
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitric oxide monooxygenation (NOM) reaction of cobalt-nitrosyl {Co(NO)}8 to CoII-nitrito {CoII(NO2−)}: base induced hydrogen gas (H2) evolution†
Sandip
Das
a,
Kulbir
a,
Somnath
Ghosh
a,
Subash
Chandra Sahoo
b and
Pankaj
Kumar
 *a
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research (IISER), Tirupati 517507, India. E-mail: pankaj@iisertirupati.ac.in
bDepartment of Chemistry, Punjab University, Chandigarh, Punjab, India
First published on 24th April 2020
Abstract
Here, we report the nitric oxide monooxygenation (NOM) reactions of a CoIII-nitrosyl complex (1, {Co-NO}8) in the presence of mono-oxygen reactive species, i.e., a base (OH−, tetrabutylammonium hydroxide (TBAOH) or NaOH/15-crown-5), an oxide (O2− or Na2O/15-crown-5) and water (H2O). The reaction of 1 with OH− produces a CoII-nitrito complex {3, (CoII-NO2−)} and hydrogen gas (H2), via the formation of a putative N-bound Co-nitrous acid intermediate (2, {Co-NOOH}+). The homolytic cleavage of the O–H bond of proposed [Co-NOOH]+ releases H2via a presumed CoIII-H intermediate. In another reaction, 1 generates CoII-NO2− when reacted with O2−via an expected CoI-nitro (4) intermediate. However, complex 1 is found to be unreactive towards H2O. Mechanistic investigations using 15N-labeled-15NO and 2H-labeled-NaO2H (NaOD) evidently revealed that the N-atom in CoII-NO2− and the H-atom in H2 gas are derived from the nitrosyl ligand and OH− moiety, respectively.
As a radical species, nitric oxide (NO) has attracted great interest from the scientific community due to its major role in various physiological processes such as neurotransmission, vascular regulation, platelet disaggregation and immune responses to multiple infections.1 Nitric oxide synthase (NOS),2 and nitrite reductase (NiR)3 enzymes are involved in the biosynthesis of NO. NOSs produce NO by the oxidation of the guanidine nitrogen in L-arginine.4 However, in mammals and bacteria, NO2− is reduced to NO by NiRs in the presence of protons, i.e., NO2− + e− + 2H+ → NO + H2O.5 Biological dysfunctions may cause overproduction of NO, and being radical it leads to the generation of reactive nitrogen species (RNS), i.e., peroxynitrite (PN, OONO−)6 and nitrogen dioxide (˙NO2),7 upon reaction with reactive oxygen species (ROS) such as superoxide (O2˙−),8 peroxide (H2O2),9 and dioxygen (O2).10 Hence, it is essential to maintain an optimal level of NO. In this regard, nitric oxide dioxygenases (NODs)11 are available in bio-systems to convert excess NO to biologically benign nitrate (NO3−).12
| NO2− + FeII + H+ ↔ NO + FeIII + OH− | (1) |
| [M–NO]n + 2OH− → [M–NO2](n−2) + H2O | (2) |
NOD enzymes generate NO3− from NO;11b,12−13 however, the formation of NO2− from NO is still under investigation. Clarkson and Bosolo reported NO2− formation in the reaction of CoIII-NO and O2.14 Nam and co-workers showed the generation of CoII-NO2− from CoIII-NO upon reaction with O2˙−.15 Recently, Mondal and co-workers reported NO2− formation in the reaction of CoII-NO with O2.16 Apart from cobalt, the formation of CuII-NO2− was also observed in the reaction of CuI-NO and O2.17 For metal-dioxygen adducts, i.e., CrIII-O2˙− and MnIV-O22−, NOD reactions led to the generation of CrIII-NO2− (ref. 18) and MnV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + NO2−,19 respectively. However, the NOD reaction of FeIII-O2˙− and FeIII-O22− with NO and NO+, respectively, generated FeIII-NO3−via FeIVO and ˙NO2.20 Ford suggested that the reaction of ferric-heme nitrosyl with hydroxide leads to the formation of NO2− and H+.12 Lehnert and co-workers reported heme-based Fe-nitrosyl complexes21 showing different chemistries due to the FeII-NO+ type electronic structures. On the other hand, Bryan proposed that the one-electron reduction of NO2− to NO in ferrous heme protein is reversible (eqn (1)).22 Also, it is proposed that excess NO in biological systems is converted to NO2− and produces one equivalent of H+ upon reaction with ˙OH.23 Previously reported reactivity of M–NOs of Fe24 with OH− suggested the formation of NO2− and one equivalent of H+, where H+ further reacts with one equivalent of OH− and produces H2O (eqn (2)).25
O + NO2−,19 respectively. However, the NOD reaction of FeIII-O2˙− and FeIII-O22− with NO and NO+, respectively, generated FeIII-NO3−via FeIVO and ˙NO2.20 Ford suggested that the reaction of ferric-heme nitrosyl with hydroxide leads to the formation of NO2− and H+.12 Lehnert and co-workers reported heme-based Fe-nitrosyl complexes21 showing different chemistries due to the FeII-NO+ type electronic structures. On the other hand, Bryan proposed that the one-electron reduction of NO2− to NO in ferrous heme protein is reversible (eqn (1)).22 Also, it is proposed that excess NO in biological systems is converted to NO2− and produces one equivalent of H+ upon reaction with ˙OH.23 Previously reported reactivity of M–NOs of Fe24 with OH− suggested the formation of NO2− and one equivalent of H+, where H+ further reacts with one equivalent of OH− and produces H2O (eqn (2)).25
Here in this report, we explore the mechanistic aspects of nitric oxide monooxygenation (NOM) reactions of the CoIII-nitrosyl complex, [(12TMC)CoIII(NO−)]2+/{Co(NO)}8 (1),15,26 bearing the 12TMC ligand (12TMC = 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane) with mono-oxygen reactive species (O2−, OH− and H2O) (Scheme 1). Complex 1 reacts with the base (OH−, tetrabutylammonium hydroxide (TBAOH)/or NaOH in the presence of 15-crown-5 as the OH− source) and generates the corresponding CoII-nitrito complex, [(12TMC)CoII(NO2−)]+ (3), with the evolution of hydrogen gas (H2) via the formation of a plausible N-bound Co-nitrous acid intermediate ([Co-NOOH]+, 2) in CH3CN at 273 K (Scheme 1, reaction (I)). Also, when 1 reacts with the oxide (O2− or Na2O in the presence of 15-crown-5), it generates the CoII-nitrito complex (3) via a probable CoI-nitro, [(12TMC)CoI(NO2−)] (4), intermediate (Scheme 1, reaction (II)); however, 1 does not react with water (Scheme 1, reaction (III)). Mechanistic investigations using 15N-labeled-15NO, D-labeled-NaOD and 18O-labelled-18OH− demonstrated, unambiguously, that the N and O-atoms in the NO2− ligand of 3 resulted from NO and OH− moieties; however, the H-atoms of H2 are derived from OH−. To the extent of our knowledge, the present work reports the very first systematic study of CoIII-nitrosyl complex reactions with H2O, OH− and O2−. This new finding presents an alternative route for NO2− generation in biosystems, and also illustrates a new pathway of H2 evolution, in addition to the reported literature.12,27
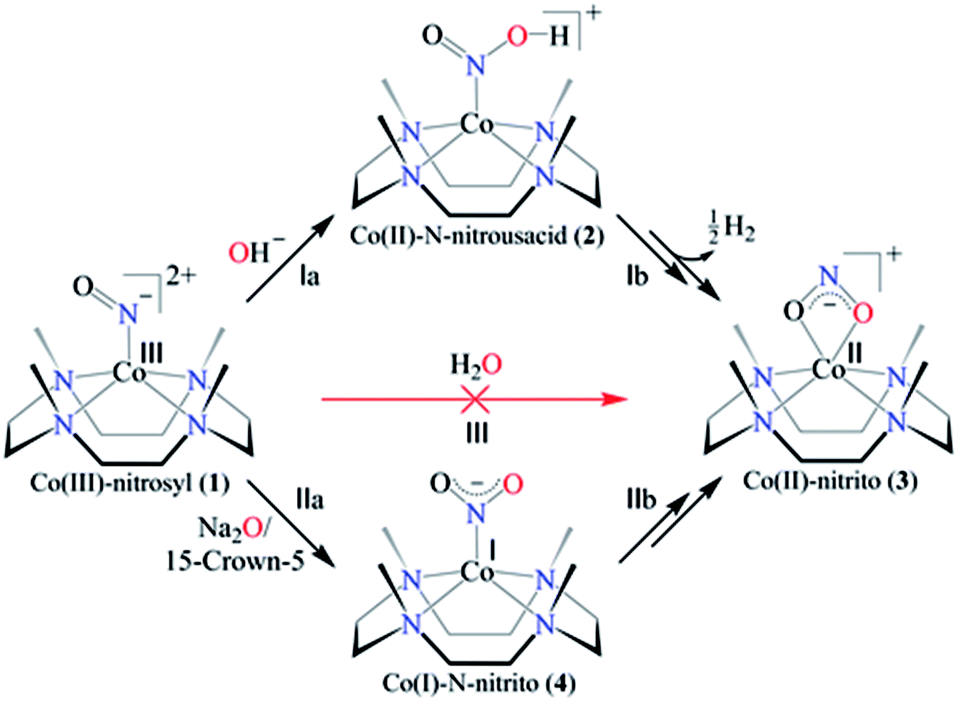 | ||
| Scheme 1 Nitric oxide monooxygenation (NOM) reactions of cobalt-nitrosyl complex (1) in the presence of a base (OH−), sodium oxide (Na2O) and water (H2O). | ||
To further explore the chemistry of [(12TMC)CoIII(NO−)]2+ (1),15,26 and the mechanistic insights of NOM reactions, we have reacted it with a base (OH−), an oxide (O2−), and water (H2O). When complex 1 was reacted with TBAOH in CH3CN, the color of complex 1 changed to light pink from dark pink. In this reaction, the characteristic absorption band of 1 (370 nm) disappears within 2 minutes (Fig. 1a; ESI, Experimental section (ES) and Fig. S1a†), producing a CoII-nitrito complex, [(12TMC)CoII(NO2−)]+ (3), with H2 (Scheme 1, reaction (Ib)), in contrast to the previous reports on base induced NOM reactions (eqn (2)).12,25,28 The spectral titration data confirmed that the ratio-metric equivalent of OH− to 1 was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (ESI, Fig. S1b†). 3 was determined to be [(12TMC)CoII(NO2−)](BF4) based on various spectroscopic and structural characterization experiments (vide infra).15,26b
:1 (ESI, Fig. S1b†). 3 was determined to be [(12TMC)CoII(NO2−)](BF4) based on various spectroscopic and structural characterization experiments (vide infra).15,26b
The FT-IR spectrum of 3 showed a characteristic peak for nitrite stretching at 1271 cm−1 (CoII-14NO2−) and shifted to 1245 cm−1 (CoII-15NO2−) when 3 was prepared by reacting 15N-labeled NO (CoIII-15NO) with OH− (Inset, Fig. 1a and Fig. S2†). The shifting of NO2− stretching (Δ = 30 cm−1) indicates that the N-atom in the NO2− ligand is derived from CoIII-15NO. The ESI-MS spectrum of 3 showed a prominent peak at m/z 333.2, [(12TMC)CoII(14NO2−)]+ (calcd m/z 333.2), which shifted to 334.2, [(12TMC)CoII(15NO2−)]+ (calcd m/z 334.2), when the reaction was performed with CoIII-15NO (Inset, Fig. 1b; ESI, Fig. S3a†); indicating clearly that NO2− in 3 was derived from the NO moiety of 1. In addition, we have reacted 1 with Na18OH (ES and ESI†), in order to follow the source of the second O-atom in 3-NO2−. The ESI-MS spectrum of the reaction mixture, obtained by reacting 1 with Na18OH, showed a prominent peak at m/z 335.2, [(12TMC)CoII(18ONO−)]+ (calcd m/z 335.2), (SI, Fig. S3b†) indicating clearly that NO2− in 3 was derived from 18OH−. The 1H NMR spectrum of 3 did not show any signal for aliphatic protons of the 12TMC ligand, suggesting a bivalent cobalt center (Fig. S4†).26b Furthermore, we have determined the magnetic moment of 3, using Evans' method, and it was found to be 4.62 BM, suggesting a high spin Co(II) metal center with three unpaired electrons (ESI† and ES).29 The exact conformation of 3 was provided by single-crystal X-ray crystallographic analysis (Fig. 2b, ESI, ES, Fig. S5, and Tables T1 and T2†) and similar to that of previously reported CoII-NO2−/MII-NO2−.15,26b Also, we have quantified the amount of nitrite (90 ± 5%), formed in the above reaction, using the Griess reagent (ESI, ES, and Fig. S6†).
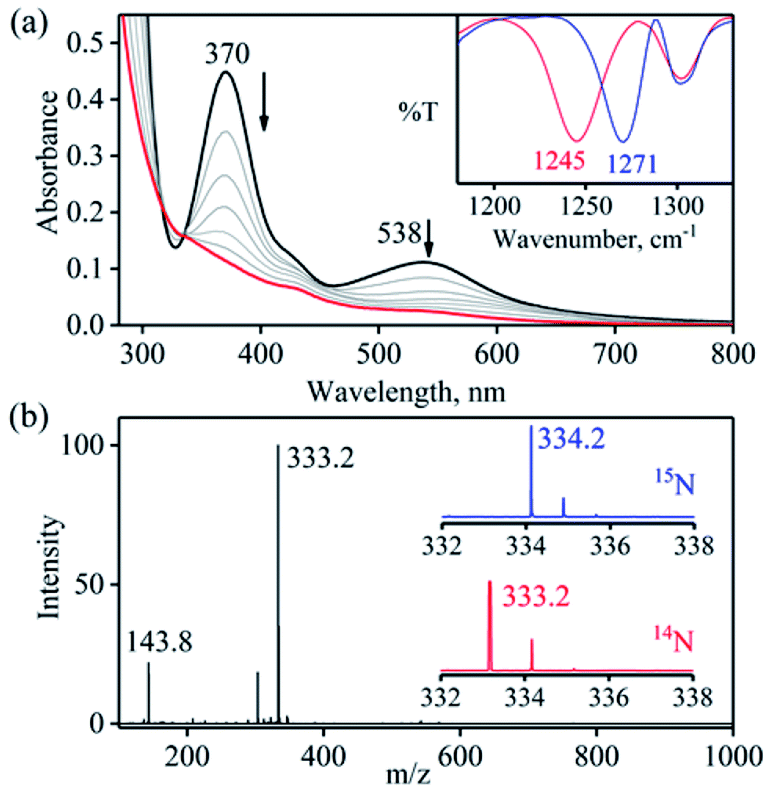 | ||
| Fig. 1 (a) UV-vis spectral changes of 1 (0.50 mM, black line) upon addition of OH− (1 equiv.) in CH3CN under Ar at 273 K. Black line (1) changed to red line (3) upon addition of OH−. Inset: IR spectra of 3-14NO2− (blue line) and 3-15NO2− (red line) in KBr. (b) ESI-MS spectra of 3. The peak at 333.2 is assigned to [(12TMC)CoII(NO2)]+ (calcd m/z 333.1). Inset: isotopic distribution pattern for 3-14NO2− (red line) and 3-15NO2− (blue line). | ||
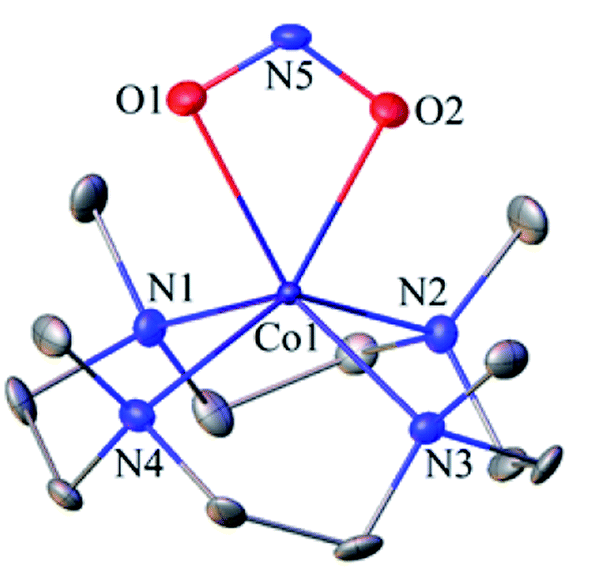 | ||
| Fig. 2 Displacement ellipsoid plot (20% probability) of 3 at 100 K. Disordered C-atoms of the TMC ring, anion and H-atoms have been removed for clarity. | ||
As is known from the literature, a metal-nitrous acid intermediate may form either by the reaction of a metal-nitrosyl with a base27 or by the metal-nitrite reaction with an acid (nitrite reduction chemistry);26b however, the products of both the reactions are different. Here, for the first time, we have explored the reaction of CoIII-nitrosyl (1) with a base. In this reaction, it is clear that the formation of CoII-nitrito would be accomplished by the release of H2 gas via the generation of a transient N-bound [Co-(NOOH)]+ intermediate (Scheme 2, reaction (II)). The formation of CoII-NO2− (3) from the [Co-(NOOH)]+ intermediate is likely to proceed by either (i) homolytic cleavage of the O–H bond and release of H2via the proposed CoIII-H transient species (CoIII-H = CoII + 1/2H2)30 (Scheme 2, reaction (III)), as reported in previous literature where the reduced cobalt, in a number of different ligand environments, is a good H+ reduction catalyst and generates H2 gas via a CoIII-H intermediate31 or (ii) heterolytic cleavage of the O–H bond and the formation of CoI-NO2− + H+.27 In the present study, we observed the formation of 3 and H2via the plausible homolytic cleavage of the NOO–H moiety of 2 as shown in Scheme 2, in contrast to the previous reports on base-induced reactions on metal-nitrosyls (eqn (3)).27 Taking together both possibilities, (i) is the most reasonable pathway for the NOM reaction of complex 1 in the presence of a base (as shown in Scheme 2, reaction (III)). And the reaction is believed to go through a CoIII-H intermediate as reported previously in CoI-induced H+ reduction in different ligand frameworks and based on literature precedence, we believe that complex 1 acts in a similar manner.31
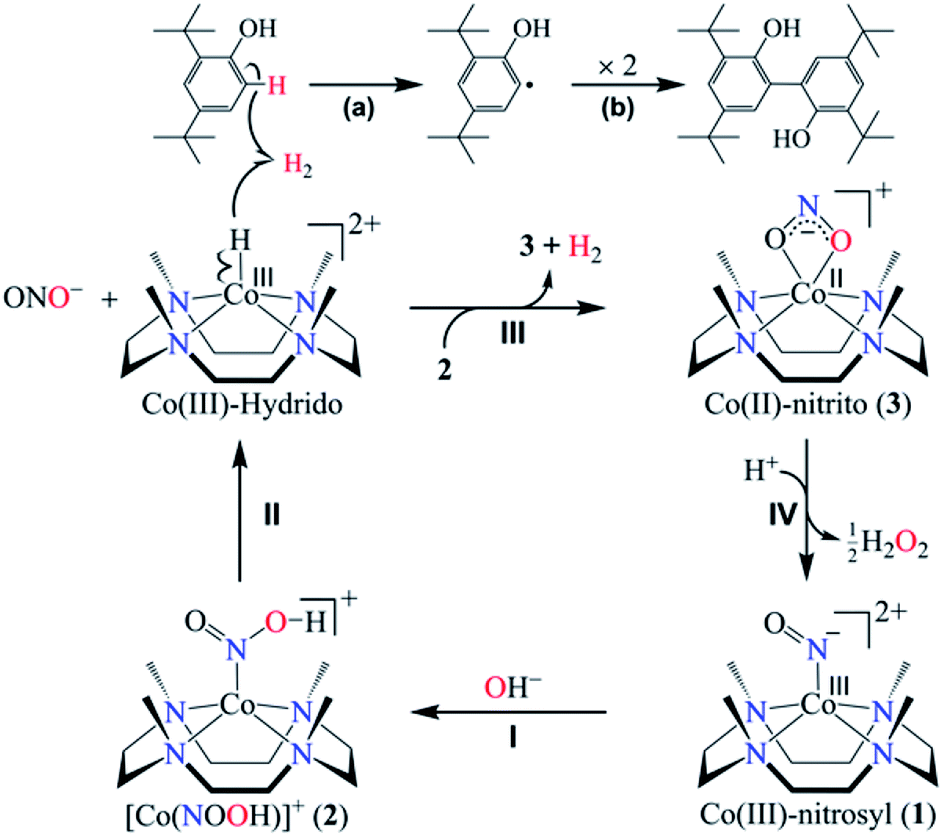 | ||
| Scheme 2 NOM reaction of complex 1 in the presence of OH−, showing the generation of CoII-nitrito (3) and H2via a Co(III)-hydrido intermediate. | ||
In contrast to an O-bound CoII-ONOH intermediate, where N–O bond homolysis of the ON-OH moiety generates H2O2 (Scheme 2, reaction (IV)),26b the N-bound [Co-(NOOH)]+ intermediate decomposes to form NO2− and a Co(III)-H transient species, arising from β-hydrogen transfer from the NOO–H moiety to the cobalt-center (Scheme 2, reaction (II)).30a,c,32 The Co(III)-hydrido species may generate H2 gas either (a) by its transformation to the Co(II)-nitrito complex (2) and H2 gas as observed in the case of CoIII-H intermediate chemistry30a,c,e−g as proposed in the chemistry of the CoI complex with H+ reduction31 and other metal-hydrido intermediates32 and also explained in O2 formation in PN chemistry17,33 or (b) by the reacting with another [Co-(NOOH)]+ intermediate (Scheme 2, reaction (III)).
Furthermore, we have confirmed the H2 formation in the NOM reaction of 1 with OH− by headspace gas mass spectrometry (Fig. 3a). Also, carrying out the reaction of 1 with NaOD leads to the formation of the [Co-(NOOD)]+ intermediate, which then transforms to a CoIII-D transient species. Further, as described above, the CoIII-D species releases D2 gas, detected by headspace gas mass spectrometry (Fig. 3b), which evidently established that H2 gas formed in the reaction of 1 with OH−. In this regard, we have proposed that in the first step of this reaction, the nucleophilic addition of OH− to {Co-NO}8 generates a transient N-bound [Co-(NOOH)]+ intermediate that is generated by an internal electron transfer to CoIII (Scheme 2, reaction (I)). By following the mechanism proposed in the case of CoIII-H,30a−c O2,15 and H2O2(ref. 26b) formation, we have proposed the sequences of the NOM reaction of 1, which leads to the generation of CoII-nitrito and H2 (Scheme 2, reaction (I)–(III) and Scheme 3). In the second step, O–H bond homolytic cleavage generates a CoIII-H transient species + NO2−via a β-hydrogen elimination reaction of the [Co-(NOOH)]+ intermediate.32 The CoIII-H intermediate may undergo the following reactions to generate H2 gas and CoII-nitrito either (a) by the natural decomposition of the CoIII-H transient species to generate H2,30a,c,e−g or (b) by the H-atom abstraction from another [Co-(NOOH)]+ intermediate (Scheme 3). Also, to validate our assumption that the reaction goes through a plausible N-bound [Co-(NOOH)]+ intermediate followed by its transformation to the CoIII-H species (vide supra), we have performed the reaction of 1 with NaOH/NaOD (in 1:1 ratio). In this reaction, we have observed the formation of a mixture of H2, D2, and HD gases, which indicates clearly that the reaction goes through the formation of CoIII-H and CoIII-D transient species via the aforementioned mechanism (Fig. 3c). This is the only example where tracking of the H atoms has confirmed the H2 generation from an N-bound NOO–H moiety as proposed for H2 formation from CoIII-H.30
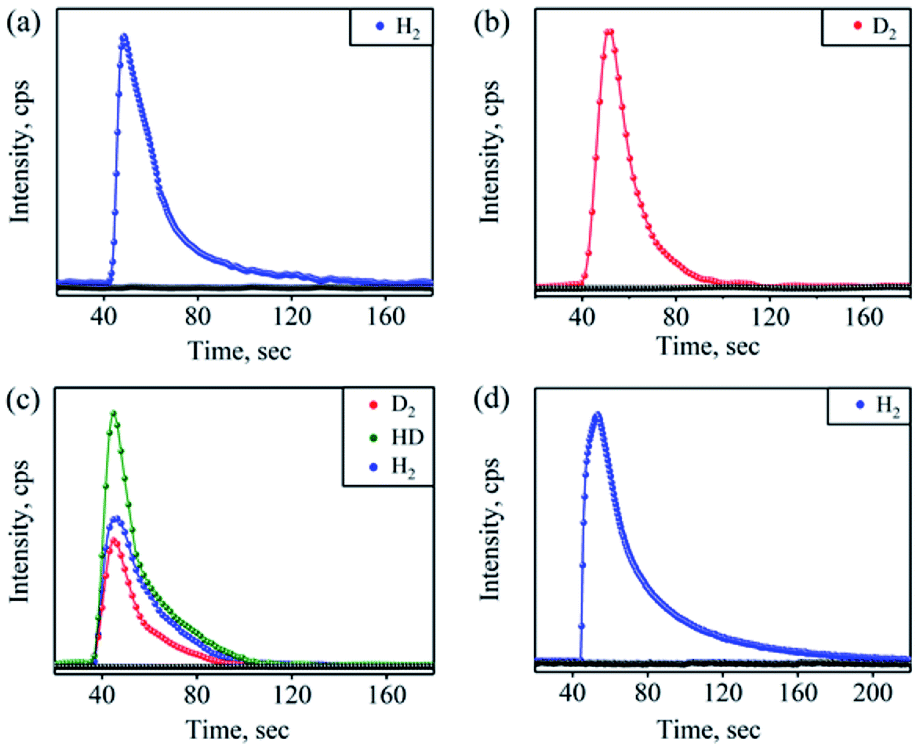 | ||
| Fig. 3 Mass spectra of formation of (a) H2 in the reaction of 1 (5.0 mM) with NaOH (5.0 mM), (b) D2 in the reaction of 1 (5.0 mM) with NaOD (5.0 mM), (c) D2, HD, and H2 in the reaction of 1 (5.0 mM) with NaOD/NaOH (1:1), and (d) H2 in the reaction of 1 (5.0 mM) with NaOH in the presence of 2,4 DTBP (50 mM). | ||
 | ||
| Scheme 3 NOM reaction of complex 1 in the presence of OH−, showing the different steps of the reaction. | ||
While, we do not have direct spectral evidence to support the formation of the transient N-bound [Co-(NOOH)]+ intermediate and its decomposition to the CoIII-H transient species via β-hydrogen transfer from the NOOH moiety to the cobalt center, support for its formation comes from our finding that the reactive hydrogen species can be trapped by using 2,4-di-tert-butyl-phenol (2,4-DTBP).34 In this reaction, we observed the formation of 2,4-DTBP-dimer (2,4-DTBP-D, ∼67%) as a single product (ESI, ES, and Fig. S7†). This result can readily be explained by the H-atom abstraction reaction of 2,4-DTBP either by [Co-(NOOH)]+ or CoIII-H, hence generating a phenoxyl-radical and 3 with H2 (Fig. 3d and Scheme 2, reaction (a)). Also, we have detected H2 gas formation in this reaction (ESI,† ES, and Fig. 3d). In the next step, two phenoxyl radicals dimerized to give 2,4-DTBP-dimer (Scheme 2c, reaction (II)). Thus, the observation of 2,4-DTBP-dimer in good yield supports the proposed reaction mechanism (Scheme 2, reaction (a) and (b)). Further, the formation of 2,4 DTBP as a single product also rules out the formation of the hydroxyl radical as observed in the case of an O-bound nitrous acid intermediate.26b
Furthermore, we have explored the NOM reactivity of 1 with Na2O/15-crown-5 (as the O2− source) and observed the formation of the CoII-nitrito complex (3) via a plausible CoI-nitro (4) intermediate (Scheme 1, reaction (IIa); also see the ESI† and ES); however, 1 was found to be inert towards H2O (Scheme 1, reaction (III); also see the ESI, ES and Fig. S8†). The product obtained in the reaction of 1 with O2− was characterized by various spectroscopic measurements.15,26b The UV-vis absorption band of 1 (λmax = 370 nm) disappears upon the addition of 1 equiv. of Na2O and a new band (λmax = 535 nm) forms, which corresponds to 3 (ESI, Fig. S9†). The FT-IR spectrum of the isolated product of the above reaction shows a characteristic peak for CoII-bound nitrite at 1271 cm−1, which shifts to 1245 cm−1 when exchanged with 15N-labeled-NO (15N16O) (ESI, ES, and Fig. S10†), clearly indicating the generation of nitrite from the NO ligand of complex 1.26b The ESI-MS spectrum recorded for the isolated product (vide supra) shows a prominent ion peak at m/z 333.1, and its mass and isotope distribution pattern matches with [(12-TMC)CoII(NO2)]+ (calc. m/z 333.1) (ESI, Fig. S11†). Also, we quantified the amount of 3 (85 ± 5%) by quantifying the amount of nitrite (85 ± 5%) using the Griess reagent test (ESI, ES, and Fig. S6†).
In summary, we have demonstrated the reaction of CoIII-nitrosyl, [(12-TMC)CoIII(NO−)]2+/{CoNO}8 (1), with mono-oxygen reactive species (O2−, OH− and H2O) (Scheme 1). For the first time, we have established the clear formation of a CoII-nitrito complex, [(12TMC)CoII(NO2−)]+ (3), and H2 in the reaction of 1 with one equivalent of OH−via a transient N-bound [Co-(NOOH)]+ (2) intermediate. This [Co-(NOOH)]+ intermediate undergoes the O–H bond homolytic cleavage and generates a CoIII-H transient species with NO2−, via a β-hydrogen elimination reaction of the [Co-(NOOH)]+ intermediate, which upon decomposition produces H2 gas. This is in contrast to our previous report, where acid-induced nitrite reduction of 3 generated 1 and H2O2via an O-bound CoII-ONOH intermediate.26b Complex 1 was found to be inert towards H2O; however, we have observed the formation of 3 when reacted with O2−. It is important to note that H2 formation involves a distinctive pathway of O–H bond homolytic cleavage in the [Co-(NOOH)]+ intermediate, followed by the generation of the proposed CoIII-H transient species (CoII + 1/2H2)30 prior to H2 evolution as described in CoI chemistry with H+ in many different ligand frameworks.31 The present study is the first-ever report where the base induced NOM reaction of CoIII-nitrosyl (1) leads to CoII-nitrito (3) with H2 evolution via an N-bound [Co-(NOOH)]+ intermediate, in contrast to the chemistry of O-bound CoII-ONOH26b, hence adding an entirely new mechanistic insight of base induced H2 gas evolution and an additional pathway for NOM reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grants-in-Aid (Grant No. EEQ/2016/000466) from SERB-DST. We acknowledge Prof. Wonwoo Nam and co-workers for the stabilization of initial complexes and providing a ground to develop new chemistry. We acknowledge Prof. K. N. Ganesh (Director, IISER Tirupati) for continuous guidance and support, special thanks to Dr Sayam Sen Gupta (IISER Kolkata), Dr E. Balaraman (IISER Tirupati) and Dr R. O. Ramabhadran (IISER Tirupati) for fruitful discussion and support. SCS thanks DST-FIST for the single crystal facility at PU.References
- (a) G. B. Richter-Addo, P. Legzdins and J. Burstyn, Chem. Rev., 2002, 102, 857–860 CrossRef CAS PubMed; (b) C. Bogdan, Nat. Immunol., 2001, 2, 907–916 CrossRef CAS PubMed; (c) L. Jia, C. Bonaventura, J. Bonaventura and J. S. Stamler, Nature, 1996, 380, 221–226 CrossRef CAS PubMed; (d) R. F. Furchgott, Angew. Chem., Int. Ed., 1999, 38, 1870–1880 CrossRef CAS; (e) L. J. Ignarro, Angew. Chem., Int. Ed., 1999, 38, 1882–1892 CrossRef CAS; (f) L. J. Ignarro, Nitric oxide: biology and pathobiology, Academic press, 2000 Search PubMed.
- (a) L. Castillo, T. C. deRojas, T. E. Chapman, J. Vogt, J. F. Burke, S. R. Tannenbaum and V. R. Young, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 193–197 CrossRef CAS PubMed; (b) R. M. Palmer, D. S. Ashton and S. Moncada, Nature, 1988, 333, 664–666 CrossRef CAS PubMed.
- (a) C. E. Sparacino-Watkins, J. Tejero, B. Sun, M. C. Gauthier, J. Thomas, V. Ragireddy, B. A. Merchant, J. Wang, I. Azarov, P. Basu and M. T. Gladwin, J. Biol. Chem., 2014, 289, 10345–10358 CrossRef CAS PubMed; (b) C. Gherasim, P. K. Yadav, O. Kabil, W. N. Niu and R. Banerjee, PLoS One, 2014, 9, e85544 CrossRef PubMed; (c) J. L. Zweier, P. Wang, A. Samouilov and P. Kuppusamy, Nat. Med., 1995, 1, 804–809 CrossRef CAS PubMed.
- (a) Q. Liu and S. S. Gross, Methods Enzymol., 1996, 268, 311–324 CAS; (b) C. Nathan and Q. W. Xie, J. Biol. Chem., 1994, 269, 13725–13728 CAS.
- (a) B. A. Averill, Chem. Rev., 1996, 96, 2951–2964 CrossRef CAS PubMed; (b) J. O. Lundberg, E. Weitzberg and M. T. Gladwin, Nat. Rev. Drug Discovery, 2008, 7, 156–167 CrossRef CAS PubMed.
- R. Radi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4003–4008 CrossRef CAS PubMed.
- C. H. Lim, P. C. Dedon and W. M. Deen, Chem. Res. Toxicol., 2008, 21, 2134–2147 Search PubMed.
- (a) S. Goldstein, J. Lind and G. Merenyi, Chem. Rev., 2005, 105, 2457–2470 CrossRef CAS PubMed; (b) P. C. Dedon and S. R. Tannenbaum, Arch. Biochem. Biophys., 2004, 423, 12–22 CrossRef CAS PubMed; (c) P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS PubMed.
- B. Kalyanaraman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11527–11528 CrossRef CAS PubMed.
- R. S. Lewis and W. M. Deen, Chem. Res. Toxicol., 1994, 7, 568–574 Search PubMed.
- (a) P. R. Gardner, A. M. Gardner, L. A. Martin and A. L. Salzman, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10378–10383 CrossRef CAS PubMed; (b) M. P. Doyle and J. W. Hoekstra, J. Inorg. Biochem., 1981, 14, 351–358 CrossRef CAS PubMed.
- P. C. Ford and I. M. Lorkovic, Chem. Rev., 2002, 102, 993–1018 CrossRef CAS PubMed.
- M. P. Schopfer, B. Mondal, D. H. Lee, A. A. Sarjeant and K. D. Karlin, J. Am. Chem. Soc., 2009, 131, 11304–11305 CrossRef CAS PubMed.
- S. G. Clarkson and F. Basolo, Inorg. Chem., 1973, 12, 1528–1534 CrossRef CAS.
- P. Kumar, Y. M. Lee, Y. J. Park, M. A. Siegler, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2015, 137, 4284–4287 CrossRef CAS PubMed.
- K. Gogoi, S. Saha, B. Mondal, H. Deka, S. Ghosh and B. Mondal, Inorg. Chem., 2017, 56, 14438–14445 CrossRef CAS PubMed.
- G. Y. Park, S. Deepalatha, S. C. Puiu, D. H. Lee, B. Mondal, A. A. Narducci Sarjeant, D. del Rio, M. Y. Pau, E. I. Solomon and K. D. Karlin, J. Biol. Inorg Chem., 2009, 14, 1301–1311 CrossRef CAS PubMed.
- A. Yokoyama, K. B. Cho, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2013, 135, 14900–14903 CrossRef CAS PubMed.
- S. Hong, P. Kumar, K. B. Cho, Y. M. Lee, K. D. Karlin and W. Nam, Angew. Chem., Int. Ed. Engl., 2016, 55, 12403–12407 CrossRef CAS PubMed.
- A. Yokoyama, J. E. Han, K. D. Karlin and W. Nam, Chem. Commun., 2014, 50, 1742–1744 RSC.
- (a) A. B. McQuarters, J. W. Kampf, E. E. Alp, M. Hu, J. Zhao and N. Lehnert, Inorg. Chem., 2017, 56, 10513–10528 CrossRef CAS PubMed; (b) V. K. K. Praneeth, F. Paulat, T. C. Berto, S. D. George, C. Näther, C. D. Sulok and N. Lehnert, J. Am. Chem. Soc., 2008, 130, 15288–15303 CrossRef CAS PubMed.
- N. S. Bryan, Free Radical Biol. Med., 2006, 41, 691–701 CrossRef CAS PubMed.
- T. A. Heinrich, R. S. da Silva, K. M. Miranda, C. H. Switzer, D. A. Wink and J. M. Fukuto, Br. J. Pharmacol., 2013, 169, 1417–1429 CrossRef CAS PubMed.
- J. H. Swinehart, Coord. Chem. Rev., 1967, 2, 385–402 CrossRef CAS.
- F. Roncaroli, L. M. Baraldo, L. D. Slep and J. A. Olabe, Inorg. Chem., 2002, 41, 1930–1939 CrossRef CAS PubMed.
- (a) P. Kumar, Y. M. Lee, L. Hu, J. Chen, Y. J. Park, J. Yao, H. Chen, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2016, 138, 7753–7762 CrossRef CAS PubMed; (b) M. A. Puthiyaveetil Yoosaf, S. Ghosh, Y. Narayan, M. Yadav, S. C. Sahoo and P. Kumar, Dalton Trans., 2019, 48, 13916–13920 RSC.
- F. Roncaroli, M. E. Ruggiero, D. W. Franco, G. L. Estiu and J. A. Olabe, Inorg. Chem., 2002, 41, 5760–5769 CrossRef CAS PubMed.
- J. H. Swinehart and P. A. Rock, Inorg. Chem., 1966, 5, 573–576 CrossRef CAS.
- (a) S. Hong, K. D. Sutherlin, J. Park, E. Kwon, M. A. Siegler, E. I. Solomon and W. Nam, Nat. Commun., 2014, 5, 5440 CrossRef CAS PubMed; (b) D. F. Evans, J. Chem. Soc., 1959, 2003–2005, 10.1039/jr9590002003; (c) D. F. Evans and D. A. Jakubovic, J. Chem. Soc., Dalton Trans., 1988, 2927–2933, 10.1039/DT9880002927.
- (a) J. L. Dempsey, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 1060–1065 CrossRef CAS PubMed; (b) V. Artero and M. Fontecave, Coord. Chem. Rev., 2005, 249, 1518–1535 CrossRef CAS; (c) J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed; (d) M. K. Sahoo, K. Saravanakumar, G. Jaiswal and E. Balaraman, ACS Catal., 2018, 8, 7727–7733 CrossRef CAS; (e) S. C. Marinescu, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15127–15131 CrossRef CAS PubMed; (f) X. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988–8998 CrossRef CAS PubMed; (g) N. Queyriaux, D. Sun, J. Fize, J. Pécaut, M. J. Field, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2020, 142, 274–282 CrossRef CAS PubMed.
- (a) V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed; (b) E. S. Andreiadis, P. A. Jacques, P. D. Tran, A. Leyris, M. Chavarot-Kerlidou, B. Jousselme, M. Matheron, J. Pecaut, S. Palacin, M. Fontecave and V. Artero, Nat. Chem., 2013, 5, 48–53 CrossRef CAS PubMed; (c) A. E. King, Y. Surendranath, N. A. Piro, J. P. Bigi, J. R. Long and C. J. Chang, Chem. Sci., 2013, 4, 1578–1587 RSC; (d) D. Basu, S. Mazumder, X. Shi, H. Baydoun, J. Niklas, O. Poluektov, H. B. Schlegel and C. N. Verani, Angew. Chem., Int. Ed., 2015, 54, 2105–2110 CrossRef CAS PubMed; (e) S. Roy, Z. Huang, A. Bhunia, A. Castner, A. K. Gupta, X. Zou and S. Ott, J. Am. Chem. Soc., 2019, 141, 15942–15950 CrossRef CAS PubMed.
- (a) A. Boddien, D. Mellmann, F. Gartner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1736 CrossRef CAS PubMed; (b) X. Yang, Dalton Trans., 2013, 42, 11987–11991 RSC.
- (a) O. A. Babich and E. S. Gould, Res. Chem. Intermed., 2002, 28, 575–583 CrossRef CAS; (b) J. W. Coddington, J. K. Hurst and S. V. Lymar, J. Am. Chem. Soc., 1999, 121, 2438–2443 CrossRef CAS; (c) S. Pfeiffer, A. C. Gorren, K. Schmidt, E. R. Werner, B. Hansert, D. S. Bohle and B. Mayer, J. Biol. Chem., 1997, 272, 3465–3470 CrossRef CAS PubMed.
- M. F. R. Mulcahy, D. J. Williams and J. R. Wilmshurst, Aust. J. Chem., 1964, 17, 1329–1341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1974616. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01572e |
| This journal is © The Royal Society of Chemistry 2020 |