
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective oxo ligand functionalisation and substitution reactivity in an oxo/catecholate-bridged UIV/UIV Pacman complex†
Bradley E.
Cowie
 a,
Iskander
Douair
b,
Laurent
Maron
b,
Jason B.
Love
*a and
Polly L.
Arnold‡
*a
a,
Iskander
Douair
b,
Laurent
Maron
b,
Jason B.
Love
*a and
Polly L.
Arnold‡
*a
aEaStCHEM School of Chemistry, The University of Edinburgh, Joseph Black Building, The King's Buildings, Edinburgh, EH9 3FJ, UK
bUniversité de Toulouse, INSA, UPS, CNRS, UMR 5215, LPCNO, 135 Avenue de Rangueil, F-31077 Toulouse, France
First published on 12th June 2020
Abstract
The oxo- and catecholate-bridged UIV/UIV Pacman complex [{(py)UIVOUIV(μ-O2C6H4)(py)}(LA)] A (LA = a macrocyclic “Pacman” ligand; anthracenylene hinge between N4-donor pockets, ethyl substituents on meso-carbon atom of each N4-donor pocket) featuring a bent UIV–O–UIV oxo bridge readily reacts with small molecule substrates to undergo either oxo-atom functionalisation or substitution. Complex A reacts with H2O or MeOH to afford [{(py)UIV(μ-OH)2UIV(μ-O2C6H4)(py)}(LA)] (1) and [{(py)UIV(μ-OH)(μ-OMe)UIV(μ-O2C6H4)(py)}(LA)] (2), respectively, in which the bridging oxo ligand in A is substituted for two bridging hydroxo ligands or one bridging hydroxo and one bridging methoxy ligand, respectively. Alternatively, A reacts with either 0.5 equiv. of S8 or 4 equiv. of Se to provide [{(py)UIV(μ-η2:η2-E2)UIV(μ-O2C6H4)(py)}(LA)] (E = S (3), Se (4)) respectively, in which the [E2]2− ion bridges the two UIV centres. To the best of our knowledge, complex A is the first example of either a d- or f-block bimetallic μ-oxo complex that activates elemental chalcogens. Complex A also reacts with XeF2 or 2 equiv. of Me3SiCl to provide [{(py)UIV(μ-X)2UIV(μ-O2C6H4)(py)}(LA)] (X = F (5), Cl (6)), in which the oxo ligand has been substituted for two bridging halido ligands. Reacting A with either XeF2 or Me3SiCl in the presence of O(Bcat)2 at room temperature forms [{(py)UIV(μ-X)(μ-OBcat)UIV(μ-O2C6H4)(py)}(LA)] (X = F (5A), Cl (6A)), which upon heating to 80 °C is converted to 5 and 6, respectively. In order to probe the importance of the bent UIV–O–UIV motif in A on the observed reactivity, the bis(boroxido)-UIV/UIV complex, [{(py)(pinBO)UIVOUIV(OBpin)(py)}(LA)] (B), featuring a linear UIV–O–UIV bond angle was treated with H2O and Me3SiCl. Complex B reacts with two equiv. of either H2O or Me3SiCl to provide [{(py)HOUIVOUIVOH(py)}(LA)] (7) and [{(py)ClUIVOUIVCl(py)}(LA)] (8), respectively, in which reactions occur preferentially at the boroxido ligands, with the μ-oxo ligand unchanged. The formal UIV oxidation state is retained in all of the products 1–8, and selective reactions at the bridging oxo ligand in A is facilitated by: (1) its highly nucleophilic character which is a result of a non-linear UIV–O–UIV bond angle causing an increase in U–O bond covalency and localisation of the lone pairs of electrons on the μ-oxo group, and (2) the presence of the bridging catecholate ligand, which destabilises a linear oxo-bridging geometry and stabilises the resulting products.
Introduction
Molecular UIII complexes are renowned for activating small molecules due their Lewis acidity, the accessibility of the UIII → UIV redox couple and in some cases, the capacity for UIII to back-donate into empty ligand-based molecular orbitals.1–3 For example, it was demonstrated in the 1980s that [UIII(η5-C5H4R)3] (R = Me, SiMe3) reacted with half an equivalent of CS2 to form [{UIV(η5-C5H4R)3}2(μ-η1:η2-CS2)] through UIII → UIV oxidation (Fig. 1A).4 Since then, a plethora of examples of low oxidation state uranium complexes for the activation of small molecules such as NO, N3−, CxHy hydrocarbons, S8 and Se have been characterised,5,6 such as a recently reported example of the use of a UIII/UIII dimer from our research group, [{UIII(OBMes2)3}2], which reacts with S8 to provide [{UV(OBMes2)3}2(μ-η2:η2-S2)2] (Fig. 1B).7 A variety of UIV–O–UIV containing complexes with many different supporting ligands have also been formed, since this is normally a thermodynamic sink. Such complexes are commonly formed either by treating 2 equiv. of a UIII complex with an oxo source such as N2O, or half an equiv. of CO2.6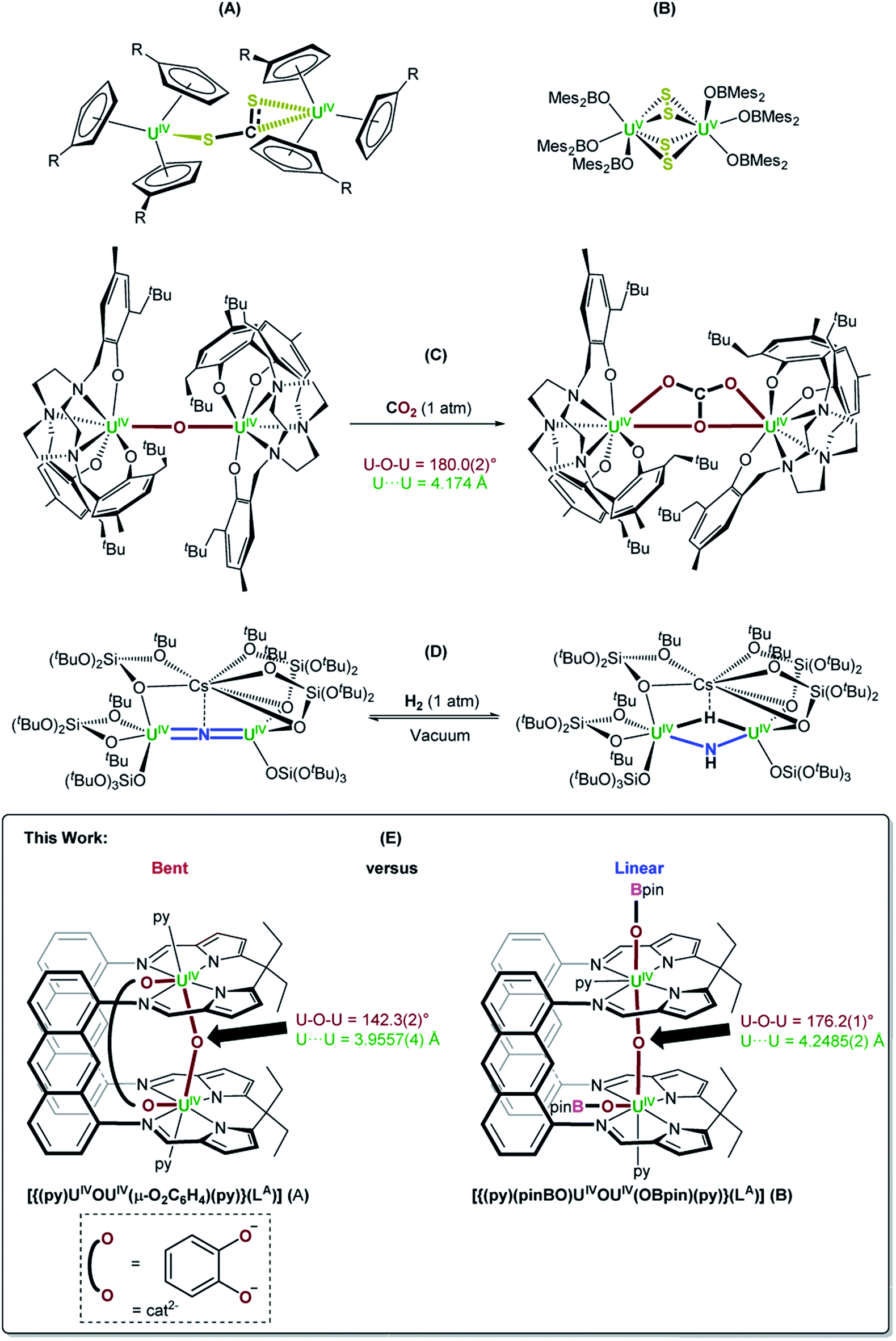 | ||
| Fig. 1 (A) and (B) Examples of CS2 and S8 activation by molecular UIII complexes, respectively.4,7 (C) and (D) Examples of functionalisation of oxo and nitride ligands bridging between two UIV centres, respectively.14,17 (E) The bent, oxo-/catecholate-bridged UIV/UIV Pacman complex, [{(py)UIVOUIV(μ-O2C6H4)(py)}(LA)] (A), and the linear, oxo-bridged bis(boroxido)-UIV/UIV Pacman complex, [{(py)(pinBO)UIVOUIV(OBpin)(py)}(LA)] (B);18 a comparison of their reactivity with a variety of small molecule reagents is presented in this work (py = pyridine). | ||
Small molecule activation by molecular UIV and UV complexes is exceedingly rare and difficult to predict or design. The UIV centre is significantly less reducing and relatively inert to further redox processes.8–11 The functionalisation of a bridging oxo ligand between two UIV centres is still very rare as the U–O bond is very strong; the single U–O bond in 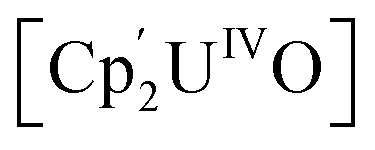 (Cp' = C5H2-1,2,4-tBu3) is 293 kJ mol−1 stronger than the double U
(Cp' = C5H2-1,2,4-tBu3) is 293 kJ mol−1 stronger than the double U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NMe bond in
NMe bond in 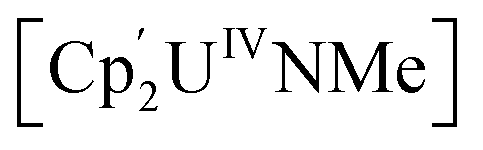 .6,12,13 However, under the right circumstances the single-atom bridged UIV–E–UIV unit can hold a privileged position. Such examples are limited to the conversion of the μ-oxo group into a bridging [CO3]− ligand by treatment with CO2, such as in the conversion of [{((Neop,MeArO)3tacn)UIV}2(μ-O)] ((Neop,MeArOH)3tacn = 1,4,7-tris(2-hydroxy-5-methyl-3-neopentylbenzyl)-1,4,7-triazacyclononane) into [{((Neop,MeArO)3tacn)UIV}2(μ-CO3)] (Fig. 1C).6,14 Further functionalisation of a UIV-coordinated, activated small molecule fragment typically requires the use of highly reactive external reagents.15 A bridging nitrido ligand in [UIVNUIV] complexes is amenable to functionalisation, such as the conversion of [Cs{UIV{OSi(OtBu)3}3}2(μ-N)] (synthesised from [UIII{OSi(OtBu)3}3]2 and CsN3)16 into [Cs{UIV{OSi(OtBu)3}3}2(μ-NH)(μ-H)] by treatment with H2 (Fig. 1D).17 The design principles that enable this reactivity from such a fragment are not yet clear.
.6,12,13 However, under the right circumstances the single-atom bridged UIV–E–UIV unit can hold a privileged position. Such examples are limited to the conversion of the μ-oxo group into a bridging [CO3]− ligand by treatment with CO2, such as in the conversion of [{((Neop,MeArO)3tacn)UIV}2(μ-O)] ((Neop,MeArOH)3tacn = 1,4,7-tris(2-hydroxy-5-methyl-3-neopentylbenzyl)-1,4,7-triazacyclononane) into [{((Neop,MeArO)3tacn)UIV}2(μ-CO3)] (Fig. 1C).6,14 Further functionalisation of a UIV-coordinated, activated small molecule fragment typically requires the use of highly reactive external reagents.15 A bridging nitrido ligand in [UIVNUIV] complexes is amenable to functionalisation, such as the conversion of [Cs{UIV{OSi(OtBu)3}3}2(μ-N)] (synthesised from [UIII{OSi(OtBu)3}3]2 and CsN3)16 into [Cs{UIV{OSi(OtBu)3}3}2(μ-NH)(μ-H)] by treatment with H2 (Fig. 1D).17 The design principles that enable this reactivity from such a fragment are not yet clear.
Here we report the remarkably different reactivity shown by two very similar UIV/UIV μ-oxo complexes of the macrocyclic “Pacman” ligand LA, Fig. 1E. Complex A [{(py)UIVOUIV(μ-O2C6H4)(py)}(LA)] is found to be reactive towards small molecules and has a catecholate ligand that bridges the U–O–U unit giving a short, yet bent U–O–U angle of 142.3(3)° and a U⋯U separation of 3.9557(4) Å. In contrast, the parent complex B [{(py)(pinBO)UIVOUIV(OBpin)(py)}(LA)] is unreactive, has a longer U⋯U separation of 4.2485(2) Å and a more ‘normal’ linear U–O–U angle of 176.2(1)° (Fig. 1E).18
Results
A. Reactivity of a bent oxo/catecholato-bridged UIV/UIV Pacman complex
We previously reported the synthesis of [{(py)UIVOUIV(μ-O2C6H4)(py)}(LA)] (A) by reaction of the bis(uranyl) Pacman complex, [{UVIO2(py)}2(LA)],19,20 with 3 equiv. of B2cat2 (Scheme 1).18 We have now found that A reacts with weak acids such as H2O and MeOH, either stoichiometrically (1 equiv.) or in excess (35 equiv. H2O or 5 equiv. MeOH) to provide [{(py)UIV(μ-OH)2UIV(μ-O2C6H4)(py)}(LA)] (1) and [{(py)UIV(μ-OMe)(μ-OH)UIV(μ-O2C6H4)(py)}(LA)] (2) as yellow solids in 73 and 54% isolated yield, respectively (Scheme 1). We have previously reported another uranium hydroxide supported by the Pacman ligand, [{UVO(OH)(py)}(H2LMe)] (H4LMe = Pacman-shaped macrocyclic Schiff-base ligand with methyl substituents on the meso-carbon atoms and a dimethylphenylene hinge). In that case it was formed by treating an oxo-lithiated uranyl(V) complex, [{UVO{OLi(py)3}(py)}(LiHLMe)], with 2 equiv. of HCl.21 In 1, the bridging oxo ligand in A has been formally substituted by two bridging hydroxo ligands in a complex that has C2h symmetry according to the 13 paramagnetically shifted resonances between 95.5 and −36.1 ppm in the 1H NMR spectrum at 300 K. In contrast, complex 2 possesses C1v symmetry with one bridging methoxy and one bridging hydroxo ligand between the two U centres. While 21 resonances are recorded in the 1H NMR spectra at either 300 K (96.65 to −35.89 ppm) or 360 K (78.48 to −27.28 ppm), the resonances for 2 are significantly sharper at elevated temperatures, suggesting dynamic behaviour of the bridging ligands in solution. The μ-OH resonance in both 1 and 2 could not be located in the 1H NMR spectra, but we note that it is close to the two paramagnetic cations and so would be significantly shifted and broadened.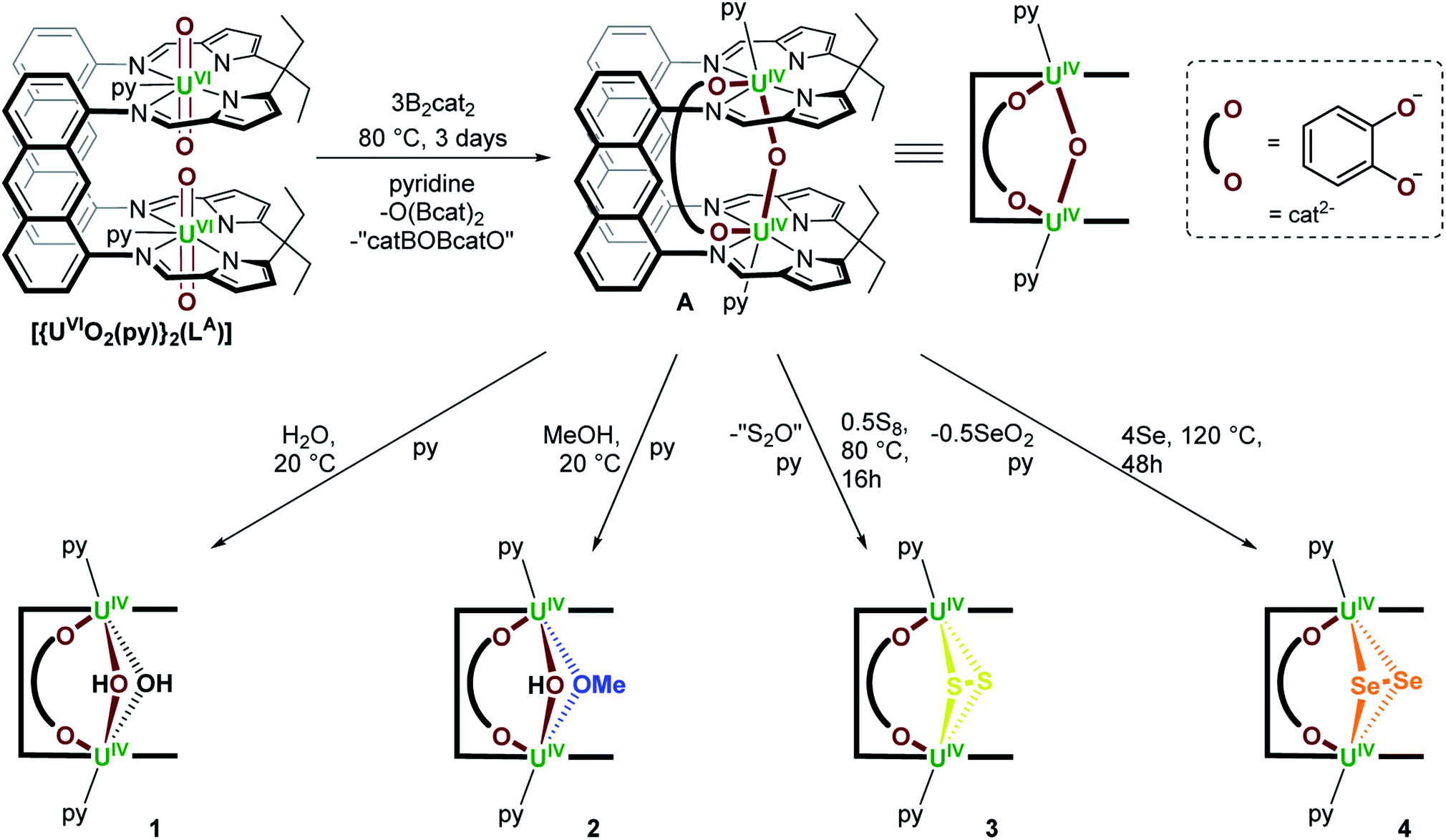 | ||
| Scheme 1 Synthesis of [{(py)UIVOUIV(μ-O2C6H4)(py)}(LA)] (A) from [{UVIO2(py)}2(LA)] and 3 equiv. of B2cat2,18 and subsequent synthesis of [{(py)UIV(μ-OH)2UIV(μ-O2C6H4)(py)}(LA)] (1), [{(py)UIV(μ-OMe)(μ-OH)UIV(μ-O2C6H4)(py)}(LA)] (2) and [{(py)UIV(μ-η2:η2-E2)UIV(μ-O2C6H4)(py)}(LA)] (E = S (3), Se (4)); py = pyridine. | ||
Complex A also reacts with 0.5 equiv. of S8 over the course of 16 hours at 80 °C to afford [{(py)UIV(μ-η2:η2-S2)UIV(μ-O2C6H4)(py)}(LA)] (3) as a brown/yellow solid in 55% isolated yield (Scheme 1); in this case the bridging oxo ligand has been substituted for an [S2]2− ligand. During this reaction, the μ-oxo ligand is likely lost as S2O, which is unstable and expected to ultimately form SO2 and S8, meaning that no change in uranium oxidation state is needed.22 Complex 3 may also be obtained by reacting A with a slight excess of CS2 (∼3 equiv.) and heating to 120 °C for 4 days. This reaction does not require any redox change in the metal or ligands if the by-product is COS. Also, no further reaction is seen with an excess of S8. The 1H NMR spectrum of 3 contains 12 resonances between 62.24 and −40.90 ppm, indicative of a UIV/UIV complex of C2h symmetry.
Additionally, A reacts with four or more equiv. of elemental selenium when heated to 125 °C for 48 hours in pyridine to provide [{(py)UIV(μ-η2:η2-Se2)UIV(μ-O2C6H4)(py)}(LA)] (4) as a red/brown solid in 43% yield (Scheme 1). The 1H NMR spectrum of 4 contains 15 resonances between 62.49 and −41.70 ppm. The formation of complex 4 involves substitution of the bridging oxo ligand in A for a bridging [Se2]2− ligand and presumably formation of Se2O as the by-product. In an attempt to prepare the mono-Se adduct [{(py)UIVSeUIV(μ-O2C6H4)(py)}(LA)], A was treated with one equivalent of the potent chalcogen atom transfer reagent, Ph3PSe.23 However no reaction occurred (see Discussion section). Furthermore, complex A does not react with elemental tellurium, P4, Ph2Se2 or Ph2Te2.
A reaction between A and XeF2, designed to target a UV/UV product containing the [FUVOUVF]6+ unit, instead forms the UIV/UIV bridging bis(fluorido) complex, [{(py)UIV(μ-F)2UIV(μ-O2C6H4)(py)}(LA)] (5), following heating the reaction mixture at 80 °C for 24 hours (Scheme 2 Path A); no reaction occurs at room temperature. Complex 5 is formed by substitution of the bridging oxo ligand in A for two bridging fluorido ligands and was isolated as a lemon yellow solid in 40% yield; the formal by-product XeO is unstable so a mixture of XeOn (n = 2 or 3) and Xe gas likely results.24 The 1H NMR spectrum of 5 at 300 K contains 11 resonances from 64.89 to −55.51 ppm, while that recorded at 360 K contains the anticipated 14 resonances (88.91 to −43.00 ppm) for a C2h symmetric product. No resonances were seen in the 19F NMR spectrum which may be due to a dynamic process rather than proximity to the paramagnetic U centres since a resonance is observed for 5A below.
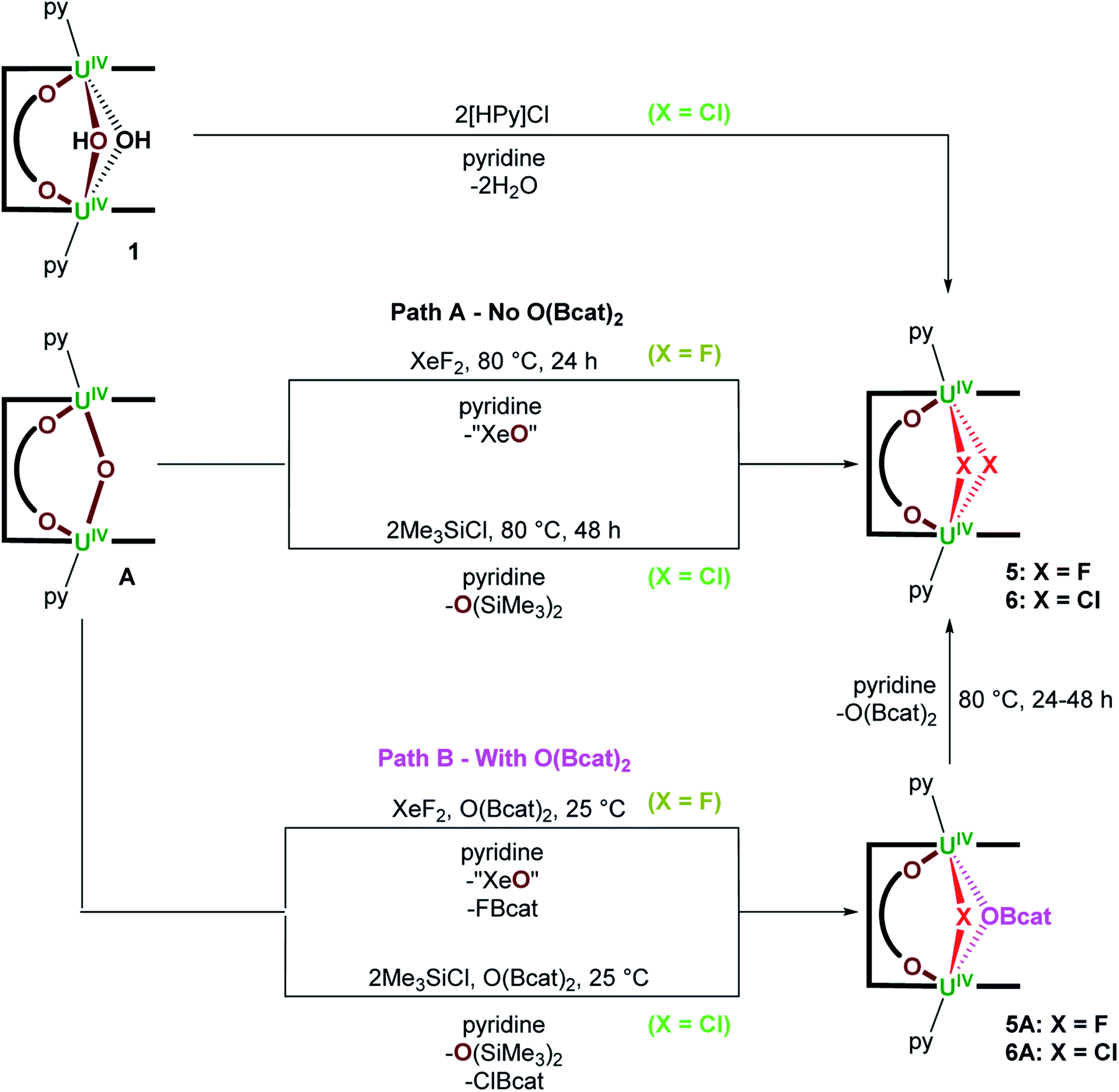 | ||
| Scheme 2 Synthesis of [{(py)UIV(μ-X)2UIV(μ-O2C6H4)(py)}(LA)] (X = F (5), Cl (6)) from (A) A and XeF2 or Me3SiCl, respectively. (B) In the presence of 1 equiv. of O(Bcat)2A reacts with XeF2 or Me3SiCl to afford [{(py)UIV(μ-X)(μ-OBcat)UIV(μ-O2C6H4)(py)}(LA)] (X = F (5A), Cl (6A)), which can be converted into 5 and 6 by heating pyridine solutions of the reaction mixtures at 80 °C for 24 and 48 hours, respectively. | ||
If the O(Bcat)2 by-product produced during the formation of A from [{UVIO2(py)}2(LA)] and 3 equiv. of B2cat2 (Scheme 1) is not removed from the product mixture, a different product is initially formed in the reaction with XeF2. From reactions of A with 1 equiv. of O(Bcat)2 and XeF2 at room temperature, the mixed–bridged complex, [{(py)UIV(μ-F)(μ-OBcat)UIV(μ-O2C6H4)(py)}(LA)] (5A; Scheme 2 Path B), can be isolated in 68% yield. In 5A the bridging oxo ligand has been substituted by a bridging fluorido and bridging catecholatoboroxido ligand. Heating 5A to 80 °C overnight results in complete conversion to the [UIV–F2–UIV] complex 5 (Scheme 2). The by-products formed alongside 5A are presumed to be FBcat and “XeO”, the former of which then reacts at higher temperature to afford 5 and O(Bcat)2 (Scheme 2). Complex 5A is characterised by a chemical shift of 141 ppm in the 19F NMR spectrum, 13 resonances located between 102.25 and −38.01 ppm in the 1H NMR spectrum at 300 K and 25 resonances between 81.60 and −42.64 ppm at 360 K. Unfortunately, no resonances are seen in the 11B NMR spectrum.
To the best of our knowledge, the conversion of a bimetallic μ-oxo complex into a bimetallic μ-fluorido complex without a change in metal oxidation state using XeF2 is unprecedented in either d- or f-block chemistry. The conversion of a μ-oxo ligand in A into two μ-fluorido ligands in 5, or μ-fluorido/μ-boroxido ligands in 5A is likely thermodynamically driven, as the coordination of two μ-X− ligands would provide increased π-donation to the UIV centres and account for the decrease in π-donation from the bent U–O–U oxo ligand (see DFT calculations in the Discussion section below).
Lastly, A reacts with 2 equiv. of Me3SiCl at 80 °C over the course of 48 hours to provide [{(py)UIV(μ-Cl)2UIV(μ-O2C6H4)(py)}(LA)] (6) as a brown/yellow solid in 57% isolated yield; O(SiMe3)2 is produced during the reaction and provides a thermodynamic driving force (Scheme 2 Path A). Complex 6 may also be accessed by treating 1 with 2 equiv. of [HPy]Cl (Scheme 2). Complex 6 possesses two bridging chlorido ligands between the two U centres and gives rise to 13 resonances in the respective 1H NMR spectrum ranging from 65.90 to −39.25 ppm. Similarly to the reactivity of A with XeF2, treating A with 2 equiv. of Me3SiCl at room temperature in the presence of O(Bcat)2 provides a new compound that shows a broad singlet in the 11B NMR spectrum at 435 ppm and 20 resonances from 100.8 to −38.30 ppm in the 1H NMR spectrum at 300 K. Based on the spectroscopic data collected, the bulk product is identified as [{(py)UIV(μ-Cl)(μ-OBcat)UIV(μ-O2C6H4)(py)}(LA)] (6A). Unfortunately, all attempts to obtain X-ray quality crystals of 6A led to the isolation of 6, as 6 is the significantly more thermodynamically stable of the two. Similarly to the formation of 5via5A, A is anticipated to react with 2 equiv. of Me3SiCl and O(Bcat)2 to provide 6A, ClBcat and O(SiMe3)2. Further heating of a pyridine solution of the generated 6A and ClBcat at 80 °C for 48 hours affords the [UIV–Cl2–UIV] complex 6 and O(Bcat)2 (Scheme 2 Path B).
The reactivity of A towards other silanes was also investigated; however, no reactions occurred with Ph2SiH2, Et3SiH, Me3SiOTf and Si2Me6 even after heating at 120 °C in pyridine for several days, and no reaction occurs when A is exposed to CO2.
B. Solid-state structures of complexes 1–6
X-ray quality crystals of 1·3THF were obtained by vapour diffusion of hexanes into a solution of 1 in THF at room temperature (Fig. 2A). The U–O(3) bond lengths of the bridging hydroxo ligands are 2.322(3) and 2.325(3) Å, and the U(1)–O(3)–U(1′) bond angle is 108.4(1)°, giving rise to a U⋯U separation of 3.7696(3) Å that is significantly contracted relative to A (3.9557(4) Å). Surprisingly, there is only one other example of a UIV/UIV complex bearing a bridging hydroxo ligand between the two metal centres that has been crystallographically characterised; the U–O(H) bond lengths in [K(2.2.2-crypt)][{((Neop,MeArO)3tacn)UIV}2(μ-O)(μ-OH)] are 2.282(3)and 2.267(3) Å (ref. 25) which are similar to those seen in 1.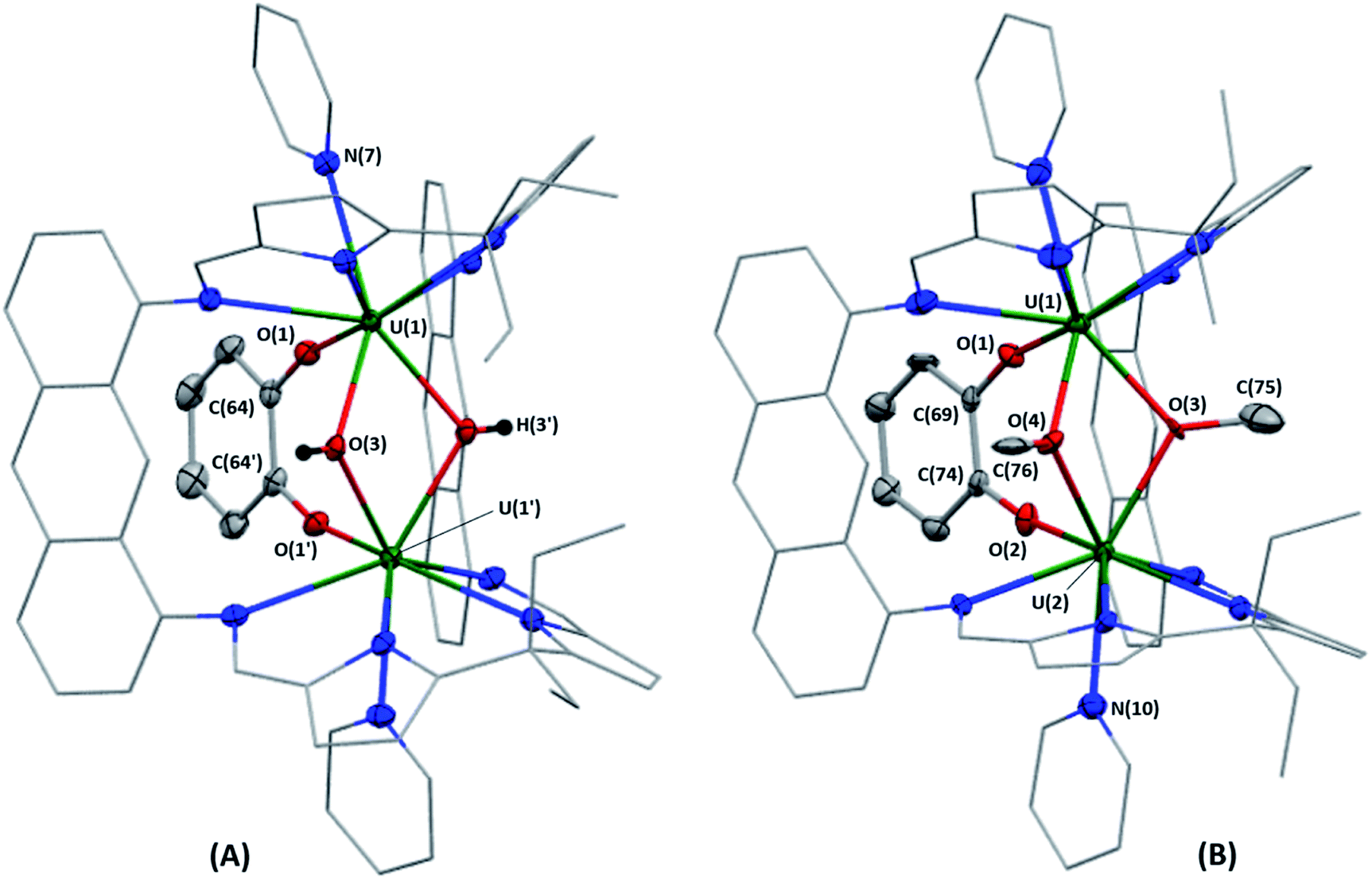 | ||
| Fig. 2 Solid-state structures of 1·3THF (A) and 2-OMe/OH·C6H6 (B). Displacement ellipsoids are drawn with 50% probability, and carbon atoms of LA and U-coordinated solvent molecules drawn wireframe. For clarity, hydrogen atoms (except for H(3) and H(3′) of 1·3THF), lattice solvent and the lower-fractional occupancy disordered components of 1·3THF (C(10)) and 2-OMe/OH·C6H6 (C(8)) are omitted. Furthermore, C(76) of 2-OMe/OH·C6H6 possesses partial occupancy (0.69). Selected bond lengths [Å] and bond angles [°] for 1·3THF: U(1)–O(1), 2.139(3); U(1)–O(3), 2.325(3); U(1)–O(3′), 2.322(3); O(1)–C(64), 1.340(5); U(1)⋯U(1′), 3.7696(3); U(1)–O(3)–U(1′), 108.4(1). Selected bond lengths [Å] and angles [°] for 2-OMe/OH·C6H6: U(1)–O(1), 2.112(4); U(1)–O(3), 2.358(5); U(1)–O(4), 2.345(4); U(2)–O(2), 2.113(4); U(2)–O(3), 2.347(4); U(2)–O(4), 2.379(4); O(1)–C(69), 1.362(7); O(2)–C(74), 1.368(7); O(3)–C(75), 1.429(9); O(4)–C(76), 1.47(1); U(1)⋯U(2), 3.7763(5); U(1)–O(3)–U(2), 106.8(2); U(1)–O(4)–U(2), 106.2(2). | ||
Complex 2 was crystallised by vapour diffusion of hexanes into a benzene solution at room temperature to afford [{(py)UIV(μ-OMe)1.69(μ-OH)0.31UIV(μ-O2C6H4)(py)}(LA)]·C6H6 (2-OMe/OH·C6H6) in which one bridging methoxy ligand is fully occupied and the other bridging ligand has partial occupancy between a methoxy and a hydroxo ligand (Fig. 2B). Unfortunately, the partially occupied hydrogen atom of the hydroxo ligand could not be located in the difference Fourier map. The U–O(3) and U–O(4) bond lengths range from 2.345(4)–2.379(4) Å, which are elongated relative to the UIV/UIV bridging alkoxy complexes K2[{UIV{OSi(OtBu)3}3}2(μ-OCH3)(μ-O)(μ-H)] (U–OMe = 2.30(1), 2.31(1) Å),26 [(tBuNON)U(OiPr)(μ-OiPr)]2 (tBuNON = O(SiMe2NtBu)2; U–OiPr = 2.33(2) Å)27 and [UIV(COT)(S2PPh2)(μ-OMe)]2 (U–OMe = 2.262(4), 2.348(4) Å).28 The bridging U(1)–O(3)–U(2) and U(1)–O(4)–U(2) bond angles are 106.8(2) and 106.2(2)°, respectively, giving rise to a U⋯U separation of 3.7763(5) Å.
Complex 3·5py crystallised by vapour diffusion of hexanes into a solution of 3 in pyridine at room temperature. In the solid-sate structure (Fig. 3A), the U–S bond lengths range from 2.782(1)–2.791(1) Å and are similar to those in [{UIV(OAr)}2(μ-η2:η2-S2)(LA)] (OAr = OC6H2-2,4,6-tBu3), which range from 2.707(3)–2.8229(8) Å,29 but are significantly shorter than those in [{UIV(TrenTIPS)}2(μ-η2:η2-S2)] (TrenTIPS = N(CH2CH2NSiiPr3)3) and [{UIV{(SiMe2NPh)3-tacn}}2(μ-η2:η2-S2)] ((Me2SiNHPh)3-tacn = 1,4,7-tris((dimethylsilyl)phenylamino)-1,4,7-triazacyclononane) which range from 2.867(1)–2.928(2) Å (ref. 30) and 2.855(2)–2.907(3) Å,31 respectively. However, the U–S bond lengths in 3 are significantly longer than those in the UIV–S–UIV complexes [{UIV{N(SiMe3)2}3}2(μ-S)] (2.640(4), 2.680(4) Å),32 [{UIV(TrenTIPS)}2(μ-S)] (2.6903(6) Å)30 and [{UIV{(SiMe2NPh)3-tacn}}2(μ-S)] (2.711(3), 2.703(3) Å),31 indicating the bridging ligand in 3 is best described as a μ-[S2]2− ligand as opposed to two μ-S2− ligands. The S–S distance in 3 is 2.108(2) Å, which is in good agreement with the aforementioned three UIV–(S22−)–UIV complexes (S–S = 2.118(3),29 2.104(2)30 and 2.105(5) Å,31 respectively). The U(1)–S(1)–U(2) and U(1)–S(2)–U(2) bond angles in 3 are 105.12(4) and 104.84(4)°, respectively, and the U(1)⋯U(2) separation is 4.4194(3) Å, which is significantly elongated relative to A and B,18 and a consequence of the larger ionic radius of sulfur relative to oxygen.33
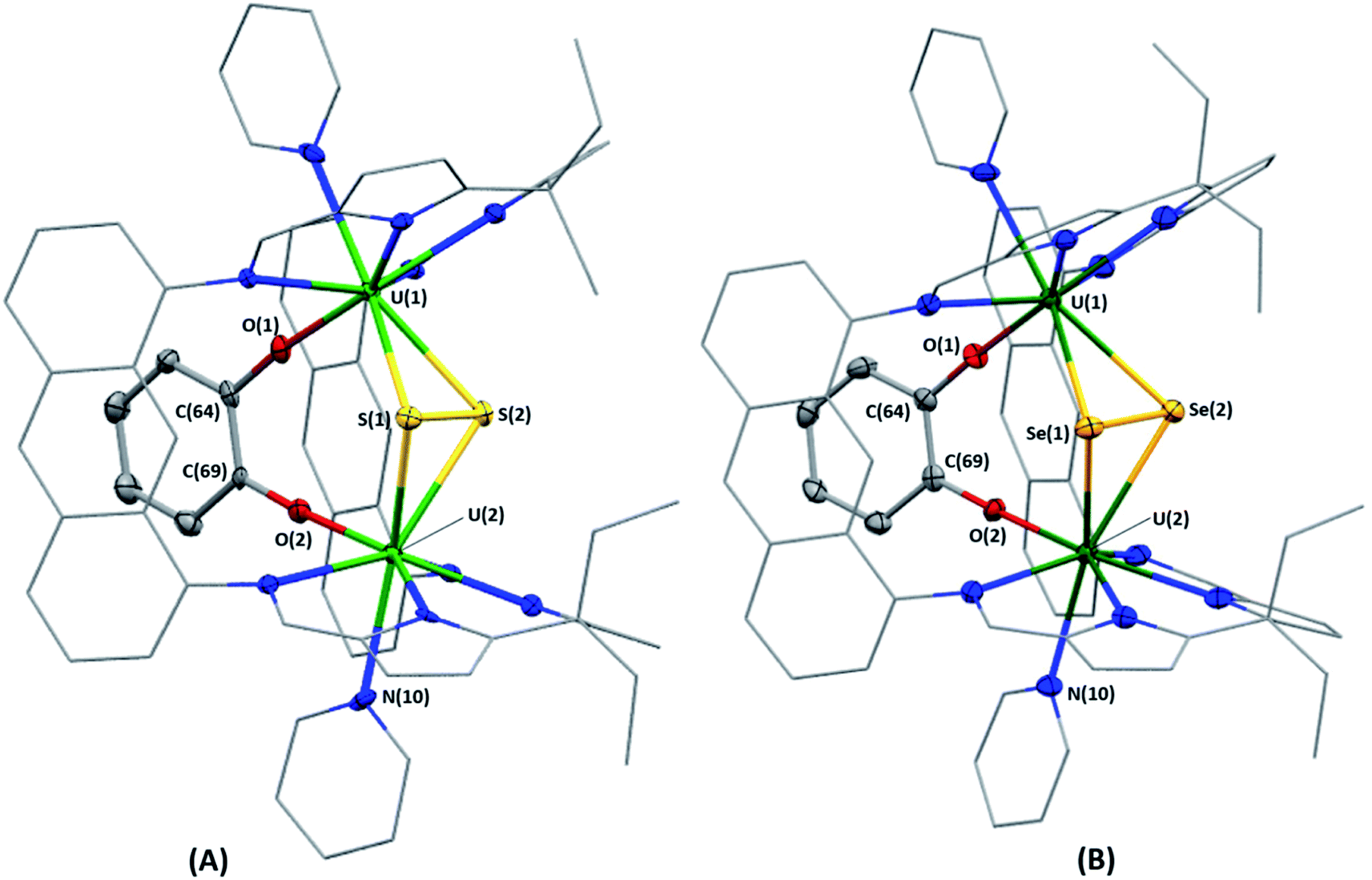 | ||
| Fig. 3 Solid-state structures of 3·5py (A) and 4·2CH2Cl2 (B). Displacement ellipsoids are drawn with 50% probability, and carbon atoms of LA and U-coordinated solvent molecules drawn wireframe. For clarity, hydrogen atoms, lattice solvent and lower-fractional occupancy disorder components of one of the U-coordinated pyridine ligands in 4·2CH2Cl2 (N(10), C(70)–C(74)) are omitted. Selected bond lengths [Å] and bond angles [°] for 3·5py (S(1)/S(2) refers to the centre of the bond between S(1) and S(2)): U(1)–S(1): 2.785(1); U(1)–S(2): 2.791(1); U(2)–S(2): 2.785(1); U(2)–S(1): 2.782(1); S(1)–S(2): 2.108(2); U(1)–O(1): 2.096(3); U(2)–O(2): 2.109(3); O(1)–C(64): 1.373(5); O(2)–C(69): 1.352(5); U(1)⋯U(2): 4.4194(3); U(1)–S(1)/S(2)–U(2): 118.0. Selected bond lengths [Å] and bond angles [°] for 4·2CH2Cl2 (Se(1)/Se(2) refer to the centroid between Se(1) and Se(2)): U(1)–Se(1): 2.9354(7); U(1)–Se(2): 2.9273(6); U(2)–Se(2): 2.9239(7); U(2)–Se(1): 2.9333(6); Se(1)–Se(2): 2.3682(9); U(1)–O(1): 2.107(4); U(2)–O(2): 2.104(4); O(1)–C(64): 1.362(7); O(2)–C(69): 1.346(7); U(1)⋯U(2): 4.5433(3); U(1)–Se(1)/Se(2)–U(2): 115.9. | ||
X-ray quality crystals of 4·2CH2Cl2 were obtained by diffusion of hexanes vapour into a solution of 4 in CH2Cl2 at room temperature, and the solid-state structure is displayed in Fig. 3B; residual electron density from highly disordered lattice solvent was removed from the structure using the “solvent mask” feature of Olex2 (84.6 electrons per unit cell, equal to 2 molecules of CH2Cl2). The U–Se bond lengths in 4 range from 2.9239(7)–2.9354(7) Å and the Se(1)–Se(2) bond length is 2.3682(9) Å; these compare well with [{((AdArO)3N)UIV}2(μ-η2:η2-Se2)(μ-dme)] (U–Se = 2.942(1)–3.079(1) Å; Se–Se = 2.377(1) Å),34 which also features a [μ-η2:η2-Se2]2− ligand between the two UIV centres. Furthermore, the U–Se bond lengths in 4 are significantly longer than those reported for [{UIV{N(SiMe3)2}3}2(μ-Se)] (2.727(2)–2.751(2) Å),32 [{((AdArO)3N)UIV(dme)}2(μ-Se)] (U–Se = 2.830(1), 2.816(1) Å), [{((tBuArO)3tacn)UIV}2(μ-Se)] ((tBuArOH)3tacn = 1,4,7-tris(3,5-di-tert-butyl-2-hydroxybenzyl)-1,4,7-triazacyclononane; 2.7188(4) Å) and [Na(dme)3]2[{((AdArO)3N)UIV(μ-Se)}2] (U–Se = 2.819(1)–2.866(1) Å)35 which feature bridging [Se]2− ligands between the two U centres, rendering the diselenide ligand in 4 best described as a bridging [Se2]2− ligand. The U(1)–Se(1)–U(2) and U(1)–Se(2)–U(2) bond angles in 4 are 101.46(2) and 101.88(2)°, respectively, and the U(1)⋯U(2) separation is 4.5433(3) Å, which similarly to 3 is significantly greater than in A.
Bright yellow X-ray quality crystals of 5·4py were grown by vapour diffusion of hexanes into a solution of 5 in pyridine at room temperature (Fig. 4A); similarly to 4·2CH2Cl2, residual electron density from highly disordered lattice solvent was removed from the structure using the “solvent mask” feature of Olex2 (90.6 electrons per unit cell, equal to approximately two molecules of pyridine or one py and one hexane molecule). While all attempts to obtain X-ray quality crystals of 5A at room temperature afforded 5, cooling a 1,2-dimethoxyethane (dme) solution of 5A to −20 °C for days provided 5A·3dme as fluorescent green needles suitable for X-ray diffraction (Fig. 4B). The U–F distances in 5 and 5A range from 2.299(3)–2.391(3) Å, which compare well with 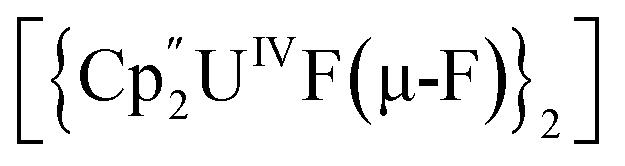 (Cp′′ = C5H3-1,3-(SiMe3)2) and
(Cp′′ = C5H3-1,3-(SiMe3)2) and 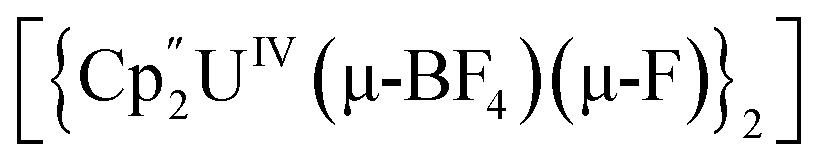 , which possess U–F bond lengths of 2.297(5) and 2.343(5) Å,36 and 2.260(5) and 2.354(5) Å, respectively.37 The U(1)–F(1)–U(2) and U(1)–F(2)–U(2) bond angles are 109.7(1)° in 5, whereas the U(1)–F(1)–U(2) and U(1)–O(3)–U(2) bond angles are 110.86(7)° and 106.53(9) in 5A, respectively. The U(1)⋯U(2) separations in 5 and 5A are 3.8349(3) and 3.8490(2) Å, respectively, which are contracted relative to A primarily due to the more acute U–X–U (X = F or OR) bond angles in 5 and 5A.
, which possess U–F bond lengths of 2.297(5) and 2.343(5) Å,36 and 2.260(5) and 2.354(5) Å, respectively.37 The U(1)–F(1)–U(2) and U(1)–F(2)–U(2) bond angles are 109.7(1)° in 5, whereas the U(1)–F(1)–U(2) and U(1)–O(3)–U(2) bond angles are 110.86(7)° and 106.53(9) in 5A, respectively. The U(1)⋯U(2) separations in 5 and 5A are 3.8349(3) and 3.8490(2) Å, respectively, which are contracted relative to A primarily due to the more acute U–X–U (X = F or OR) bond angles in 5 and 5A.
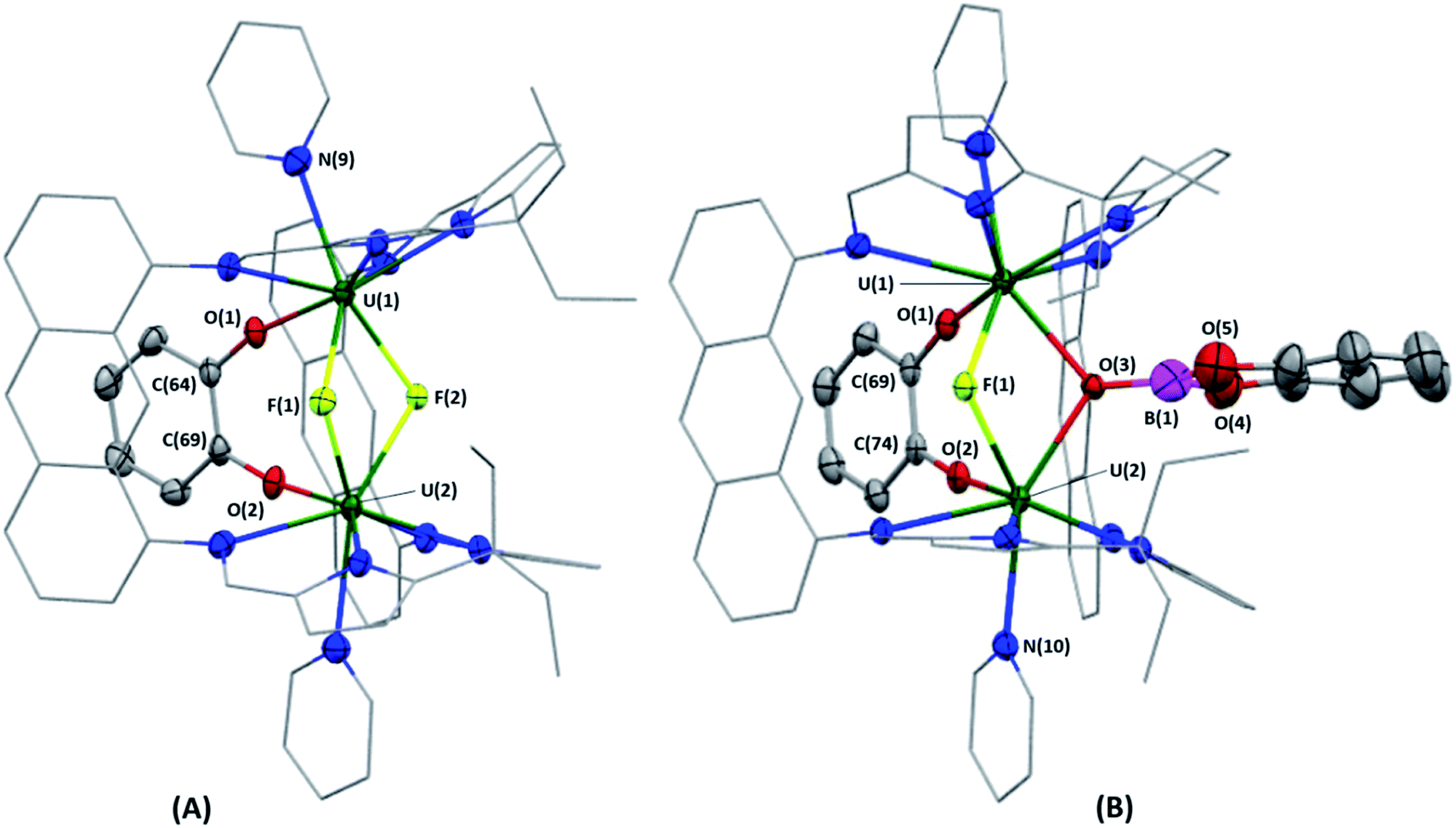 | ||
| Fig. 4 Solid-state structure of 5·4py (A) and 5A·3dme (B). Displacement ellipsoids are drawn at 50% probability, and carbon atoms of LA and U-coordinated solvent molecules are drawn wireframe. For clarity, hydrogen atoms and lattice solvent are omitted. Selected bond lengths [Å] and bond angles [°] for 5·4py: U(1)–F(1), 2.391(3); U(2)–F(1), 2.299(3); U(1)–F(2), 2.307(3); U(2)–F(2), 2.382(3); U(1)–O(1), 2.120(4); U(2)–O(2), 2.106(4); O(1)–C(64), 1.347(6); O(2)–C(69), 1.362(6); U(1)⋯U(2), 3.8349(3); U(1)–F(1)–U(2), 109.7(1); U(1)–F(2)–U(2), 109.7(1). Selected bond lengths [Å] and bond angles [°] for 5A·3dme: U(1)–O(1), 2.106(2); U(2)–O(2), 2.106(2); O(1)–C(69), 1.360(4); O(2)–C(74), 1.355(4); U(1)–F(1), 2.300(2); U(2)–F(1), 2.374(2); U(1)–O(3), 2.429(2); U(2)–O(3), 2.373(2); O(3)–B(1), 1.379(7); O(4)–B(1), 1.407(7); O(5)–B(1), 1.431(7); U(1)⋯U(2), 3.8490(2); U(1)–F(1)–U(2), 110.86(7); U(1)–O(3)–U(2), 106.53(9). | ||
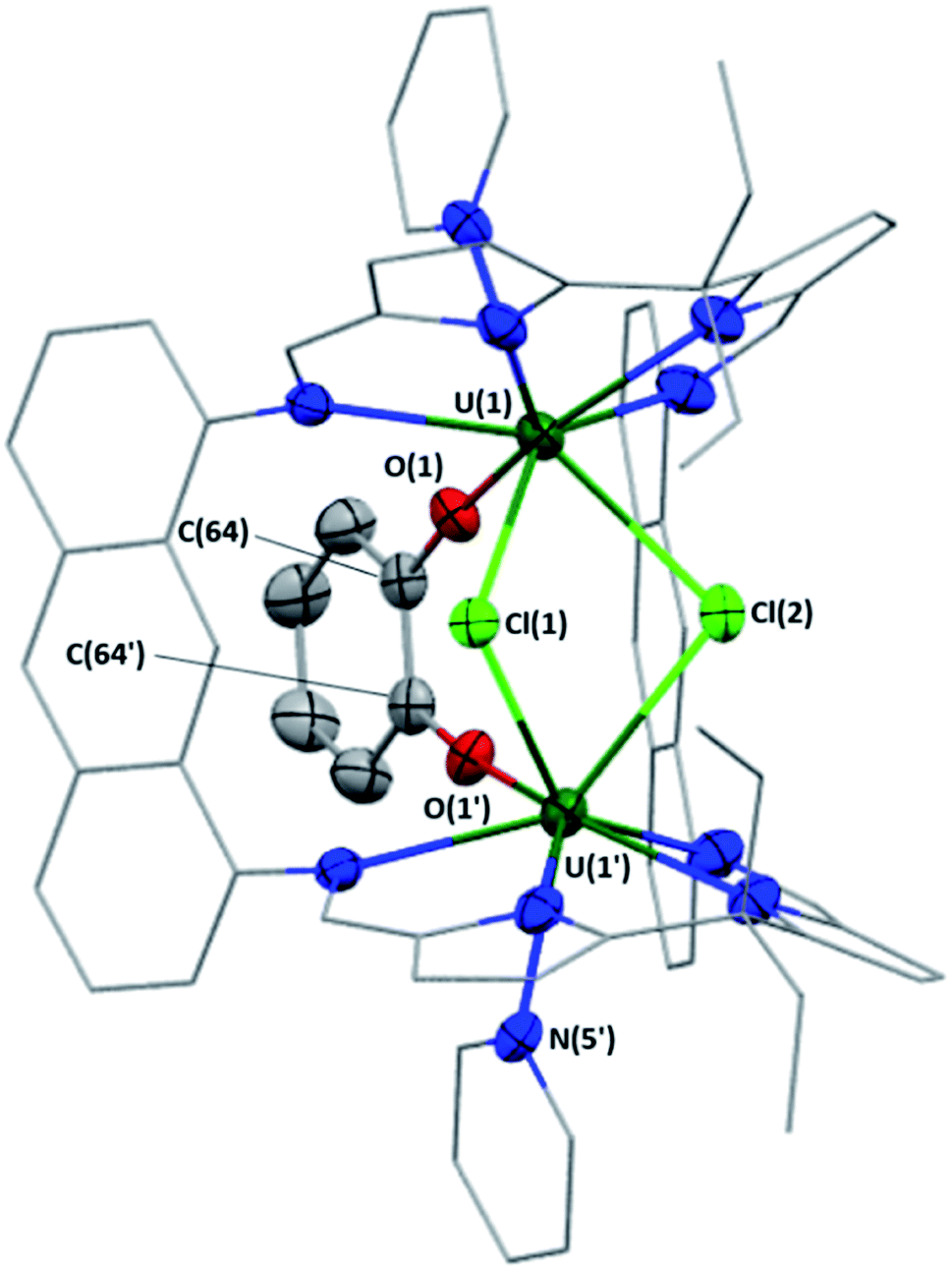 | ||
| Fig. 5 Solid-state structure of 6. Displacement ellipsoids are drawn at 50% probability, carbon atoms of LA and U-coordinated solvent molecules are drawn wireframe. For clarity, hydrogen atoms and the lower-fractional occupancy disorder component of 6 (C(8) and one of the U-coordinated pyridine ligands, N(5), C(59)–C(63)) are omitted. Selected bond lengths [Å] and bond angles [°] for 6: U(1)–Cl(1), 2.826(1); U(1)–Cl(2), 2.808(1); U(1)–O(1), 2.091(3); O(1)–C(64), 1.367(5); U(1)⋯U(1′), 4.1681(1); U(1)–Cl(1)–U(1′), 95.03(5); U(1)–Cl(2)–U(1′), 95.86(5). | ||
X-ray quality crystals of 6 were obtained by vapour diffusion of hexanes into a pyridine solution of 6 at room temperature (Fig. 5); residual electron density from highly disordered lattice solvent was removed from the structure using the “solvent mask” feature of Olex2 (393.3 electrons per unit cell, equal to approximately one pyridine and seven hexane molecules). The U–Cl bond lengths range from 2.808(1)–2.826(1) Å, which compare well with [{Mes2(p-OMePh)corrole}UIV(μ-Cl)(dme)]2 (U–Cl = 2.873(2), 2.840(1) Å),38 but are elongated relative to [UIV{N(SiMe2tBu)2}{N(SiMe2tBu)(SiMetBuCH2-κ2-N,C)}(μ-Cl)]2 (U–Cl = 2.799(2) Å)39 and [(iPrNON)UIVCl(μ-Cl)]2 (iPrNON = O{SiMe2N(C6H3-2,6-iPr2)}2; U–Cl = 2.754(3) Å),40 all of which being UIV/UIV complexes exhibiting a bridging-bis(chlorido) structural motif. The U(1)–Cl(1)–U(1′) and U(1)–Cl(2)–U(1′) bond angles are 95.03(5) and 95.86(5)°, respectively, and the U(1)⋯U(1′) separation is 4.1681(1) Å.
The U–O bond lengths to the μ-oxo ligand, U–O–U bond angles and U⋯U separations are 2.090(2) Å, 142.3(3)° and 3.9557(4) Å in bent A, respectively, and 2.139(2)/2.112(2) Å, 176.2(1)° and 4.2485(2) Å in linear B, respectively (Table 1).18 In comparison, [{((Neop,MeArO)3tacn)UIV}2(μ-O)] and [{{(AdArO)3N}UIV}(μ-O)(μ-dme)] ((AdArO)3N = tris(2-hydroxy-3-adamantyl-5-methylbenzyl)amine; Ad = adamantyl), which exhibit reactivity towards CO2 at the μ-oxo have U–O bond lengths of 2.0869(2) and 2.1036(2) Å, U–O–U bond angles of 180.0(2)° and U⋯U separations of 4.174 and 4.207 Å, respectively.14,41 Therefore, while the U–O bond lengths compare relatively well with each other, the U–O–U bond angle and U⋯U separation in linear B are more similar to the reactive UIV/UIV bridging oxo complexes than in A.
| a X refers to the bridging ligand between the two U centres. | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Complex number | A | B | 1 | 2 | 3 | 4 | 5 | 5A | 6 |
| Identity of central atom(s) X | O | O | 2 × OH | OH, OMe | (S2) | (Se2) | 2 × F | F, OBcat | 2 × Cl |
| U⋯U [Å] | 3.9557(4) | 4.2485(2) | 3.7696(3) | 3.7763(5) | 4.4194(3) | 4.5433(3) | 3.8349(3) | 3.8490(2) | 4.1681(1) |
| U–X [Å] | 2.090(2) | 2.139(2), 2.112(2) | 2.322(3), 2.325(3) | 2.345(4), 2.358(5), 2.347(4), 2.379(4) | 2.785(1), 2.791(1), 2.782(1), 2.785(1) | 2.9354(7), 2.9273(6), 2.9333(6), 2.9239(7) | 2.391(3), 2.307(3), 2.299(3), 2.382(3) | X = F: 2.300(2), 2.374(2). X = OBcat: 2.429(2), 2.373(2) | 2.826(1), 2.808(1) |
| U(1)–O(1) [Å] | 2.128(3) | N/A | 2.139(3) | 2.112(4) | 2.096(3) | 2.107(4) | 2.120(4) | 2.106(2) | 2.091(3) |
| U(2)–O(2) [Å] | N/A | N/A | N/A | 2.113(4) | 2.109(3) | 2.104(4) | 2.106(4) | 2.106(2) | N/A |
| C–O(1) [Å] | 1.340(6) | N/A | 1.340(5) | 1.362(7) | 1.373(5) | 1.362(7) | 1.347(6) | 1.360(4) | 1.367(5) |
| C–O(2) [Å] | N/A | N/A | N/A | 1.368(7) | 1.352(5) | 1.346(7) | 1.362(6) | 1.355(4) | N/A |
| X–X [Å] | N/A | N/A | N/A | N/A | 2.108(2) | 2.3682(9) | N/A | N/A | N/A |
| U–X–U [°] | 142.3(3) | 176.2(1) | 108.4(1) | 106.2(2), 106.8(2) | 105.12(4), 104.84(4) | 101.46(2), 101.88(2) | 109.7(1), 109.7(1) | X = F: 110.86(7). X = OBcat: 106.53(9) | 95.03(5), 95.86(5) |
The U–O bond lengths between the U centres and the bridging dianionic catecholate ligand in complexes 1–6 range from 2.091(3)–2.139(3) Å. In addition, the C–O bond lengths within the catecholate ligand range from 1.340(5)–1.373(5) Å. Both of these are similar to those found for complex A (U(1)–O(1)/U(1′)–O(1′) = 2.128(3) Å; C(64)–O(1)/C(64′)–O(1′) = 1.340(6) Å),18 supporting the assignment of complexes 1–6 as two UIV centres bridged by a dianionic catecholate. The U⋯U separations in complexes 1–6 range from 3.7696(3) Å in 1 to 4.5433(3) Å in 4 (Table 1). This wide variation is a result of the ability of the U centre to move out of the N4-plane towards the endocyclic cavity. This out-of-plane distance was found to be the greatest for complex 1, in which U(1) and U(2) are displaced towards the centre of the molecule by 0.791 Å from their respective N4-donor planes, and the smallest for complex 4, in which the out-of-plane distances for U(1) and U(2) are 0.531 and 0.524 Å. For comparison, the non-catecholate bridged UIV/UIV Pacman complexes B (0.235, 0.467 Å), [{(py){(py)catBO}UIVOUIV(OBcat)(py)}(LA)] (0.262, 0.454 Å), [{(py)(Ph2HSiO)UIVOUIV(OSiHPh2)(py)}(LA)] (0.27 Å), [{(THF)(Ph2HSiO)UIVOUIV(OSiHPh2)(THF)}(LA)] (0.283 Å), [{(ArO)UIV(μ-η2:η2-S2)UIV(OAr)}(LA)] (0.067 Å) and [{(ArO)UIVSUIV(OAr)}(LA)] (0.034, 0.097 Å; Ar = C6H2-2,4,6-tBu3) exhibit significantly less out-of-plane distances of the U centres.18,29 Such ligand flexibility, in combination with the tethering bidentate catecholate ligand allows for coordination of a wide variety of X-ligand units between the UIV centres. Finally, the U–Nimine, U–Npyridine and U–Npyrrolide bond lengths range from 2.533(5)–2.662(3), 2.564(5)–2.645(3) and 2.441(4)–2.508(3) Å, respectively, and compare well with previously published UIV-Pacman complexes.18,42
C. Reactivity of the linear oxo-bridged UIV/UIV Pacman complex, B
The μ-oxo group in A is very bent, U–O–U = 142.3(3)° and its substitution by larger ligating atoms forming U–S or U–Se bonds, which would normally be expected to form a weaker bond to an oxophilic uranium(IV) centre, lead us to hypothesise that the structural constraints imposed by the (μ-O2C6H4) ligand renders the μ-oxo group reactive. To test this, we compared the reactivity of the analogous UIV/UIV bis(pinacolatoboroxido) complex, [{(py)(pinBO)UIVOUIV(OBpin)(py)}(LA)] (B), which does not possess a bridging catecholate ligand, and exhibits a near linear U–O–U bonding.18Reactions of B with XeF2, H2O and Me3SiCl were carried out, and the products identified in situ. While B decomposes to [UVIO2(py)(H2LA)] and a mixture of unidentifiable species when treated with XeF2, it reacts cleanly with 2 equiv. of H2O to provide [{(py)HOUIVOUIVOH(py)}(LA)] (7), as determined by 1H NMR spectroscopy, giving rise to thirteen resonances between 48.23 and −34.34 ppm, diagnostic of C2h symmetry. The 11B NMR spectrum indicated that HOBpin was the sole by-product of the reaction (Scheme 3). In fact, treating B with 10 equiv. of H2O produces 7 and 2 equiv. of HOBpin as the sole products, even after 24 hours at room temperature, indicating that the boroxido ligands are the preferred site of reaction over the bridging oxo ligand. Treating B with a slight excess of Me3SiCl (3 equiv.) provides [{(py)ClUIVOUIVCl(py)}(LA)] (8), as determined by X-ray crystallography (see ESI†), and Me3SiOBpin as a reaction by-product (as determined by 11B NMR spectroscopy). Unreacted Me3SiCl, and Me3SiOBpin and O(SiMe3)2 in an approximate 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio were observed in the 29Si_INEPT NMR spectrum, indicating that the bridging oxo ligand in B possesses some reactivity towards Me3Si+, and a mixture of paramagnetic species were observed by 1H NMR spectroscopy, a likely result of isomerisation of the coordinated chlorido ligands from axial to equatorial coordination sites (see ESI†). Conversely to treating B with 10 equiv. of H2O, treating B with 10 equiv. of Me3SiCl yields a mixture of products, with a nearly 1:1 ratio of Me3SiOBpin:O(SiMe3)2 (as observed by 29Si NMR spectroscopy), verifying that the boroxido and bridging oxo ligands in B are reactive towards Me3Si+.
:1 ratio were observed in the 29Si_INEPT NMR spectrum, indicating that the bridging oxo ligand in B possesses some reactivity towards Me3Si+, and a mixture of paramagnetic species were observed by 1H NMR spectroscopy, a likely result of isomerisation of the coordinated chlorido ligands from axial to equatorial coordination sites (see ESI†). Conversely to treating B with 10 equiv. of H2O, treating B with 10 equiv. of Me3SiCl yields a mixture of products, with a nearly 1:1 ratio of Me3SiOBpin:O(SiMe3)2 (as observed by 29Si NMR spectroscopy), verifying that the boroxido and bridging oxo ligands in B are reactive towards Me3Si+.
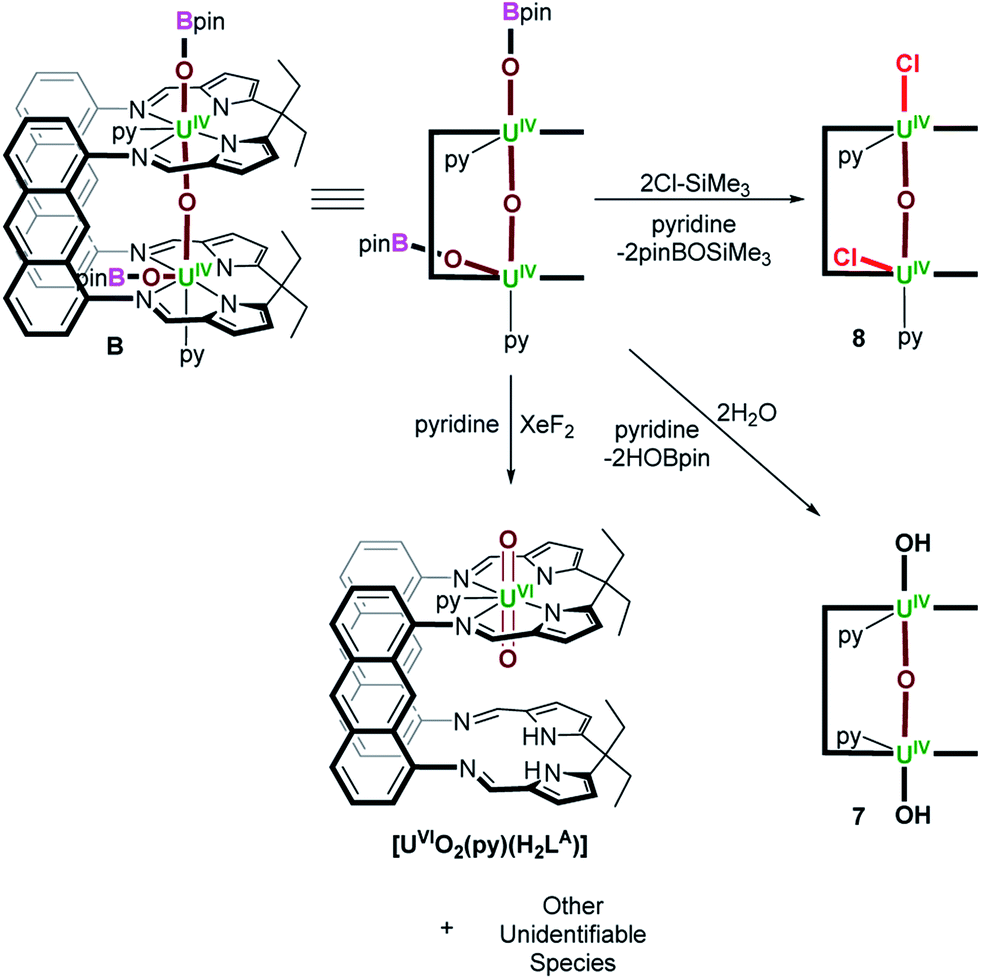 | ||
| Scheme 3 Reactivity of [{(py)(pinBO)UIVOUIV(OBpin)(py)}(LA)] (B) with XeF2, 2 equiv. of H2O and 2 equiv. of Me3SiCl, providing [UVIO2(py)(H2LA)] and a mixture of unidentifiable species, [{(py)HOUIVOUIVOH(py)}(LA)] (7) and [{(py)ClUIVOUIVCl(py)}(LA)] (8), respectively. | ||
Discussion
Complexes 1–6 are synthesised from [{(py)UIVOUIV(μ-O2C6H4)(py)}(LA)] (A) and H2O, MeOH, S8, Se, XeF2 and Me3SiCl, respectively, in which the bridging oxo ligand within the bent UIV–O–UIV core in A (U–O–U = 142.3(3)°) undergoes either functionalisation or substitution. In contrast, [{(py)(pinBO)UIVOUIV(OBpin)(py)}(LA)] (B), which retains a linear UIV–O–UIV structure motif (U–O–U = 176.2(1)°) reacts with H2O and Me3SiCl to provide complexes 7 and 8, respectively, in which reactions have occurred at the axially coordinated boroxido ligands as opposed to the μ-oxo ligand.The reactivity of A with H2O, MeOH, XeF2 and Me3SiCl to form complexes 1, 2, 5 and 6 likely occurs by initial nucleophilic attack of the μ-oxo group on the δ+ entity of each small molecule (i.e. H+, Xe2+ and Me3Si+ in H2O/MeOH, XeF2 and Me3SiCl, respectively), by analogy with previous work that concluded that a nucleophilic μ-oxo group can be generated by reducing steric protection and enabling flexibility. It was noted that [{(Neop,MeArO)3tacn}UIII], [{(tBuArO)mes}UIII] ((tBuArOH)3mes = 1,3,5-trimethyl-2,4,6-tris(2,4-di-tert-butyl-hydroxybenzyl)methylbenzene) and [{(AdArO)3N}UIII] react with CO2 to provide the bridging [CO3]2− complexes, [{((Neop,MeArO)3tacn)UIV}2(μ-CO3)], [{{(tBuArO)mes}UIV}2(μ-CO3)] and [{{(AdArO)3N}UIV}(μ-CO3)], respectively, via the initial formation of a bridging [UIV–O–UIV] complex followed by nucleophilic attack of the bridging oxo on CO2.14,41 Alternatively, [{(Ad,tBuArO)3tacn}UIII] ((Ad,tBuArOH)3tacn = 1,4,7-tris(3-adamantyl-5-tert-butyl-2-hydroxybenzyl)-1,4,7-triazacyclononane) and [{(tBuArO)3tacn}UIII] react with CO2 to provide [{(Ad,tBuArO)3tacn}UIV(η1-OCO)] and [{{(tBuArO)3tacn}UIV}2(μ-O)], respectively, in which no further reactivity of either the [(η1-OCO)]1− or [μ-O]2− ligands with CO2 was observed.43,44 The use of either the structurally more flexible (RArOH)3mes or (RArOH)3N ligand backbones, or sterically less demanding neo-pentyl substituents on the phenolate donors provides a more accessible, and therefore more reactive μ-oxo ligand, whereas the structurally more rigid (RArOH)3tacn ligand backbone in combination with the more sterically demanding tert-butyl or adamantyl substituents on the phenolate donors provide protection for the μ-oxo ligand and quenches its reactivity. Here, the reactivity of A is enabled by the bridging dianionic catecholate ligand, which enforces a less sterically protected and bent U–O–U bond angle (142.3(3)°), and therefore more accessible and reactive bridging oxo ligand.18 The reactivity observed can be attributed to nucleophilic attack by this oxo.
To the best of our knowledge, reactions involving A and elemental sulfur and selenium to provide complexes 3 and 4 are the first examples of any d- or f-block bimetallic μ-oxo complex possessing the ability to activate elemental chalcogens, which in both cases typically requires access to low valent, highly reducing metal precursors such as UIII,7,29–32,34 YbII,45,46 VI/VIII (ref. 47 and 48) and NiI.49 This reactivity sheds light on how uranium reactivity differs from the d-block and remainder of the f-block elements, and how complexes containing groups considered inert by convention can be manipulated for productive transformations.
Reactions involving A and S8 or Se may proceed by one of two ways. First, similarly to the formation of complexes 1, 2, 5 and 6, complex A may be reacting with S8 and Se via initial nucleophilic attack of the bridging oxo ligand on the small molecule substrates. The [UIV–E–UIV] complex (E = S or Se), [{((AdArO)3N)UIV(dme)}2(μ-E)], formed from [((AdArO)3N)UIII(dme)] and either 0.125 equiv. of S8 or 1 equiv. of selenium, have been shown to react further with either 0.125 or 0.375 equiv. of S8, or 1 or 3 equiv. of selenium to yield [{((AdArO)3N)UIV}2(μ-E2)(μ-dme)x] (E = X, x = 0; E = Se, x = 1) and [{((AdArO)3N)UIV}2(μ-η2:η2-E2)2], respectively. It was reasoned that: (1) the bridging [E]2− ligand in [{((AdArO)3N)UIV(dme)}2(μ-E)] is highly nucleophilic; (2) there is reduced steric protection of the μ-E ligand through the use of a flexible amine-tethered ligand backbone; (3) the chalcogens have a propensity to catenate.34 Alternatively, the bridging catecholate ligand could stabilise transient UIII centres to enable a reductive activation pathway given that it may possess three different canonical forms; (A) a dianionic catecholate, (B) a monoanionic 1,2-semiquinone, and (C) a neutral 1,2-benzoquinone (Fig. 6). Given that strongly reducing metals are typically required for the activation of elemental chalcogens,34 resonance structures (B) and (C) could be operative in order to provide access to UIII centres, which would be sufficient for S8 or Se activation (see Fig. 1B for an example of S8 activation by a UIII/UIII complex). It is possible that S8/Se activation may be proceeding via metal-based reactivity of a transient UIII/UIII or UIII/UIV complex whereby short-lived monoanionic 1,2-semiquinone or neutral 1,2-benzoquinone resonance forms of the (μ-O2C6H4) ligand provides an extra 1 to 2 electrons to the metal centres. Given that A reacts with H2O, MeOH, XeF2 and Me3SiCl by nucleophilic attack of the μ-oxo ligand, and does not react with P4, CO or CO2, which may require redox changes in the U–cat–U (cat = catecholate) unit, we believe that the operative route to the formation of complexes 3 and 4 is by the former pathway, in which nucleophilic attack of the bridging oxo ligand on either S8 or Se occurs initially.
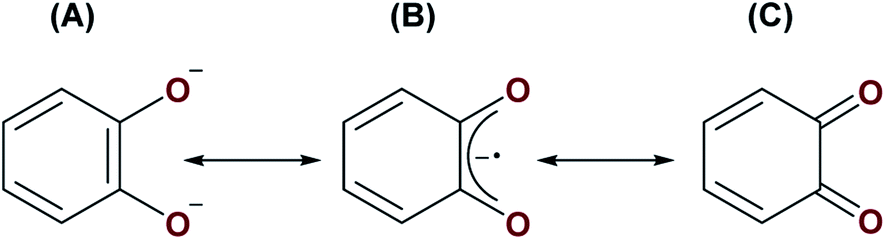 | ||
| Fig. 6 Three possible resonance structures of the catecholate ligand in A: (A) a dianionic catecholate, (B) a monoanionic 1,2-semiquinone, and (C) neutral 1,2-benzoquinone. | ||
An additional factor enabling conversion of A into complexes 1–6 is that the μ-oxo ligand may reside in a strained geometry due to the presence of a bridging catecholate ligand. This could be released upon ejection of the oxo group, with the new ligand bridges providing a more thermodynamically stable geometry.
The most helpful information comes from comparing the reactions of bent oxo A with its linear analogue B. While B reacts with H2O and Me3SiCl to give 7 and 8, respectively, these reactions only take place at the axially coordinated boroxido ligands and not at the μ-oxo ligand. Overall, these results suggest that the bridging catecholate ligand plays a vital role in the formation/stabilisation of 1–6.
In order to understand the high reactivity of complex A, DFT calculations (B3PW91) were carried out and the bonding situation analysed. The optimised geometry is in excellent agreement with the experimental one with the U–O bonds 2.10 Å (vs. 2.09 Å, Table 1), the U–O(cat) 2.13 Å (vs. 2.13 Å) and the U–O–U angle 144° (vs. 142°). Furthermore, the calculated spin density on the complexes A and B concurs with the +IV oxidation state (2.13 e− on each uranium centre, Fig. S53†). Both indicate the suitability of the computational method to describe such a system. By scrutinising the molecular orbitals, it has been possible to locate two molecular orbitals that describe the U–O–U bonding interaction (Fig. 7).
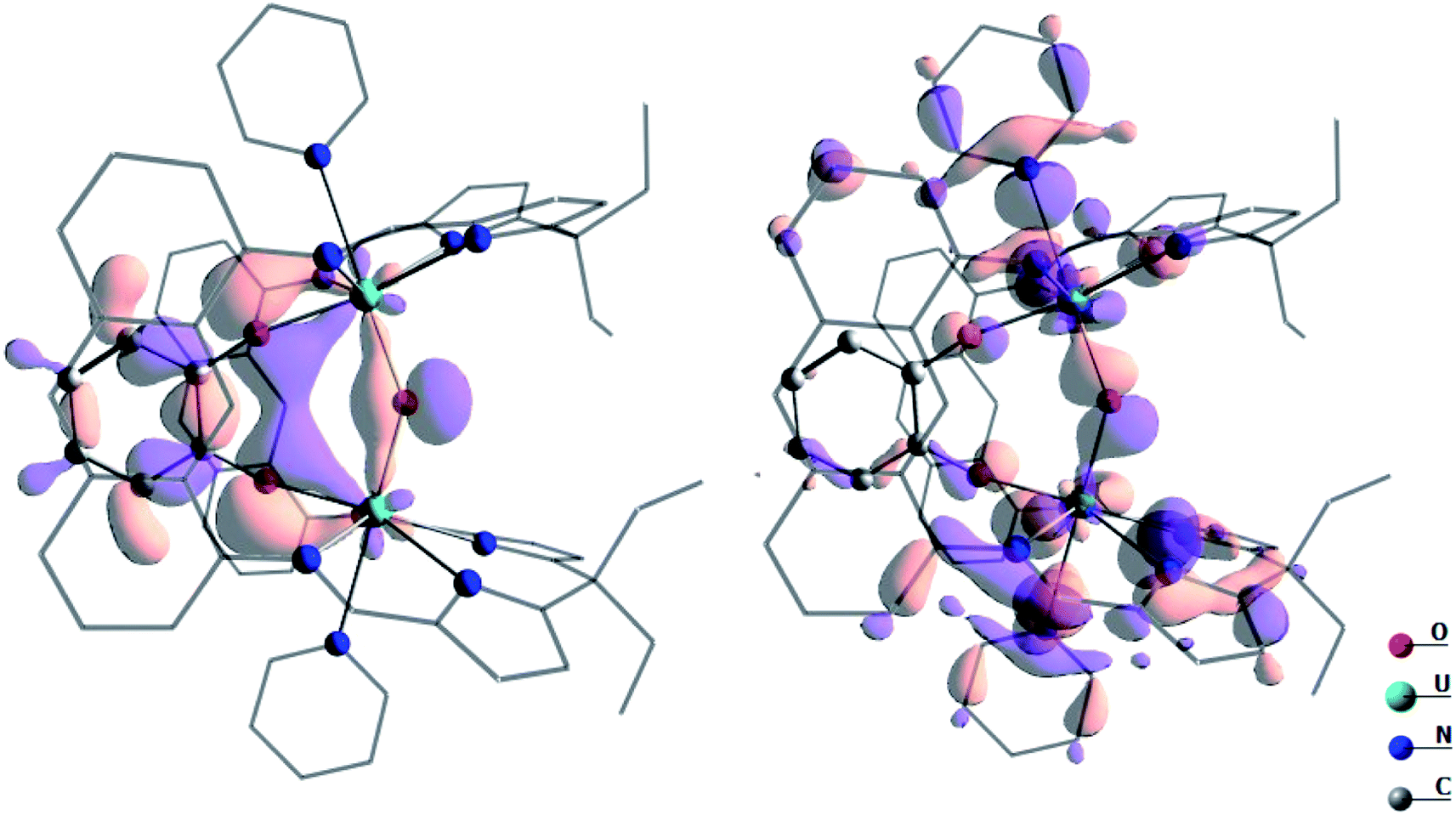 | ||
| Fig. 7 Depiction of the two molecular orbitals describing the U–O bonding in complex A. These two orbitals are 2.42 eV lower in energy than the SOMO. | ||
These bonding interactions involve a sp hybrid orbital on the bridging O (91%) and a df hybrid from U (9%). Natural Bonding Orbital (NBO) analysis shows a similar bonding description, except that the bonding interaction is found to be pure donation from two sp lone pairs on the bridging O towards empty df orbitals on U (donation of 198 kcal mol−1, 90% σ and 10% π, at the second order donor–acceptor; i.e. donation of electron density where there is no bond currently). Interestingly the Wiberg Indexes (WBI) are 0.89 for the U–O bonds (0.82 for the U–O(cat) ones) indicating strong covalent contributions to the bonding. Therefore, the U–O interactions in complex A are found to be covalent and overlap-driven. A similar analysis was carried out for the linear oxo complex B and the WBI of the U–O bond is slightly lower (0.86). In this case, the bond appears to be even more polarised toward O in complex B (97%) than in A. At the NBO level, a donation from sp orbital on O to an spdf orbital on U is found (134 kcal mol−1 at the second order donor–acceptor which is 70% σ and 30% π). For comparison, the U–O WBI for a bent-oxo calculated intermediate in [{((MeArO)3mes)UIV}2(μ-O)], is 0.84. This intermediate is proposed to form during the reaction between [((MeArO)3mes)UIII] and CO2 to yield [{((MeArO)3mes)UIV}2(μ-CO3)].50 However, the U–O WBI for [{((Neop,MeArO)3tacn)UIV}2(μ-O)], is an isolable linear oxo complex, is only 0.55.51 Therefore, the covalency increases with the bending of the U–O–U bond angle. The presence of the two covalent U–O bonds induces the localisation of two lone pairs on O (as in a water molecule) and therefore the bending of the structure. The localisation of the lone pairs at the oxo (and its bent structure) helps categorise the reaction of the oxo with small molecules as nucleophilic since the lone pairs are ready to overlap with an incoming empty orbital, such as for example with water or methanol, and from which point the oxo is easily protonated.
The presence of the catecholate ligand is also important electronically. Indeed, as can be seen on the highest energy SOMO (Fig. 8) of the system, which is mainly a linear combination of non-bonding f orbitals on the uranium centres, the π system of the catecholate stabilises the complex by counterbalancing the reduced level of π donation from the oxo.
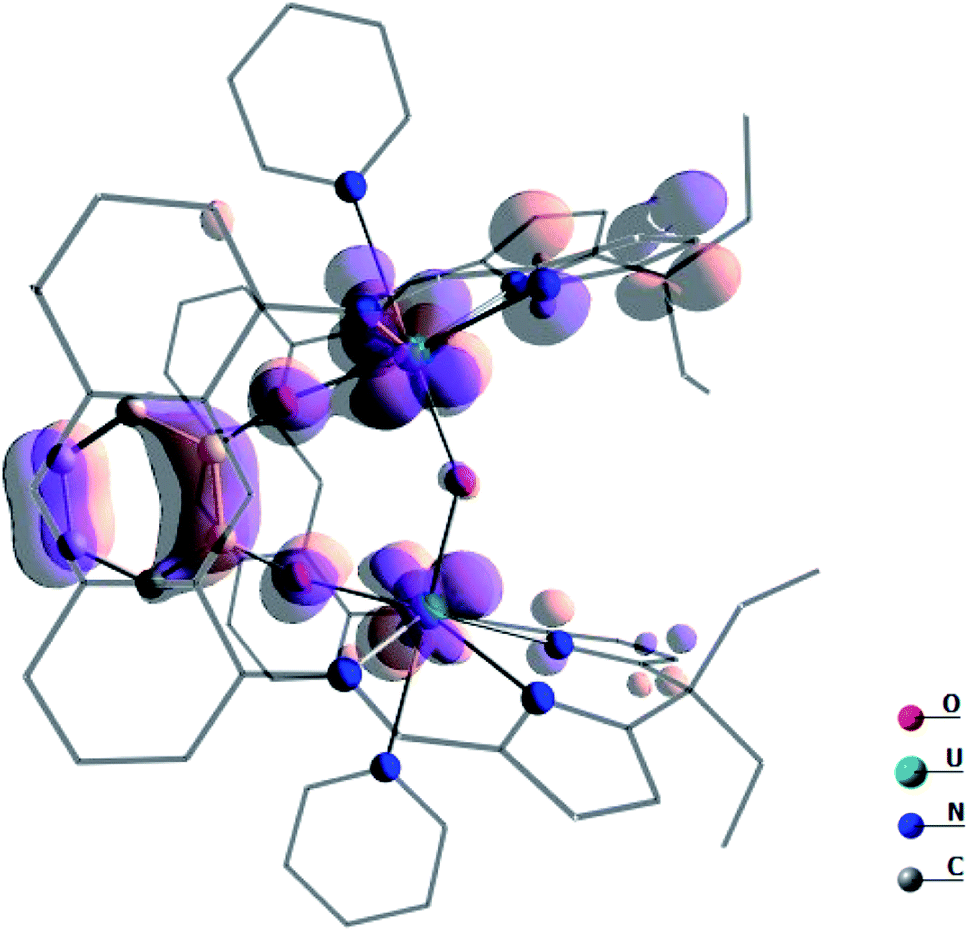 | ||
| Fig. 8 Highest energy SOMO in complex A. | ||
The propensity for [S2]2− and [Se2]2− to displace O2− during the formation of 3 and 4 is remarkable given the differences in U–O and U–S bond dissociation energies, which are 758(13)52 and 510(63) kJ mol−1,53 respectively (a value for the U–Se bond dissociation energy could not be found in the literature, but is anticipated to be even lower in energy). In light of the degree of covalency in U–μ-oxo bonding in A, this reversal in anticipated reactivity may be attributed to an increase in covalency and orbital overlap during coordination of the diatomic [X2]2− ligands to the U centres versus a monoatomic X2− ligand. This is further highlighted by a lack of reactivity between A and Ph3PSe, which possess PSe and PO (in the anticipated by-product) bond dissociation energies of 364(10) and 589(1) kJ mol−1,54 respectively. The inability for Ph3PSe to transfer a single Se2− ligand to A is a consequence of a decrease in orbital overlap between the U centres and an [Se]2− ligand versus [O]2−.
As was previously noted, the lack of reaction between A and CO2 is somewhat surprising, given the propensity for the [UIV–O–UIV] complexes [{((Neop,MeArO)3tacn)UIV}2(μ-O)] and [{{(AdArO)3N}UIV}2(μ-O)(μ-dme)] to react with CO2 to form [UIV–(CO3)–UIV] products. While the U–O bond lengths to the bridging oxo ligand in A are similar to those in the U(tacn) complexes, the U–O–U bond angles in these latter compounds are linear and the U⋯U separations are significantly longer and are similar those found in complex B. Furthermore, the longest U⋯U separation observed for complexes 1–6 is 4.5433(3) Å, whereas they are 5.275,14 6.277 and 5.253 Å in [{((Neop,MeArO)3tacn)UIV}2(μ-CO3)], [{{(AdArO)3N}UIV(dme)}2(μ-CO3)] and [{((tBuArO)3mes)UIV}2(μ-CO3)] respectively. (Note: [{((tBuArO)3mes)UIV}2(μ-CO3)] was formed from [((tBuArO)3mes)UIII] and CO2 and no isolable UIV/UIV μ-oxo intermediate was obtained.)41 Therefore, while the μ-oxo ligand in A is both highly nucleophilic and potentially more sterically approachable due to the bent U–O–U angle caused by the μ-catecholate ligand, we suggest the lack of reactivity between A and CO2 is because the short U⋯U distance is constrained by the bridging catecholate ligand.
Compounds with U–O–U bond angles similarly acute to A include K2[{OUV(μ-O)2UVO}(LMe)] (U⋯U = 3.3795(5) Å; U–O–U = 107.5(2), 104.5(2)°), K2[{O2UVI(μ-O)UVIO2}(LMe)] (U⋯U = 3.9762(4) Å; U–O–U = 136.4(3)°)55 and the calculated [{(THF)UIVOUIV(THF)}(LMe)]2+ (U⋯U = 3.819 Å; U–O–U = 134.0°),56 all of which feature a smaller Pacman ligand that makes use of a phenylene hinge between the two N4-donor pockets as opposed to an anthracenyl hinge. The complex K2[{Me3SiOUIV(μ-O)2UIVOSiMe3}(LMe)] was not crystallographically characterised but would be anticipated to possess similarly acute U–O–U bond angles.55 The oxo reactivity of these synthesised complexes has not been probed.
Conclusions
The substitution of two monodentate anionic ancillary O-ligands in the linear, oxo-bridged bis(boroxido)-UIV/UIV Pacman complex, [{(py)(pinBO)UIVOUIV(OBpin)(py)}(LA)] (B) with the small bite-angle catecholate ligand in A causes the bending of the μ-oxo UIVOUIV unit, and results in an increase in nucleophilicity of the oxo group. This enables a wide range of reactions that either functionalise or substitute the O atom with another functional group, including some softer ligands such as S and Se which would normally not be expected to be thermodynamically capable of this displacement. These latter transformations are to the best of our knowledge unprecedented in both d- and f-block chemistry. All of the uranium products from these reactions maintain the +4 oxidation state for both uranium centres. Computational analyses of the selective reactivity seen at the bridging oxo ligand in A confirms its highly nucleophilic character. There is an increase in covalency within the U–O bonds and localisation of the lone pairs of the μ-oxo ligand caused by bending the UIV–O–UIV angle. The bridging catecholate (μ-O2C6H4) ligand also stabilises the resulting products of these reactions by providing additional electron donation to the U centres in order to counter balance decreased π-donation from the bridging X-ligands (X = OH, OMe, S2, Se2, Cl, F, OBcat). Altogether, the generation of a bent [UIV–O–UIV] unit should enable oxo reactivity without changes in formal oxidation state. The U(III) complexes that are so famous for small molecule activation by reductive routes are also very difficult to re-reduce to close a hypothetical reaction cycle. Thus, there may be interesting new opportunities for catalytic small molecule transformations that do not require additional redox additives to achieve turnover.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the University of Edinburgh, the EPSRC (Grants EP/N022122/1 and EP/M010554/1), the European Commission Directorate General and Actinet JRC Userlab (ACTINET-I3-CP-CSA-JRP-232631) and the Natural Sciences and Engineering Research Council of Canada for an NSERC Post-Doctoral Fellowship (BEC). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 740311). Additional discussion, analysis, and writing of this manuscript (PLA) was supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division at the Lawrence Berkeley National Laboratory under Contract DE-AC02-05CH1123. LM is a senior member of the Institut Universitaire de France and the Humboldt Foundation and the Chinese Academy of Science are acknowledged for support. CalMip is also gratefully thanked for a generous grant of computing time.References
- G. Aitken, N. Hazari, A. S. P. Frey, F. G. N. Cloke, O. Summerscales and J. C. Green, Dalton Trans., 2011, 40, 11080–11088 RSC.
- S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed.
- L. Maron, O. Eisenstein and R. A. Andersen, Organometallics, 2009, 28, 3629–3635 CrossRef CAS.
- J. G. Brennan, R. A. Andersen and A. Zalkin, Inorg. Chem., 1986, 25, 1756–1760 CrossRef CAS.
- L. Barluzzi, M. Falcone and M. Mazzanti, Chem. Commun., 2019, 55, 13031–13047 RSC.
- P. L. Arnold and Z. R. Turner, Nat. Rev. Chem., 2017, 1, 1–16 CrossRef.
- P. L. Arnold, L. Puig-Urrea, J. A. L. Wells, D. Yuan, F. L. Cruickshank and R. D. Young, Dalton Trans., 2019, 4894–4905 RSC.
- P. L. Arnold, Chem. Commun., 2011, 47, 9005–9010 RSC.
- W. J. Evans and S. A. Kozimor, Coord. Chem. Rev., 2006, 250, 911–935 CrossRef CAS.
- D. E. Morris, R. E. Da Re, K. C. Jantunen, I. Castro-Rodriguez and J. L. Kiplinger, Organometallics, 2004, 23, 5142–5153 CrossRef CAS.
- S. Cotton, in Lanthanide and Actinide Chemistry, ed. D. Woolins, B. Crabtree, D. Atwood and G. Meyer, John Wiley & Sons, West Sussex, UK, 2006, pp. 145–153 Search PubMed.
- N. Barros, D. Maynau, L. Maron, O. Eisenstein, G. Zi and R. A. Andersen, Organometallics, 2007, 26, 5059–5065 CrossRef CAS.
- G. Zi, L. Jia, E. L. Werkema, M. D. Walter, J. P. Gottfriedsen and R. A. Andersen, Organometallics, 2005, 24, 4251–4264 CrossRef CAS.
- A.-C. Schmidt, A. V. Nizovtsev, A. Scheurer, F. W. Heinemann and K. Meyer, Chem. Commun., 2012, 48, 8634–8636 RSC.
- B. M. Gardner, J. C. Stewart, A. L. David, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9265–9270 CrossRef CAS PubMed.
- C. Camp, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed.
- M. Falcone, L. N. Poon, F. F. Tirani and M. Mazzanti, Angew. Chem., Int. Ed., 2018, 57, 3697–3700 CrossRef CAS PubMed.
- B. E. Cowie, G. S. Nichol, J. B. Love and P. L. Arnold, Chem. Commun., 2018, 54, 3839–3842 RSC.
- Q.-J. Pan, S. O. Odoh, G. Schreckenbach, P. L. Arnold and J. B. Love, Dalton Trans., 2012, 41, 8878–8885 RSC.
- P. L. Arnold, G. M. Jones, Q.-J. Pan, G. Schreckenbach and J. B. Love, Dalton Trans., 2012, 41, 6595–6597 RSC.
- P. L. Arnold, A.-F. Pécharman and J. B. Love, Angew. Chem., Int. Ed., 2011, 50, 9456–9458 CrossRef CAS PubMed.
- R. Steudel and Y. Steudel, Eur. J. Inorg. Chem., 2004, 3513–3521 CrossRef CAS.
- J. E. McDonough, A. Mendlratta, J. J. Curley, G. C. Fortman, S. Fantasla, C. C. Cummins, E. V. Rybak-Aklmova, S. P. Nolan and C. D. Hoff, Inorg. Chem., 2008, 47, 2133–2141 CrossRef CAS PubMed.
- D. S. Brock and G. J. Schrobilgen, J. Am. Chem. Soc., 2011, 133, 6265–6269 CrossRef CAS PubMed.
- A.-C. Schmidt, F. W. Heinemann, W. W. Lukens and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed.
- M. Falcone, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2019, 141, 9570–9577 CrossRef CAS PubMed.
- C. E. Hayes, Y. Sarazin, M. J. Katz, J.-F. Carpentier and D. B. Leznoff, Organometallics, 2013, 32, 1183–1192 CrossRef CAS.
- T. Arliguie, M. Blug, P. L. Floch, N. Mézailles, P. Thuéry and M. Ephritikhine, Organometallics, 2008, 27, 4158–4165 CrossRef CAS.
- P. L. Arnold, C. J. Stevens, N. L. Bell, R. M. Lord, J. M. Goldberg, G. S. Nichol and J. B. Love, Chem. Sci., 2017, 8, 3609–3617 RSC.
- B. M. Gardner, D. M. King, F. Tuna, A. J. Wooles, N. F. Chilton and S. T. Liddle, Chem. Sci., 2017, 8, 6207–6217 RSC.
- C. Camp, M. A. Antunes, G. Garcia, I. Ciofini, I. C. Santos, J. Pécaut, M. Almeida, J. Marçalo and M. Mazzanti, Chem. Sci., 2014, 5, 841–846 RSC.
- J. L. Brown, G. Wu and T. W. Hayton, Organometallics, 2013, 32, 1193–1198 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- S. M. Franke, F. W. Heinemann and K. Meyer, Chem. Sci., 2014, 5, 942–950 RSC.
- O. P. Lam, F. W. Heinemann and K. Meyer, Chem. Sci., 2011, 2, 1538–1547 RSC.
- J. W. W. Lukens, S. M. Beshouri, L. L. Blosch, A. L. Stuart and R. A. Andersen, Organometallics, 1999, 18, 1235–1246 CrossRef.
- P. B. Hitchcock, M. F. Lappert and R. G. Taylor, J. Chem. Soc., Chem. Commun., 1984, 1082–1084 RSC.
- A. L. Ward, H. L. Buckley, W. W. Lukens and J. Arnold, J. Am. Chem. Soc., 2013, 135, 13965–13971 CrossRef CAS PubMed.
- C. A. P. Goodwin, F. Tuna, E. J. L. McInnes and D. P. Mills, Eur. J. Inorg. Chem., 2018, 2356–2362 CrossRef CAS.
- C. E. Hayes, R. H. Platel, L. L. Schafer and D. B. Leznoff, Organometallics, 2012, 31, 6732–6740 CrossRef CAS.
- O. P. Lam, S. C. Bart, H. Kameo, F. W. Heinemann and K. Meyer, Chem. Commun., 2010, 46, 3137–3139 RSC.
- B. E. Cowie, J. M. Purkis, J. Austin, J. B. Love and P. L. Arnold, Chem. Rev., 2019, 119, 10595–10637 CrossRef CAS PubMed.
- I. Castro-Rodriguez and K. Meyer, J. Am. Chem. Soc., 2005, 127, 11242–11243 CrossRef CAS PubMed.
- I. Castro-Rodriguez, H. Nakai, L. N. Zakharov, A. L. Rheingold and K. Meyer, Science, 2004, 305, 1757–1759 CrossRef CAS PubMed.
- D. Werner, X. Zhao, S. P. Best, L. Maron, P. C. Junk and G. B. Deacon, Chem.–Eur. J., 2017, 23, 2084–2102 CrossRef CAS PubMed.
- A. Y. Kornienko, T. J. Emge and J. G. Brennan, J. Am. Chem. Soc., 2001, 123, 11933–11939 CrossRef CAS PubMed.
- S. K. Bose, K. Geetharani, V. Ramkumar, B. Varghese and S. Ghosh, Inorg. Chem., 2010, 49, 2881–2888 CrossRef CAS PubMed.
- H.-M. Wu, Y.-H. Chang, Y.-F. Tsai, K.-F. Hsu, G.-H. Lee and H.-F. Hsu, Dalton Trans., 2015, 44, 4468–4473 RSC.
- U. Chakraborty, F. Urban, B. Mühldorf, C. Rebreyend, B. de Bruin, N. van Velzen, S. Harder and R. Wolf, Organometallics, 2016, 35, 1624–1631 CrossRef CAS.
- L. Castro, O. P. Lam, S. C. Bart, K. Meyer and L. Maron, Organometallics, 2010, 29, 5504–5510 CrossRef CAS.
- A.-C. Schmidt, F. W. Heinemann, C. E. Kefalidis, L. Maron, P. W. Roesky and K. Meyer, Chem.–Eur. J., 2014, 20, 13501–13506 CrossRef CAS PubMed.
- J. Marçalo and J. K. Gibson, J. Phys. Chem. A, 2009, 113, 12599–12606 CrossRef PubMed.
- C. C. L. Pereira, C. J. Marsden, J. Marçalo and J. K. Gibson, Phys. Chem. Chem. Phys., 2011, 13, 12940–12958 RSC.
- Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- G. M. Jones, P. L. Arnold and J. B. Love, Angew. Chem., Int. Ed., 2012, 51, 12584–12587 CrossRef CAS PubMed.
- J. Yao, X.-J. Zheng, Q.-J. Pan and G. Schreckenbach, Inorg. Chem., 2015, 54, 5438–5449 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1984900–1984907. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02297g |
| ‡ Current address: Department of Chemistry, University of California, Berkeley, CA 94720, USA; Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA94720, USA. E-mail: pla@berkeley.edu |
| This journal is © The Royal Society of Chemistry 2020 |