
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall molecule-mediated co-assembly of amyloid-β oligomers reduces neurotoxicity through promoting non-fibrillar aggregation†
Hao
Liu
a,
Chengyuan
Qian
a,
Tao
Yang
b,
Yanqing
Wang
d,
Jian
Luo
a,
Changli
Zhang
e,
Xiaohui
Wang
 *ac,
Xiaoyong
Wang
b and
Zijian
Guo
*c
*ac,
Xiaoyong
Wang
b and
Zijian
Guo
*c
aCollege of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing 211816, P. R. China. E-mail: wangxhui@njtech.edu.cn
bKey Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, Nanjing, 210093, P. R. China
cState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing, 210093, P. R. China. E-mail: zguo@nju.edu.cn
dInstitute of Environmental Toxicology and Environmental Ecology, Yancheng Teachers University, Yancheng 224007, P. R. China
eSchool of Environmental Science, Nanjing Xiaozhuang College, Nanjing 211171, P. R. China
First published on 22nd June 2020
Abstract
Amyloid-β (Aβ) oligomers, particularly low molecular weight (LMW) oligomers, rather than fibrils, contribute very significantly to the onset and progression of Alzheimer's Disease (AD). However, due to the inherent heterogeneity and metastability of oligomers, most of the conventional anti-oligomer therapies have indirectly modulated oligomers' toxicity through manipulating Aβ self-assembly to reduce oligomer levels, which are prone to suffering from the risk of regenerating toxic oligomers from the products of modulation. To circumvent this disadvantage, we demonstrate, for the first time, rational design of rigid pincer-like scaffold-based small molecules with blood–brain barrier permeability that specifically co-assemble with LMW Aβ oligomers through directly binding to the exposed hydrophobic regions of oligomers to form non-fibrillar, degradable, non-toxic co-aggregates. As a proof of concept, treatment with a europium complex (EC) in such a structural mode can rescue Aβ-mediated dysfunction in C. elegans models of AD in vivo. This small molecule-mediated oligomer co-assembly strategy offers an efficient approach for AD treatment.
Introduction
The ‘toxic Aβ oligomer’ hypothesis has attracted more and more attention as a way of overcoming the dilemma of amyloid-β (Aβ) plaque-directed therapeutics for Alzheimer's Disease (AD), in which all phase III clinical trials have failed, even though the levels of Aβ plaques in the brain can be significantly reduced.1,2 The hypothesis asserts that Aβ oligomers rather than fibrils might be the central player in AD pathogenesis and have been recognized as the most toxic species with strong correlation with disease symptoms.3,4 Importantly, the oligomeric species can be produced in the earliest stages of AD and even appear as early as ∼10–15 years before the clinical symptoms.5 Therefore, Aβ oligomers seem to be the most promising targets for disease-modifying therapies of AD particularly at the early presymptomatic stage. However, the oligomers are heterogeneous and metastable in nature, which has seriously restricted the development of anti-oligomer therapies. To date, the majority of therapeutic approaches have focused on indirect modulation of oligomers' toxicity through immunotherapy6 and manipulating Aβ self-assembly,7,8 including inhibition of oligomerization,9,10 acceleration of fibril formation,11,12 redirection of Aβ into off-pathway aggregates,13–16 and disaggregation of oligomers.17 However, the generated Aβ monomers or fibrils in such treatments might function as reservoirs for oligomers, which probably lead to recurring neurotoxicity.Multicomponent co-assembly of peptides is an emerging supramolecular strategy, which presents an opportunity to irreversibly alter the properties of the self-assemblies of individual peptides via co-assembly.18 Considering the causative link between the structure of oligomers and their toxicity,19,20 this strategy would be effective to reduce oligomers' toxicity through forming co-aggregates that are structurally distinct from Aβ self-aggregates. Recently, a few polymers and peptides have plausibly succeeded in co-assembling with Aβ into non-toxic co-aggregates, depending on their abundant binding sites for protein interactions.21–27 However, the deficient oligomer specificity limits their inhibitory effect on oligomers' toxicity, which hinders their further utilization in vivo. Alternatively, only very few small molecules have exhibited high specificity for oligomers derived from Aβ peptide.28–31 Nevertheless, none have enabled co-assembly with Aβ oligomers to form non-toxic co-aggregates, probably due to the lack of principle-based rational design of multicomponent co-assemblies of peptides. Despite being challenging, small molecule-mediated co-assembly with Aβ oligomers would be reasonably attainable to diminish oligomer-mediated toxicity, given the many advantages of small molecules, such as cheap and facile preparation, controllable oligomer specificity, tunable biocompatibility and blood–brain barrier (BBB) permeability.8
Here we propose a rationally designed small molecule-mediated Aβ co-assembly that co-assembles Aβ oligomers and small molecular probes into non-toxic co-aggregates based on intermolecular interactions. Under the guidance of this concept, a pincer-like europium(III) complex (EC) with rigid configuration was constructed as a representative (Fig. 1A). The EC shows excellent oligomer specificity through binding to the exposed hydrophobic regions of oligomers as the oligomer specific binding sites that are responsible for both toxicity and aggregation propensity of oligomers. Based on the specific recognition and directional interactions, the EC is able to accelerate and promote Aβ aggregation into non-fibrillar EC–Aβ co-aggregates, which are almost non-toxic in neuroblastoma cells and degradable in microglial cells through upregulating autophagy upon phagocytosis. Strikingly, the EC can also ameliorate Aβ-mediated toxicity in Caenorhabditis elegans (C. elegans) models of AD through co-assembly in vivo. Our work for the first time establishes a small molecule-mediated Aβ oligomer co-assembly that can irreversibly diminish oligomers' toxicity as a potential therapeutic approach for AD treatment.
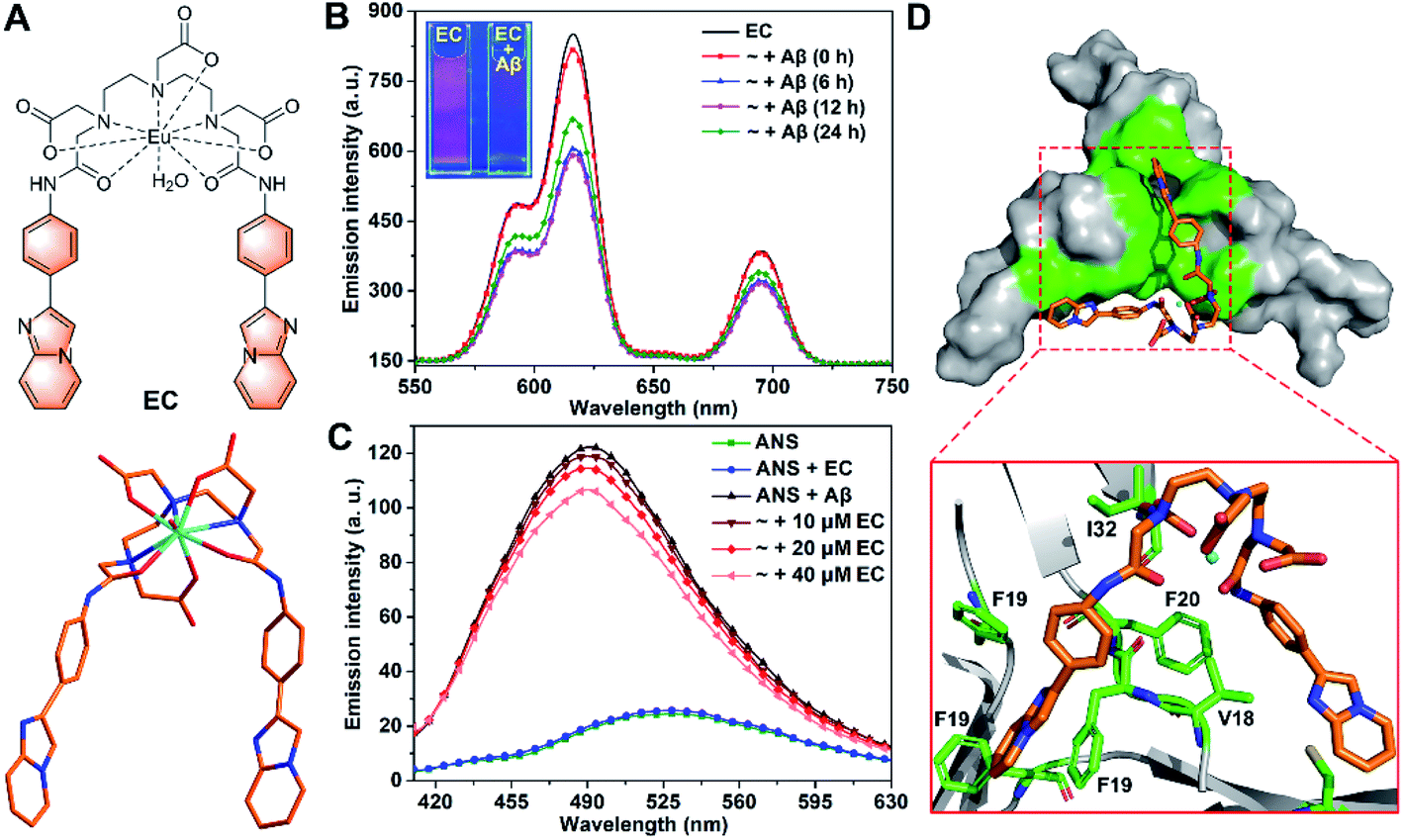 | ||
| Fig. 1 (A) Structural formula of the EC (top) and optimized structure of the EC at the B3LYP/6-31G* level (bottom). (B) Time-resolved luminescence spectra of the EC (20 μM, λex = 300 nm) in the absence and presence of Aβ40 species (20 μM) with different pre-incubation times (0, 6, 12, and 24 h) in buffer (20 mM Tris–HCl, 150 mM NaCl, 4‰ v/v DMSO, pH 7.4). Inset shows the luminescence images of EC solutions with or without Aβ oligomers under a UV lamp at 254 nm. (C) Fluorescence spectra of ANS (20 μM, λex = 380 nm) in the absence and presence of Aβ40 oligomers (20 μM) with or without 10, 20, and 40 μM EC in buffer (20 mM Tris–HCl, 150 mM NaCl, 4‰ v/v DMSO, pH 7.4). (D) Lowest energy docking model of the EC with Aβ trimer (PDB ID: 5HOX) in surface (top) and cartoon (bottom) representations. The amino acid residues interacting with the EC are highlighted in green. | ||
Results and discussion
Design of the EC with oligomer specificity
Both specific recognition and directional interactions are important parameters for small molecules to achieve co-assembly with peptides.32 First, given that the exposure of hydrophobic regions to solvents has been recognized as a unique structural feature of Aβ oligomers in contrast to other counterparts, the specific recognition of small molecules for oligomers can be identified through testing their binding affinity to such solvent-exposed hydrophobic patches.28,33 6-Iodo-2-(4-dimethylamino)phenylimidazo[1,2-a]pyridine (IMPY), a single-photon emission computed tomography imaging probe for amyloid plaques,34 has exhibited favorable pharmacokinetic properties with good BBB permeability in vivo.35 Inspired by these findings, an aromatic 4-(imidazo[1,2-a]pyridin-2-yl)aniline (IPA) group was selected as the targeting group in view of the binding affinity of its scaffold to the central hydrophobic regions of Aβ. On the other hand, rigid configuration would be necessary for the small molecules to consolidate the directional interactions with mutable Aβ oligomer and to stabilize the co-assembled structure. For this purpose, a diethylenetriaminepentaacetate (DTPA)-based Eu(III) chelating center was chosen as a bridge to link two IPA groups to form a rigid pincer-like complex, the EC (Fig. 1A). The chelating center with multiple coordinate bonds can endow the EC with high stability and rigidity. The optimized structure of the EC based on quantum mechanical calculations at the B3LYP/6-31G* level shows that two IPA groups as pincer arms are firmly restricted by the chelating center (Fig. 1A). Another reason for the selection of the Eu(III) center is that the luminescence of Eu(III) can be used to verify the oligomer specificity of the EC. Finally, the Aβ toxicity could be significantly suppressed by the proposed co-assembly system because of the coverage of the hydrophobic motifs of oligomers.33,36 The synthetic route and characterization of the EC are described in Scheme S1 and Fig. S1 (ESI).† Because Lipinski's rules do not necessarily apply to metallodrugs, the BBB permeability of the EC was evaluated from the quantity of Eu in the mouse brain by inductively coupled plasma mass spectrometry (ICP-MS) after i.v. injection. The amount of Eu rapidly increased to its peak level at 10 min after dosing, followed by a relatively slow decrease (Fig. S2†), suggesting that the EC is able to cross the BBB.Next, the luminescence responses of the EC for Aβ species were tested, which would provide useful evidence for the selectivity of the EC for oligomers. Different Aβ aggregates were prepared via pre-incubation for corresponding times according to the aggregation states recognized by the growth kinetics curve in the Thioflavin T (ThT) fluorescence assay using the dye ThT as a fluorescence probe for fibrils (Fig. S3†).37,38 Similar excitation and absorption spectra of the EC consolidate the energy transfer from IPA groups as antennae to Eu3+ ions (Fig. S4†).39 Consequently, three characteristic emission bands of europium were observed at 592, 615, and 695 nm upon excitation of the EC at 300 nm by time-resolved luminescence measurements, which can be assigned to the transition from the 5D0 excited state to the 7F1, 7F2, and 7F4 ground states of Eu3+, respectively (Fig. 1B).40 The emission intensity of the EC remarkably decreased in the presence of abundant low molecular weight (LMW) oligomers with pre-incubation for 6 and 12 h, while the luminescence of the EC partially recovered in the presence of Aβ species with pre-incubation for 24 h, in which the amount of LMW oligomers decreased with conversion into high molecular weight (HMW) oligomers and protofibrils. In contrast, a slight decrease of EC luminescence was observed in the presence of Aβ without any pre-incubation (0 h), which contains few LMW oligomers (vide infra). The results indicate that the luminescence response of the EC depends on the amount of LMW oligomers. Interestingly, such a luminescence change of the EC was clearly visible under a UV lamp (254 nm), where bright red emission was almost quenched by LMW oligomers (inset of Fig. 1B). Furthermore, the stronger luminescence of the EC in D2O than in H2O both in the presence and absence of Aβ oligomers indicates that the coordinated water still binds to Eu3+ (Fig. S5†),39,41 thereby excluding the possibility of coordination between Eu3+ and Aβ. In the presence of LMW oligomers, non-covalent interactions between IPA groups and hydrophobic regions of Aβ may take place to favor the photoelectron transfer from the amino acid residues to the IPA groups,42 thereby preventing the energy transfer from the antenna to Eu3+ and eventually quenching EC luminescence. Therefore, IPA-involved high affinity binding to LMW oligomers would dominantly contribute to the selectivity of the EC for LMW oligomers.
To validate the binding mode of the EC with Aβ oligomers, an 8-anilinonaphthalene-1-sulfonate (ANS) fluorescence competition assay was carried out. ANS has been shown to bind to solvent-exposed hydrophobic patches of Aβ oligomers with remarkable fluorescence enhancement and a blue shift of its maximum emission wavelength.19 As shown in Fig. 1C, the oligomer-induced strong fluorescence of ANS decreased with addition of an increasing amount of the EC, indicating that the EC may displace ANS to bind with the hydrophobic regions of oligomers, because the EC per se hardly affects the fluorescence of ANS. To further understand the structural features of the EC for the selective binding to LMW Aβ oligomers, docking studies were performed using a well-known Aβ17-36 trimer as a working model (PDB ID: 5HOX).43 In the trimer, a large hydrophobic surface consists of hydrophobic residues L17, F19, and V36 of three β-hairpins packing against the residues A21, I32, and L34 of the adjacent chains, which should be the oligomer specific binding motif, although it may not completely represent the structure of full-length Aβ oligomers. In the lowest energy conformation of the adduct, the EC adopting a slightly twisted geometry is positioned in the hydrophobic domain mainly through π–π stacking interactions between the planar IPA groups and the side chains of F19 residues (Fig. 1D), while the DTPA group also forms intermolecular CH⋯O interaction with the I32 side chain upon binding. Moreover, the EC displayed the lowest binding free energy (ΔG = −43.39 kJ mol−1) and inhibition constant (Ki = 24.85 nM) for the trimer than for both the monomer and fibrils through thermodynamics analysis (Table S1†), indicating the specificity of the EC for the oligomeric conformation of Aβ, which could be dominantly attributed to the pincer-like configuration decorated with hydrophobic motif-binding ligands.
The EC dramatically promotes Aβ aggregation
On the basis of the LMW oligomer specificity of the EC, we next investigated the effect of the EC on Aβ aggregation. In this work, the ThT fluorescence assay is unable to monitor the aggregation of Aβ in the presence of the EC, since the EC can significantly enhance the ThT emission intensity with a red shift of the emission wavelength in both the presence and absence of Aβ (Fig. S6†). Alternatively, because there is no absorption of the EC itself at 405 nm (Fig. S4†), the turbidity assay was employed to measure the absorbance/turbidity at 405 nm (A405) of the Aβ solution, which can reflect the degree of Aβ aggregation.44,45 Moreover, the time course of Aβ40 self-aggregation by the turbidity assay was similar to that of the ThT fluorescence assay, adopting a sigmoidal mode that consists of a lag phase, an elongation phase, and a plateau phase, as reported in previous studies (Fig. S3†).37,38 Accordingly, the time-dependent turbidities of the Aβ40 solution in the presence of different concentrations of the EC were determined. As shown in Fig. 2A, the resulting kinetic absorption curves were obtained from data fitting with a sigmoidal equation (eqn (S1)†),46 while the time at half completion of the aggregation process (t50) and the elongation rate constant (k) were calculated (Table S2†). Compared with Aβ40 self-aggregation, the EC induced a slight increase of turbidity of the solution by ∼2-fold and a minor change in the values of t50 and k after incubation at concentrations of 5 and 10 μM. Noticeably, significant enhancement of turbidity with a relatively short t50 (4.94 h) and fast aggregation rate (k = 0.66 h−1) was observed after addition of 20 μM EC. Moreover, 40 μM EC had a similar effect on the turbidity with 20 μM EC, but longer t50 (10.95) and slower aggregation rate (k = 0.44 h−1). These results clearly indicate that the EC can not only accelerate the aggregation of Aβ40 through shortening the lag phase and elongation rate, but also boost the level of Aβ40 aggregates. Besides Aβ40, the effect of the EC on Aβ42 aggregation was also measured by the turbidity assay (Fig. 2B). Due to the additional two C-terminal residues, Aβ42 can enter into the plateau phase within a short time more quickly than Aβ40. The EC showed a weaker promotive effect on the aggregation of Aβ42 than of Aβ40, probably owing to the ultra-fast self-aggregation kinetics of Aβ42. Nevertheless, the EC-induced acceleration at the early stage of Aβ42 aggregation can be obviously observed, similar to that of Aβ40. Moreover, as the potential binding site of the EC, the central hydrophobic stretches are responsible for the aggregation in both Aβ40 and Aβ42.47 Thus, it is suggested that the EC may interact with Aβ42 in a similar mechanism to that with Aβ40. Accordingly, Aβ40 was used in most of the experiments in this work and is cheaper and seems more favorable for clarifying the mechanism of action of the EC under our experimental conditions.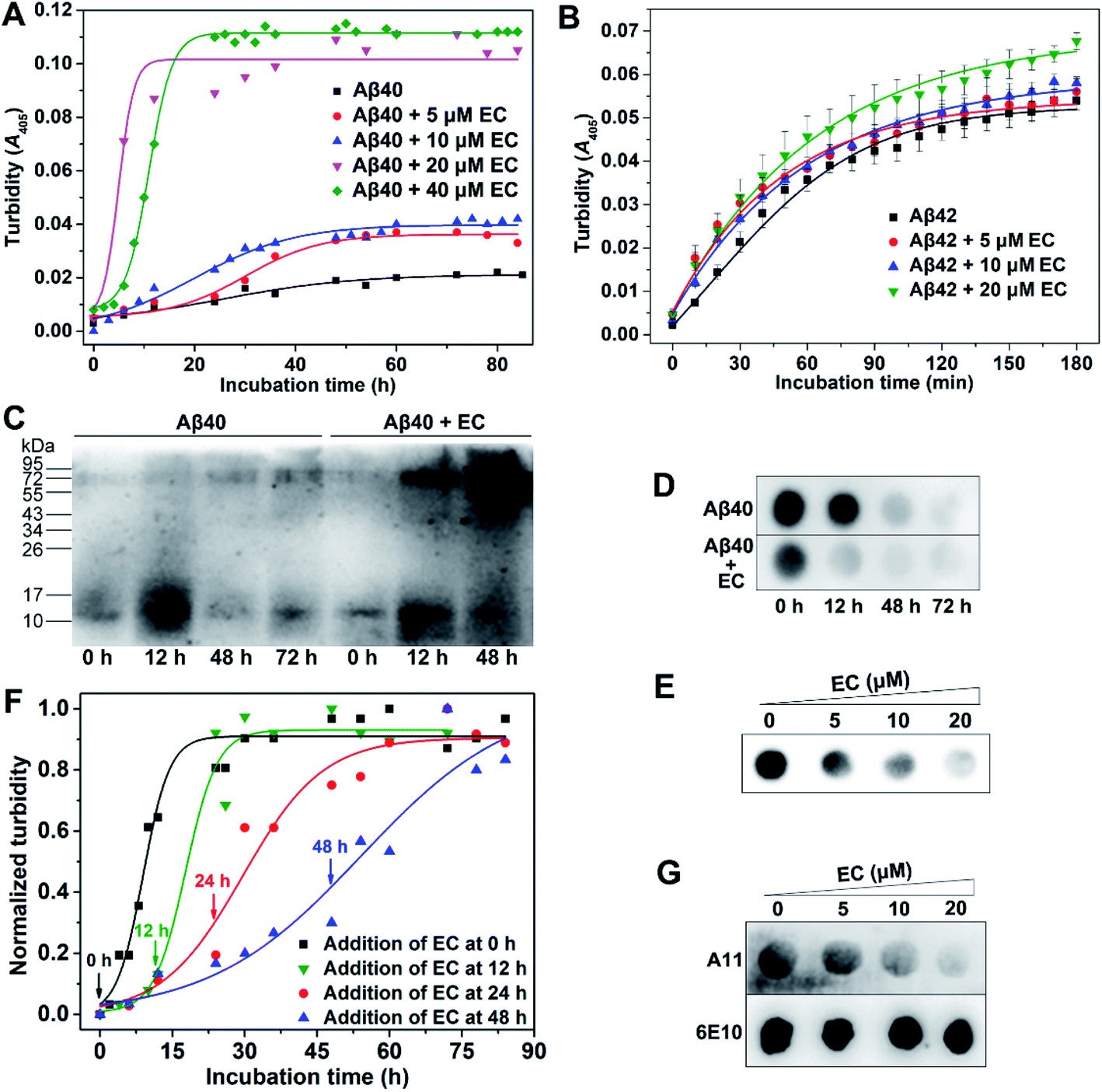 | ||
| Fig. 2 (A) Representative profile of the aggregation kinetics of Aβ40 (20 μM) in the presence of 0, 5, 10, 20, or 40 μM EC in buffer (20 mM Tris–HCl, 150 mM NaCl, 4‰ v/v DMSO, pH 7.4) at 37 °C by the turbidity assay. (B) The aggregation kinetics of Aβ42 (10 μM) in the presence of 0, 5, 10, or 20 μM EC in buffer (20 mM Tris–HCl, 150 mM NaCl, 4‰ v/v DMSO, pH 7.4) at 37 °C by the turbidity assay. Error bars indicate ±s.d. (n = 5 independent experiments). (C) Western blot analysis of the time-dependent effect of the EC (20 μM) on Aβ40 (20 μM) aggregation using anti-Aβ antibody 6E10. (D) Dot blot analysis of the time-dependent effect of the EC (20 μM) on soluble Aβ40 species using 6E10 antibody. (E) Dot blot analysis of the concentration-dependent effect of the EC (20 μM) on soluble Aβ40 species after incubation for 24 h using 6E10 antibody. (F) Representative profile of turbidity-monitored aggregation kinetics of Aβ40 (20 μM) when the EC was added at different time points indicated by the arrows. (G) Detection of Aβ oligomers by dot blot probed with oligomer-specific antibody A11. Total Aβ40 levels were detected by 6E10 antibody. | ||
To confirm the promotion of the EC on Aβ aggregation, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by western blotting using anti-Aβ antibody 6E10 was employed in a time-dependent manner. As shown in Fig. 2C, in the absence of the EC, few LMW oligomers, notably dimers and trimers (10–17 kDa), were observed at the start of Aβ aggregation, accompanied by a remarkable increase at the early stage of the lag phase within 12 h due to the propensity for Aβ self-aggregation, which obviously decreased after 48 h of incubation in the elongation phase. Meanwhile, the larger aggregates (>70 kDa) of Aβ increased. In contrast, a higher amount of large aggregates was observed in the presence of the EC (20 μM) as early as 12 h, whereas the LMW oligomers almost disappeared at 48 h. This result is consistent with those of turbidity assays. A similar result was also obtained from the western blot analysis of the concentration-dependent effect of the EC on Aβ aggregation (Fig. S7†). Dot blot assays using 6E10 were further performed. The total amounts of Aβ in whole solutions for different incubation times are almost identical (Fig. S8†). In contrast, the soluble Aβ species in the supernatant of the incubating solutions dramatically decreased at 12 h in the presence of the EC (20 μM), compared with that of the Aβ self-aggregation solution (Fig. 2D), indicating that the EC induced more aggressive aggregation of Aβ into insoluble species. Identical to the results of turbidity and western blot assays, the EC exhibited the most efficient promotion of Aβ aggregation at 20 μM with the lowest level of soluble Aβ aggregates in the supernatant (Fig. 2E).
The EC preferentially targets soluble LMW Aβ oligomers
We next examined whether the EC targets oligomers to promote Aβ aggregation. First, the promotion effect of the EC on Aβ aggregation upon addition of the complex at different time points after the start of Aβ aggregation was measured. As shown in Fig. 2F, the EC exhibited similar acceleratory efficacy when added at initiation (0 h) and in the middle of the lag phase (12 h) of Aβ aggregation. In contrast, the efficacy gradually decreased when added at the late stage of the lag phase (24 h) and elongation phase (48 h) of Aβ aggregation, which was confirmed by t50, k, and the lag time (tlag) values (Table S3†). LMW oligomers already existed at the start of Aβ aggregation and obviously increased within 12 h (Fig. 1B and 2C). Instead, HMW oligomers and insoluble large aggregates including protofibrils and fibrils can be recognized as the dominant constituents at 24 h and 48 h of Aβ aggregation, respectively (Fig. 2C and S3†). Given its binding selectivity, it is reasonable that the EC can still preferentially target LMW oligomers in the dynamic process of Aβ aggregation. As a consequence, the lag phase of Aβ aggregation was significantly shortened with reduced tlag, thereby accelerating the Aβ aggregation.The binding behavior of the EC with oligomers during Aβ aggregation was further confirmed by the dot blot assay using Aβ oligomer-specific antibody A11 that selectively recognizes soluble Aβ oligomers.48 6E10 was also used to verify equal sample loading. As shown in Fig. 2G, A11-sensitive oligomers gradually decreased with the increasing concentration of the EC and were almost diminished in the presence of 20 μM EC upon incubation for 24 h. In contrast, no change in the signal intensity was observed for the samples stained with 6E10. These results support the view that the EC can target oligomers and promote the conversion of oligomers into large conformers, which cannot be recognized by the A11 antibody.
The EC rapidly converts oligomers into non-fibrillar aggregates
To further study the morphology of EC-induced Aβ aggregates, transmission electron microscopy (TEM) analysis was carried out. The images of Aβ samples with or without the EC after incubation for 24 h at 37 °C are shown in Fig. 3A. Consistent with the result of ThT-monitored aggregation kinetics (Fig. S3†), short and slender string-like aggregates including HMW oligomers and protofibrils were seen in the solution of Aβ40 in the absence of the EC, which gradually decoupled in the samples with an increasing concentration of the EC. Instead, more and more amorphous aggregates can be observed after incubating Aβ40 with the EC. While 20 μM EC totally remodeled the fibrillary species into massive smear-like disordered aggregates. Similar morphological changes can be obtained upon co-incubation of the EC with fresh Aβ or 12 h-incubated Aβ for 72 or 60 h, respectively (Fig. S9†). The effect of the EC (20 μM) on the size distribution of Aβ40 was further tested by dynamic light scattering (DLS). Both samples with and without the EC were pre-incubated to their plateau phases in order to obtain the EC-generated aggregates and mature fibrils as dominant species, respectively. As shown in Fig. 3B, the mean hydrodynamic radius of the EC-treated Aβ sample was around 380 nm, which was much smaller than that of Aβ fibrils (∼600 nm). These observations suggest that the EC not only promotes Aβ aggregation but also converts LMW oligomers into non-fibrillar aggregates instead of fibrils.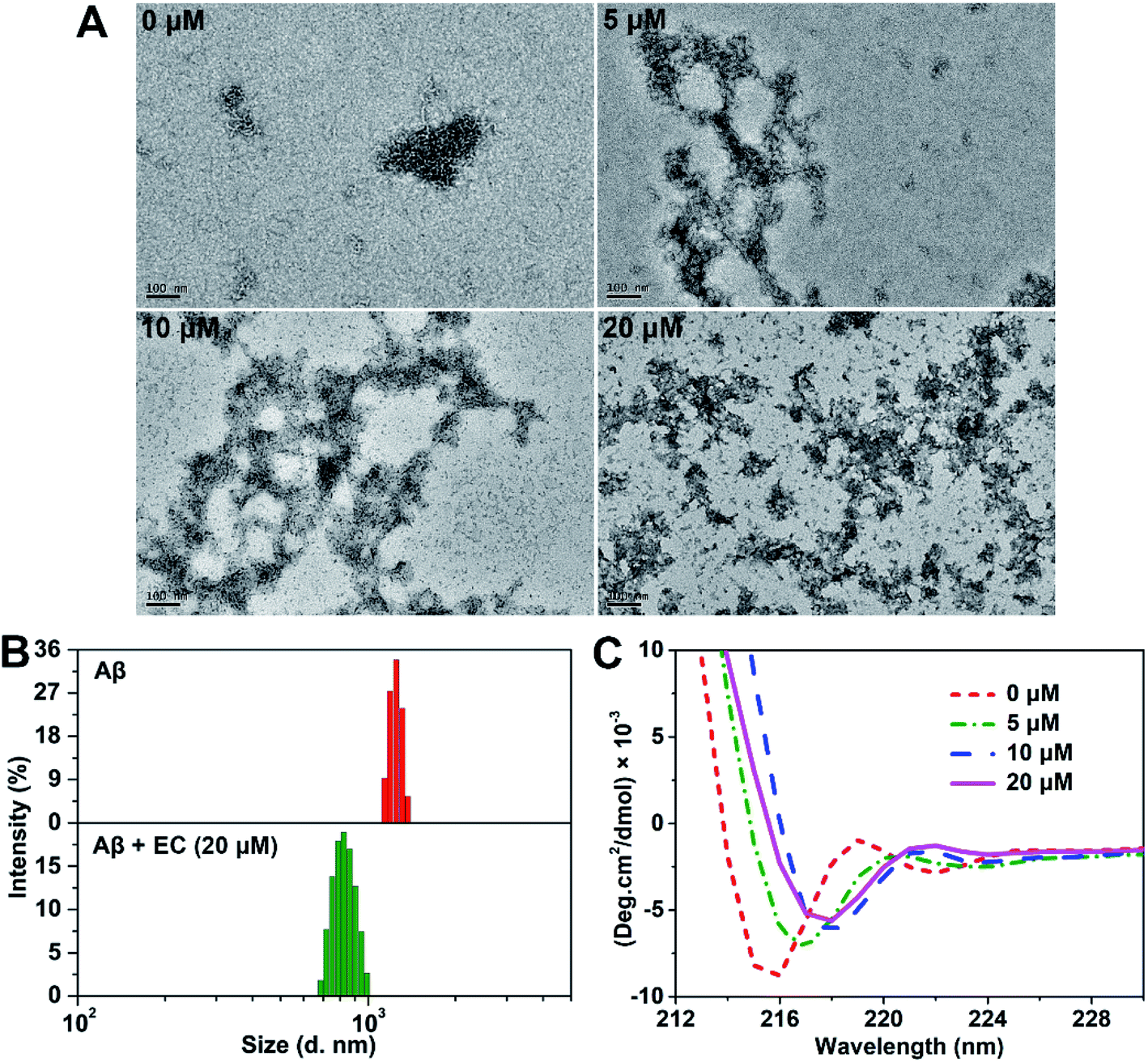 | ||
| Fig. 3 (A) Morphological analysis of Aβ40 aggregates in the presence of 0, 5, 10, or 20 μM EC by TEM (scale bar = 100 nm). (B) DLS analysis of Aβ40 fibrils and EC-induced Aβ aggregates. (C) CD spectra of Aβ40 (20 μM) in the absence or presence of different concentrations of the EC (20 μM) after incubation for 24 h. All samples were incubated in a buffer (20 mM Tris–HCl, 150 mM NaCl, 4‰ v/v DMSO, pH 7.4) at 37 °C. | ||
Circular dichroism (CD) spectroscopy was used to evaluate the influence of EC-induced conversion of Aβ aggregates on the characteristic β-sheet conformation.49 The negative band (∼195 nm) of the random coil structure for the 0 h sample did not appear, probably resulting from the influence of the formed LMW oligomers under our experimental conditions during data collection. The typical β-sheet structure characterized by one single minimum at ∼216 nm,50 however, was observed after incubation of Aβ for 12 h, which would belong to the oligomers produced in the middle of the lag phase. A significant decrease of the β-sheet structure can be seen in the presence of the EC (20 μM) after 12 h of co-incubation (Fig. S10†). Moreover, EC treatment can also reduce the negative band (∼216 nm) with red shifts in a concentration-dependent manner (Fig. 3C), which was substantially consistent with the results of the above EC-promoted Aβ aggregation experiments. Taking into account all the observations, we deduce that EC-mediated promotion of Aβ aggregation can lead to the formation of non-fibrillar aggregates with weak β-sheet conformation, which are structurally distinct from Aβ fibrils.
Molecular mechanism of EC-mediated Aβ co-aggregation
We then investigated the microscopic molecular mechanism of EC-mediated promotion of Aβ aggregation. A growing body of kinetic research has revealed that Aβ aggregation is a supramolecular self-assembly process undergoing multiple mechanisms substantially divided into three categories: nucleation and fragmentation processes, growth processes, and dissociation processes.51,52 In comparison, the nucleation and fragmentation processes are determinative factors for the aggregation levels and rate, which can be classified into primary pathways and secondary pathways based on their dependencies on the concentrations of free monomers and of existing aggregates, respectively.53 The addition of preformed seeds can accelerate Aβ aggregation via secondary nucleation pathways.9,12,13,25 Accordingly, a pre-seeded assay was performed through monitoring turbidity. As shown in Fig. 4A, addition of preformed fibers as seeds (10%, v/v) to an unseeded Aβ40 solution accelerated the aggregation by shortening the t50 from 5.40 ± 0.82 to 3.80 ± 0.47 h, confirming the dominant role of the secondary process in Aβ40 self-fibrillation. Since the EC can promote Aβ aggregation to form non-fibrillar aggregates through targeting LMW oligomers, EC-treated samples are presumed unable to seed the fibrillization of unfolded monomers. However, to our surprise, EC-induced non-fibrillar aggregates can also promote Aβ aggregation to an even higher extent than EC-untreated seeds, yielding a lower t50 of 2.35 ± 0.16 h. The result implies that EC-promoted Aβ aggregation may also mainly adopt a secondary nucleation pathway for formation of non-fibrillar aggregates. To verify this speculation, aggregation kinetics depending on the initial Aβ40 concentration with or without the EC were further investigated (Fig. 4B and S11†). The initial concentration dependence of t50 can be described by the scaling exponent, γ, from the power law, t50 ∼ [Aβ]γ, which is commonly used to predict the dominant nucleation mechanism.54,55 In this regard, a completely monomer-independent secondary nucleation pathway is characterized by a value of γ = −0.5, whereas a fully monomer-dependent process yields an exponent of γ = −1.5.38,51 A linear relationship was obtained when the power law was plotted as a double logarithmic plot, in which the slope is equal to γ (inset of Fig. 4B). In the case of Aβ40 itself with a concentration interval of 10–25 μM, the scaling exponent was found to be −0.80, suggesting that the nucleation process would depend on both monomers and aggregates, which is indicative of surface-catalyzed secondary nucleation on existing aggregates.53,56 In contrast, the presence of the EC resulted in an exponent of γ = −0.49, strongly implying the saturated secondary nucleation mechanism in accordance with the poor dependence on monomers. The results suggest that the assembly of EC-involved hetero-oligomers may dominate the further aggregation of Aβ.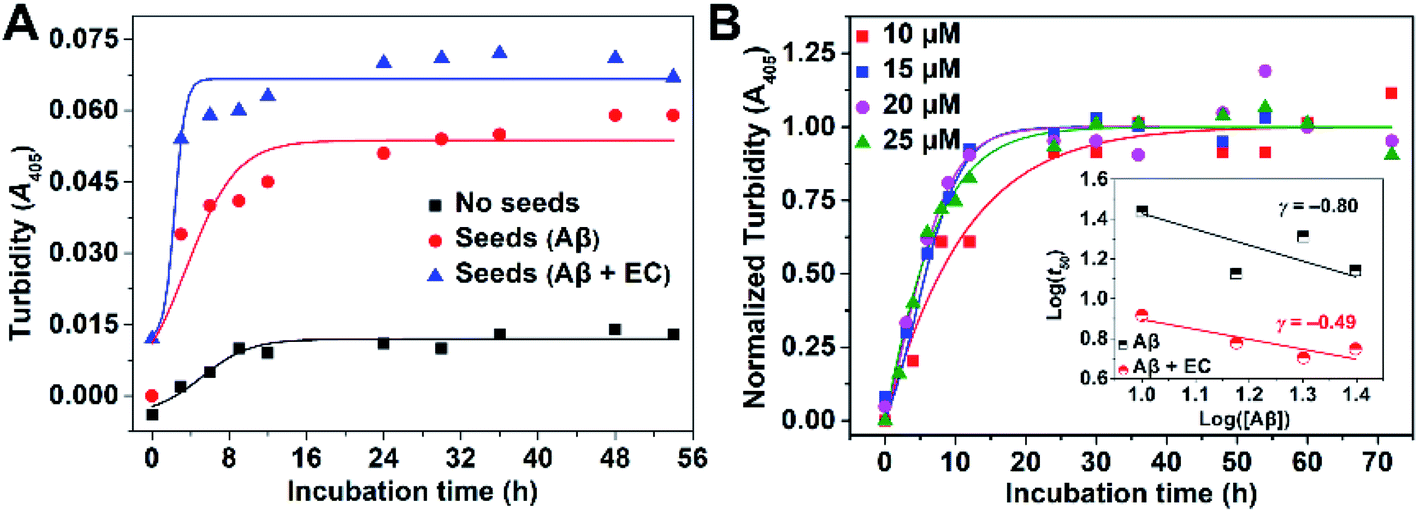 | ||
| Fig. 4 (A) Representative profile of seed-catalyzed aggregation kinetics of Aβ40 (20 μM) in the absence and presence of preformed seeds (10%, v/v) with or without the EC by the turbidity assay. (B) Representative profile of turbidity-monitored aggregation kinetics of Aβ40 with different initial concentrations (10, 15, 20, or 25 μM) in buffer (20 mM Tris–HCl, 150 mM NaCl, 4‰ v/v DMSO, pH 7.4) at 37 °C. The inset shows the logarithm of the time at half completion of the aggregation (t50) as a function of logarithm of initial Aβ concentration. The scaling exponent can be obtained by linear fitting. | ||
To gain deeper insight into EC-mediated assembly processes of Aβ, we performed computational simulations of the binding of the EC with two Aβ trimers via molecular docking. As shown in Fig. S12 (ESI),† the EC is anchored in the middle of two trimers by noncovalent interactions with the hydrophobic domain of the trimers, including π–π stacking interactions between IPA groups and the side chains of F19 residues as well as intermolecular CH⋯O interactions between DTPA group and L17, L34, and V36. In addition, lower ΔG (−45.98 kJ mol−1) and Ki (8.75 nM) can be obtained for this triadduct than for the above-mentioned biadduct of the EC with Aβ trimer (Table S1†). The results imply that the EC may be able to co-assemble with the Aβ trimer in alternate binding mode. The rigid scaffold of the EC may play a critical role in maintaining the stability of EC–Aβ co-aggregates, which was verified by the effect of the ligand of the EC, L-3H, on Aβ aggregation. The result demonstrated that L-3H with a flexible configuration can hardly influence Aβ aggregation (Fig. S13†), suggesting the indispensability of the rigidity of small molecules for co-assembly with Aβ oligomers.
Taken together, we proposed a mechanistic model as depicted in Fig. 5 for EC-mediated co-assembly of LMW oligomers. The π–π stacking interactions and hydrogen bonding between the EC and the exposed hydrophobic motifs of oligomers trigger the bicomponent co-assembly in the alternate binding mode to disrupt Aβ self-assembly and rapidly convert Aβ into EC-containing non-fibrillar architectures. Given the seed-like feature of the peculiar co-aggregates, the secondary nucleation mechanism may govern the co-assembly, that is, the preformed co-aggregates can act as seeds to catalyze formation of secondary EC-involved hetero-oligomers, ultimately promoting Aβ aggregation with greater degrees than Aβ self-assembly through accelerating secondary nucleation and elongation processes.
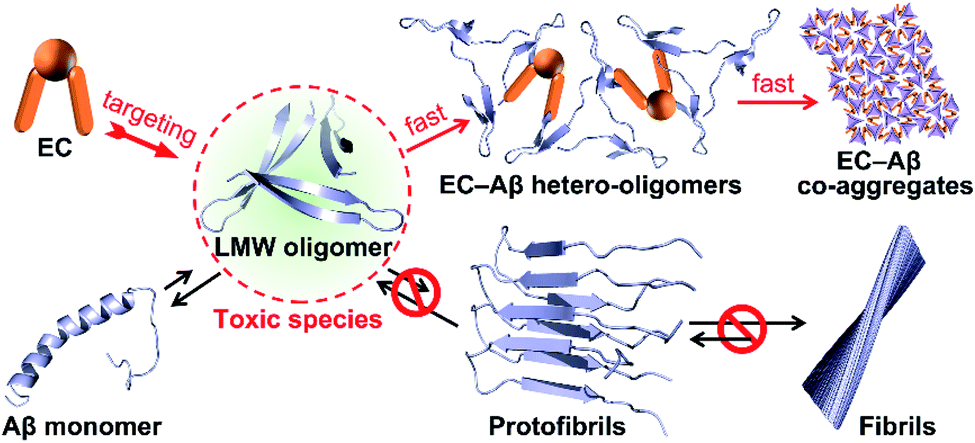 | ||
| Fig. 5 Proposed mechanism of EC-mediated co-assembly of Aβ oligomers. The co-assembly blocks Aβ self-aggregation and redirects Aβ fibrils into non-fibrillar EC–Aβ co-aggregates. | ||
EC-mediated co-assembly of Aβ oligomers reduces cellular toxicity
To evaluate whether EC-mediated co-assembly of Aβ oligomers influences Aβ-induced toxicity, 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed to determine the cell viability upon co-incubation of SH-SY5Y neuroblastoma cells with Aβ or/and different concentrations of the EC. More than 88% cell survival was still observed for cells treated with an EC concentration of 40 μM (Fig. 6A), indicating the minimal toxicity of the compound per se under such conditions. In contrast, the incubation of cells with Aβ oligomers resulted in an obvious reduction of cell viability to ca. 69%. The neurotoxicity, however, was significantly suppressed in the presence of the EC in a dose-dependent manner (Fig. 6B), which is positively related to the capacity of the EC for promoting Aβ aggregation at different concentrations. More than 96% cell survival can be observed for cells treated with up to 20 μM EC (Fig. 6B, **P < 0.01), suggesting that the co-assembly with coverage of hydrophobic regions of oligomers by the EC can form EC–Aβ co-aggregates with subtle toxicity, thereby diminishing the oligomers' toxicity in the cell culture medium.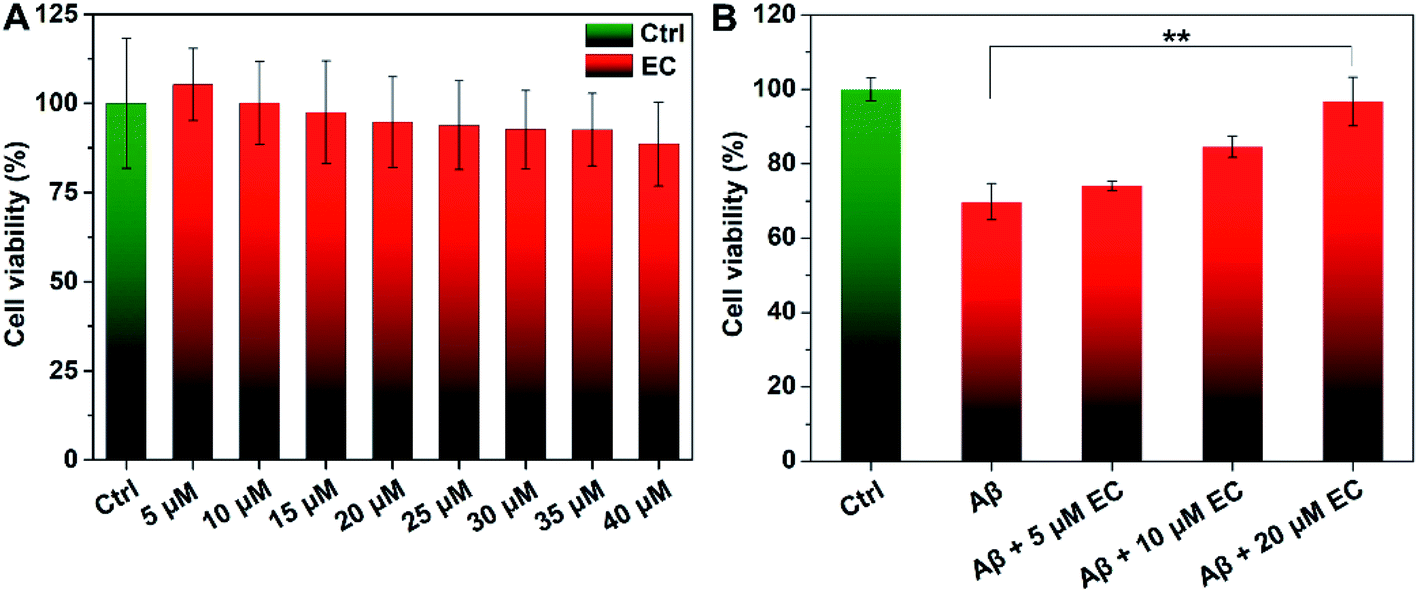 | ||
| Fig. 6 Viability (%) of SH-SY5Y cells upon incubation with different concentrations of the EC in the absence (A) or presence (B) of Aβ (40 μM). The data was normalized and calculated as a percentage of untreated cells only containing 4‰ DMSO as a control. Error bars indicate ±s.d. (n = 5 independent experiments). **P < 0.01 (Student's t-test). | ||
Phagocytosis of EC-mediated co-aggregates by microglia cells
Microglia-dependent phagocytosis is one of the major mechanisms of Aβ clearance.57,58 Previous studies suggested that the alteration of Aβ conformation would facilitate the removal of Aβ by glial cells.59–61 Given the unique structure of EC–Aβ co-aggregates, the phagocytosis of the preformed aggregates in murine microglial cells (BV-2) was investigated by confocal laser scanning microscopy (CLSM). Immunofluorescence analysis of BV-2 cells with anti-Aβ antibody 6E10 showed that a much higher level of Aβ uptake was observed from the cells incubated with co-aggregates for 4 h, compared to those incubated with Aβ fibrils, in which the fluorescence signals were mainly located outside the cells (Fig. 7A). By contrast, only an extremely faint fluorescence signal can be found around EC-treated cells in the absence of Aβ (Fig. S14†). The statistic results of fluorescent intensity in the cytosolic region also exhibited better internalization of EC–Aβ co-aggregates (Fig. 7B, ***P < 0.001), suggesting that the co-aggregates are more easily phagocytosed by microglial cells than Aβ fibrils, which was consistent with the result of dot blot analysis of cell lysates (Fig. 7C). Despite being controversial, autophagy has been plausibly recognized as the crucial pathway to degrade and clear the phagocytosed Aβ.57 In order to evaluate the autophagy effect of Aβ aggregates on BV-2 cells, Acridine Orange (AO), a pH-sensitive fluorescence probe for autophagy,62 was employed to stain the cells, followed by imaging using CLSM. AO is a cell-permeable green fluorophore that can accumulate in acidic vesicular organelles (AVOs) and convert into its protonated type with red fluorescence.63 Thus, the red-to-green fluorescence intensity ratio of AO can be used to quantify the volume of AVOs, which increases during the cellular autophagy. As shown in Fig. 7D, the brightest red fluorescence was observed in the co-aggregate-treated cells after incubation for 4 h, compared to that of DMSO-treated and Aβ40 fibril-treated cells. The quantification of fluorescence in the region of whole cells also revealed the higher red/green signal ratio of co-aggregate-treated cells than of Aβ40 fibril-treated cells (Fig. 7E, ***P < 0.001). Based on the above findings, EC-mediated Aβ co-aggregates might be able to recover the phagocytosis of microglia, subsequently upregulating the autophagy of the cells to facilitate their degradation and clearance.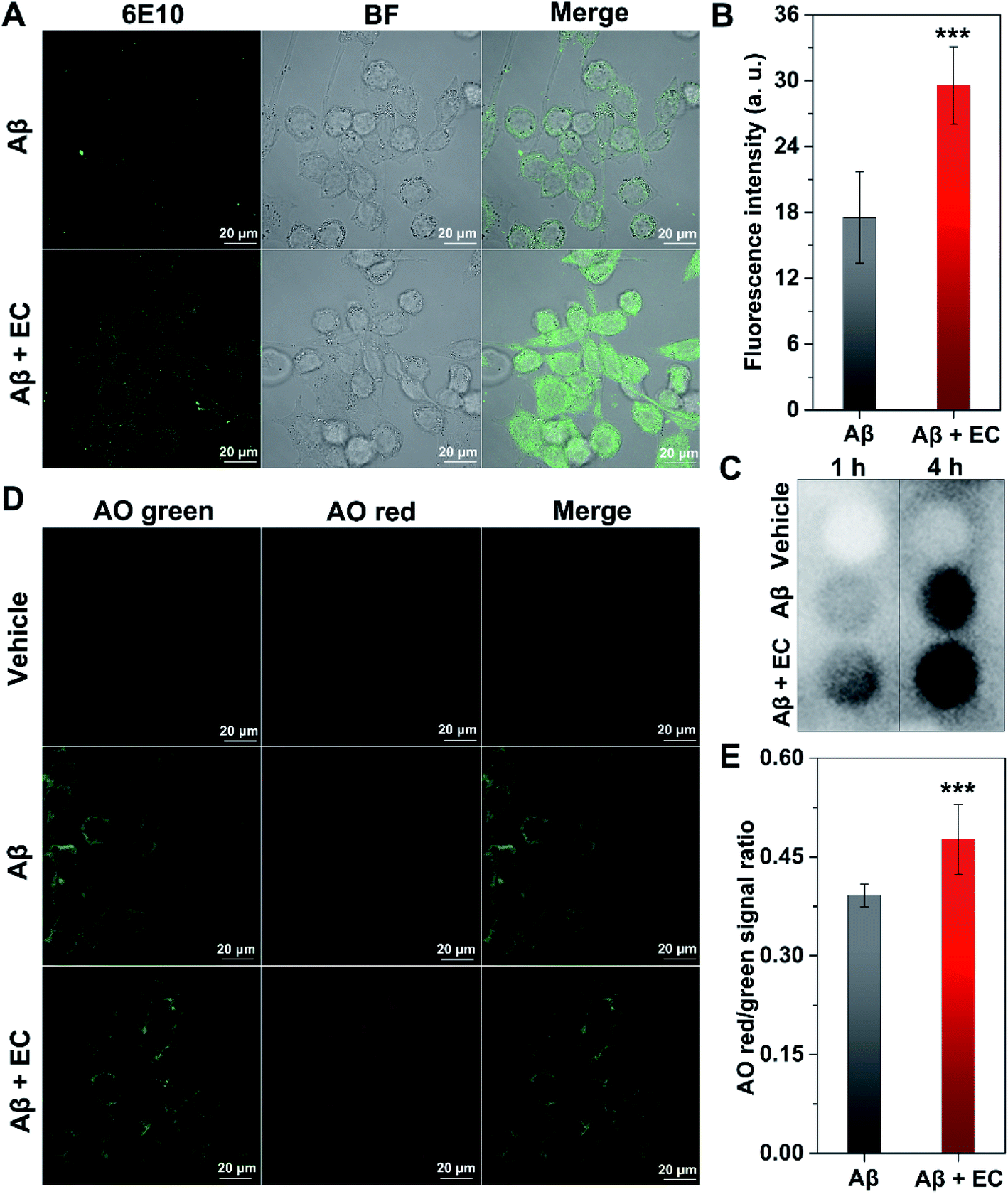 | ||
| Fig. 7 (A) Immunofluorescence imaging of BV-2 cells treated with Aβ (10 μM) in the absence and presence of the EC (10 μM) for 4 h. (B) The fluorescence intensity in the internal regions of cells of A was quantified with ZEN microscope software. More than 20 cells were counted under each condition and data were analyzed by Student's t-test (***P < 0.001). (C) Dot blot probed with 6E10 antibody to evaluate the uptake of preformed Aβ aggregates in the absence and presence of the EC in BV-2 cells. (D) Acridine Orange (AO) staining of BV-2 cells treated with Aβ (10 μM) in the absence and presence of the EC (10 μM) for 4 h. Green fluorescence was observed at Ex 488 nm, Em 500 to 560 nm; red fluorescence was acquired with Ex 488 nm, Em 600 to 700 nm using a Zeiss confocal microscope. (E) Quantification of the red/green intensity ratio in the cytosolic region of cells of (D). The red/green signal ratio was calculated using ZEN microscope software. More than 30 cells were counted under each condition and data were analyzed by Student's t-test (***P < 0.001). | ||
The EC rescues Aβ-mediated dysfunction in C. elegans models of AD
We further examined the effects of EC-mediated co-assembly of toxic Aβ42 oligomers in a transgenic Caenorhabditis elegans (C. elegans) model of AD in vivo, in which the human Aβ gene was expressed.64C. elegans strain CL4176 can express Aβ42 in body wall muscle cells, which accumulates into a toxic deposit and results in progressive paralysis of the worms.65 Accordingly, we exposed the CL4176 worms to different concentrations (20 and 40 μM) of the EC and measured their rate of paralysis, which is indicative of the effect of the EC on Aβ-associated fitness of the worms. Given the preferential interactions between the EC and LMW Aβ oligomers, worms at the L2 larval stage of the life cycle where Aβ aggregates have not formed yet53 were selected with the temperature increased from 15 °C to 25 °C to induce Aβ42 expression. In contrast to untreated worms, significant delays of the Aβ42-induced paralysis rate to a similar extent were observed in the CL4176 worms fed with 20 and 40 μM EC (Fig. 8A), which is consistent with the results of the concentration-dependent promotive effect of the EC on Aβ aggregation. Conversely, no paralysis can be found in the wild-type worm model CL802 that does not express Aβ in both the absence and presence of the EC (Fig. 8A). The effect of the EC on the motility of CL4176 is visually shown in Fig. 8B and Movies S1–S3 (ESI).† After living at 25 °C for 44 h, most of the untreated worms were paralyzed in the straight line shape, but only moved the head in situ. In contrast, some EC-treated worms can still sinusoidally move in the characteristic “C” curve just like the movements of wild-type C. elegans in both the presence and absence of the EC (Fig. S15, Movies S4 and S5†).66 These results indicated that the EC is able to effectively rescue Aβ42-induced behavioral dysfunction of C. elegans in vivo.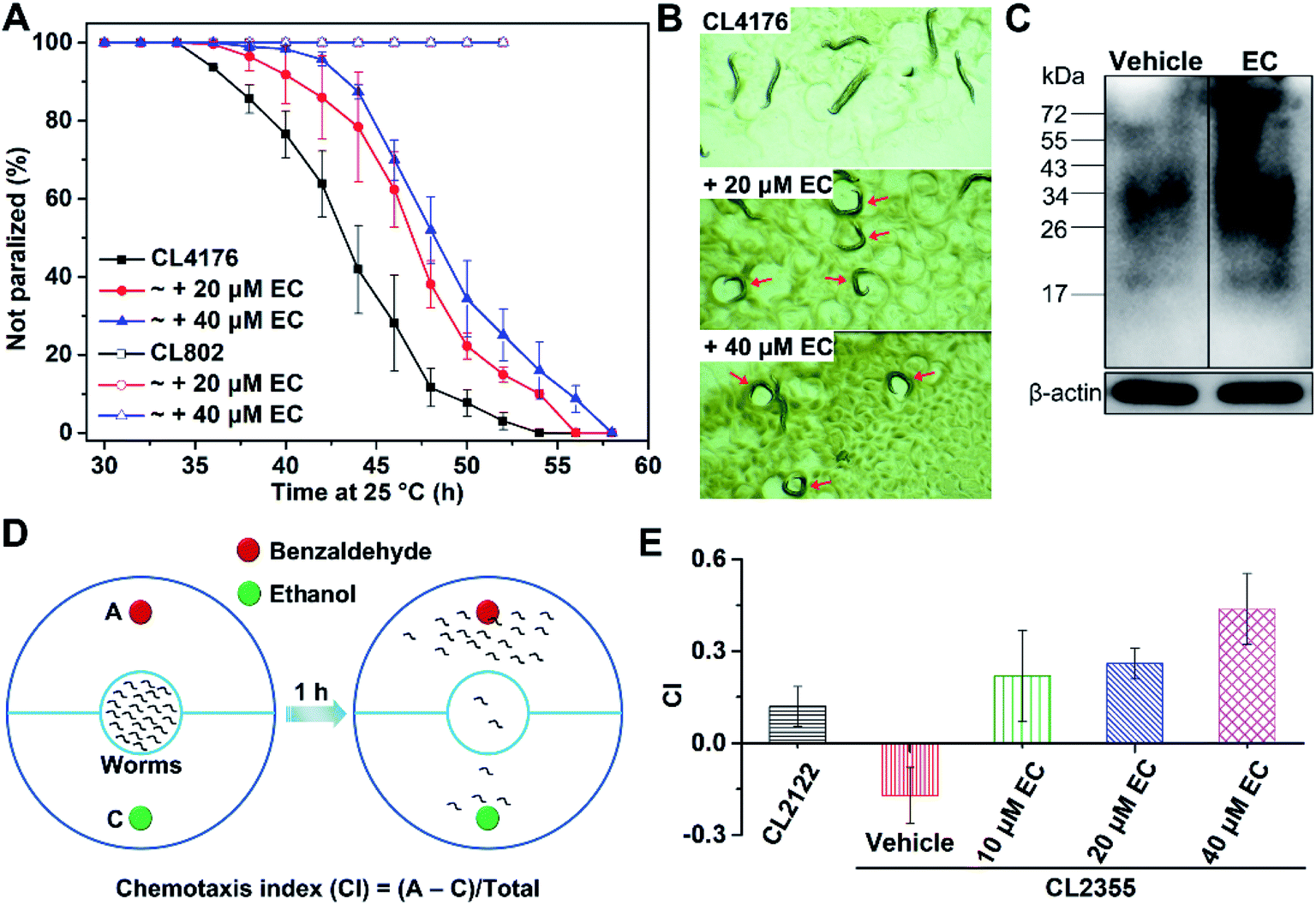 | ||
| Fig. 8 (A) Measurements of the effect of different concentrations of the EC on the Aβ42-induced paralysis in the transgenic C. elegans strain CL4176. The wild-type worm model CL802 that does not express Aβ was used as a control strain. (B) The motion modalities of C. elegans CL4176 upon treatment with DMSO (4‰, v/v) or different concentrations of the EC after living at 25 °C for 44 h. (C) Western blot analysis of Aβ extracted from C. elegans CL4176 in the absence and presence of the EC using 6E10 antibody. β-Actin was used as the internal reference to ensure equal protein loading. (D) Schematic diagram of the chemotaxis behavior assay. Culture plates were divided into two semicircles. Each semicircle was labeled either as an attractant (A) or control odorant (C). Worms were placed in the inner circle at the center of the plate and counted on each semicircle after 1 h. Individuals that did not cross out of the inner circle were not counted. A chemotaxis index (CI) was calculated using the equation CI = (A (worms in A) − C (worms in C))/total (total of scored worms) to indicate the chemotaxis behavior of worms. (E) Measurements of the effect of different concentrations of the EC on Aβ toxicity-induced defects in neuron-controlled chemotaxis behavior of transgenic C. elegans strain CL2355. The wild-type worm model CL2122 that does not express Aβ was used as a control strain. Error bars in (A) and (E) indicate mean ± s.d. (n = 3 independent experiments). | ||
We further investigated the level of Aβ aggregates in EC-treated and untreated worms by western blot assay using anti-Aβ antibody 6E10. Compared with untreated C. elegans, a higher amount of large aggregates (>34 kDa) was observed from the EC-treated worms, indicating that the EC is also able to promote Aβ aggregation via co-assembly in vivo (Fig. 8C). These findings would suggest that the EC-delayed paralysis rate of transgenic C. elegans is mainly attributed to the inhibitory effect of co-assembly between the EC and Aβ42 on Aβ-induced toxicity through promoting the conversion of toxic Aβ42 oligomers into non-toxic co-aggregates.
Besides the paralysis phenotype reflecting the toxicity of Aβ aggregates to muscle cells, a behavior phenotype of the transgenic strain (CL2355) that express Aβ42 in neuronal cells has also been commonly used to investigate the neuroprotective effect of the candidates against Aβ-induced defects in neuron-controlled chemotaxis behavior.67,68 The chemotaxis behavior can be evaluated by measuring the chemotaxis index (CI), that is, the fraction of worms that are able to arrive at the location of the attractants.68 To investigate the effect of the EC on the chemotaxis behavior, we chose benzaldehyde as an attractant and ethanol as a control (Fig. 8D). In comparison with the control strain CL2122 that does not express Aβ, the untreated CL2355 showed a quite low CI value, due to the Aβ-induced defect in chemotaxis behavior (Fig. 8E). However, EC feeding of the transgenic worms significantly reversed the reduced CI in a concentration-dependent manner. These results suggest that the EC can also improve the chemotaxis behavior of transgenic C. elegans through attenuating Aβ42-induced toxicity in vivo.
Conclusions
We have described a rational design of small molecules that irreversibly suppress oligomers' toxicity through co-assembly with Aβ oligomers. A pincer-like scaffold combining a rigid center as a linker and two pendants with high binding affinity to the hydrophobic regions of Aβ has been proposed, which is expected to directly bind to the exposed hydrophobic patches of Aβ oligomers that are responsible for toxicity and aggregation. As a proof of concept, a europium complex, EC, in such a structural mode demonstrated high affinity for LMW oligomers and can rapidly promote Aβ aggregation, but convert the peptides into non-fibrillar aggregates with weak β-sheet conformation, according to the spectrometry, kinetics studies, electrophoresis, and morphology analysis. Completely distinct from the reported off-pathway products that are totally seeding-incompetent, EC-mediated non-fibrillar aggregates exhibited strong ability to accelerate Aβ aggregation as seeds. The data imply that the EC would co-assemble with Aβ oligomers to form EC–Aβ co-aggregates, which are structurally distinct from Aβ self-aggregates. We have also found that the co-aggregates are almost non-toxic and prone to degradation by glial cells via up-regulating autophagy. Importantly, the EC can effectively rescue Aβ-mediated dysfunction in C. elegans models of AD in vivo owing to the bicomponent co-assembly. Since Eu3+ does not participate in the interactions between the EC and Aβ oligomers, such a pincer-like scaffold would also be available to other hydrophobic motif-binding groups for constructing Aβ oligomer-specific small molecules but not limited to lanthanide complexes when linked by proper rigid groups. Thus, this ‘small molecule-mediated co-assembly of oligomers’ strategy representing a powerful anti-oligomer therapy would provide a new direction for design of small molecule-based drug-like leads in the treatment of AD. Further structural studies to elucidate the conformation of co-aggregates, particularly EC–Aβ42 co-assemblies, and the screening of this class of compounds on human Aβ-expressing rodent models are currently in progress.Ethical statement
All animal experiments were conducted in accordance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals” and were approved by the Institutional Animal Care and Use Committee of the MARC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the financial support from the National Natural Science Foundation of China (Grants: 21771105 and 21301090), the Natural Science Foundation of Jiangsu Province (Grant: BK20170103), the Natural Science Foundation of the Jiangsu Higher Education Institutions (Grant: 17KJB150018), and the Six Talent Peaks Project in Jiangsu Province (Grant: SWYY-043).References
- S. Makin, Nature, 2018, 559, S4–S7 CrossRef CAS PubMed.
- I. Benilova, E. Karran and B. De Strooper, Nat. Neurosci., 2012, 15, 349–357 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- B. Zott, M. M. Simon, W. Hong, F. Unger, H.-J. Chen-Engerer, M. P. Frosch, B. Sakmann, D. M. Walsh and A. Konnerth, Science, 2019, 365, 559–565 CrossRef CAS PubMed.
- R. J. Perrin, A. M. Fagan and D. M. Holtzman, Nature, 2009, 461, 916–922 CrossRef CAS PubMed.
- I. Klyubin, D. M. Walsh, C. A. Lemere, W. K. Cullen, G. M. Shankar, V. Betts, E. T. Spooner, L. Jiang, R. Anwyl and D. J. Selkoe, Nat. Med., 2005, 11, 556–561 CrossRef CAS PubMed.
- S. J. C. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Chem. Soc. Rev., 2017, 46, 310–323 RSC.
- S. Ayala, P. Genevaux, C. Hureau and P. Faller, ACS Chem. Neurosci., 2019, 10, 3366–3374 CrossRef CAS PubMed.
- S. Kumar, A. Henningknechtel, I. Chehade, M. Magzoub and A. D. Hamilton, J. Am. Chem. Soc., 2017, 139, 17098–17108 CrossRef CAS PubMed.
- Y. Jiang, X. Jiang, X. Shi, F. Yang, Y. Cao, X. Qin, Z. Hou, M. Xie, N. Liu, Q. Fang, F. Yin, W. Han and Z. Li, iScience, 2019, 17, 87–100 CrossRef CAS PubMed.
- J. Bieschke, M. Herbst, T. Wiglenda, R. P. Friedrich, A. Boeddrich, F. Schiele, D. Kleckers, J. M. L. D. Amo, B. Gruning and Q. Wang, Nat. Chem. Biol., 2012, 8, 93–101 CrossRef CAS PubMed.
- R. Limbocker, S. Chia, F. S. Ruggeri, M. Perni, R. Cascella, G. T. Heller, G. Meisl, B. Mannini, J. Habchi and T. C. T. Michaels, Nat. Commun., 2019, 10, 225 CrossRef CAS PubMed.
- D. E. Ehrnhoefer, J. Bieschke, A. Boeddrich, M. Herbst, L. Masino, R. Lurz, S. Engemann, A. Pastore and E. E. Wanker, Nat. Struct. Mol. Biol., 2008, 15, 558–566 CrossRef CAS PubMed.
- J. Bieschke, J. Russ, R. P. Friedrich, D. E. Ehrnhoefer, H. J. Wobst, K. Neugebauer and E. E. Wanker, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7710–7715 CrossRef CAS PubMed.
- A. R. A. Ladiwala, J. C. Lin, S. S. Bale, A. M. Marcelinocruz, M. Bhattacharya, J. S. Dordick and P. M. Tessier, J. Biol. Chem., 2010, 285, 24228–24237 CrossRef CAS PubMed.
- T. Arai, D. Sasaki, T. Araya, T. Sato, Y. Sohma and M. Kanai, ChemBioChem, 2014, 15, 2577–2583 CrossRef CAS PubMed.
- A. R. A. Ladiwala, J. S. Dordick and P. M. Tessier, J. Biol. Chem., 2011, 286, 3209–3218 CrossRef CAS PubMed.
- D. M. Raymond and B. L. Nilsson, Chem. Soc. Rev., 2018, 47, 3659–3720 RSC.
- S. Campioni, B. Mannini, M. Zampagni, A. Pensalfini, C. Parrini, E. Evangelisti, A. Relini, M. Stefani, C. M. Dobson and C. Cecchi, Nat. Chem. Biol., 2010, 6, 140–147 CrossRef CAS PubMed.
- A. R. A. Ladiwala, J. Litt, R. S. Kane, D. Aucoin, S. O. Smith, S. Ranjan, J. Davis, W. E. Van Nostrand and P. M. Tessier, J. Biol. Chem., 2012, 287, 24765–24773 CrossRef CAS PubMed.
- S. R. Chowdhury, M. Agarwal, N. Meher, B. Muthuraj and P. K. Iyer, ACS Appl. Mater. Interfaces, 2016, 8, 13309–13319 CrossRef CAS PubMed.
- Y. Song, P. N. Cheng, L. Zhu, E. G. Moore and J. S. Moore, J. Am. Chem. Soc., 2014, 136, 5233–5236 CrossRef CAS PubMed.
- Z. Xu, S. Jia, W. Wang, Z. Yuan, B. J. Ravoo and D. Guo, Nat. Chem., 2019, 11, 86–93 CrossRef CAS PubMed.
- L. Zhu, Y. Song, P. N. Cheng and J. S. Moore, J. Am. Chem. Soc., 2015, 137, 8062–8068 CrossRef CAS PubMed.
- O. Szczepankiewicz, B. Linse, G. Meisl, E. Thulin, B. Frohm, C. S. Frigerio, M. T. Colvin, A. C. Jacavone, R. G. Griffin and T. P. J. Knowles, J. Am. Chem. Soc., 2015, 137, 14673–14685 CrossRef CAS PubMed.
- M. Suzuki, T. Takahashi, J. Sato, M. Mie, E. Kobatake and H. Mihara, ChemBioChem, 2010, 11, 1525–1530 CrossRef CAS PubMed.
- E. A. Fradinger, B. H. Monien, B. Urbanc, A. Lomakin, M. Tan, H. Li, S. M. Spring, M. M. Condron, L. Cruz, C.-W. Xie, G. B. Benedek and G. Bitan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14175–14180 CrossRef CAS PubMed.
- C. L. Teoh, D. Su, S. Sahu, S. Yun, E. Drummond, F. Prelli, S. Lim, S. Cho, S. Ham and T. Wisniewski, J. Am. Chem. Soc., 2015, 137, 13503–13509 CrossRef CAS PubMed.
- G. Lv, A. Sun, P. Wei, N. Zhang, H. Lan and T. Yi, Chem. Commun., 2016, 52, 8865–8868 RSC.
- Y. Li, D. Xu, A. Sun, S. Ho, C. Poon, H. Chan, O. T. W. Ng, K. K. L. Yung, H. Yan and H. Li, Chem. Sci., 2017, 8, 8279–8284 RSC.
- P. J. Salveson, S. Haerianardakani, A. Thuy-Boun, S. Yoo, A. G. Kreutzer, B. Demeler and J. S. Nowick, J. Am. Chem. Soc., 2018, 140, 11745–11754 CrossRef CAS PubMed.
- B. O. Okesola and A. Mata, Chem. Soc. Rev., 2018, 47, 3721–3736 RSC.
- M. Beeg, M. Stravalaci, M. Romeo, A. D. Carra, A. Cagnotto, A. Rossi, L. Diomede, M. Salmona and M. Gobbi, J. Biol. Chem., 2016, 291, 6958–6966 CrossRef CAS PubMed.
- H. Fu, M. Cui, P. Tu, Z. Pan and B. Liu, Chem. Commun., 2014, 50, 11875–11878 RSC.
- M. Kung, C. Hou, Z. Zhuang, B. Zhang, D. M. Skovronsky, J. Q. Trojanowski, V. M. Y. Lee and H. F. Kung, Brain Res., 2002, 956, 202–210 CrossRef CAS.
- B. Mannini, R. Cascella, M. Zampagni, M. Van Waardeverhagen, S. Meehan, C. Roodveldt, S. Campioni, M. Boninsegna, A. Penco and A. Relini, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12479–12484 CrossRef CAS PubMed.
- M. G. Savelieff, S. Lee, Y. Liu and M. H. Lim, ACS Chem. Biol., 2013, 8, 856–865 CrossRef CAS PubMed.
- G. Meisl, J. B. Kirkegaard, P. Arosio, T. C. T. Michaels, M. Vendruscolo, C. M. Dobson, S. Linse and T. P. J. Knowles, Nat. Protoc., 2016, 11, 252–272 CrossRef CAS PubMed.
- X. Wang, H. Chang, J. Xie, B. Zhao, B. Liu, S. Xu, W. Pei, N. Ren, L. Huang and W. Huang, Coord. Chem. Rev., 2014, 273, 201–212 CrossRef.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- X. Wang, T. Yang, J. Luo, L. Yang and C. Yao, Chem. Commun., 2015, 51, 8185–8188 RSC.
- E. A. Weitz, J. Y. Chang, A. H. Rosenfield and V. C. Pierre, J. Am. Chem. Soc., 2012, 134, 16099–16102 CrossRef CAS PubMed.
- A. G. Kreutzer, I. L. Hamza, R. K. Spencer and J. S. Nowick, J. Am. Chem. Soc., 2016, 138, 4634–4642 CrossRef CAS PubMed.
- G. V. De Ferrari, M. Canales, I. Shin, L. Weiner, I. Silman and N. C. Inestrosa, Biochemistry, 2001, 40, 10447–10457 CrossRef CAS PubMed.
- M. Necula, R. Kayed, S. Milton and C. G. Glabe, J. Biol. Chem., 2007, 282, 10311–10324 CrossRef CAS PubMed.
- M. Necula, L. Breydo, S. Milton, R. Kayed, W. E. van der Veer, P. Tone and C. G. Glabe, Biochemistry, 2007, 46, 8850–8860 CrossRef CAS PubMed.
- W. Kim and H. Hecht, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15824–15829 CrossRef CAS PubMed.
- R. Kayed, E. Head, J. L. Thompson, T. M. Mcintire, S. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef CAS PubMed.
- N. Lorenzen, S. B. Nielsen, A. K. Buell, J. D. Kaspersen, P. Arosio, B. S. Vad, W. Paslawski, G. Christiansen, Z. Valnickovahansen and M. Andreasen, J. Am. Chem. Soc., 2014, 136, 3859–3868 CrossRef CAS PubMed.
- T. Yang, X. Wang, C. Zhang, X. Ma, K. Wang, Y. Wang, J. Luo, L. Yang, C. Yao and X. Wang, Chem. Commun., 2016, 52, 2245–2248 RSC.
- S. I. A. Cohen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, J. Mol. Biol., 2012, 421, 160–171 CrossRef CAS PubMed.
- J. Habchi, S. Chia, C. Galvagnion, T. C. T. Michaels, M. M. J. Bellaiche, F. S. Ruggeri, M. Sanguanini, I. Idini, J. R. Kumita and E. Sparr, Nat. Chem., 2018, 10, 673–683 CrossRef CAS PubMed.
- J. Habchi, P. Arosio, M. Perni, A. R. Costa, M. Yagiutsumi, P. D. Joshi, S. K. R. Chia, S. I. A. Cohen, M. B. Muller and S. Linse, Sci. Adv., 2016, 2, e1501244 CrossRef PubMed.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef CAS PubMed.
- G. Meisl, X. Yang, E. Hellstrand, B. Frohm, J. B. Kirkegaard, S. I. A. Cohen, C. M. Dobson, S. Linse and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9384–9389 CrossRef CAS PubMed.
- C. Dammers, M. Schwarten, A. K. Buell and D. Willbold, Chem. Sci., 2017, 8, 4996–5004 RSC.
- R. A. Nixon, Nat. Med., 2013, 19, 983–997 CrossRef CAS PubMed.
- J. M. Tarasoffconway, R. O. Carare, R. S. Osorio, L. Glodzik, T. Butler, E. Fieremans, L. Axel, H. Rusinek, C. Nicholson and B. V. Zlokovic, Nat. Rev. Neurol., 2015, 11, 457–470 CrossRef CAS PubMed.
- Q. Luo, Y. Lin, P. Yang, Y. J. Wang, G. Qi, Z. Qiao, B. Li, K. Zhang, J. Zhang and L. Wang, Nat. Commun., 2018, 9, 1802 CrossRef PubMed.
- Y. Zhao, J. Cai, Z. Liu, Y. Li, C. Zheng, Y. Zheng, Q. Chen, H. Chen, F. Ma and Y. An, Nano Lett., 2019, 19, 674–683 CrossRef PubMed.
- K. Debnath, N. R. Jana and N. R. Jana, ACS Biomater. Sci. Eng., 2019, 5, 390–401 CrossRef CAS.
- D. J. Klionsky, F. C. Abdalla, H. Abeliovich, R. T. Abraham, A. Acevedoarozena, K. Adeli, L. Agholme, M. Agnello, P. Agostinis and J. A. Aguirreghiso, Autophagy, 2012, 8, 445–544 CrossRef CAS PubMed.
- W. Fu, X. Li, X. Lu, L. Zhang, R. Li, N. Zhang, S. Liu, X. Yang, Y. Wang and Y. Zhao, Cell Death Dis., 2017, 8, e3086 CrossRef PubMed.
- Y. Guan, Z. Du, N. Gao, Y. Cao, X. Wang, P. Scott, H. Song, J. Ren and X. Qu, Sci. Adv., 2018, 4, eaao6718 CrossRef PubMed.
- C. D. Link, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9368–9372 CrossRef CAS PubMed.
- C. D. Link, Exp. Gerontol., 2006, 41, 1007–1013 CrossRef CAS PubMed.
- E. F. Fang, Y. Hou, K. Palikaras, B. A. Adriaanse, J. S. Kerr, B. Yang, S. Lautrup, M. M. Hasanolive, D. Caponio and X. Dan, Nat. Neurosci., 2019, 22, 401–412 CrossRef CAS PubMed.
- R. Keowkase, N. Shoomarom, W. Bunargin, W. Sitthithaworn and N. Weerapreeyakul, Biomed. Pharmacother., 2018, 107, 656–664 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00392a |
| This journal is © The Royal Society of Chemistry 2020 |