
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licencemeta-Selective C–H functionalisation of aryl boronic acids directed by a MIDA-derived boronate ester†
Alexander F.
Williams
,
Andrew J. P.
White
,
Alan C.
Spivey
 * and
Christopher J.
Cordier‡
*
* and
Christopher J.
Cordier‡
*
Department of Chemistry, Imperial College London White City Campus, Molecular Sciences Research Hub, 80 Wood Lane, London, W12 0BZ, UK. E-mail: a.c.spivey@imperial.ac.uk
First published on 3rd March 2020
Abstract
N-Methyliminodiacetic acid (MIDA) boronates are boronic acid derivatives which are stable to reduction, oxidation and transmetalation. This has led to their widespread use as boronic acid protecting groups (PGs) and in iterative cross-couplings. We describe herein the development of a novel MIDA derivative that acts in a dual manner, as a protecting group and a directing group (DG) for meta C(sp2)–H functionalisation of arylboronic acids. Palladium catalysed C–H alkenylations, acetoxylations and arylations are possible, at room temperature and under aerobic conditions. Deprotection to reveal the functionalised boronic acids is rapid and allows for full recovery of the DG. The technique allows the facile diversification of aryl boronic acids and their subsequent use in a range of reactions or in iterative processes.
Introduction
In recent years, directed C–H functionalisation has rapidly progressed from a new concept to an indispensable synthetic technique.1 Directing groups (DGs) have emerged as powerful tools to achieve these transformations, often over-riding the inherent steric or electronic bias of the substrate.2 While numerous methods for the directed ortho C(sp2)–H functionalisation of aryl systems have been developed, directed, meta-selective processes remain relatively underexplored.3 The distal nature of the meta C–H bond, its proximity to the ortho and para positions and often unfavourable electronic properties pose unique challenges. In 2012, Yu was able to address these challenges with the seminal design of U-shaped DGs for the meta alkenylation of hydrocinnamic acid and toluene derivatives.4Numerous DG designs have since been developed, expanding on Yu's work.3d,5 However, the DGs are often bespoke for a particular substitution process, require stepwise syntheses and attachment via functional groups (FGs) which are themselves non-ideal for subsequent derivatisation (e.g. amides).6 Consequently, the synthetic versatility of the DGs is limited. A single, more general, meta-selective DG, tethered via a synthetically versatile FG would reduce the current need for many individually bespoke systems (Scheme 1).
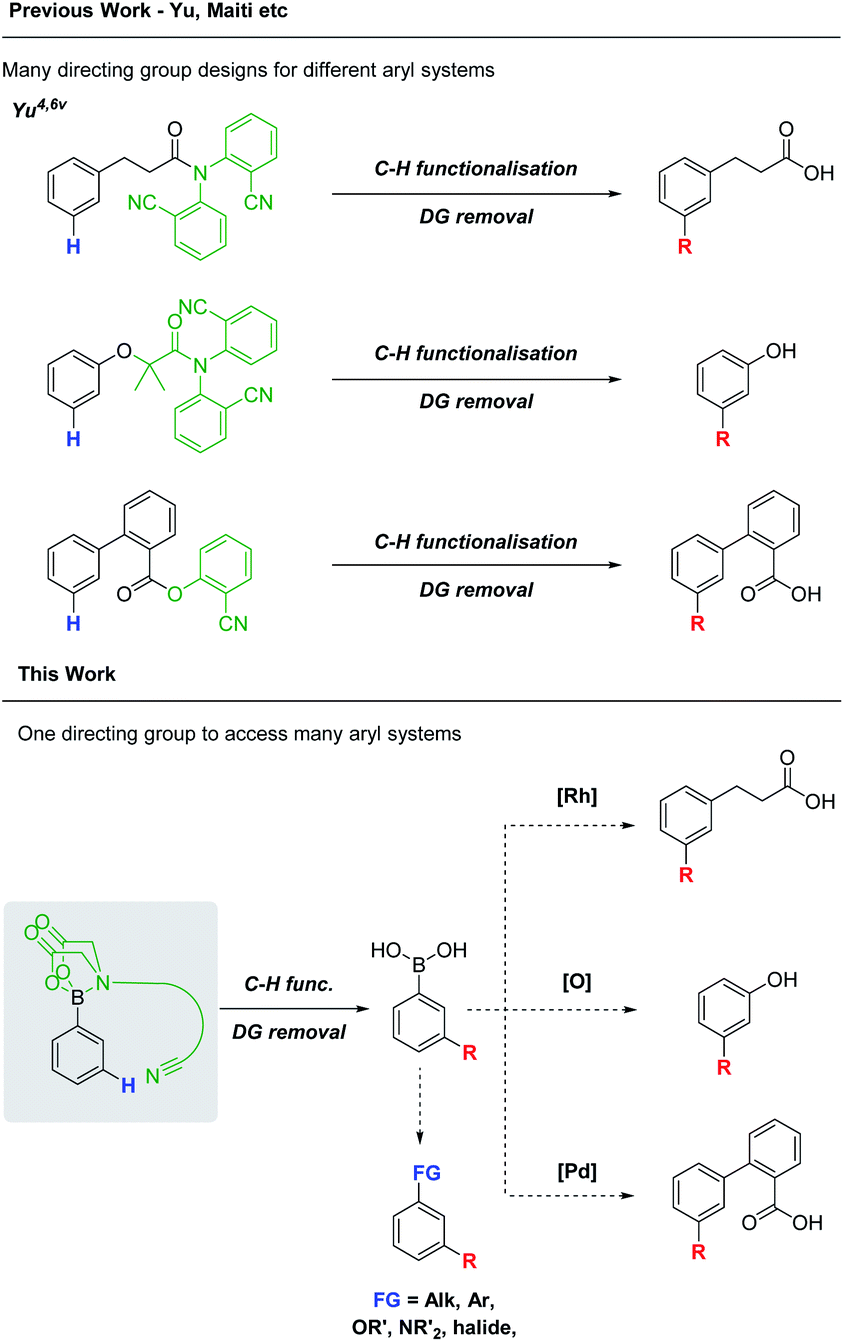 | ||
| Scheme 1 A comparison between previously developed, bespoke meta-DGs and a general boron-tethered meta-DG. | ||
We identified the boronic acid FG as an attractive foundation for such a meta-selective DG. Boronic acids are ubiquitous in organic synthesis, with a wealth of transformations available for their derivatisation.7,8 Their propensity to undergo transmetalation precludes their direct use in metal catalysed C–H functionalisations however.9,10 Selective transition metal insertion into the desired, comparatively inert, C–H bond over the C–B bond must therefore be engineered to allow boronic acids to be exploited as the foundation of the envisioned DG.
Such selectivity has been demonstrated by Suginome et al., who developed anthranilamide (aam) and 2-(1H-pyrazole-3-yl)aniline (pza) boronate DGs which enable ortho-directed C–H functionalisations.11 Using these DGs, selective C–H silylations, alkynylations and borylations are possible using iridium, ruthenium or rhodium catalysts, with retention of the protected boronic acid FGs.
N-Methyliminodiacetic acid (MIDA) boronate esters, popularised by Burke et al., similarly suppress B → M transmetalation, allowing the selective Suzuki–Miyaura coupling of boronic acids in the presence of masked MIDA boronates, under anhydrous conditions.12 This reactivity profile, combined with their ease of introduction and deprotection, and the fact that N-tethered moieties are held in enforced proximity to the parent boronic acid by the rigid bicyclic system, make the MIDA boronate an attractive scaffold upon which to base a DG.13
We describe herein the development of a novel MIDA derivative for the meta selective C(sp2)–H functionalisation of aryl boronic acids. The MIDA-DG is simple to attach and remove, with quantitative recovery possible. The DG enables Pd-catalysed C–H functionalisation of aryl boronic acids, at room temperature and under aerobic conditions, with retention of the boronic acid FG. On deprotection, the functionalised boronic acids can be derivatised into numerous other FGs using established boronic acid chemistry. Lastly, we demonstrate the utility of MIDA-DG boronates by using iterative C–H functionalisation/cross-coupling procedures to access diverse polysubstituted aromatic systems.
When considering the design of a MIDA-tethered DG, two general principles were identified: that MIDA boronates are unstable towards strongly basic reagents and nucleophilic solvents,12b and that electron-withdrawing groups adjacent to the B–N bond were anticipated to weaken the strength of this interaction, causing the MIDA ligand to become more labile.
Consequently, DG design focused on a cyanophenol moiety, attached via an O-alkyl linker to the nitrogen of the iminodiacetic acid.
The aryl ether linkage was anticipated to impart a minimal detrimental effect on the MIDA B–N bond, while allowing for a cyclophane-like macrocyclic transition state to be accessed.3d,4
A number of DGs were synthesised and screened using conditions based on those reported for meta C–H alkenylation (Fig. 1).3d,4,5 Product formation was initially observed with the simple 2-cyanophenol based system 2, as well as ethyl cinnamate 1, formed after deprotection and oxidative Heck coupling. Changes to the 2-cyanophenol motif, investigating sterically biasing the direction of the nitrile DG (3 and 4), increasing its Lewis basicity (5), or varying the nitrile position (6) did not lead to improved performance. However, increasing the length of the linker from ethyl to propyl (7) resulted in a doubling of the yield and a concomitant drop in formation of by-product 1.
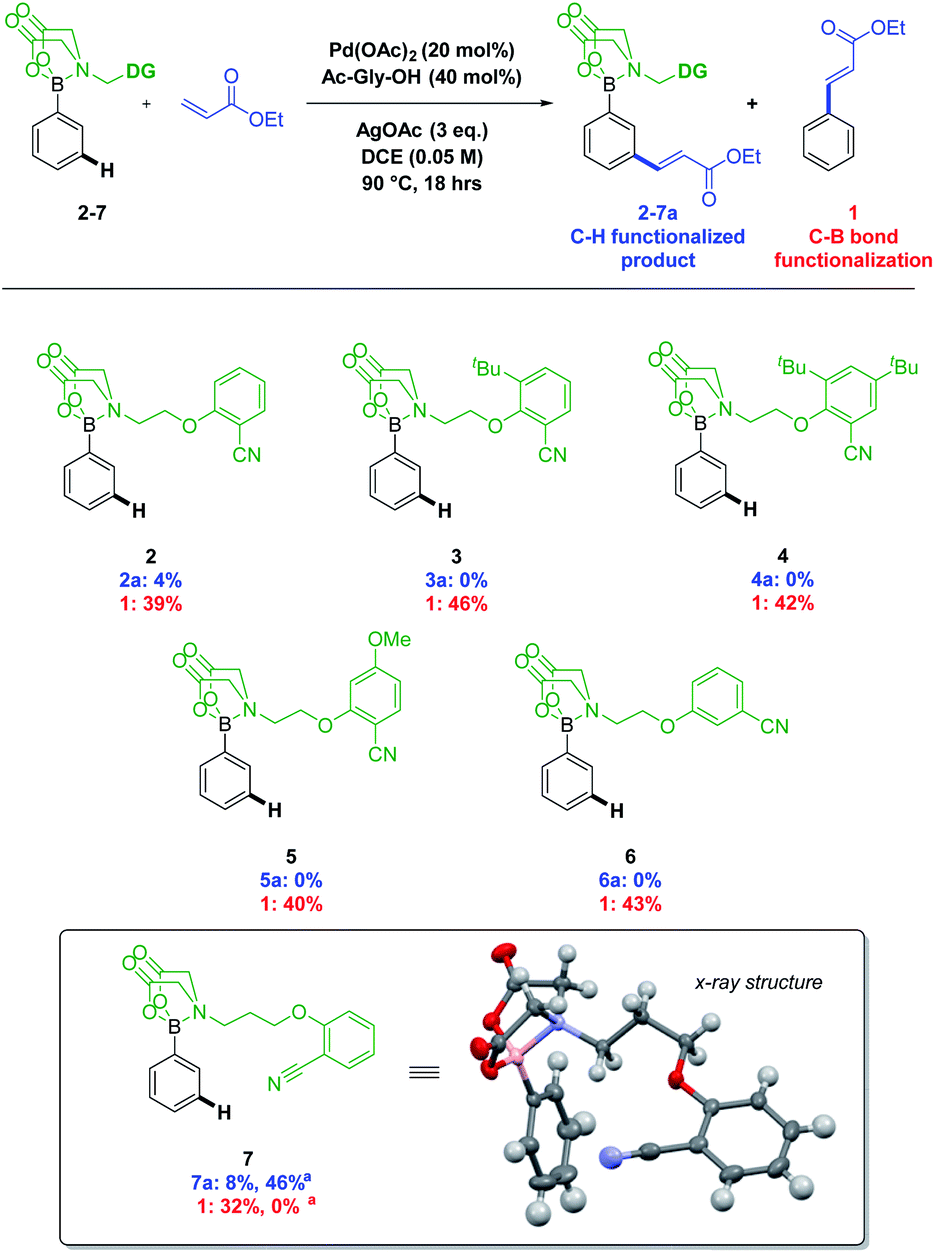 | ||
| Fig. 1 Screening of DG designs against C–H alkenylation conditions. Reactions performed on 0.05 mmol scale, yields calculated using 1H NMR against a CH2Br2 internal standard. a HFIP (0.1 M), AgOAc (1.0 eq.), 50 °C used with 7, resulting in 46% yield as a mixture of 32% meta (7a), 6% para, 5% di meta–meta and 3% di meta–para isomers. | ||
Switching solvent to HFIP, a privileged solvent in C–H functionalisation protocols,14,15 and lowering temperature to 50 °C, led to a further increase in yield (Fig. 1, 7a), along with the formation of para and di-functionalised products. A design of experiment (DOE) approach was taken to optimise the conditions (see ESI†). The optimisation process led to the surprising discovery that functionalisation occurred at room temperature, which is unusual for a transition metal catalysed C–H functionalisation process (Scheme 2, middle).6u
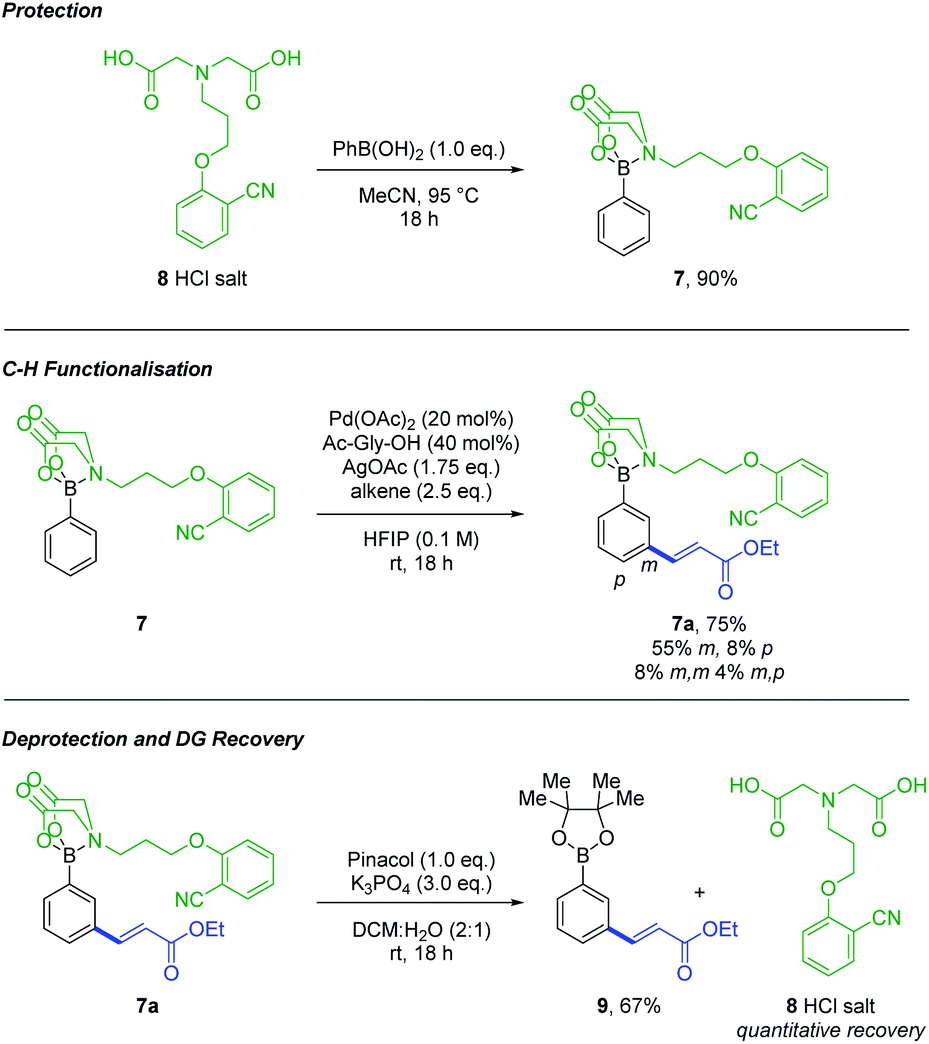 | ||
| Scheme 2 MIDA-DG 8 allows protection of boronic acids, functionalisation via C–H insertion and is simple to deprotect with quantitative recovery. | ||
The MIDA-DG boronate 7 can easily be accessed from the MIDA-DG 8 simply by heating with phenylboronic acid (Scheme 2). All MIDA-DG boronates synthesised thus far have been bench-stable, free-flowing solids, stable for extended periods (>6 months) and to column chromatography using silica gel. Lastly, the MIDA-DG salt 8 is fully recoverable and reusable after deprotection. An unmodified phenylboronic acid MIDA ester was submitted to the optimised conditions, yielding no alkenylated product and indicating the necessity of the directing motif for achieving reactivity (see ESI†).
A 3 × 3 array was devised to investigate the reaction scope, incorporating electron-donating to electron-withdrawing substituents in the ortho, meta and para positions (Fig. 2, left). Yields varied from 33% to 79%, with electron-withdrawing ester groups (16a–18a) requiring longer reaction times. Ortho substituents gave a more complex mixture of regioisomers, as the meta positions are no longer equivalent or blocked. It appears that electron-withdrawing groups strengthen the PG ability of the MIDA-DG, while the opposite was true for electron-donating substituents, especially the para-methoxy analogue (12a), where significant degradation occurred. This may reflect donation of electron density weakening the B–N dative bond. The regioselectivity could be increased by employing a more sterically encumbered Ac-Val-OH ligand in place of Ac-Gly-OH (14ab).
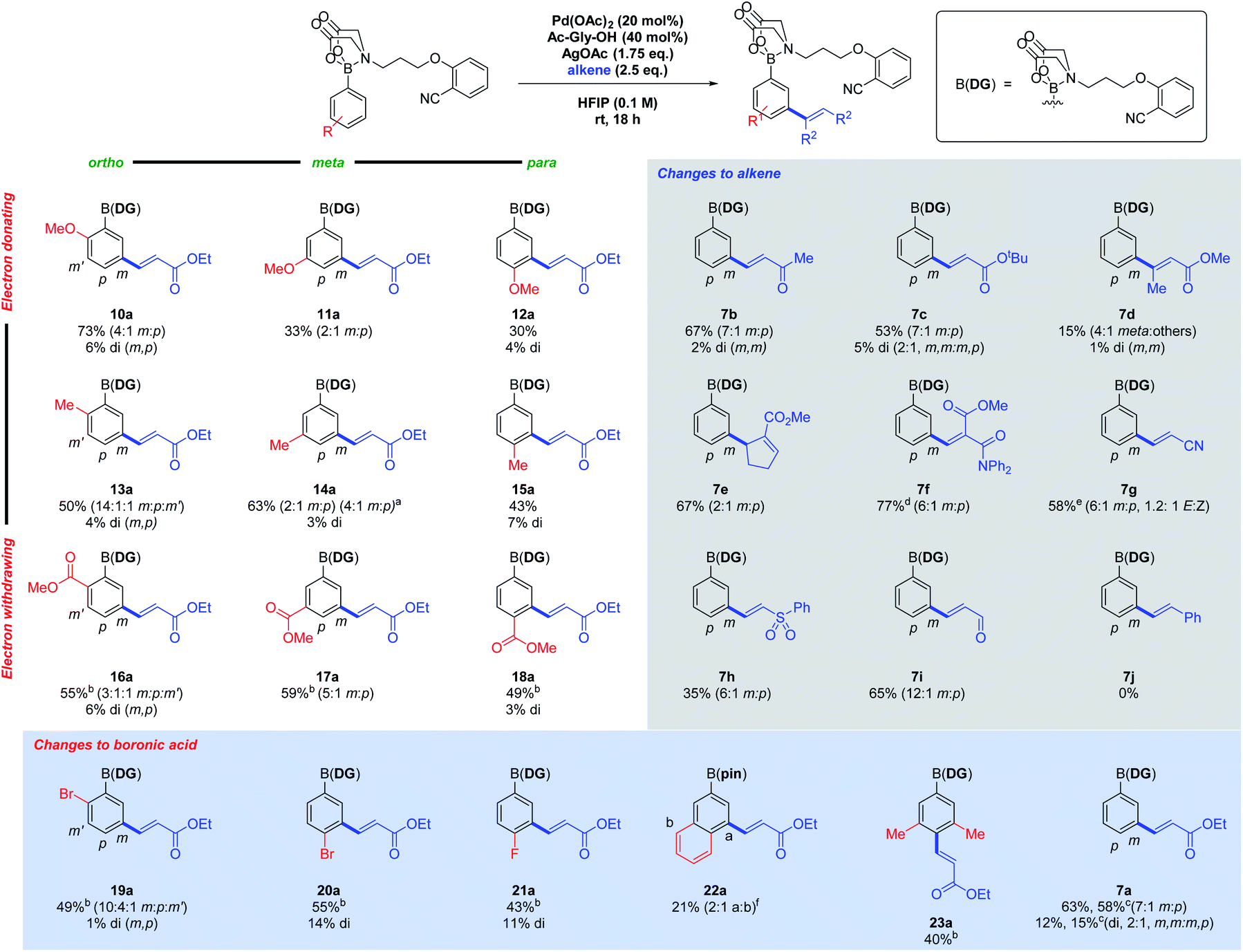 | ||
| Fig. 2 The scope of the C–H alkenylation reaction. Reactions performed on 0.3 mmol scale, isolated yields. a Ac-Val-OH (40 mol%), 40 °C used. b Reaction time 48 h. c Average yield of 4 and 3 mmol scale reactions. d 1.2 eq. of alkene used. e 2.0 eq. alkene used, reaction time 48 h. f Reported yield is over 2 steps following synthesis of the pinacol boronate ester. | ||
Other functionalised aryl boronic acids were also investigated. Bromo (19a and 20a) and fluoro (21a) substituents are tolerated under the conditions, with selective C–H functionalisation occurring in preference to oxidative insertion. The alkenylated products provide interesting handles for further SNAr or cross-coupling chemistry. 2-Naphthylboronic acid (22a) gave two regioisomers at the 4- and 8-positions, with the former being the major product. 3,5-Dimethyl analogue 23a gave a good yield of the para product selectively. The reaction is amenable to scale-up to gram scale (7ac), with quantitative recovery of the DG after further functionalisations (see Scheme 4).
The scope of the alkene coupling partner was next investigated (Fig. 2, right). Ketone (7b), nitrile (7g), sulfone (7h) and aldehyde (7i), FGs were all suitable alkene activating groups. Competition between the acrylonitrile and the MIDA-DG 7 for catalyst co-ordination led to extended reaction times. A cyclic alkene was tolerated, yielding only one alkene regioisomer (7e). A 1,1-heterodisubstituted alkene featuring a diphenylamide was successful (7f), demonstrating the tolerance of secondary amide FGs and that fewer equivalents of alkene can be utilised. N,N-Dimethylacrylamide, styrene (7j) and diethyl vinylphosphonate were unreactive under the standard conditions.
In all cases, the alkenylated products were converted to the pinacol esters using a one-pot procedure (see ESI†), allowing unambiguous characterisation of products.
Nitrile DGs have shown activity in C(sp2)–H acetoxylations, using a suitable oxidant to access a putative PdIV intermediate.5 Building upon this, meta selective C–H acetoxylations were successfully achieved using PhI(OAc)2 as oxidant. Using a 3-methylboronic acid MIDA-DG ester 14 model system, a DOE approach was again applied to optimise the reaction conditions (see ESI†). The scope of the reaction was then investigated using the same 3 × 3 grid to assess the effects of sterics and electronics on yield and regioselectivity (Fig. 3).
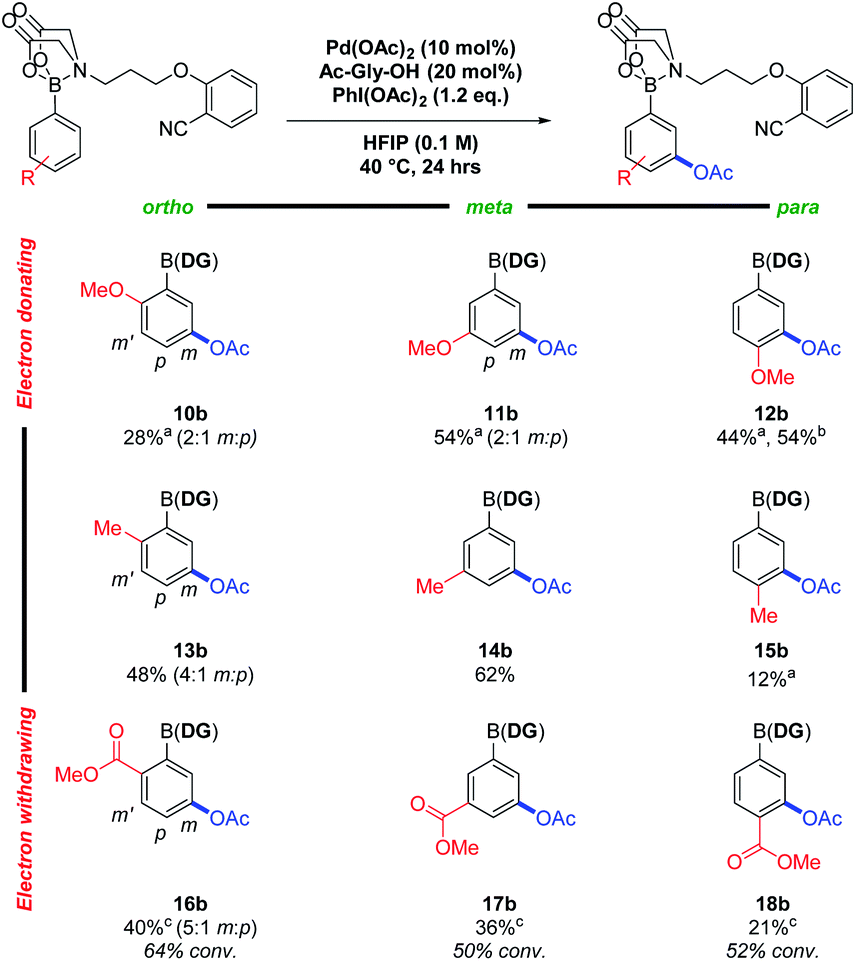 | ||
| Fig. 3 Scope of the C–H acetoxylation reaction. Reactions were performed on a 0.2 mmol scale, isolated yields. a Reactions performed at room temperature. b Reaction performed at room temperature, for 18 h, on 0.9 mmol scale. c Reaction time 48 h. | ||
Electron-donating groups (10b–12b) again resulted in increased degradation – lowering the reaction temperature from 40 °C to room temperature reduced this and led to an increase in yield. Increased sensitivity to steric bulk was observed relative to the alkenylation reactions particularly with methyl substituents (13b–15b), potentially due to the involvement of a sterically encumbered hexacoordinate PdIV intermediate. The rate was also reduced when using electron-withdrawing groups (16b–18b) and full conversions were not achieved. With the phenylboronic acid MIDA-DG ester 7, 70% yield (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 meta:para) was obtained (see ESI†).
:1 meta:para) was obtained (see ESI†).
The success of the MIDA-DG in achieving C–H alkenylations and acetoxylations, led us to perform a brief screen of C–H arylation conditions.16 Gratifyingly, the transformation was successful, allowing access to biphenyl product 24via the selective C–H coupling of one organoboron species in the presence of another (Scheme 3).
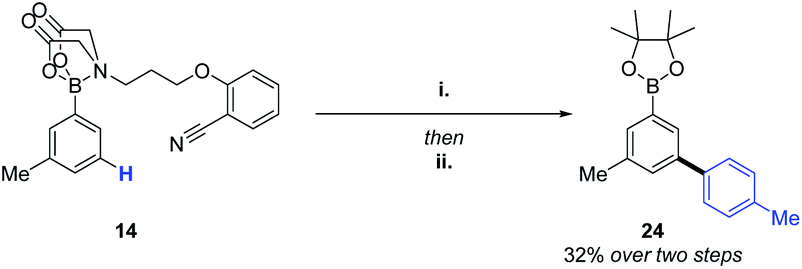 | ||
| Scheme 3 (i) Pd(OAc)2 (40 mol%), Ac-Gly-OH (40 mol%), 4-methylphenylboronic acid pinacol ester (2.0 eq.), Ag2CO3 (2.0 eq.), Cs2CO3 (2.0 eq.), HFIP (0.1 M), rt, 18 h, 52%. (ii) Pinacol (1.0 eq.), K3PO4 (3.0 eq.), H2O, THF, DCM, rt, 18 h, 62%. | ||
Removal of the DG is facile and can be performed using NaOH, or K3PO4, with quantitative recovery of the MIDA-DG 8 (Scheme 4, top).17
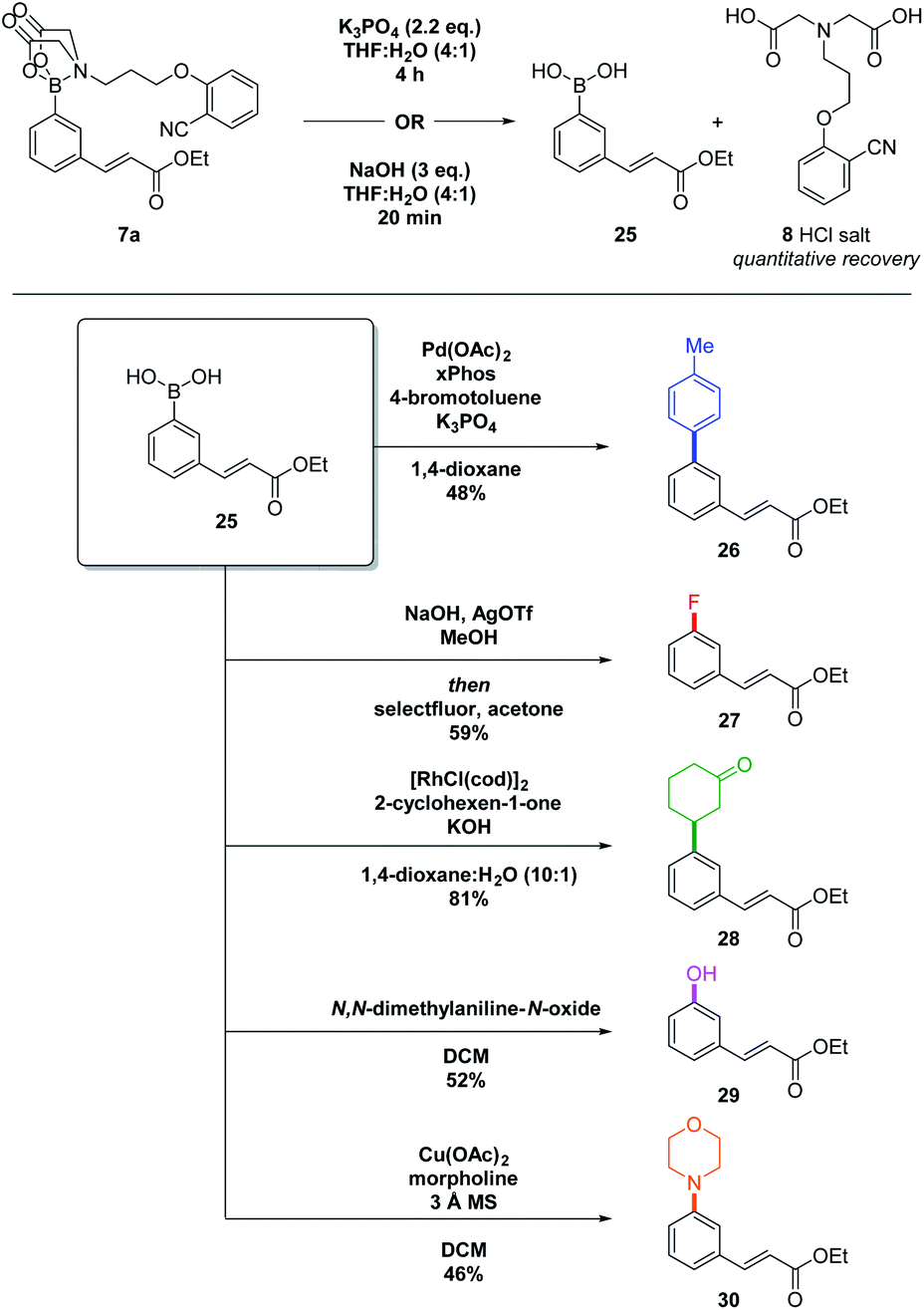 | ||
| Scheme 4 Deprotection and derivatisation of C–H functionalised aryl MIDA-DG boronate. The boronic acid was used directly after deprotection without purification. All yields given are over two steps. | ||
The deprotected boronic acid can subsequently be used in numerous transformations (Scheme 4). Suzuki–Miyaura (→26), Chan–Lam (→30) and Rh-catalysed conjugate addition reactions (→28) all proceeded well. A chemoselective oxidation using N,N-dimethylaniline-N-oxide (→29) and a fluorination reaction (→27) also achieved good yields over the two-steps. The telescoped deprotection–functionalisation procedure allows access to a broad range of motifs, including those that would be difficult to access via alternative methods.
Burke et al. were able to develop an automated iterative synthesis platform using the unusual properties of MIDA boronates on silica.12e Pleasingly, MIDA-DG boronates display similar properties, allowing for their facile purification using the ‘catch and release’ method. Reaction mixtures can be loaded directly onto silica and washed with Et2O to remove organic by-products, while the MIDA-DG-containing components remain ‘caught’. After washing, MIDA-DG boronates can be selectively ‘released’ using a more polar solvent.
In this manner, an iterative C–H functionalisation/cross-coupling procedure was envisaged, enabling access to poly-substituted aromatic systems, in just three catalytic steps using the ‘catch and release’ purifications (Scheme 5). To demonstrate the feasibility of this approach, the hetero-tetrasubstituted arene 31 was expediently accessed using this method. Importantly, the procedures appear to be amenable to automation using the platform developed by Burke.
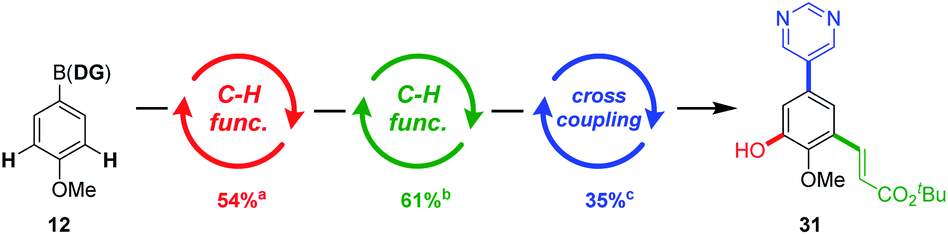 | ||
| Scheme 5 An iterative C–H functionalisation/cross-coupling procedure allows access to 1,3,5-trisubstituted aromatic systems. a Pd(OAc)2 (10 mol%), Ac-Gly-OH (20 mol%), PhI(OAc)2 (1.2 eq.), HFIP (0.1 M), rt, 18 h. b Pd(OAc)2 (20 mol%), Ac-Gly-OH (40 mol%), tert-butyl acrylate (2.5 eq.), AgOAc (1.75 eq.), HFIP (0.1 M), rt, 18 h. c Pd(OAc)2 (10 mol%), xPhos (20 mol%), 5-bromopyrimidine (1.0 eq.), K3PO4 (3.0 eq.), 1,4-dioxane:H2O (4:1), 75 °C, 18 h. | ||
Conclusions
In conclusion, a new MIDA derivative 8 has been developed which condenses to aryl boronic acids, enabling their meta-selective C–H functionalisation at room temperature. Installation of the DG and subsequent C–H functionalisation are operationally simple and use commercially available reagents. Removal of the DG is fast, occurs under mild conditions and provides access to synthetically useful aryl boronic acids with quantitative recovery of the MIDA-DG 8. Finally, the technique enables iterative C–H functionalisation/cross-coupling pathways and holds the potential for future automation. Studies to expand the breadth of transformations possible with the MIDA-DG 8 are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from Imperial College London, the EPSRC (PhD Studentship, A. F. W.), and the Royal Society for a University Research Fellowship (C. J. C.). We thank Dr Li-Jie Cheng for early prototype work, Alexander Dudnik for helpful discussions during the concept stage, Ben Deadman for assistance performing DOE optimisation and Peter Haycock for NMR assistance.Notes and references
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- For selected reviews on directed C–H functionalisation see: (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (b) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (c) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (d) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC; (e) G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391–4397 CrossRef CAS PubMed; (f) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- For selected reviews on meta selective C–H functionalisations see: (a) J. Yang, Org. Biomol. Chem., 2015, 13, 1930–1941 RSC; (b) C. G. Frost and A. J. Paterson, ACS Cent. Sci., 2015, 1, 418–419 CrossRef CAS PubMed; (c) A. Dey, S. Agasti and D. Maiti, Org. Biomol. Chem., 2016, 14, 5440–5453 RSC; (d) A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 10820–10843 CrossRef CAS PubMed. For recent examples of non-covalently bound meta-selective DGs see: (e) H. J. Davis, M. T. Madalina and R. J. Phipps, J. Am. Chem. Soc., 2016, 138, 12759–12762 CrossRef CAS PubMed; (f) H. J. Davis, G. R. Genov and R. J. Phipps, Angew. Chem., Int. Ed., 2017, 56, 13351–13355 CrossRef CAS PubMed; (g) Z. Zhang, K. Tanaka and J.-Q. Yu, Nature, 2017, 543, 538–542 CrossRef CAS PubMed.
- D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed.
- Y. Ping, L. Wang, Q. Ding and Y. Peng, Adv. Synth. Catal., 2017, 359, 3274–3291 CrossRef CAS.
- For nitrile based meta directing groups bound via carbonyl linkages see: (a) Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344–355 CrossRef CAS PubMed; (b) Y. Deng and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 888–891 CrossRef CAS PubMed; (c) R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215–220 CrossRef CAS PubMed; (d) M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760–5763 CrossRef CAS PubMed; (e) S. Li, H. Ji, L. Cai and G. Li, Chem. Sci., 2015, 6, 5595–5600 RSC; (f) S. Li, L. Cai, H. Ji, L. Yang and G. Li, Nat. Commun., 2016, 7, 10443–10450 CrossRef PubMed; (g) L. Fang, T. G. Saint-Denis, B. L. H. Taylor, S. Ahlquist, K. Hong, S. Liu, L. Han, K. N. Houk and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 10702–10714 CrossRef CAS PubMed; (h) S. Maity, E. Hoque, U. Dhawa and D. Maiti, Chem. Commun., 2016, 52, 14003–14006 RSC; (i) H.-J. Xu, Y. Lu, M. E. Farmer, H.-W. Wang, D. Zhao, Y.-S. Kang, W.-Y. Sun and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 2200–2203 CrossRef CAS PubMed. For nitrile based meta directing groups bound via silyl linkages see: (j) S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778–18781 CrossRef CAS PubMed; (k) T. Patra, R. Watile, S. Agasti, T. Naveen and D. Maiti, Chem. Commun., 2016, 52, 2027–2030 RSC; (l) R.-J. Mi, Y.-Z. Sun, J.-Y. Wang, J. Sun, Z. Xu and M.-D. Zhou, Org. Lett., 2018, 20, 5126–5129 CrossRef CAS PubMed. For nitrile based meta directing groups bound via sulfonyl linkages see: (m) G. Yang, P. Lindovska, D. Zhu, J. Kim, P. Wang, R.-Y. Tang, M. Movassaghi and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 10807–10813 CrossRef CAS PubMed; (n) G. Yang, D. Zhu, P. Wang, R.-Y. Tang and J.-Q. Yu, Chem.–Eur. J., 2018, 24, 3434–3438 CrossRef CAS PubMed; (o) M. Bera, A. Maji, S. K. Sahoo and D. Maiti, Angew. Chem., Int. Ed., 2015, 54, 8515–8519 CrossRef CAS PubMed; (p) A. Maji, B. Bhaskararao, S. Singha, R. B. Sunoj and D. Maiti, Chem. Sci., 2016, 7, 3147–3153 RSC; (q) A. Modak, A. Mondal, R. Watile, S. Mukherjee and D. Maiti, Chem. Commun., 2016, 52, 13916–13919 RSC; (r) L. Zhang, C. Zhao, Y. Liu, J. Xu, X. Xu and Z. Jin, Angew. Chem., Int. Ed., 2017, 56, 12245–12249 CrossRef CAS PubMed; (s) A. Modak, T. Patra, R. Chowdhury, S. Raul and D. Maiti, Organometallics, 2017, 36, 2418–2423 CrossRef CAS; (t) M. Bera, S. Agasti, R. Chowdhury, R. Mondal, D. Pal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 5272–5276 CrossRef CAS PubMed. For nitrile based meta directing groups bound via phosphonate linkages see: (u) M. Bera, S. K. Sahoo and D. Maiti, ACS Catal., 2016, 6, 3575–3579 CrossRef CAS. For nitrile based meta directing groups bound via ether linkages see: (v) H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567–7571 CrossRef CAS PubMed.
- D. G. Hall, Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine, John Wiley & Sons, Ltd, 1st edn, 2006 Search PubMed.
- J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31–55 CAS.
- B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2116–2119 CrossRef CAS PubMed.
- A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2013, 52, 7362–7370 CrossRef CAS PubMed.
- For Suginome's work on ortho directing groups for boronic acids see: (a) H. Ihara and M. Suginome, J. Am. Chem. Soc., 2009, 131, 7502–7503 CrossRef CAS PubMed; (b) H. Ihara, A. Ueda and M. Suginome, Chem. Lett., 2011, 40, 916–918 CrossRef CAS; (c) H. Ihara, M. Koyanagi and M. Suginome, Org. Lett., 2011, 13, 2662–2665 CrossRef CAS PubMed; (d) T. Yamamoto, A. Ishibashi and M. Suginome, Chem. Lett., 2017, 46, 1169–1172 CrossRef CAS; (e) T. Yamamoto, A. Ishibashi and M. Suginome, Org. Lett., 2017, 19, 886–889 CrossRef CAS PubMed.
- For highlights of Burke's work on MIDA boronates see: (a) E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716–6717 CrossRef CAS PubMed; (b) E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 14084–14085 CrossRef CAS PubMed; (c) D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961–6963 CrossRef CAS PubMed; (d) E. M. Woerly, J. Roy and M. D. Burke, Nat. Chem., 2014, 6, 484–491 CrossRef CAS PubMed; (e) J. Li, S. G. Ballmer, E. P. Gillis, S. Fujii, M. J. Schmidt, A. M. E. Palazzolo, J. W. Lehmann, G. F. Morehouse and M. D. Burke, Science, 2015, 347, 1221–1226 CrossRef CAS PubMed.
- J. Li and M. D. Burke, J. Am. Chem. Soc., 2011, 133, 13774–13777 CrossRef CAS PubMed.
- I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 88 CrossRef CAS.
- S. K. Sinha, T. Bhattacharya and D. Maiti, React. Chem. Eng., 2019, 4, 244–253 RSC.
- L. Wan, N. Dastbaravardeh, G. Li and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 18056–18059 CrossRef CAS PubMed.
- J. A. Gonzalez, O. M. Ogba, G. F. Morehouse, N. Rosson, K. N. Houk, A. G. Leach, P. H.-Y. Cheong, M. D. Burke and G. C. Lloyd-Jones, Nat. Chem., 2016, 8, 1067–1075 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1961105. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00230e |
| ‡ Present address: Grow Biotech, Rothamsted Enterprise, West Common, Harpenden, Hertfordshire, AL5 2JQ, UK, E-mail: chris.cordier@growbiotech.com. |
| This journal is © The Royal Society of Chemistry 2020 |