem. Soc., 1989, 111, 3958–3961 CrossRef CAS.
- M.-L. Wang and H.-S. Wu, Chem. Eng. Sci., 1991, 46, 509–517 CrossRef CAS.
- C. Mukherjee, T. Kitanosono and S. Kobayashi, Chem. - Asian J., 2011, 6, 2308–2311 CrossRef CAS PubMed.
- R. B. Cody, J. A. Laramée and H. D. Durst, Anal. Chem., 2005, 77, 2297–2302 CrossRef CAS PubMed.
- R. G. Cooks, Z. Ouyang, Z. Takats and J. M. Wiseman, Science, 2006, 311, 1566–1570 CrossRef CAS PubMed.
- M.-Z. Huang, C.-H. Yuan, S.-C. Cheng, Y.-T. Cho and J. Shiea, Annu. Rev. Anal. Chem., 2010, 3, 43–65 CrossRef CAS PubMed.
- A. Ray, T. Bristow, C. Whitmore and J. Mosely, Mass Spectrom. Rev., 2018, 37, 565–579 CrossRef CAS PubMed.
- S. Yu, E. Crawford, J. Tice, B. Musselman and J.-T. Wu, Anal. Chem., 2009, 81, 193–202 CrossRef CAS PubMed.
- R. Kubec, R. B. Cody, A. J. Dane, R. A. Musah, J. Schraml, A. Vattekkatte and E. Block, J. Agric. Food Chem., 2010, 58, 1121–1128 CrossRef CAS PubMed.
- L. K. Ackerman, G. O. Noonan and T. H. Begley, Food Addit. Contam., Part A, 2009, 26, 1611–1618 CrossRef CAS PubMed.
- A. H. Grange, Environ. Forensics, 2008, 9, 127–136 CrossRef CAS.
- A. H. Grange, Environ. Forensics, 2008, 9, 137–143 CrossRef CAS.
- A. H. Grange, Environ. Forensics, 2009, 10, 183–195 CrossRef CAS.
- A. H. Grange, Rapid Commun. Mass Spectrom., 2013, 27, 305–318 CrossRef CAS PubMed.
- D. S. Cho, S. C. Gibson, D. Bhandari, M. E. McNally, R. M. Hoffman, K. D. Cook and L. Song, Rapid Commun. Mass Spectrom., 2011, 25, 3575–3580 CrossRef CAS PubMed.
- K. Manabe, Y. Mori and S. Kobayashi, Tetrahedron, 1999, 55, 11203–11208 CrossRef CAS.
- K. Manabe and S. Kobayashi, Synlett, 1999, 1999, 547–548 CrossRef.
- K. Manabe, Y. Mori, T. Wakabayashi, S. Nagayama and S. Kobayashi, J. Am. Chem. Soc., 2000, 122, 7202–7207 CrossRef CAS.
- S. Kobayashi and T. Wakabayashi, Tetrahedron Lett., 1998, 39, 5389–5392 CrossRef CAS.
- K. Manabe and S. Kobayashi, Org. Lett., 1999, 1, 1965–1967 CrossRef CAS.
- Y. Mori, K. Kakumoto, K. Manabe and S. Kobayashi, Tetrahedron Lett., 2000, 41, 3107–3111 CrossRef CAS.
- M. Kokubo, C. Ogawa and S. Kobayashi, Angew. Chem., Int. Ed., 2008, 47, 6909–6911 CrossRef CAS PubMed.
- S. Kobayashi, M. Kokubo, K. Kawasumi and T. Nagano, Chem. - Asian J., 2010, 5, 490–492 CrossRef CAS PubMed.
- C. Bolm, M. Ewald, M. Felder and G. Schlingloff, Chem. Ber., 1992, 125, 1169–1190 CrossRef CAS.
- C. Bolm, M. Zehnder and D. Bur, Angew. Chem., Int. Ed. Engl., 1990, 29, 205–207 CrossRef.
- S. Ishikawa, T. Hamada, K. Manabe and S. Kobayashi, J. Am. Chem. Soc., 2004, 126, 12236–12237 CrossRef CAS PubMed.
- T. Kitanosono, M. Sakai, M. Ueno and S. Kobayashi, Org. Biomol. Chem., 2012, 10, 7134 RSC.
- U. Schneider, M. Ueno and S. Kobayashi, J. Am. Chem. Soc., 2008, 130, 13824–13825 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic/analytical procedures, characterization of materials, and detailed technical information. See DOI: 10.1039/d0sc00021c |
| This journal is © The Royal Society of Chemistry 2020 |