
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic reaction-induced phase separation in tunable, adaptive covalent networks†
Katie M.
Herbert‡
 a,
Patrick T.
Getty‡
a,
Neil D.
Dolinski
a,
Jerald E.
Hertzog
b,
Derek
de Jong
c,
James H.
Lettow
a,
Joy
Romulus
d,
Jonathan W.
Onorato
d,
Elizabeth M.
Foster
d and
Stuart J.
Rowan
*abe
a,
Patrick T.
Getty‡
a,
Neil D.
Dolinski
a,
Jerald E.
Hertzog
b,
Derek
de Jong
c,
James H.
Lettow
a,
Joy
Romulus
d,
Jonathan W.
Onorato
d,
Elizabeth M.
Foster
d and
Stuart J.
Rowan
*abe
aPritzker School of Molecular Engineering, University of Chicago, Chicago, IL 60637, USA. E-mail: stuartrowan@uchicago.edu
bDepartment of Chemistry, University of Chicago, Chicago, IL 60637, USA
cThe University of Chicago Laboratory Schools, 1362 E. 59th St., Chicago, IL 60637, USA
dDepartment of Macromolecular Science and Engineering, Case Western Reserve University, 2100 Adelbert Road, Cleveland, OH 44106, USA
eChemical Science and Engineering Division and Center for Molecular Engineering, Argonne National Laboratory, 9700 S. Cass Ave., Lemont, IL 60434, USA
First published on 1st May 2020
Abstract
A series of catalyst-free, room temperature dynamic bonds derived from a reversible thia-Michael reaction are utilized to access mechanically robust dynamic covalent network films. The equilibrium of the thiol addition to benzalcyanoacetate-based Michael-acceptors can be directly tuned by controlling the electron-donating/withdrawing nature of the Michael-acceptor. By modulating the composition of different Michael-acceptors in a dynamic covalent network, a wide range of mechanical properties and thermal responses can be realized. Additionally, the reported systems phase-separate in a process, coined dynamic reaction-induced phase separation (DRIPS), that yields reconfigurable phase morphologies and reprogrammable shape-memory behaviour as highlighted by the heat-induced folding of a predetermined structure.
Introduction
Dynamic covalent networks (DCNs), also known as covalent adaptable networks (CANs), are polymeric networks that incorporate dynamic covalent chemistries (DCCs) which enable the formation of functional, responsive systems.1–7 Under ideal conditions, these dynamic covalent bonds undergo bond exchange without the encumbrance of unwanted side reactions.8,9 The incorporation of a range of dynamic chemistries such as transesterification,10–12 Diels–Alder reactions,13–16 disulfide bonds,17–19 and boronic esters20–22 into materials has given researchers access to adaptive materials that exhibit self-healing, reprocessibility, stress relaxation, adhesion, and shape-memory properties.23,24 The thia-Michael (tM) reaction, typically described as the base- or nucleophile-catalyzed addition of a thiol across an α,β-unsaturated carbonyl compound, has been a staple synthetic technique in polymer chemistry and materials science for decades.25 While its reputation as a versatile reaction is well established, its ability to act as a reversible bond has only recently begun to be investigated in the realm dynamic polymer networks.5,26–31In 2016, Konkolewicz and coworkers demonstrated that thiol-acrylate-based polymer networks are thermally-reversible at elevated temperatures (90 °C), affording such properties as healing and malleability.26 The same group also reported pH-responsive tM systems in which thiol-maleimide adducts were shown to be reversible under basic conditions, again leading to healable films.27 Using an alternative approach, Ishibashi and Kalow employed a Meldrum's acid derivative capable of reversibly crosslinking siloxane polymers containing pendant thiol groups without the use of a catalyst.28 They showed that the resulting silicone network could be reprocessed via compression molding (150 °C, 9 tons, 15 minutes) at least ten times without significant loss of mechanical properties, demonstrating the viability of a catalyst-free approach to dynamic tM polymer networks. While these approaches elegantly demonstrate the utility of tM adducts as reversible bonds for responsive, reprocessable materials, they rely on basic conditions and/or high operating temperatures to access the desired dynamic behaviour, potentially limiting the application scope.
An interesting subset of the tM reaction with the potential to expand the capabilities of previously reported systems is the thiol addition to benzalcyanoacetate Michael-acceptors, Fig. 1. Pioneering small molecule studies by Taunton,32 Anslyn,33 and Houk34 have shown that thiol addition to such Michael acceptors requires no catalyst and is dynamic at room temperature. The latter fact caught our attention as one class of dynamic covalent networks that has received little attention are those in which the bonds are dynamic at room temperature.13,35 In addition, it has been shown that the presence of electron-donating or electron-withdrawing species on the β-phenyl ring alters the kinetics and thermodynamics of the reaction without drastically modifying the reactive site.33 The ability to tune the strength of interactions has been well established as a means to control materials properties in non-covalent systems such as Lewis pairs36 and metal–ligand interactions.37,38 As such, these modular benzalcyanoacetates are a promising moiety for the preparation of tailorable materials. Despite these favourable properties, there have been very limited studies using this class of dynamic covalent bond in polymeric systems. The amide-based derivative, benzalcyanoacetamide, has been explored by Schubert, Hager and co-workers in organic solvent and in bulk polymeric systems. However, the reported benzalcyanoacetamide networks utilized basic conditions (specifically using 1,8-diazabicyclo(5.4.0)undec-7-ene, DBU) which ultimately led to the irreversible decomposition of the thiol component into H2S gas.29 It was later discovered that the same benzalcyanoacetamide motif could be used without degradation by eliminating the DBU in a bulk polymer system.30 The acetate-based acceptors, on the other hand, have been largely set aside presumably due to their reported instability in the presence of water.33 Preliminary results by Anslyn showed that the benzalcyanoacetate acceptors are indeed capable of participating in thiol-Michael addition although the aqueous conditions applied in those experiments resulted in hydrolysis of the benzalcyanoacetate through a retro-Knoevenagel reaction.33
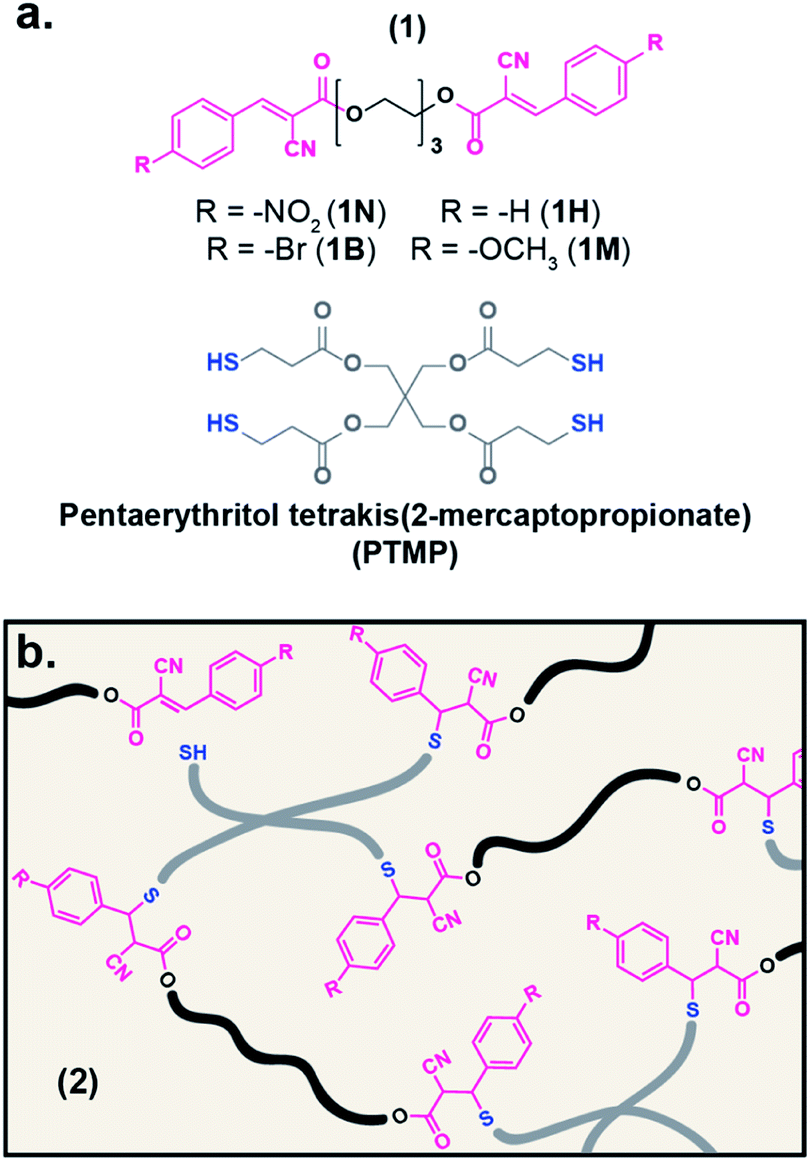 | ||
| Fig. 1 (a) Chemical structures of ditopic electrophiles (1) and tetrathiol crosslinker, PTMP. (b) Illustration depicting the network formation of electrophiles associated/dissociated with PTMP (2). | ||
Bonds that undergo active exchange at room temperature represent an attractive avenue for tuning materials properties at ambient conditions. However, without some form of reinforcement, these systems would likely flow freely and lack robust mechanical properties. One potential avenue of reinforcement, widely utilized in polymeric systems, is reaction/polymerization induced phase separation (RIPS/PIPS), whereby the evolution of the polymeric structure creates regions of dissimilar viscoelastic properties (i.e. viscosity, crosslink density, insolubility) that constitute the various phases throughout the films.39–42 It is important to note that there have been recent reports of phase separated DCNs,43–48 in which the materials primarily rely on dissimilar polymeric blocks to induce phase separation and an enhancement in mechanical properties is observed with such systems.
With the goal of harnessing the tunable, catalyst-free, room temperature, dynamic nature of the benzalcyanoacetate-based tM reaction in polymeric systems, this report first explores the dynamic bonding of the thiol addition to small molecule benzalcyanoacetate controls in organic media. Inspired by the apparent tunability of the tM reaction in solution, the investigation expands into bulk polymeric films based on the benzalcyanoacetate motif. These dynamic tM films show a wide range of mechanical properties, which is related to both the nature of the dynamic bond and the fact that these films exhibit phase separated morphologies, in a process termed here dynamic reaction-induced phase separation (DRIPS). The results demonstrate the ability of this tM reaction to tailor materials properties in adaptable networks, highlighting their DRIPS morphologies and shape memory behaviour.
Results and discussion
To probe a range of these tM dynamic bonds, a series of four benzalcyanoacetate compounds were synthesized (Fig. 2a). Each compound was differentiated by the electron-withdrawing/-donating capabilities of the substituent on the para-position of the β-phenyl ring (mono-X), namely nitro- (mono-N), bromo- (mono-B), hydrogen- (mono-H), and methoxy-substituted (mono-M).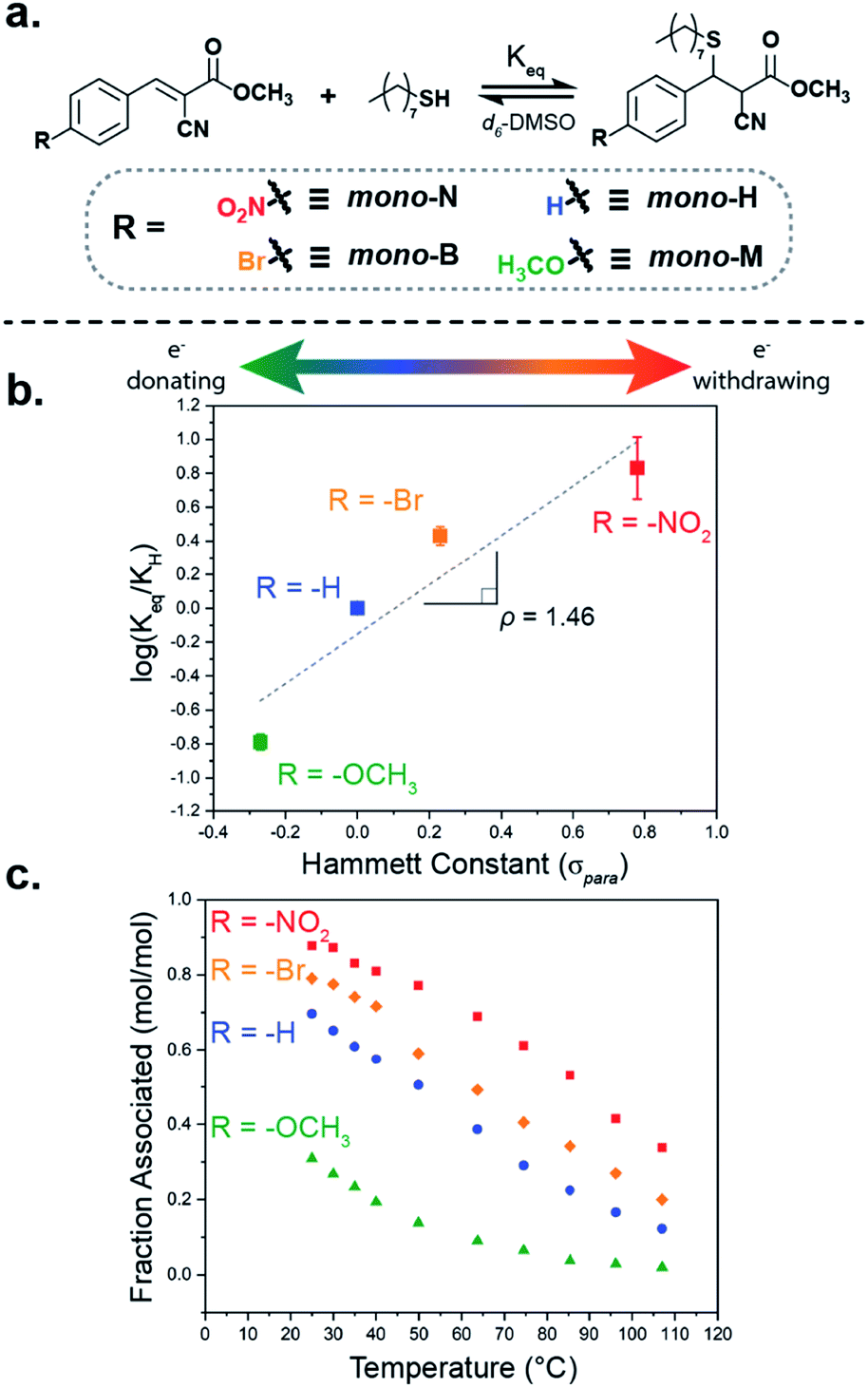 | ||
| Fig. 2 (a) Schematic of benzalcyanoacetate electrophile equilibrium reaction with 1-octanethiol, (b) Hammett plot of equilibrium constants at room temperature in d6-DMSO, and (c) equilibrium position of association as a function of temperature for mono-N (red), mono-B (orange), mono-H (blue), and mono-M (green) derivatives at 100 mM concentrations in d6-DMSO. | ||
The benzalcyanoacetate species were synthesized via a Knoevenagel condensation of methyl cyanoacetate with the corresponding benzaldehyde. To compare these Michael-acceptors to previous examples, the benzalcyanoacetate species were titrated with 1-octanethiol in anhydrous deuterated dimethyl sulfoxide (d6-DMSO) at room temperature following NMR titration procedures and considerations outlined by Thordarson.49 The titration experiments determined the equilibrium constant (Keq) of the mono-X acceptors in d6-DMSO to be ca. 470, 160, 60, and 10 M−1, respectively (Fig. S2 and 3, Table S2†). Consistent with Anslyn's previous work, a Hammett plot of the equilibrium constants (Fig. 2b) reveals a positive trend, with ρ = 1.46 suggesting a system whose equilibrium properties are dictated by the electronic nature of the β-phenyl substituent.33 As an interesting note, the apparent downward curvature of the data shown in the Hammett plot was also observed by Anslyn and suggests there may be variability in the overall mechanism of dynamic exchange depending on the electronic nature of the Michael acceptor.
To further investigate the impact of the electronic substituent on bond formation, the equilibrium position of equimolar (100 mM) mixtures of thiol and mono-X was measured as a function of temperature in d6-DMSO from ambient temperatures to ca. 110 °C (Fig. 2c). As expected, the fraction of the associated tM adduct observed decreased with increasing temperature, while maintaining the overall equilibrium trend (mono-N > mono-B > mono-H > mono-M) throughout the temperature range. Additionally, evaluation of these equilibrium values in a van't Hoff plot shows that the differences in bonding are primarily enthalpic (Fig. S5 and Table S3†). The thermal response of the thia-Michael equilibrium in these small molecule models anticipates two key properties in networks composed of these species: (1) a thermoresponsive material whose mechanical properties adapt to temperature and (2) a variation in total adduct formation at room temperature corresponding to a tunable effective crosslink density. As such, utilization of these electronically different Michael-acceptors in dynamic networks, should allow for direct control over the effective crosslinking of the networks, enabling the precise tuning of their properties.
To expand the small molecule studies to network systems, a series of ditopic acceptor compounds (1, Fig. 1a) were prepared with nitro- (1N), bromo- (1B), hydrogen- (1H), and methoxy-substituted (1M) benzalcyanoacetate moieties on the chain ends of a triethylene glycol core.50 A tetrathiol crosslinker, pentaerythritol tetrakis(2-mercaptopropionate) (PTMP), and 1 were mixed in chloroform and cast onto a Teflon® dish to yield a film of the resultant tM network (2, Fig. 1b) after drying. The dried films were then compression molded at elevated temperature (10 kPSI, 70–90 °C), resulting in robust polymeric films approximately 400 μm thick. For consistency amongst the prepared networks, the ratio of reactive sites (thiol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :alkene) was kept constant to a 1:1 ratio. The resulting samples are denoted 2Nx, 2Bx, 2Hx, and 2Mx to indicate the use of 1N, 1B, 1H, and 1M, respectively, to form the network. The x relates the mole percent (mol%) of each acceptor site (x = 100 or 50 for hybrid networks) relative to the thiol groups.
:alkene) was kept constant to a 1:1 ratio. The resulting samples are denoted 2Nx, 2Bx, 2Hx, and 2Mx to indicate the use of 1N, 1B, 1H, and 1M, respectively, to form the network. The x relates the mole percent (mol%) of each acceptor site (x = 100 or 50 for hybrid networks) relative to the thiol groups.
The tM networks were studied via Raman spectroscopy to assess the degree of formation of the thia-Michael linkages within the bulk network, specifically focusing on the nitrile regime (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch, 2260–2200 cm−1). The spectra of the neat acceptors (1N, 1B, 1H, and 1M) reveal a single peak at ca. 2226 cm−1 resulting from the stretching vibration of the conjugated CN bond. Once a thiol adds to the acceptor across the double bond, conjugation is lost and the CN signal shifts to higher frequencies, allowing for direct monitoring of adduct formation in the solid state (Fig. 3). In agreement with the model small molecule studies, the amount of adduct formed within the networks is highly dependent on the electronic nature of the β-phenyl substituent, with the more electron-withdrawing acceptors in 2N100 reacting almost completely with the thiols (Fig. 3a, ca. 92% reacted by peak maximum), the electron-neutral groups in 2H100 displaying a middling amount of adduct formation (Fig. 3c, ca. 85% reacted by peak maximum), while the electron-donating compounds in 2M100 exhibit a significant amount of unreacted acceptor species (Fig. 3d, ca. 24% reacted by peak maximum). It is worthwhile noting however, that 2B100 has an anomalously high relative concentration of unreacted species (Fig. 3b, only ca. 58% reacted by peak maximum) attributed to sample crystallinity as discussed below.
N stretch, 2260–2200 cm−1). The spectra of the neat acceptors (1N, 1B, 1H, and 1M) reveal a single peak at ca. 2226 cm−1 resulting from the stretching vibration of the conjugated CN bond. Once a thiol adds to the acceptor across the double bond, conjugation is lost and the CN signal shifts to higher frequencies, allowing for direct monitoring of adduct formation in the solid state (Fig. 3). In agreement with the model small molecule studies, the amount of adduct formed within the networks is highly dependent on the electronic nature of the β-phenyl substituent, with the more electron-withdrawing acceptors in 2N100 reacting almost completely with the thiols (Fig. 3a, ca. 92% reacted by peak maximum), the electron-neutral groups in 2H100 displaying a middling amount of adduct formation (Fig. 3c, ca. 85% reacted by peak maximum), while the electron-donating compounds in 2M100 exhibit a significant amount of unreacted acceptor species (Fig. 3d, ca. 24% reacted by peak maximum). It is worthwhile noting however, that 2B100 has an anomalously high relative concentration of unreacted species (Fig. 3b, only ca. 58% reacted by peak maximum) attributed to sample crystallinity as discussed below.
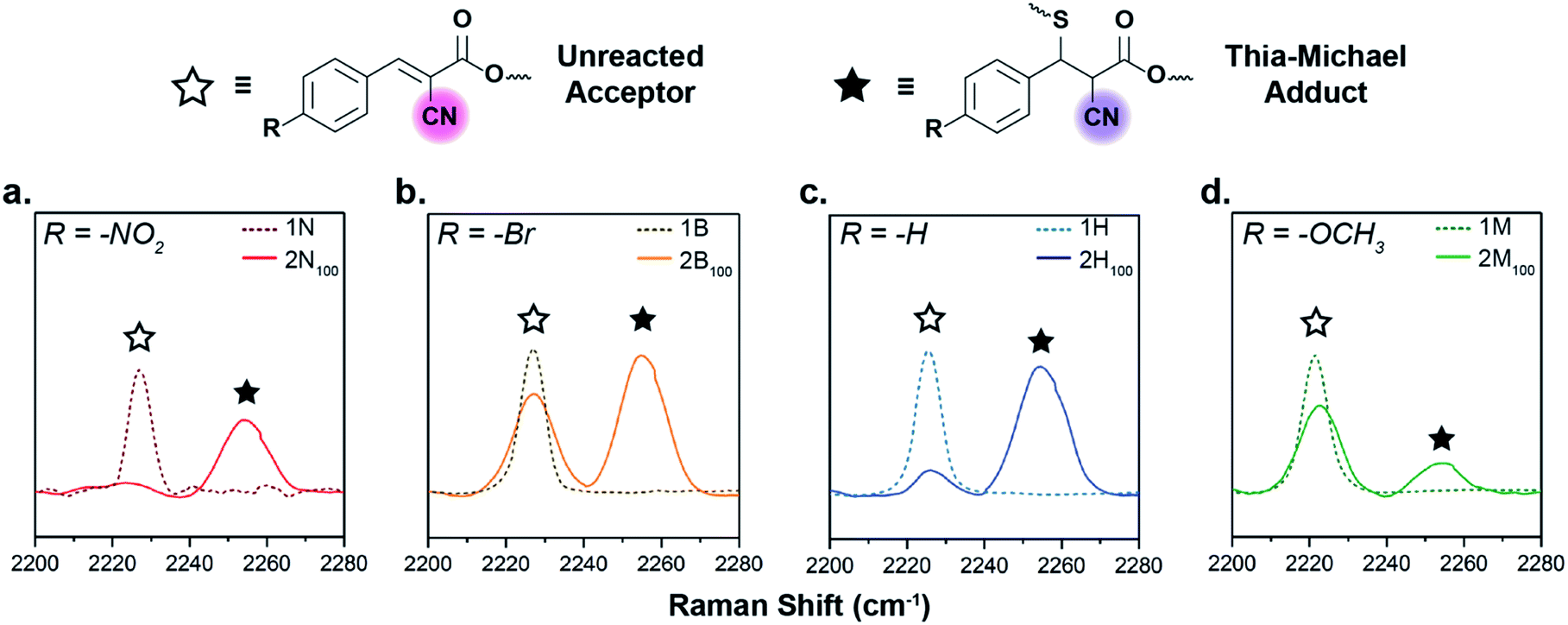 | ||
| Fig. 3 Raman spectra of CN vibrational stretches for the neat electrophiles (dotted lines) overlaid with the respective thia-Michael networks (solid lines). (a) 1N and 2N100, (b) 1B and 2B100, (c) 1H and 2H100, and (d) 1M and 2M100. | ||
In addition to the shift in the CN stretch, there is a corresponding decrease in the thiol peak intensity at 2570 cm−1 as a result of the PTMP reactive groups participating in the crosslinking reaction (Fig. S6A†). As the reduction in the amount of free thiols could equally be attributed to disulfide formation within the network, the spectra were compared to a control sample crosslinked via disulfide bonds; however, no notable disulfide bond was observed in the tM films (Fig. S6B†).
Given that the extent of tM linkages should directly influence the mechanical properties of the networks, a series of experiments was conducted to elucidate the bulk properties of the tM systems. To measure the response of the tM films to uniaxial tension, rectangular strips of 2N100 and 2H100 were cut from the pressed films and uniaxially deformed at a rate of 10 mm min−1 in ambient conditions. Unfortunately, neither 2B100 nor 2M100 formed films suitable for tensile testing (likely a result of film crystallinity/inadequate crosslinking) and were therefore excluded from further mechanical tests. As expected, films formed using different acceptors exhibited distinctive mechanical properties at room temperature (Fig. 4a), with 2N100 displaying the highest stress at break (20 ± 3 MPa) and very little ductility (strain at break = 1.5 ± 0.3%). Conversely, 2H100 exhibited a much lower ultimate strength (0.17 ± 0.03 MPa), but stretched to more than 700% of its original length. In an effort to better assess the impact of the different Michael acceptors on mechanical properties, a series of hybrid materials, 2N50B50, 2N50H50, 2N50M50, were also prepared, and resulted in robust films across the series. Interestingly, the hybrid material tensile properties were found to correlate strongly with the equilibrium binding studies, with 2N50B50 > 2N50H50 > 2N50M50 in terms of ultimate strength, and the opposite trend for overall sample ductility, which can be attributed in part to higher degrees of effective crosslinking with increasingly electron-withdrawing groups. It should be noted that this increase in crosslinking also increases the Tg of materials, leading to the stiff, relatively brittle character of 2N100 and 2N50B50 at room temperature. To assess the impact of adding weaker bonds in the hybrid materials (particularly in the case of 2N50M50) control studies using 2N50 samples were carried out. While 2N50 and 2N50M50 samples displayed similar thermal properties (i.e. Tg), 2N50 displayed significantly enhanced yield strength compared to the 2N50M50 film, suggesting that the weak-bonding 1M species have a larger effect on the properties than simply diluting the concentration of the 1N species (Fig S9†).
 | ||
| Fig. 4 (a) Stress–strain curves (strain rate = 10 mm min−1, 25 °C), (b) dynamic mechanical analysis (DMA; temperature ramp rate = 3 °C min−1, frequency = 1 Hz, tensile geometry) and (c) shear rheology (temperature ramp rate = 3 °C min−1, frequency = 2 Hz, parallel plate geometry) curves for 2N100 (red), 2N50B50 (dashed orange), 2N50H50 (dashed blue), 2N50M50 (dashed green), and 2H100 (blue). | ||
As temperature was shown to strongly influence the extent of adduct formation in the model studies, the effect of temperature on mechanical behaviour was evaluated. As the moduli of these materials span several orders of magnitude, a combination of methods, namely dynamic mechanical analysis (DMA) and shear rheology, was used to describe their thermomechanical properties. The results of the DMA measurements (Fig. 4b) revealed the glass transition temperature (Tg, as defined by loss modulus [E′′] peak, Fig. S10†) as 47.5 °C and 8.9 °C, for 2N100 and 2H100, respectively, in accordance with the distinct mechanical properties of the two networks at room temperature. In the hybrid networks, the combination of crosslinks within the system enabled a systematic tuning of Tg with 2N50B50, 2N50H50, and 2N50M50 exhibiting Tg at 39, 29 and 21 °C, respectively. Following this glass transition, a significant difference in performance within the plateau regime was also observed. Where 2N100 exhibited a clear plateau on the order of 106 Pa, no plateau was observed in the 2H100 film (under these experimental conditions). The hybrid materials followed the same trend observed in the tensile studies, with 2N50B50 and 2N50H50 behaving more similarly to 2N100, yielding plateau behaviour above Tg while 2N50M50 did not exhibit a plateau.
To more accurately probe the performance of these materials at higher temperatures, shear rheology experiments were carried out. As can be seen in Fig. 4c, 2N100 maintained a plateau up to ca. 125 °C whereas 2H100 was weaker by two orders of magnitude and began its second transition at ca. 90 °C. The hybrid materials followed expected trends with 2N50B50 exhibiting a slightly higher plateau modulus and second transition temperature (ca. 180 kPa until ca. 122 °C) than 2N50H50 (ca. 110 kPa until ca. 117 °C). 2N50M50 lacked a stable plateau regime and demonstrated a second transition at ca. 101 °C. Thus, these initial evaluations demonstrate the ability to tailor both thermal and mechanical properties of dynamic networks through the use of dynamic bonds with different equilibrium behaviours.
Reflecting on the equilibrium adduct formation as a function of temperature, it is curious to note that certain samples exhibit a plateau (under these experimental conditions) at elevated temperatures. Flory–Stockmayer theory predicts that a system consisting of di- and tetrafunctional monomers, such as the 2N100 network, would percolate above ca. 58% conversion (for a tetrafunctional monomer with a stoichiometric amount of a difunctional partner). Recalling the NMR temperature studies (Fig. 2c), this would imply that the 2N100 film should lose connectivity at temperatures above ∼75 °C assuming a homogeneous network construction. To investigate this seeming contradiction, differential scanning calorimetry (DSC) was carried out on all the films. Interestingly, the DSC traces confirmed the DMA experiments with all the networks exhibiting two distinct thermal transitions. While the trend in the lower transition temperatures (assigned as a Tg) is consistent with that observed in the DMA, there is also a broad second transition at much higher temperatures (>125 °C) (Fig. 5 and S11†), suggesting the presence of multiple phases in these dynamic films. The lone exception to dual transitions in the DSC is the 2B100 film which exhibited multiple phase transitions at elevated temperatures, which are attributed to the cold-crystallization of the electrophile. This phase behaviour explains the previously discussed anomalies for the 2B100 films, namely that crystallization of the electrophile results in the relatively high amount of unreacted Michael acceptor and the lack of robust film formation. Modulated DSC experiments on 2N100 samples verify the lower temperature thermal transition has a high degree of reversing character, consistent with a Tg while the higher transition shows little to no reversing character, suggesting a more complex phase behaviour (Fig. S12†). The non-reversing character of the upper transition may potentially be explained by ‘trapped’ tM bonds in a densified hard phase possibly formed by highly reacted tM adduct oligomers crashing out of the solution or melt. As the matrix softens and the bonds are able to exchange, they rapidly dissociate in accordance with temperature dependent small molecule studies in Fig. 2c.
 | ||
| Fig. 5 Differential scanning calorimetry (DSC) curves for 2N100 (red), 2N50B50 (dashed orange), 2N50H50 (dashed blue), and 2N50M50 (dashed green), 2H100 (blue). | ||
The dual phase transition behaviour was also observed in the DSC traces for the hybrid systems, with Tg values of these films tracking with equilibrium constant and higher values observed for networks formed from more thermodynamically stable dynamic bonds. Additionally, the onset of the lower temperature transition in the hybrid films reflects an approximate Fox-like blend of the glass transitions of the homo-Michael acceptor films (Fig. S13†). A similar trend was observed for the higher temperature transition, although the breadth of the transition in the hybrid films makes the precise transition temperature difficult to accurately compare.
As the thermal properties indicated a dual phase system, atomic force microscopy (AFM) of as-cast films (to avoid surface features transferred by compression molding) was undertaken to further elucidate the microphase morphology of the networks (Fig. 6). Although the high Tg of 2N100 prevented differentiation of hard/soft regions, AFM phase images of both 2H100 and 2M100 clearly show dual-phase materials composed of a soft matrix (attributed to the lower thermal transition) with hard, circular inclusions (attributed to the higher thermal transition) (Fig. 6a and b). We hypothesize that while dynamic bonds exist in both phases, access to the dynamic exchange in either phase is restricted to the corresponding phase transition temperature. As such, in both cases, the connectivity of the overall film is primarily dictated by the dynamic behaviour of the soft phase, leading to the lack of a plateau above Tg for the 2H100 and to weak films in the case of 2M100. The hybrid films, on the other hand, revealed more complex morphologies, indicative of a combination of phase separation phenomena (Fig. 6c and d). For example, 2N50H50 reveals concentric phase development in which a continuous hard phase contains encapsulated softer phases, leading to a percolated network of the higher thermal transition phase, which explains the resulting plateau regime in the DMA above Tg. Similar phase behaviour was also observed in the case of 2N50B50 (Fig. S15†). In contrast, the 2N50M50 shows circular regions of two different phases (bright and dark phases in Fig. 6d inset and S16†) that are both distinct from the soft continuous phase, leading to a lack of plateau behaviour at elevated temperatures and the low yield strength in tension at room temperature.
 | ||
| Fig. 6 Atomic force micrographs showing phase image, phase color scheme (bottom, dark = soft phase, light = hard phase). (a) 2H100, (b) 2M100, (c) 2N50H50, (d) 2N50M50 (top right corner of each image recolored to highlight shape outlines, not scaled to phase). Scale bar = 1 μm. | ||
As a result of the phase separated morphology and the presence of a plateau in the DMA, it was hypothesized that films formed via the dynamic reaction-induced phase separation (DRIPS) process should exhibit reprogrammable shape memory behaviour. In a representative example, the shape memory behaviour of the 2N50H50 film was evaluated using force-controlled DMA shape memory experiments. A single temporary shape was imparted to the sample at 60 °C and fixed upon cooling below Tg to room temperature, where the applied force was released (Fig. 7a). 2N50H50 demonstrated an ideal fixing ratio of 100%, showing no change in strain after the temporary shape was set (maximum strain, εm, and unloading strain, εu = 0.365). Upon reheating, the sample recovered to 90% of its original shape (recovery strain, εr = 0.0371), with the residual 10% likely lost on account of creep within the sample that occurred throughout the shape memory process (a phenomenon common to dynamic covalent materials). The ability to reprogram this behaviour was demonstrated by fixing a new permanent shape (Fig. 7b, Shape 1) in the film at elevated temperatures (90 °C), allowing the continuous stiff phase to relax and maintain Shape 1 upon cooling. Carrying out the previous conditions to impart a new temporary shape (Fig. 7b, Shape 2) into the material, once again demonstrated a 100% fixing ratio of Shape 2 followed by a 95% recovery back to the relaxed Shape 1 ((εm,2 − εr,2/εm,2 − εr,1) × 100).
 | ||
| Fig. 7 Force-controlled dynamic mechanical analysis (DMA) experiments demonstrating (a) one-way and (b) reprogrammable shape memory behaviour of 2N50H50. εm = maximum strain; εu = unloading strain; εr = recovery strain; sample size = ca. 10 mm × 4 mm × 0.5 mm (length × width × thickness). | ||
To further demonstrate the shape memory behaviour of these materials a sheet of 2N50H50 was prepared for a demonstration of self-folding. The capacity to trigger stimuli-responsive folding has attracted interest, for example, as a means to control soft robotics systems that can move, bend, and propel themselves through space.51 Thus, by using the reprogrammable shape memory property of films formed via DRIPS, it was possible to program larger, complex architectures through simply folding (creasing) a flat film to a structure, such as the airplane in Fig. 8, that self-folds upon heating. This was accomplished by creasing a 2N50H50 film at elevated temperatures (over a hot plate at 105 °C) and subsequently cooling to room temperature, thereby programming the permanent shape into the folded geometry (Fig. 8a and S17†). The film was unfolded just above Tg (ca. 45 °C), cooled to room temperature, and fixed into a flat, temporary geometry that maintained the creases from folding, similar to those observed in a standard paper airplane (Fig. 8b). Upon heating again to >45 °C, the sample folded itself into a “plastic” airplane (Fig. 8c, see ESI for Video†).
 | ||
| Fig. 8 Demonstration of reprogrammable shape memory behaviour of 2N50H50. (a) The programmed airplane shape fixed at 105 °C and cooled to room temperature, (b) same sample flattened into its temporary shape at room temperature, (c) time-lapse photos of the film folding into the programmed airplane shape upon exposure to heat. | ||
Another potentially interesting property of dynamic reaction-induced phase separation (DRIPS) is the ability to thermally reprocess materials after casting to yield materials with new morphologies and properties via melt processing. This is distinct from standard RIPS/PIPS of network materials where the reaction/polymerization is irreversible. The incorporation of dynamic thia-Michael bonds into the network creates a fully reversible, tunable environment that allows the films to be reprocessed and reprogrammed. To demonstrate the impact of melt processing conditions on DRIPS, a solution cast sample of 2N50H50 was annealed at 200 °C (above the upper transition) for 30 minutes then either cooled slowly (1 °C min−1) or quenched in liquid nitrogen (LN2). This thermal treatment resulted in a drastically different phase structure and mechanical properties as compared to the non-annealed films (processed between thermal transitions) shown in Fig. 6. Annealing above the upper transition of 2N50H50 followed by slow cooling enables the formation of a much higher percentage of the hard phase which forms continuously throughout the material, resulting in a glassy, brittle film at room temperature (Fig. 9a and c) whereas quenching the material disrupted the continuity of the hard phase, yielding a soft, viscoelastic solid (Fig. 9b and d). This development of the phases was confirmed by DSC of a single sample cooled at various rates, demonstrating increased amounts of the high temperature phase with slower cooling rates (Fig. S18†). Additionally, the cooling rate directly impacts the lower thermal transition as well, with slower cooling rates (more hard phase) increasing the temperature of the lower transition, which is typical for glass transitions in, somewhat analogous, semi-crystalline polymer samples. These findings were further supported by shear rheology experiments in which a 2N100 sample was thermally cycled at elevated temperatures (60–110 °C). Each cycle resulted in a steady increase in storage modulus, presumably a consequence of the growth of hard phase segments during heating and cooling cycles (Fig. S19†).
 | ||
| Fig. 9 Change in morphology of 2N50H50 after annealing at 200 °C then slow cooling (1 °C min−1) to room temperature ((a) photograph and (c) AFM image) or quenching in liquid nitrogen ((b) photograph and (d) AFM image). Scale bar = 5 μm. Phase gradient applies to both (c) and (d). | ||
Conclusions
In summary, the combination of the tetrathiol PTMP with ditopic benzalcyanoacetate Michael acceptors grants access to dynamic networks with a wide range of thermomechanical properties. Furthermore, the electronic nature of the substituent on the acceptors has a direct effect on the equilibrium reaction of the thiols to the benzalcyanoacetate moieties. These differences in equilibrium can be used to directly manipulate the thermomechanical properties of these materials. Interestingly, even though these tM-adducts are dynamic at room temperature, it is possible to access mechanically robust films on account of a dynamic reaction-induced phase separation (DRIPS) process. In fact, the phase separation is critical to the properties of these materials and lends a complimentary handle for tuning the material properties. The phase-separated morphology of the films combined with their dynamic covalent chemistry gives rise to creaseable materials exhibiting reprogrammable shape memory. This behaviour was highlighted by the self-folding of a “paper/plastic airplane” structure along thermally-programmed folds. Ongoing research seeks to better understand the phase separation of these materials and to explore the potential of these easy-to-access, low temperature dynamic networks in a variety of applications including multi-stage low-temperature adhesives and self-folding actuators.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Division of Materials Research of the NSF (Award #1609076) and in part by NIST contract 60NANB15D077, the Center for Hierarchical Materials Design (CHiMaD). N.D.D. thanks the Pritzker School of Molecular Engineering for support through a postdoctoral fellowship. The authors thank Dr Phillip Griffin and Dr Justin Jureller for their helpful discussions. The authors also thank Arvin Sookezian for donating the disulfide network material used in the control Raman experiments. Parts of this work were carried out at the Soft Matter Characterization Facility and at the Materials Research Science and Engineering Center (MRSEC NSF DMR-1420709) at the University of Chicago.References
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- C. N. Bowman and C. J. Kloxin, Angew. Chem. Int. Ed., 2012, 51, 4272–4274 CrossRef CAS PubMed.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2010, 10, 14–27 CrossRef PubMed.
- P. Chakma and D. Konkolewicz, Angew. Chem. Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed.
- S. Mukherjee, J. J. Cash and B. S. Sumerlin, Dyn. Covalent Chem., 2017, 321–358 Search PubMed.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem. Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- H. Zhang, C. Cai, W. Liu, D. Li, J. Zhang, N. Zhao and J. Xu, Sci. Rep., 2017, 7, 11833 CrossRef PubMed.
- J. L. Self, N. D. Dolinski, M. S. Zayas, J. Read de Alaniz and C. M. Bates, ACS Macro Lett., 2018, 7, 817–821 CrossRef CAS.
- P. Reutenauer, E. Buhler, P. J. Boul, S. J. Candau and J.-M. Lehn, Chem.–Eur. J., 2009, 15, 1893–1900 CrossRef CAS PubMed.
- P. M. Imbesi, C. Fidge, J. E. Raymond, S. I. Cauët and K. L. Wooley, ACS Macro Lett., 2012, 1, 473–477 CrossRef CAS.
- B. J. Adzima, H. Alan Aguirre, C. J. Kloxin, T. F. Scott and C. N. Bowman, Macromolecules, 2008, 41, 9112–9117 CrossRef CAS PubMed.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS PubMed.
- B. T. Michal, E. J. Spencer and S. J. Rowan, ACS Appl. Mater. Interfaces, 2016, 8, 11041–11049 CrossRef CAS PubMed.
- B. T. Michal, C. A. Jaye, E. J. Spencer and S. J. Rowan, ACS Macro Lett., 2013, 2, 694–699 CrossRef CAS.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS.
- A. P. Bapat, D. Roy, J. G. Ray, D. A. Savin and B. S. Sumerlin, J. Am. Chem. Soc., 2011, 133, 19832–19838 CrossRef CAS PubMed.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2018, 80, 39–93 CrossRef CAS.
- Q. Zhao, H. J. Qi and T. Xie, Prog. Polym. Sci., 2015, 49–50, 79–120 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2013, 26, 724–744 CrossRef.
- B. Zhang, Z. A. Digby, J. A. Flum, P. Chakma, J. M. Saul, J. L. Sparks and D. Konkolewicz, Macromolecules, 2016, 49, 6871–6878 CrossRef CAS.
- P. Chakma, L. H. Rodrigues Possarle, Z. A. Digby, B. Zhang, J. L. Sparks and D. Konkolewicz, Polym. Chem., 2017, 8, 6534–6543 RSC.
- J. S. A. Ishibashi and J. A. Kalow, ACS Macro Lett., 2018, 7, 482–486 CrossRef CAS.
- N. Kuhl, R. Geitner, R. K. Bose, S. Bode, B. Dietzek, M. Schmitt, J. Popp, S. J. Garcia, S. van der Zwaag, U. S. Schubert and M. D. Hager, Macromol. Chem. Phys., 2016, 217, 2541–2550 CrossRef CAS.
- N. Kuhl, R. Geitner, J. Vitz, S. Bode, M. Schmitt, J. Popp, U. S. Schubert and M. D. Hager, J. Appl. Polym. Sci., 2017, 134, 44805 CrossRef.
- N. Van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Angew. Chem. Int. Ed., 2020, 59, 3609–3617 CrossRef CAS PubMed.
- I. M. Serafimova, M. A. Pufall, S. Krishnan, K. Duda, M. S. Cohen, R. L. Maglathlin, J. M. McFarland, R. M. Miller, M. Frödin and J. Taunton, Nat. Chem. Biol., 2012, 8, 471 CrossRef CAS PubMed.
- Y. Zhong, Y. Xu and E. V. Anslyn, Eur. J. Org Chem., 2013, 2013, 5017–5021 CrossRef CAS.
- E. H. Krenske, R. C. Petter and K. N. Houk, J. Org. Chem., 2016, 81, 11726–11733 CrossRef CAS PubMed.
- A. Rekondo, R. Martin, A. Ruiz De Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- F. Vidal, J. Gomezcoello, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2019, 141, 15963–15971 CrossRef CAS PubMed.
- M. S. Menyo, C. J. Hawker and J. H. Waite, Soft Matter, 2013, 9, 10314–10323 RSC.
- S. C. Grindy, R. Learsch, D. Mozhdehi, J. Cheng, D. G. Barrett, Z. Guan, P. B. Messersmith and N. Holten-Andersen, Nat. Mater., 2015, 14, 1210–1216 CrossRef CAS PubMed.
- R. J. J. Williams, B. A. Rozenberg and J.-P. Pascault, in Polymer Analysis Polymer Physics. Advances in Polymer Science, Springer, Berlin, Heidelberg, 1997, vol. 128, pp. 95–156 Search PubMed.
- K. Yamanaka, Y. Takagi and T. Inoue, Polymer, 1989, 30, 1839–1844 CrossRef CAS.
- A. H. Torbati, H. B. Nejad, M. Ponce, J. P. Sutton and P. T. Mather, Soft Matter, 2014, 10, 3112–3121 RSC.
- Y. Liu, J. Appl. Polym. Sci., 2013, 127, 3279–3292 CrossRef CAS.
- L. Zhang, L. Chen and S. J. Rowan, Macromol. Chem. Phys., 2017, 218, 1600320 CrossRef.
- M. Guerre, C. Taplan, R. Nicolaÿ, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2018, 140, 13272–13284 CrossRef CAS PubMed.
- J. J. Lessard, G. M. Scheutz, S. H. Sung, K. A. Lantz, T. H. Epps and B. S. Sumerlin, J. Am. Chem. Soc., 2020, 142, 283–289 CrossRef CAS PubMed.
- X. Chen, L. Li, T. Wei and J. M. Torkelson, Macromol. Chem. Phys., 2019, 220, 1900083 CrossRef.
- X. Chen, L. Li and J. M. Torkelson, Polymer, 2019, 178, 121604 CrossRef CAS.
- R. G. Ricarte, F. Tournilhac and L. Leibler, Macromolecules, 2019, 52, 432–443 CrossRef CAS.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- World Intellectual Property Organization International Bureau, WO 2010/056452 A2, 2009, 32.
- A. Kotikian, C. Mcmahan, E. C. Davidson, J. M. Muhammad, R. D. Weeks, C. Daraio and J. A. Lewis, Sci. Robot., 2019, 4, eaax7044 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary results and video. See DOI: 10.1039/d0sc00605j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |