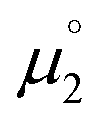DOI:
10.1039/D0SC00699H
(Edge Article)
Chem. Sci., 2020,
11, 4138-4149
Aromaticity and sterics control whether a cationic olefin radical is resistant to disproportionation†
Received
6th February 2020
, Accepted 28th March 2020
First published on 30th March 2020
Abstract
We elucidate why some electron rich-olefins such as tetrathiafulvalene (TTF) or paraquat (1,1′-dimethyl-4,4′-bipyridinylidene) form persistent radical cations, whereas others such as the dimer of N,N′-dimethyl benzimidazolin-2-ylidene (benzNHC) do not. Specifically, three heterodimers derived from cyclic (alkyl) (amino) carbenes (CAAC) with N,N′-dimethyl imidazolin-2-ylidene (NHC), N,N′-dimethyl imidazolidin-2-ylidene (saNHC) and N-methyl benzothiazolin-2-ylidene (btNHC) are reported. Whereas the olefin radical cations with the NHC and btNHC are isolable, the NHC compound with a saturated backbone (saNHC) disproportionates instead to the biscation and olefin. Furthermore, the electrochemical properties of the electron-rich olefins derived from the dimerization of the saNHC and btNHC were assessed. Based on the experiments, we propose a general computational method to model the electrochemical potentials and disproportionation equilibrium. This method, which achieves an accuracy of 0.07 V (0.06 V with calibration) in reference to the experimental values, allows for the first time to rationalize and predict the (in)stability of olefin radical cations towards disproportionation. The combined results reveal that the stability of heterodimeric olefin radical cations towards disproportionation is mostly due to aromaticity. In contrast, homodimeric radical cations are in principle isolable, if lacking steric bulk in the 2,2′ positions of the heterocyclic monomers. Rigid tethers increase accordingly the stability of homodimeric radical cations, whereas the electronic effects of substituents seem much less important for the disproportionation equilibrium.
Introduction
Electron-rich olefins are an exciting class of organic redox systems with potentially three stable redox states1–3 and are popular reductants in organic synthesis.4–9 Exciting applications are in particular associated with the open-shell redox state with one unpaired electron, which allows for intriguing conductive and photochemical properties. The arguably most relevant derivative, tetrathiafulvalene (TTF 1, Fig. 1), its saturated congener 2,10,11 as well as the benzannulated derivative 3 even have been called “the brick and mortar” of organic materials. They are now commonly applied in switches, solar cells, or organic field-effect transistors (OFETs).12–14 Very similar redox properties are found for olefins derived from biscationic paraquat (methyl viologen, respectively; 1,1′-dimethyl-4,4′-bipyridinylidene) 4, the related ortho-derivative (1,1′-dimethyl-2,2′-bipyridinylidene) 5, the dimethylaminopyridine (DMAP) dimer 6,15 as well as 4,4′-bipyrylene 7.16–20
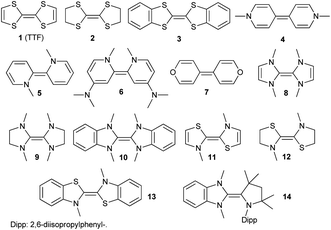 |
| | Fig. 1 Homo- (1–13) and heterodimeric (14) electron-rich olefins. | |
The aza-analogues of TTF, i.e. tetraaminoethylenes or enetetramines, originate from the formal dimerization of unsaturated- (NHC, 8), saturated- (saNHC, 9) or benzannulated- (benzNHC, 10) N-heterocyclic carbenes (NHCs). Other examples comprise thiazolin-2-ylidenes (11, 12, 13, Fig. 1).21,22 However, harnessing the exceptional electronic properties of NHC-derived electron-rich olefins for organic electronics remained challenging due to the undesired dissociation into the free carbenes (“Wanzlick's equilibrium”).23–30 Whereas 8 is kinetically unstable towards dissociation,31–3310 stands in equilibrium with its monomers at room temperature.34,35
Thus, we introduced electron-rich triazaolefins.36 Intriguingly, heterodimer 14, resulting from formal dimerization of a cyclic (alkyl) (amino) carbene (CAAC)37 with a benzNHC, did not dissociate even upon heating to 100 °C and showed similar redox potentials as TTF (1: E1,2 = +0.32 V, E2,3 = −0.08 V vs. Fc/Fc+).14 Paralleling the rich chemistry of TTF derivatives, subsequent studies were directed at introducing bridges in order to obtain mixed-valent compounds and singlet biradicaloids.38–41 Other investigations explored complementary synthetic approaches42–45 or focused on other heterocycles as building blocks.46–48 In combination with further related reports,49–52 a considerable variety of such two-stage redox systems are now available. For applications as organic materials, it is highly desirable to predict or at least rationalize the redox potentials of these scaffolds. One of the authors suggested that the electronic properties of carbene heterodimers could be understood by the π-acceptor properties of the related free carbenes.47 Yet, it is not understood which combinations of carbenes afford isolable radical cations upon removal of one electron. It has been proposed that the delocalization of the radical, heteroatom effects, or steric bulk might be important.17 Surprisingly and in opposition to the very stable radical cations derived from TTF, the benzNHC dimer 10 does not give a persistent radical cation. Instead, only a two-electron oxidation is observed in the cyclic voltammetry (CV) experiment with E1,2 = −0.92 V vs. Fc/Fc+.21 The electrochemical properties of 9 have not been reported according to our knowledge.
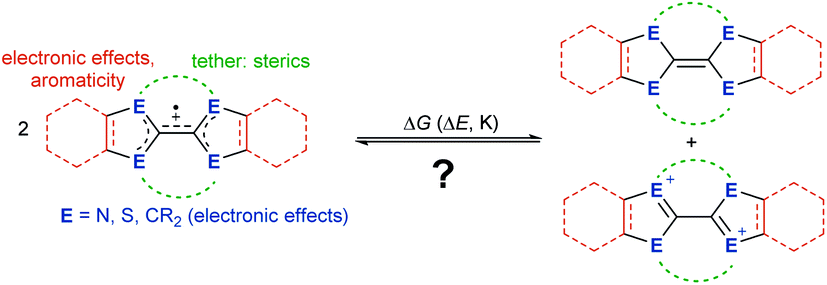
Herein, we show which factors determine whether a cationic olefin radical is stable towards disproportionation into the parent olefin and biscation. Thereby, we elucidate how electronic effects of the heteroatoms within the heterocycles, aromaticity, tethers and sterics influence the disproportionation equilibrium (Fig. 2). Based on the experimental properties of newly and previously synthesized electron-rich olefins, we propose a convenient and general concept to understand, predict and model in silico this disproportionation equilibrium.
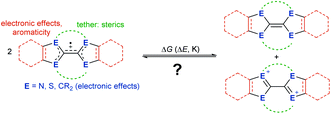 |
| | Fig. 2 What controls whether cationic olefin radicals disproportionate? ΔG, Gibbs free energy; ΔE, electrochemical potential; K, equilibrium constant. | |
Results and discussion
Synthesis
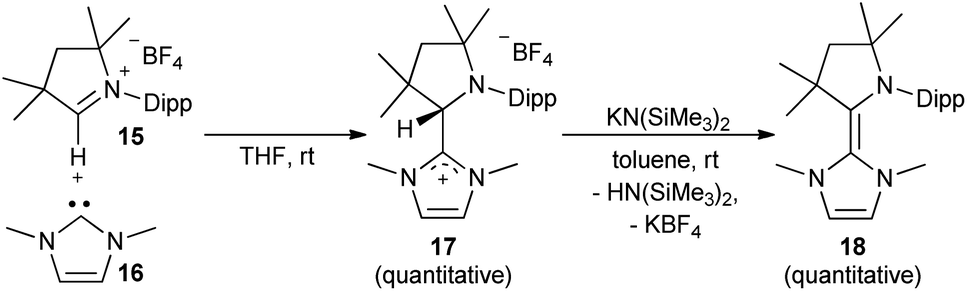
The synthesis of the CAAC–NHC heterodimer 18 followed the synthetic approach chosen for 14.36 The cyclic iminium salt 15 reacted swiftly at room temperature with 16 to give the colorless addition product 17 (Scheme 1). Subsequent deprotonation by potassium hexamethyldisilazide (KHMDS) afforded 18 quantitatively as a yellow solid.
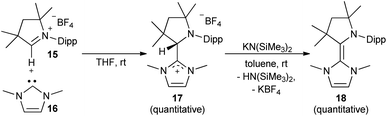 |
| | Scheme 1 Formation of CAAC–NHC adduct 17 and deprotonation to 18. | |
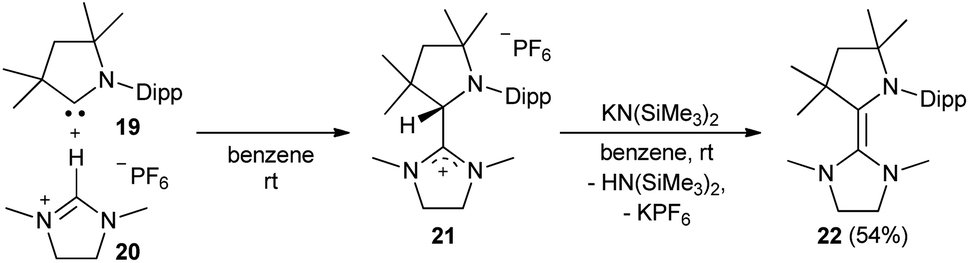
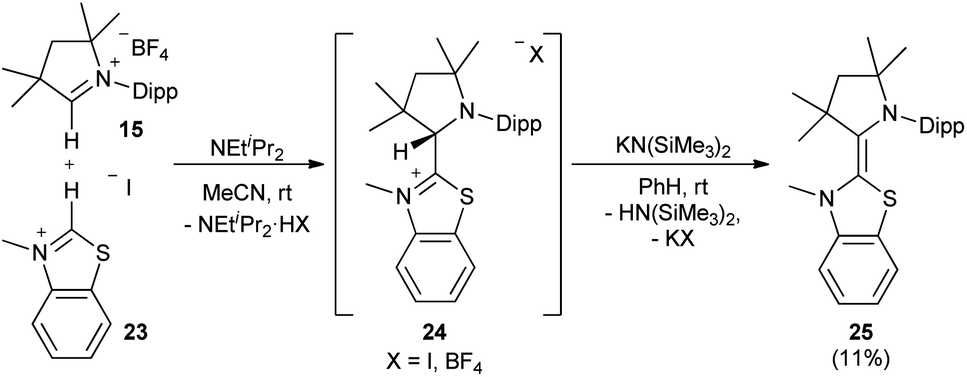
Also the addition of CAAC 19 to the imidazolinium salt 20 gave 21 (Scheme 2). Subsequent deprotonation afforded the saturated heterodimer 22 after workup in 54% overall yield. These two synthetic approaches were not successful for the sulfur-containing derivative 25, where instead the homodimer of the btNHC (13) formed irreversibly. However, slow addition of Hünig's base to 15 in presence of excess of 23 led to the formation of the desired addition product 24 as a mixture with residual 15 and 13, where the latter could be removed during the workup. Treatment with KHMDS gave then, after crystallization at −35 °C, 25 as a yellow, crystalline solid (Scheme 3).53
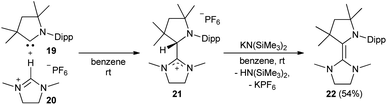 |
| | Scheme 2 Formal CH insertion of 19 and deprotonation afforded 22. | |
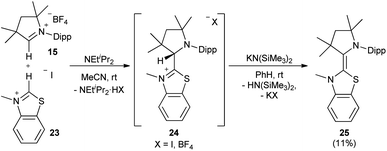 |
| | Scheme 3 Heterodimer 25 was synthesized using Hünig's base. | |
Electrochemical properties
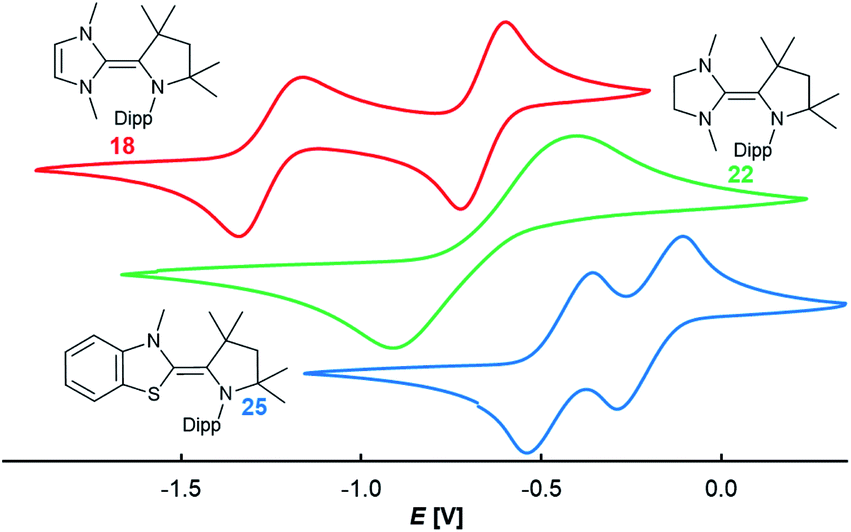
In order to elucidate the electrochemical properties of these two-stage redox-systems, CV experiments were performed (Fig. 3). All redox events were found to be reversible (Fig. S14–S16†). The CAAC–NHC dimer 18 showed two sequential one-electron oxidations giving rise to the stable radical cation 18rad (E2,3 = −1.26 V vs. Fc/Fc+ in THF) and the biscation 18biscat (E1,2 = −0.67 V vs. Fc/Fc+ in THF). Surprisingly, 22 featured only one redox wave (E1,2 = −0.69 V). The large separation of the two halfwaves of ΔE = 0.64 V is indicative of a high reorganization energy and hence in this particular case of a two-electron oxidation. The btNHC derivative 25 showed two redox events (E2,3 = −0.50 V, E1,2 = −0.24 V; vs. Fc/Fc+ in THF), which can be assigned to the oxidation of the olefin to the radical cation 25rad and the biscation 25biscat, respectively. The halfwave potentials E1,2 and E2,3 for 25 are shifted to more positive potential in relation to the CAAC–NHC heterodimer 18.
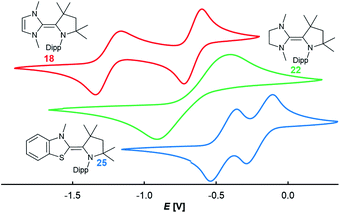 |
| | Fig. 3 CV in 0.1 M nBu4NPF6, 100 mV s−1, vs. Fc/Fc+ in THF (18, red, E1,2 = −0.67 V, E2,3 = −1.27 V; 22, green, E1,2 = −0.69 V; 25, blue, E1,2 = −0.24 V, E2,3 = −0.50 V). | |
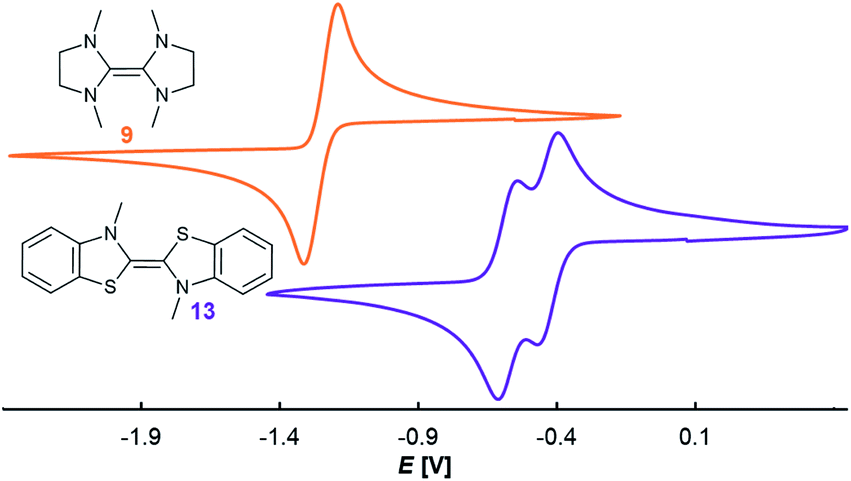
Comparison with the redox potentials of the CAAC–benzNHC dimer 14 (E1,2 = −0.55 V; E2,3 = −0.89 V vs. Fc/Fc+ in THF)54 reveals furthermore a shift to positive potential upon substitution of the methylamino group by sulfur.36 However, the separation ΔE of the two redox potentials, i.e. the stability of the heterodimeric radical 25rad towards disproportionation, is less affected by the heteroatom substitution (14: ΔE = 0.34 V; 25: ΔE = 0.26 V). In light of the structural analogies between the homo- (9, 13) and heterodimers (18, 25) derived from saNHC and btNHC, we also measured the CV for the two homodimers 9 and 13 (Fig. 4).55 Indeed, 9 showed only a two-electron oxidation wave in dimethylformamide (E1,2 = −1.26 V vs. Fc/Fc+),56 while homodimer 13 featured two reversible waves separated by ΔE = 0.15 V in acetonitrile.57 Unfortunately, no CV could be obtained for 9 in acetonitrile. However, a survey of reported potentials of electron-rich olefins (Table S1†) gratifyingly revealed that solvation effects on the separation of the two redox waves, subject of research herein, typically amount to less than 0.1 V.58
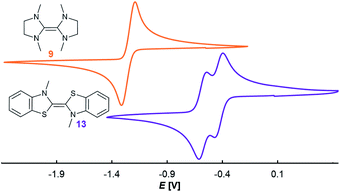 |
| | Fig. 4 CV in 0.1 M nBu4NPF6, 100 mV s−1, vs. Fc/Fc+ (9, orange, E1,2 = −1.27 V in DMF; 13, purple, E1,2 = −0.45 V, E2,3 = −0.60 V in MeCN). | |
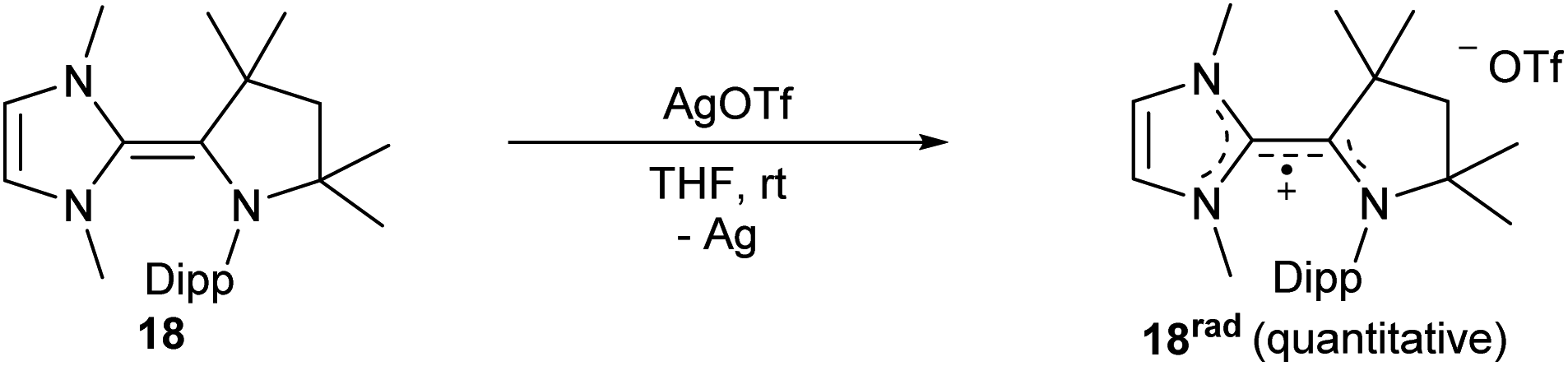
Radical cation generation: oxidation of heterodimer 18
Following the electrochemical studies, we aimed for the isolation of the radical cation 18rad. Treatment of 18 with silver triflate led indeed to the quantitative formation of deeply red colored 18rad (Scheme 4).
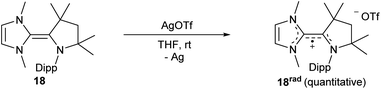 |
| | Scheme 4 Synthesis of radical cation 18rad through oxidation with AgOTf. | |
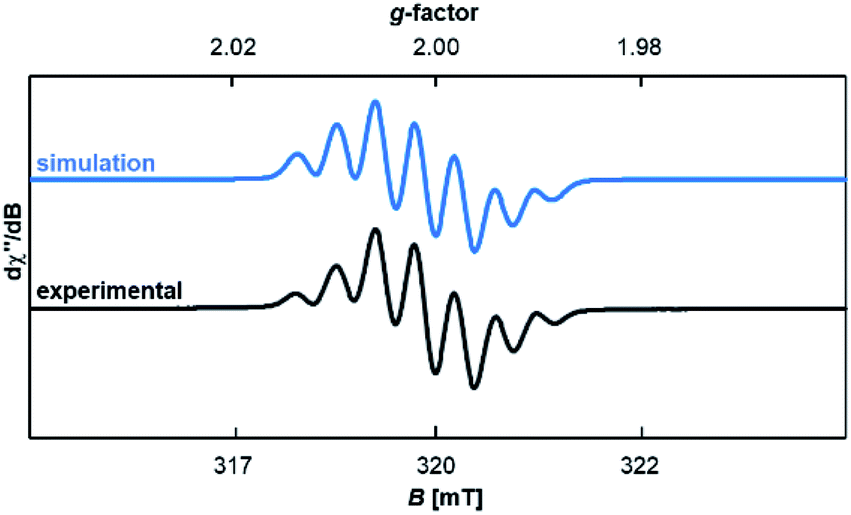
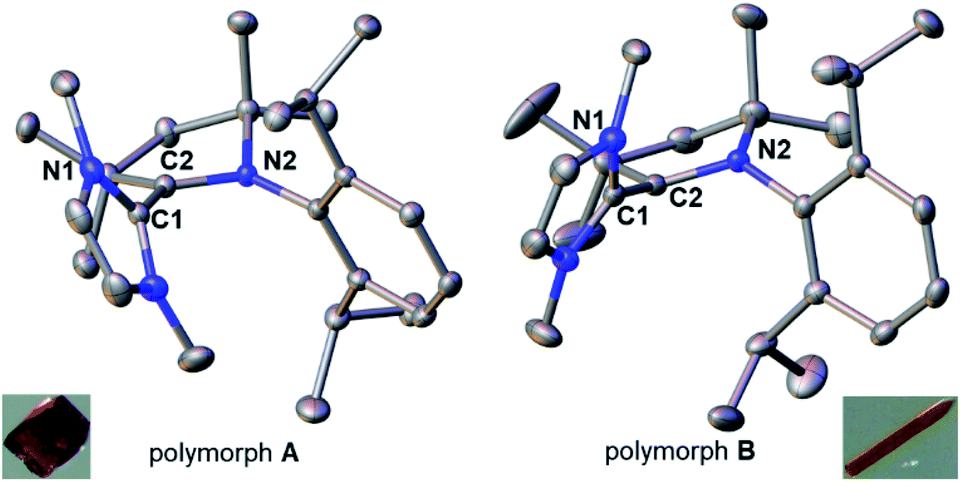
Compound 18rad features an EPR spectrum in THF at room temperature with a g-value of 2.0019 and hyperfine coupling to three non-equivalent nitrogen atoms (a1 = 12.2 MHz; a2 = 13.0 MHz; a3 = 14.1 MHz; Fig. 5). To further corroborate the structure of the radical cation 18rad, single crystals were obtained by vapor diffusion of diethyl ether into a saturated solution in tetrahydrofuran. Single crystals of two polymorphs A (red) and B (orange) were obtained from the same batch (Fig. 6). These two polymorphs showed similar structural parameters as had been reported for 14rad.36 The C1–C2 bonds [A: 1.446(2) Å; B: 1.449(3) Å] are significantly longer than olefinic double bonds [1: 1.349(3) Å;5914: 1.346(2) Å],36 but comparable in length to the C–C bond reported for 14rad [1.439(3) Å].36 Strikingly however, the dihedral angle between the two carbene moieties differed considerably for the two polymorphs [N1–C1–C2–N2 for A: 117.2(2)°; N1–C1–C2–N2 for B: 50.3(3)°]. We conclude that a discussion of solid-state dihedral angles should be taken with caution.
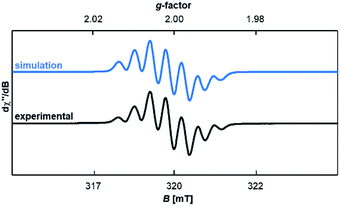 |
| | Fig. 5 Experimental and simulated EPR spectra of 18rad in THF. | |
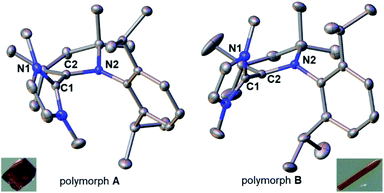 |
| | Fig. 6 Solid-state structures and colors of the two polymorphs A (left) and B (right) of the radical cation in 18rad. Ellipsoids are shown at the 50% probability level; hydrogen atoms and triflate anions are omitted for clarity. Selected bond lengths [Å] and dihedral angle [°] for A: C1–C2 1.446(2), C1–N1 1.386(2), C2–N2 1.359(2); N1–C1–C2–N2 117.2(2)°. Selected bond lengths [Å] and dihedral angle [°] for B: C1–C2 1.449(3), C1–N1 1.396(2), C2–N2 1.357(2); N1–C1–C2–N2 50.3(3)°. | |
Computations: what makes a cationic radical persistent?
Generally, “potential inversion” (i.e., a seemingly two-electron redox process) is opposed to a “normal ordering” of well-behaved one-electron redox events.
A two-electron transformation occurs in a two-stage redox system, if the addition of the second electron proceeds with greater ease than for the first (Scheme 5).60 Despite continuous improvement in the recent decades,61 the solution phase thermodynamical data of chemical reactions comprising charge transfer remain often challenging to predict accurately.62 Indeed, we did not obtain a satisfying correlation between experiments and computations using computationally efficient implicit solvation for the two-stage redox systems studied herein (Fig. S28†). Contrarily, conceptual DFT (density functional theory),63 which relies on the energy of the frontier orbitals, seems an attractive alternative. Indeed, conceptual DFT has been applied to predict electrochemical potentials of one-stage redox systems (eqn (1)).64
| | 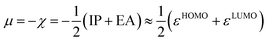 | (1) |
 |
| | Scheme 5 Stepwise reduction to give the electron-rich olefin. | |
There, the chemical potential μ of a compound corresponds to the Mulliken electronegativity χ.65 The mean of the vertical ionization potential (IP) and electron affinity (EA), which defines the Mulliken electronegativity, can be expressed by the frontier orbital energies.66–68 The ionization potential is related with the eigenvalue, i.e. the energy of the highest occupied molecular orbital (HOMO) εHOMO. Correspondingly, the electron affinity is related to the energy of the singly occupied molecular orbital (SOMO) after addition of one electron (i.e., the vertical electron affinity). The latter value can be sometimes approximated by the energy of the lowest unoccupied molecular orbital (LUMO) or the energy of the lowest unoccupied β-orbital in case of a radical, respectively. Pearson suggested that eqn (1) should be replaced by eqn (2) for an “oxidized” compound without significant ionization potential.69 The value of the chemical potential is then equivalent to the electron affinity (eqn (2)).
In order to model the electrochemical properties of two-stage redox systems, we propose the following method: reconsidering the redox processes depicted in Scheme 5, the chemical potential 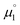 for the biscations, which are very difficult to be further oxidized, should be solely related with their electron affinity according to eqn (2). Differently, the chemical potential
for the biscations, which are very difficult to be further oxidized, should be solely related with their electron affinity according to eqn (2). Differently, the chemical potential 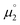 for the reduction of the cationic radicals should be approximated by eqn (1). Importantly, the cationic radicals will only be resistant towards disproportionation, if the reductive formation of the radical cation FROM the biscation
for the reduction of the cationic radicals should be approximated by eqn (1). Importantly, the cationic radicals will only be resistant towards disproportionation, if the reductive formation of the radical cation FROM the biscation 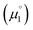 is more favorable than further reduction TO the olefin
is more favorable than further reduction TO the olefin 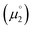 . Consequently, a persistent radical cation is formed (Δμ° > 0), if the first reduction to give the radical cation
. Consequently, a persistent radical cation is formed (Δμ° > 0), if the first reduction to give the radical cation 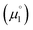 is more facile than the second reduction to the olefin
is more facile than the second reduction to the olefin 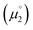 .
.
The difference of eqn (1) and (2) (eqn (3)–(5)) should hence describe the separation of the two redox waves E1,2 and E2,3 (ΔE) in experimental CVs.
| | 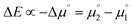 | (3) |
| | 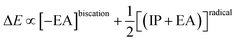 | (4) |
| | 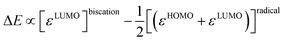 | (5) |
The chemical potential Δμ (unit: electronvolt eV) is the molecular equivalent to the molar Gibbs free energy ΔG and both values are interconvertible using Avogadros's constant and the elementary charge of an electron (i.e., Faraday's constant F). Accordingly, it is directly connected with the electrochemical potential ΔE (unit: volt V) as well as the equilibrium constant K of a reaction via Nernst's equation (eqn (6); RT/zF, Nernst factor; R, ideal gas constant; T, temperature; z, number of electrons; F, Faraday constant). A negative sign for Δμ hence indicates an exergonic reaction, i.e. disproportionation of the cationic radical to the olefin and biscation.
| | 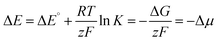 | (6) |
We performed the quantum chemical calculations on the B3LYP-D3BJ/def2-TZVPP//B3LYP-D3BJ/def2-SVP level of theory. Thereby, we were aiming at a straightforward computational protocol of general applicability. As discussed above, solvation has sometimes an influence on the redox potentials.56 A literature survey reveals nevertheless that it affects the electronic coupling of electron-rich olefins by less than 0.1 V (cf. Table S1†). Furthermore note that most electrochemical data have been reported in various solvents due to solubility, stability and resolution challenges.
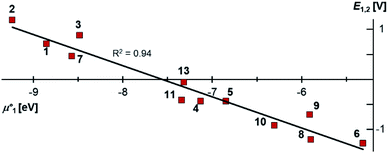
Indeed, the first oxidation step E1,2 for the homodimers was well (R2 = 0.94) reproduced by calculating the chemical potential 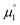 using eqn (2) (Fig. 7). A worse fit was obtained when approximating the electron affinity with the respective LUMO energies (Fig. S19;†R2 = 0.88). Accordingly, no satisfying correlation was obtained with eqn (1) (Fig. S20;†R2 = 0.60). The second redox step E2,3 was well (R2 = 0.93) reproduced with eqn (1) (Fig. 8), where the energies of the α-HOMO and β-LUMO were used for the ionization potential and the electron affinity, respectively.70 Modelling ΔE of the two halfwaves showed an equally good agreement with the experimental data (Fig. 9; R2 = 0.94). Also here, reasonable results were only (Fig. S22–S24†) obtained when using eqn (2) for
using eqn (2) (Fig. 7). A worse fit was obtained when approximating the electron affinity with the respective LUMO energies (Fig. S19;†R2 = 0.88). Accordingly, no satisfying correlation was obtained with eqn (1) (Fig. S20;†R2 = 0.60). The second redox step E2,3 was well (R2 = 0.93) reproduced with eqn (1) (Fig. 8), where the energies of the α-HOMO and β-LUMO were used for the ionization potential and the electron affinity, respectively.70 Modelling ΔE of the two halfwaves showed an equally good agreement with the experimental data (Fig. 9; R2 = 0.94). Also here, reasonable results were only (Fig. S22–S24†) obtained when using eqn (2) for 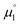 and eqn (1) for
and eqn (1) for 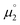 . Impressively, the accuracy (mean absolute deviation MAD) of the computational predictions is within 0.06 V deviation from the experiment (0.07 V if including the further examples discussed below; Table S2†). The calculations predict a large separation for TTF 1 (ΔE = 0.4 V; Δμ° = 0.36 eV) and saTTF 2 (ΔE = 0.5 V; Δμ° = 0.45 eV) and two-electron processes for the NHC- (8), saNHC- (9) and benzNHC (10) homodimers (ΔE = 0 V; Δμ°< 0). The same trend is obtained for the heterodimers 14, 18, 22, 25 (Fig. S26†).
. Impressively, the accuracy (mean absolute deviation MAD) of the computational predictions is within 0.06 V deviation from the experiment (0.07 V if including the further examples discussed below; Table S2†). The calculations predict a large separation for TTF 1 (ΔE = 0.4 V; Δμ° = 0.36 eV) and saTTF 2 (ΔE = 0.5 V; Δμ° = 0.45 eV) and two-electron processes for the NHC- (8), saNHC- (9) and benzNHC (10) homodimers (ΔE = 0 V; Δμ°< 0). The same trend is obtained for the heterodimers 14, 18, 22, 25 (Fig. S26†).
 |
| | Fig. 7 The experimental halfwave potentials E1,2 of the homodimers are linearly correlated with the calculated chemical potential 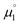 according to eqn (2). E1,2 is given vs. SCE. according to eqn (2). E1,2 is given vs. SCE. | |
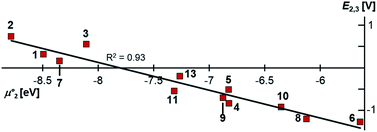 |
| | Fig. 8 The experimental halfwave potentials E2,3 of the homodimers are linearly correlated with the calculated chemical potential 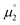 according to eqn (1). E2,3 is given vs. SCE. according to eqn (1). E2,3 is given vs. SCE. | |
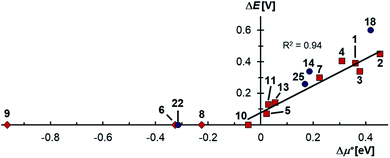 |
| | Fig. 9 The thermodynamic stability towards disproportionation of the cationic radicals (ΔE) is linearly correlated with the difference of the calculated chemical potentials in the gas phase 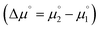 . Heterodimers are given in blue circles, homodimers are given in red squares and diamonds. Heterodimers and homodimers given in red diamonds are not included in the linear regression. . Heterodimers are given in blue circles, homodimers are given in red squares and diamonds. Heterodimers and homodimers given in red diamonds are not included in the linear regression. | |
Aromaticity
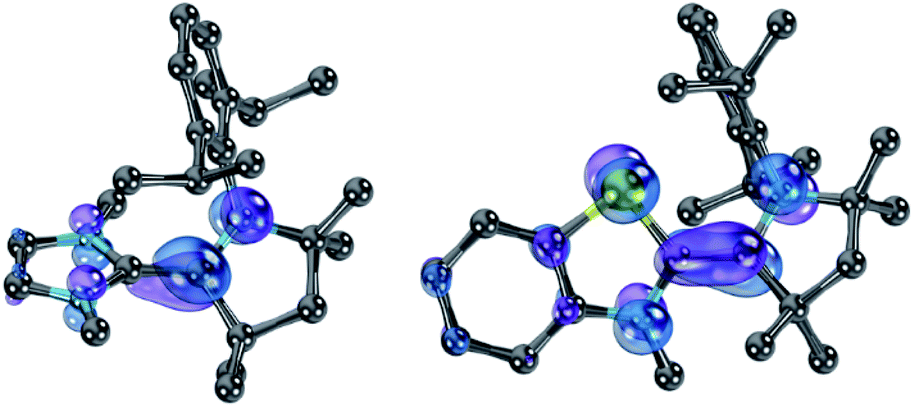
Nevertheless, ΔE was both experimentally and computationally slightly larger for the hetero- than for the homodimers. We attribute this to a systematic influence of the CAAC. We found previously that the spin density in 14rad is principally located on the CAAC.36 The same is true for 18rad and 25rad (Fig. 10). This suggests that the aromaticity of the NHC moieties is more reduced in the second reduction step from the radical cations to the olefins 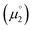 than in the first step from the biscations to the cationic radicals
than in the first step from the biscations to the cationic radicals 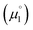 . Consequently, we had a look at the biscations' nucleus-independent chemical shift (NICS) of the NHC derived carbene rings.71 NICS is a convenient and straightforward method to approximately assess the aromaticity of planar π rings. The NICS0zz value relates to the calculated out-of-plane (“zz”) part of the isotropic negative chemical NMR shift of a dummy hydrogen atom in the center of an aromatic ring. This method seems well suitable for the molecules studied herein due to the two diastereotopic faces of the aromatic rings.72 The more negative the chemical shift of the dummy atom, the “more aromatic” is the carbene entity.
. Consequently, we had a look at the biscations' nucleus-independent chemical shift (NICS) of the NHC derived carbene rings.71 NICS is a convenient and straightforward method to approximately assess the aromaticity of planar π rings. The NICS0zz value relates to the calculated out-of-plane (“zz”) part of the isotropic negative chemical NMR shift of a dummy hydrogen atom in the center of an aromatic ring. This method seems well suitable for the molecules studied herein due to the two diastereotopic faces of the aromatic rings.72 The more negative the chemical shift of the dummy atom, the “more aromatic” is the carbene entity.
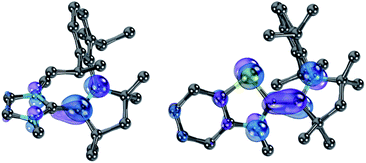 |
| | Fig. 10 SOMOs of 18rad (left) and 25rad (right). | |
Truly, the NICS0zz values for the carbene moieties connected to the CAACs in the biscations 14biscat, 18biscat, 25biscat as well as non-aromatic 22biscat predict that increasing aromaticity (i.e. a more negative NICS0zz value) increases the stability towards disproportionation (Fig. 11). The same order is found using experimentally determined aromaticity descriptors (Fig. S29†)73–76 as well as NICS scans dissecting σ-contributions (Fig. S30–S32†).72c The aromaticity effect can be explained by the loss of aromatic stabilization upon reduction to the olefins. Apparently, for cationic radicals where the spin density is mainly located on the CAAC moiety, a large aromatization energy associated with the other heterocycle renders the second reduction 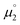 unfavorable and hence prevents overall the disproportionation to the olefin and biscation. For the homodimers,77 the aromaticity seems to be almost irrelevant to the stability of the radical cations as evidenced by the large separation ΔE obtained for both aromatic 1 (ΔE = 0.39 V)4 as well as aliphatic 2 (ΔE = 0.45 V).78 Accordingly, the aromatization energy, which originates in the oxidation of the olefin to its biscation, does not seem to have an influence on the (in)stability of the homodimeric radical cations towards disproportionation.
unfavorable and hence prevents overall the disproportionation to the olefin and biscation. For the homodimers,77 the aromaticity seems to be almost irrelevant to the stability of the radical cations as evidenced by the large separation ΔE obtained for both aromatic 1 (ΔE = 0.39 V)4 as well as aliphatic 2 (ΔE = 0.45 V).78 Accordingly, the aromatization energy, which originates in the oxidation of the olefin to its biscation, does not seem to have an influence on the (in)stability of the homodimeric radical cations towards disproportionation.
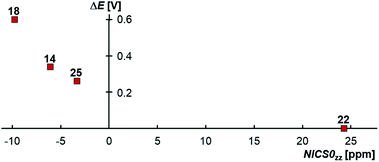 |
| | Fig. 11 CAAC-carbene heterodimeric radical cations, where the spin density is mainly located on the CAAC moiety, form isolable radical cations due to the aromaticity of the carbene heterocycles as modeled by NICS0zz. Correction for σ-effects affords a NICS0π,zz value of +1.0 ppm for 22.72c | |
Steric- and heteroatom effects
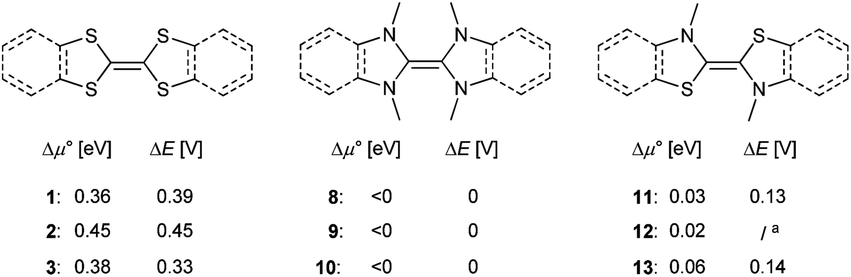
When comparing TTF (1) with its saturated (2) or benzannulated (3) congener, only a marginal difference was observed between the experimentally determined separation of the redox waves ΔE and the predicted chemical potentials Δμ° (Δμ° = 0.36, 0.45, 0.38 V; Fig. 12, left). The dimers 8, 9 and 10 are not stable towards disproportionation and the calculated redox potentials Δμ° are consequently all negative (Fig. 12, middle). The thiazolin-2-ylidene derivatives 11, 12, and 13 are borderline cases (Fig. 12, right).
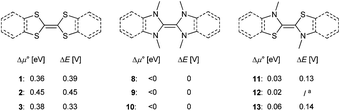 |
| | Fig. 12 Extending the aromatic system does only have a very small influence on the stability of the homodimeric radical cations towards disproportionation (calculated: Δμ°; experimentally determined: ΔE). aTo the best of our knowledge, 12 has not been reported in the literature. | |
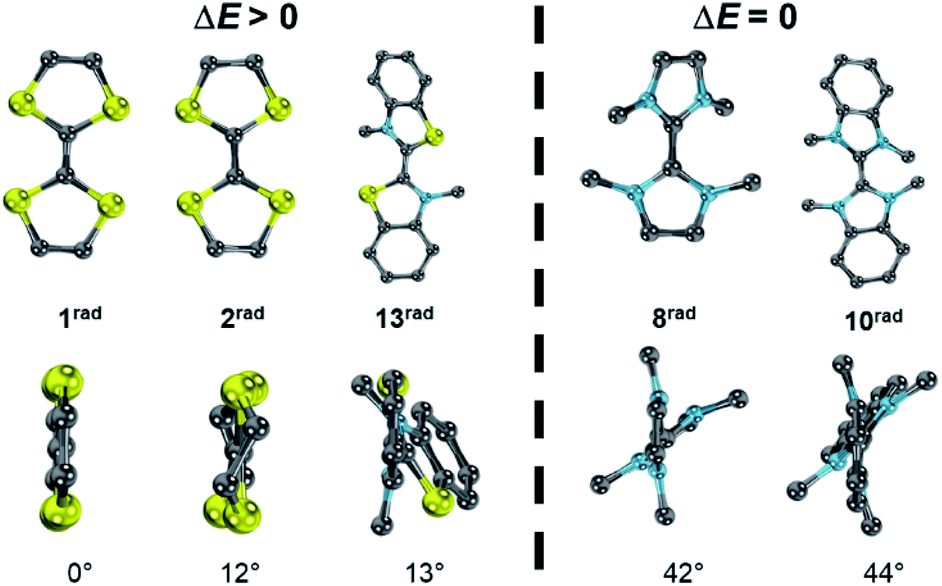
Although the two separate potentials E1,2, E2,3 (cf.Fig. 7 and 8) vary considerably, the stability towards disproportionation of the radical cations is also here not considerably affected. However, it is intriguing to note that all compounds with a large ΔE value (1, 2, 3, 4, 7) do not feature alkyl substituents in the 2,2′ position of the parent carbenes. The compounds with one substituent (5, 11, 12, 13) show moderate ΔE values, while the separation between the two halfwaves is very small for the 2,2′ disubstituted heterocycles (Fig. S27†). For example, 1rad and 2rad with sulfur atoms in the 2,2′ positions form persistent radical cations (1rad: ΔE = 0.39 V; 2rad: ΔE = 0.45 V).4,78 Notably, the two heterocycles in these radicals are essentially coplanar (1rad: 0°; 2rad: 12°). Likewise, 13rad with a calculated dihedral angle of only 13° forms a stable radical cation (ΔE = 0.15 V, vide supra). Contrarily, the diaminocarbene derived radical cations 8rad and 10rad with significantly larger dihedral angles of 42° and 44° disproportionate (Fig. 13). This observation can be qualitatively understood by enhanced stabilization of a delocalized radical cation, but also quantitatively using eqn (5). An enhanced twist of the radical cations leads in all cases to an increase of the ionization potential in reference to the electron affinity of the biscations. Accordingly, Δμ° becomes negative and the radical cations disproportionate (ΔE = 0 V). Contrarily, for compounds without strong structural rearrangement, the ionization potential of the radical cations is larger than the electron affinity of the biscations. This allows for an overall positive Δμ° and hence a stable radical cation (ΔE > 0 V). Following the computational predictions, we find that the steric bulk in the 2,2′ position favors a twisting of the cationic radical olefins. Therefore, they disproportionate to the parent olefins and biscations.
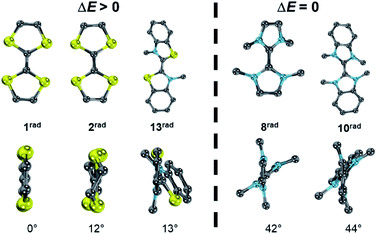 |
| | Fig. 13 Optimized structural parameters of radicals 1rad, 2rad, 8rad, 10rad, and 13rad. Side view illustrates the dihedral angle S–C–C–S′ or N–C–C–N′, respectively. | |
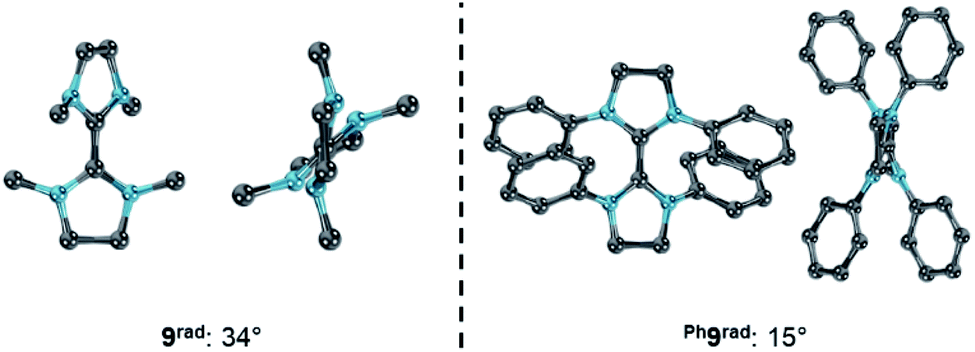
At first sight contradicting this analysis, the replacement of the methyl substituents in 9 by phenyl groups leads to the isolable purple radical cation Ph9rad upon one electron oxidation as evidenced by a signal in the EPR spectrum.79,80 In lack of detailed electrochemical data, we resynthesized Wanzlick's dimer Ph9 and measured the CV (Fig. S11 and S12†).81 Notably, the CV of Ph9 showed two overlapping, quasi-reversible redox waves with E1,2 = −0.63 V and E2,3 = −0.73 V vs. Fc/Fc+ (ΔE = 0.10 V).82 This is in reasonable agreement with the previously reported equilibrium constant of K = 25 with K = [radical-cation]2/([olefin] [biscation]).80 Intriguingly, the computationally predicted difference of the chemical potentials for Ph9 of Δμ° = −0.09 eV is very different to 9 (Δμ° = −0.96 eV) and borderline in respect to forming a persistent radical cation (cf.Fig. 8). Examining the optimized structural parameters of the cationic radicals 9rad and Ph9rad (Fig. 14) reveals a considerably larger dihedral angle for 9rad (34°) in reference to Ph9rad (15°). This indicates counter-intuitively that the “bulky” phenyl groups are better accommodated in the planar than in the twisted conformation due to a coplanar orientation. Accordingly, the planar conformation of Ph9rad is stabilized and hence prevents the disproportionation of the cationic radical. We conclude that Ph9 is a challenging borderline case, where the computational analysis predicts and rationalizes the surprising resistance of the cationic radical to disproportionate very well.
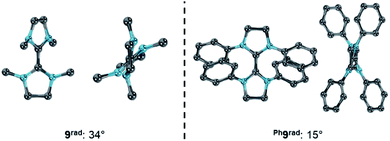 |
| | Fig. 14 Optimized structural parameters of radicals 9rad (left) and Ph9rad (right). Side view illustrates the dihedral angle N–C–C–N′. | |
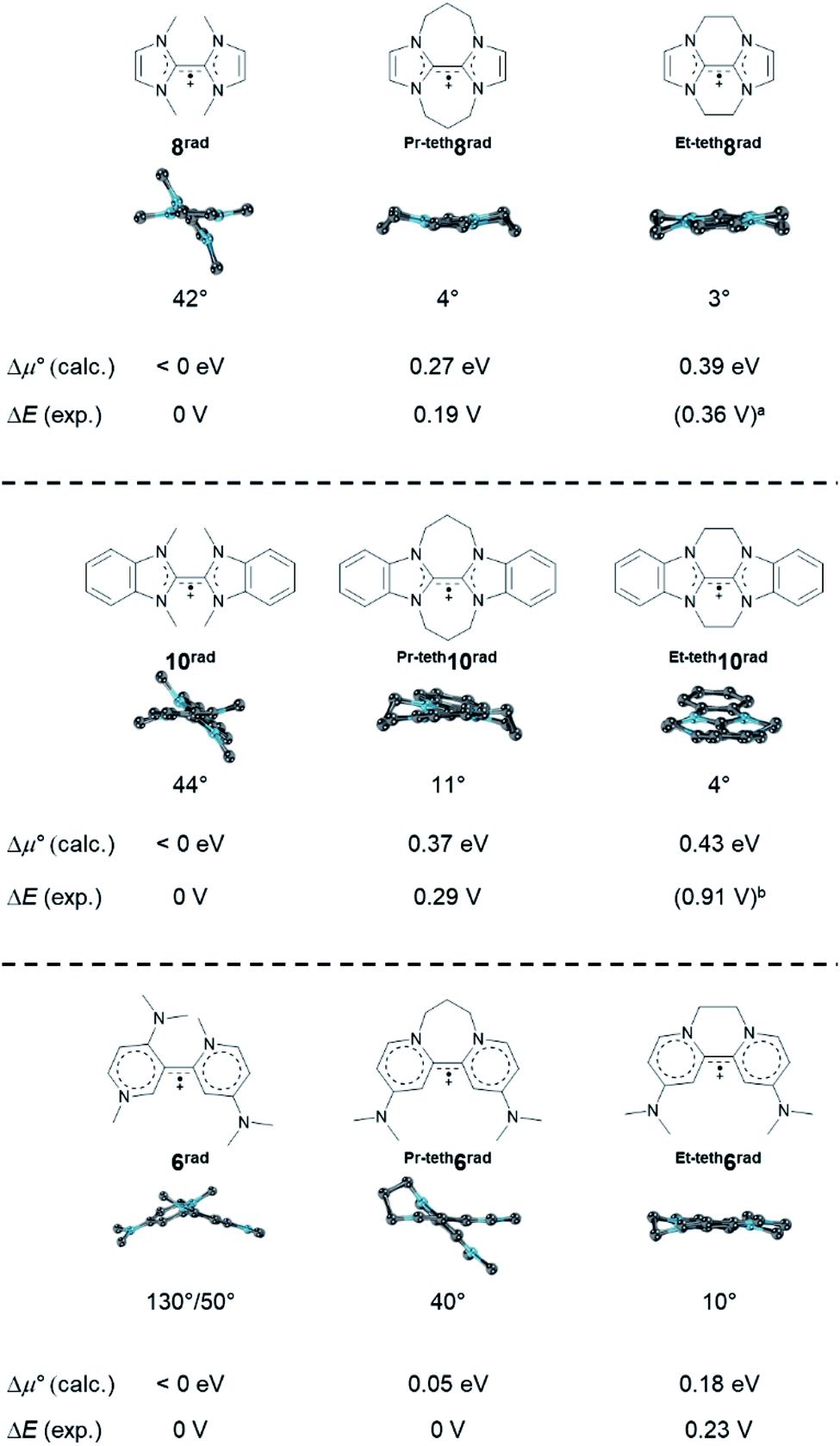
Inspired by the unexpected effect of the phenyl substituents on stability of the radical cation Ph9rad towards dissociation, we eventually investigated tethers in the 2,2′ positions of the homodimers (Fig. 15). We find that tethers33,83 increase the stability towards disproportionation. For example, whereas the disproportionation of the bisNHC radical 8rad is predicted to be exergonic (Δμ° < 0 eV), Pr-teth8rad (Δμ° = 0.27 eV) and Et-teth8rad (Δμ° = 0.39 eV) should be persistent radicals. Indeed, this is also what had been found experimentally (ΔE = 0, 0.19, 0.36 V). The same trend was predicted for 10rad as well as the dimethylaminopyridine derivatives 6rad with only one tether. Also here, the calculated potentials are in excellent agreement with the experimental data (e.g., Et-teth6rad: Δμ° = 0.18 eV; ΔE = 0.23 V).7,15 Intriguingly, the resistance of the radicals to disproportionate is also here controlled by the twist of the cationic olefin radical. Untethered and hence unperturbed 10rad shows for instance a twist of 44°, whereas the comparable long propylene linker allows for 11° and the shorter ethylene linker for only 4°. We conclude that tethers enforce a more planar, rigid structure which increases the resistance of the radical cations towards disproportionation due to enhanced resonance stabilization.
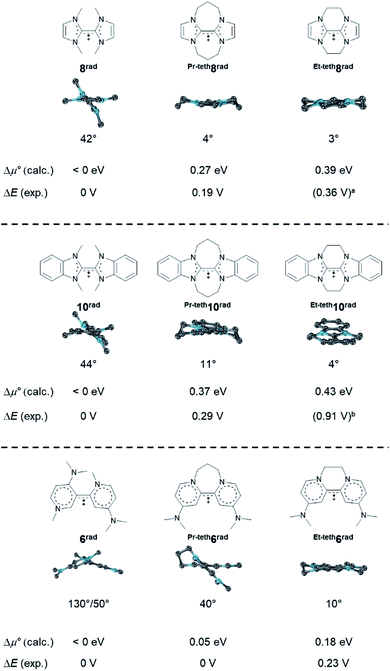 |
| | Fig. 15 Optimized structural parameters of radicals 6rad, Et-teth6rad, Pr-teth6rad, 8rad, Et-teth8rad, Pr-teth8rad, 10rad, Et-teth10rad and Pr-teth10rad as well as calculated and experimental CV parameters. Side view illustrates the dihedral angle N–C–C–N′. Two conformers of comparable energy (ΔG < 1 kcal mol−1) were calculated for Et-teth8rad, Pr-teth8rad, Et-teth10rad, and Pr-teth10rad; given values relate to the average. aTo the best of our knowledge, Et-teth8 has not been reported. Instead, the potential for mixed ethylene/propylene bridged Et,Pr-teth8 is given.84bHünig and coworkers question the position of E2,3.84,85 | |
Conclusion
Although key for the design of organic electronics, it has been unclear for decades why some electron-rich olefins such as tetrathiafulvalene (TTF) or bipyridinylidenes form persistent and isolable radical cations, whereas others such as the enetetramine derived from dimerized N,N′-dimethyl imidazolidin-2-ylidenes (saNHC; “Wanzlick's dimer”) do not. Herein, we showed by a combined experimental and computational investigation that sterics and aromaticity determine the stability of cationic olefin radicals towards disproportionation into the parent olefins and biscations. More specifically, we reported three electron-rich carbene heterodimers derived from a cyclic (alkyl) (amino) carbene (CAAC) in combination with saturated- (saNHC) and unsaturated N-heterocyclic carbenes (NHCs) as well as benzothiazolin-2-ylidene (btNHC). In order to pinpoint the differences between heterodimers and their homodimeric congeners, we also investigated the electrochemical properties of three previously reported homodimers. Importantly, the heterodimer derived from the saturated NHC does not form a persistent radical cation in opposition to the other heterodimers. We then proposed a computational method based on conceptual density functional theory to understand and predict the stability of olefin radical cations towards disproportionation. This method predicts the experimentally determined values with an accuracy of 0.07 V. We demonstrated that the persistence of CAAC-derived heterodimeric radical cations is due to aromaticity, which we suggest exploiting further in the future. Contrarily, the persistence of homodimer-derived cationic radicals is controlled almost exclusively by the steric bulk in the 2,2′ positions of the parent carbenes, which we suggest deliberately harnessing as well. Neither the introduction of electron-attracting nor electron-donating groups leads to considerably increased stability towards disproportionation. Tethers however have a huge beneficial effect, because they prevent a tilt of the carbene moieties, thus enforcing enhanced resonance stabilization of the olefin radical cation. The positive effect of sulfur atoms in the 2,2′ position as present in TTF consequently is not electronic in nature. Instead, substituents in the 2,2′ position sterically destabilize the cationic olefin radical through enforcement of a twisted conformation with reduced resonance stabilization.
Experimental section
All reactions were carried out under an atmosphere of dry dinitrogen, either in an MBraun dinitrogen filled glovebox or using standard Schlenk techniques. 1H and 13C NMR spectra were recorded on JEOL ECX 270, JEOL ECX 400 or Bruker Avance III HD 600 MHz instruments operating at 269.71 MHz, 399.79 MHz and 600.13 MHz for 1H and at 67.82 MHz, 100.62 MHz and 150.90 MHz for 13C, respectively and at a probe temperature of 25 °C. The solvent residual signals were used as an internal reference for the 1H NMR and 13C NMR spectra. 1H NMR multiplicities are abbreviated as follows: s = singlet, d = doublet, dd = double of doublets, t = triplet, q = quartet, spt = septet, m = multiplet. All coupling constants J are given in Hz. Solvents were purified using a two-column solid-state purification system (Glass Contour System, Irvine, CA). Hexanes, toluene and benzene were stored over a mirror of potassium; all other solvents were stored over activated molecular sieves. Deuterated NMR solvents were obtained dry and packaged under argon and stored over activated molecular sieves or a mirror of potassium (C6D6). Melting points were determined using a Bibby Scientific SMP10 melting point apparatus. Elemental analyses were obtained using Euro EA 3000 (Euro Vector) and EA 1108 (Carlo-Erba) elemental analyzers. The solution EPR spectrum were recorded on a JEOL continuous wave spectrometer JESFA200 equipped with an X-band Gunn diode oscillator bridge, a cylindrical mode cavity, and a helium cryostat. The spectrum was obtained on a freshly prepared solution of 5 mM compound 18rad in THF and simulated using the eviewX and esimX programs written by E. Bill (Max-Planck-Institut für Kohlenforschung, Mülheim an der Ruhr, Germany). The EPR spectrum was measured under following conditions: microwave frequency ν = 8.953 GHz, modulation width 1.0 mT, microwave power = 0.1 mW, modulation frequency = 100 kHz, time constant = 0.1 s. Electrochemical measurements were carried out at room temperature under inert atmosphere with an Autolab Type-III potentiostat. They were recorded in 0.1 M nBu4NPF6 solution in THF using a glassy carbon working electrode and platinum electrodes as counter and reference electrodes. Ferrocene was added as internal standard and all measurements were referenced as described in the literature. Electrochemical potentials referenced vs. the Fc/Fc+ redox couple were converted to potentials references vs. SCE as suggested by Connelly and Geiger (Fc/Fc+: 0.40 V vs. SCE in MeCN; Fc/Fc+: 0.45 V vs. SCE in DMF; Fc/Fc+: 0.56 V vs. SCE in THF).86 Electrochemical potentials referenced vs. Ag/AgNO3 (0.01 M) and Ag/AgCl, KCl (saturated) were converted to potentials referenced vs. SCE (Ag/AgNO3 (0.01 M): 0.30 V vs. SCE; Ag/AgCl, KCl (saturated): −0.045 V vs. SCE).87,88 1,3-Dimethyl-1H-imidazol-3-ium iodide, 3-methylbenzo[d]-thiazol-3-ium iodide 23, the cyclic iminium salt 15 (1-(2,6-diisopropyl-phenyl)-2,2,4,4-tetramethyl-3,4-dihydro-2H-pyrrol-1-ium tetrafluoroborate), and free CAAC 19 were synthesized according to literature procedures.37a,89–92 1,3-Dimethylimidazolinium hexafluorophosphate 20 was synthesized as described in the literature93 but using NH4PF6 instead of NH4BF4. All other reagents were obtained from commercial sources and used as is without further purification. All DFT calculations were performed using ORCA 4.0.1.94,95 The geometry optimizations were performed at the B3LYP-D3BJ/def2-SVP level of theory96–99 and applying Grimme's D3 dispersion correction100 with Becke–Johnson damping.101,102 The RIJCOSX103 approximation in combination with the def2/J auxiliary basis set was used in order to save computation time. Tighter than default convergence criteria were chosen for both the optimization (tightopt) of the structural parameters as well as the scf iterations (tightscf) and more accurate than default grid values (Grid5, FinalGrid6) were used. All reported optimized structures were verified as true minima by the absence of negative eigenvalues in the harmonic vibrational frequency analysis. The energies of all structures were corrected by single-point calculations on the B3LYP-D3BJ/def2-TZVPP level of theory. For the calculation of 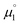 , the energies of the α-HOMO and the β-LUMO for the optimized structural parameters of the cationic radicals were used. For the calculation of
, the energies of the α-HOMO and the β-LUMO for the optimized structural parameters of the cationic radicals were used. For the calculation of 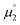 , the vertical electron affinity was used, i.e. the energy of the SOMO upon addition of one electron for the optimized structural parameters of the biscations.
, the vertical electron affinity was used, i.e. the energy of the SOMO upon addition of one electron for the optimized structural parameters of the biscations.
saNHC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) saNHC olefin 9
saNHC olefin 9
This compound was synthesized according to a modified literature procedure. 10 was deprotonated using an equimolar amount KHMDS in benzene. Filtration and removal of volatiles afforded 9 in quantitative yield. The analytical data were consistent with the literature.104
saNHCsaNHC olefin Ph9
This compound was synthesized according to a modified literature procedure.105N,N′-Diphenylethylenediamine was prepared as reported,106 but purified by column chromatography (EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane/1:5, Rf = 0.8). The cyclization and subsequent deprotonation was performed according to the literature procedure for aryl-substituted saturated NHC olefins.107 The analytical data were consistent with the values in the literature.26
:hexane/1:5, Rf = 0.8). The cyclization and subsequent deprotonation was performed according to the literature procedure for aryl-substituted saturated NHC olefins.107 The analytical data were consistent with the values in the literature.26
btNHC=btNHC olefin13
btNHC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) btNHC olefin 13 was synthesized according to a modified literature procedure108 by deprotonation of the benzothiazolium salt 23 using Hünig's base in MeCN. The ammonium salt was removed by reaction with tripotassium phosphate in Et2O.
btNHC olefin 13 was synthesized according to a modified literature procedure108 by deprotonation of the benzothiazolium salt 23 using Hünig's base in MeCN. The ammonium salt was removed by reaction with tripotassium phosphate in Et2O.
3-Dimethylimidazolin-2-ylidene 16
This compound was synthesized according to a modified literature procedure.109 1,3-Dimethyl-1H-imidazol-3-ium iodide (2.00 g, 8.93 mmol, 1.0 eq.) and KHMDS (1.75 g, 8.75 mmol, 1.0 eq.) were combined in a Schlenk flask and suspended in Et2O (15 mL). The mixture was stirred for 20 min at room temperature. The supernatant solution was separated from the precipitated potassium salt via a filter cannula. Volatiles were removed in vacuo to afford the carbene 16 as a pale yellow oil, which was stored at −40 °C. The low yield of only 45% (390 mg) is due to the volatility of 16 under reduced pressure. The analytical data were consistent with the values in the literature.109
CAAC–NHC HBF4 salt 17
The salt 15 (1.50 g, 4.0 mmol) and 16 (409 mg, 4.22 mmol, 1.05 eq.) were combined in a Schlenk flask and suspended in THF (20 mL). The mixture was stirred for 12 h at room temperature. The supernatant solution was removed via a filter cannula and the residue was washed with THF (30 mL) until the powder was colorless. The solid was dried in vacuo to afford 17 in quantitative yield (1.88 g). Mp.: 201 °C. 1H NMR (600 MHz, CD3CN) δ = 7.38 (s, 1H, NCH imidazole), 7.28 (dd, J1 = 7.7 Hz, J2 = 2.0 Hz, 1H, m-ArCH Dipp), 7.25 (t, J = 7.7 Hz, 1H, p-ArCH Dipp), 7.22 (d, J = 2.0 Hz, 1H, NCH imidazole), 7.12 (dd, J1 = 7.3 Hz, J2 = 2.0 Hz, 1H, m-ArCH Dipp), 5.02 (s, 1H, NCH pyrrolidine), 4.26 (s, 3H, NCH3), 3.55 (s, 3H, NCH3), 3.45 (spt, J = 6.8 Hz, 2H, 2x CH(CH3)2iPr), 2.43 (d, J = 13.9 Hz, 1H, CH2 pyrrolidine), 2.29 (d, J = 13.9 Hz, 1H, CH2 pyrrolidine), 1.72 (s, 3H, NCCH3), 1.37 (d, J = 6.8 Hz, 3H, CHCH3iPr), 1.36 (s, 3H, NCCH3), 1.31 (d, J = 6.6 Hz, 3H, CHCH3iPr), 1.25 (s, 3H, NCHCCH3), 1.18 (d, J = 6.8 Hz, 3H, CHCH3iPr), 0.96 (s, 3H, NCHCCH3), 0.48 (d, J = 6.8 Hz, 3H, CHCH3iPr) ppm. 13C NMR (101 MHz, CD3CN) δ = 152.2 (NCN), 151.0 (ArC Dipp), 148.2 (ArC Dipp), 137.8 (ArC Dipp), 129.1 (ArCH Dipp), 127.2 (ArCH Dipp), 125.9 (ArCH Dipp), 73.4 (CCHN), 67.7 (NC(CH3)2), 55.2 (CH2 pyrrolidine), 44.6 (NCC(CH3)2), 38.7 (NCH3), 37.8 (NCH3), 33.5 (CH3 pyrrolidine), 31.7 (CH3 pyrrolidine), 30.2 (CH3 pyrrolidine), 28.9 (CHCH3iPr), 28.4 (CHCH3iPr), 27.1 (CH3 pyrrolidine), 25.9 (CHCH3iPr), 25.8 (CHCH3iPr), 25.6 (CHCH3iPr), 24.3 (CHCH3iPr) ppm. Elemental analysis for C25H40F4N3B·0.6THF: calcd: C 64.19; H 8.81; N 8.20. Found: C 64.01; H 8.69; N 8.06%.
CAACNHC olefin 18
The salt 17 (1.50 g, 3.19 mmol) and KHMDS (624 mg, 3.13 mmol, 0.98 eq.) were combined in a Schlenk flask. Toluene (15 mL) was added and it was stirred for 12 h at room temperature. The supernatant solution was separated from the potassium salt via a filter cannula. The solvent was removed in vacuo and the residue was washed with hexanes. The pale yellow solid was dried in vacuo to afford 18 in 80% yield (970 mg). Mp.: 63 °C. 1H NMR (270 MHz, C6D6) δ = 7.13–7.07 (m, 3H, ArCH Dipp), 5.71 (d, J = 2.1 Hz, 1H, NCH imidazoline), 5.58 (d, J = 2.1 Hz, 1H, NCH imidazoline), 3.55 (spt, J = 6.7 Hz, 2H, 2x CH(CH3)2iPr), 2.52 (s, 3H, NCH3), 1.84 (s, 2H, CH2 pyrrolidine), 1.69 (s, 6H, NC(CH3)2), 1.39 (s, 3H, NCH3), 1.37 (d, J = 6.7 Hz, 6H, CH(CH3)2iPr), 1.28 (d, J = 6.8 Hz, 6H, CH(CH3)2iPr), 1.13 (s, 6H, NCC(CH3)2) ppm. 13C NMR (68 MHz, C6D6) δ = 150.1 (NCN), 143.2 (ArC Dipp), 142.7 (ArC Dipp), 129.7 (ArCH Dipp), 129.4 (ArCH Dipp), 127.4 (ArC Dipp), 126.8 (CCN), 124.9 (ArCH Dipp), 63.0 (NC(CH3)2), 58.4 (CH2 pyrrolidine), 48.1 (NCH imidazoline), 44.0 (NCH imidazoline), 42.5 (NCC(CH3)2), 31.1 (NCH3), 29.9 (NCH3), 28.9 (CHCH3iPr), 25.7 (CHCH3iPr), 24.7 (CHCH3iPr) ppm (three CH/CH3 signals are superimposed). Elemental analysis for C25H39N3: calcd: C 78.69; H 10.30; N 11.01. Found: C 78.16; H 10.35; N 10.61%.
CAAC–NHC radical 18rad
Silver trifluoromethanesulfonate (AgOTf) (134 mg, 0.52 mmol, 1.0 eq.) and 18 (200 mg, 0.52 mmol, 1.0 eq.) were suspended in THF (5 mL). The mixture instantly turned deep maroon. The mixture was filtered and the solvent was removed in vacuo to afford 18rad as a red solid in quantitative yield (276 mg). Single crystals suitable for X-ray analysis were obtained by slow diffusion of Et2O into a saturated THF solution at room temperature.
CAAC–NHC PF6 salt 21
The carbene 19 (395 mg, 1.38 mmol, 1.2 eq.) and 1,3-dimethylimidazolinium hexafluorophosphate (20) (281 mg, 1.15 mmol, 1.0 eq.) were suspended in benzene (10 mL). The mixture was stirred for 16 h at room temperature, filtered, the residue washed with benzene (2 × 2 mL) and dried in vacuo to give 390 mg of the crude heterodimer. This compound was used as is for the next step without further purification. 1H NMR (270 MHz, CD3CN): δ = 7.28–7.20 (m, 3H, ArCH Dipp), 4.77 (s, 1H, NCH pyrrolidine), 3.81–3.58 (m, 5H, CH(CH3)2iPr and H2C–CH2 imidazoline overlapping), 3.31 (spt, J = 6.5 Hz, 1H, CH(CH3)2iPr), 2.98 (s, 3H, NCH3), 2.33 (d, J = 13.9 Hz, 1H, CH2 pyrrolidine), 2.23 (d, J = 13.9 Hz, 1H, CH2 pyrrolidine), 1.68 (s, 3H, NCCH3), 1.32 (d, J = 6.5 Hz, 6H, CH(CH3)2iPr), 1.31 (s, 3H, NCCH3), 1.22 (d, J = 6.8 Hz, 3H, CHCH3iPr), 1.17 (s, 3H, NCHCCH3), 1.16 (s, 3H, NCHCCH3), 1.04 (d, J = 6.8 Hz, 3H, CHCH3iPr) ppm.
CAACsaNHC olefin 22
The salt 21 was suspended in benzene (2 mL). A solution of KHMDS (132 mg, 0.663 mmol, 0.9 eq. referring to the heterodimer salt) was added under stirring at room temperature and solids were then filtered off. Removal of volatiles afforded 22 in overall 54% yield (238 mg). Mp.: 95 °C. 1H NMR (400 MHz, C6D6) δ = 7.18–7.15 (m, 1H, m-ArCH Dipp, superimposed by solvent), 7.12–7.11 (m, 1H, p-ArCH Dipp), 7.10 (d, J = 1.7 Hz, 1H, m-ArCH Dipp), 3.54 (spt, J = 6.8 Hz, 2H, CH(CH3)2iPr), 2.69–2.60 (m, 4H, H2C–CH2 imidazolidine), 2.58 (s, 3H, NCH3), 1.89 (s, 2H, CH2 pyrrolidine), 1.73 (s, 6H, NC(CH3)2), 1.63 (s, 3H, NCH3), 1.44 (d, J = 6.7 Hz, 6H, CH(CH3)2iPr), 1.33 (d, J = 6.7 Hz, 6H, CH(CH3)2iPr), 1.19 (s, 6H, NCC(CH3)2) ppm. 13C NMR (151 MHz, C6D6) δ = 150.4 (NCN), 142.5 (ArC Dipp), 141.3 (ArC Dipp), 131.4 (CCN), 127.3 (ArCH Dipp), 124.5 (ArCH Dipp), 62.5 (NC(CH3)2), 58.0 (CH2 pyrrolidine), 54.6 (CH2 imidazolidine), 53.8 (CH2 imidazolidine), 47.5 (NCH3), 45.6 (NCH3), 42.6 (NCC(CH3)2), 30.9 (CH3 pyrrolidine), 29.7 (CH3 pyrrolidine), 29.0 (CH(CH3)2iPr), 25.8 (CH(CH3)2iPr), 24.6 (CH(CH3)2iPr) ppm. Elemental analysis for C25H41N3: calcd: C 78.27; H 10.77; N 10.95. Found: C 78.35; H 10.85; N 10.70%.
CAACbtNHC olefin 25
The salt 15 (534 mg, 1.43 mmol, 1.0 eq.) and 3-methylbenzo[d]thiazol-3-ium iodide (23) (594 mg, 2.15 mmol, 1.5 eq.) were suspended in MeCN. Diisopropylethylamine (278 mg, 2.15 mmol, 1.5 eq.) was added dropwise over the course of 20 min at room temperature. The red suspension was stirred for 8 h and the volatiles were removed in vacuo. Dry K3PO4 (3.04 g, 14.3 mmol, 10 eq.) and Et2O (20 mL) were added. The mixture was stirred for 24 h and filtered. The yellow solid was washed with Et2O (3 × 5 mL) and extracted with MeCN (4 × 5 mL). The solvent was evaporated. The yellow solid was suspended in benzene (10 mL), stirred for 10 min, filtered and dried (3×). A solution of KHMDS (285 mg, 1.43 mmol, 1.0 eq.) in benzene (2 mL) was then added to a suspension of the yellow solid in benzene (5 mL) at room temperature. The mixture was stirred for 1 h and the volatiles were removed in vacuo. The heterodimer was extracted with hexanes (3 × 3 mL). The solution was concentrated to 2 mL, filtered, and cooled to −35 °C upon which crystals formed. The mother liquor was decanted after 16 h and the yellow crystals were washed with cold hexanes (2 × 1 mL). Drying in vacuo afforded 25 in 11% yield (67 mg). Mp.: 190–191 °C. 1H NMR (400 MHz, C6D6) δ = 7.25 (dd, J1 = 8.3 Hz, J2 = 6.9 Hz, 1H, p-ArCH Dipp), 7.16–7.14 (m, 2H, m-ArCH Dipp, superimposed by solvent), 6.88 (dd, J1 = 9.3 Hz, J2 = 7.7 Hz, 2H, ArCH benzothiazoline), 6.79 (dd, J1 = 8.8 Hz, J2 = 7.4 Hz, 1H, ArCH benzothiazoline), 6.66 (dd, J1 = 8.8 Hz, J2 = 7.4 Hz, 1H, ArCH benzothiazoline), 3.40 (spt, J = 6.9 Hz, 2H, 2x CH(CH3)2iPr), 2.73 (s, 3H, NCH3), 1.78 (s, 2H, CH2 pyrrolidine), 1.64 (s, 6H, NC(CH3)2), 1.47 (d, J = 6.9 Hz, 6H, CH(CH3)2iPr), 1.26 (d, J = 6.9 Hz, 6H, CH(CH3)2iPr), 1.13 (s, 6H, NCC(CH3)2) ppm. 13C NMR (101 MHz, C6D6) δ = 152.5 (NCS), 150.3 (ArCS), 148.6 (ArCN benzothiazoline), 138.2 (ArC Dipp), 137.3 (ArC Dipp), 128.4 (ArCH, superimposed by solvent), 125.1 (ArCH), 124.8 (ArCH), 124.7 (ArCH), 121.2 (ArCH), 117.4 (ArCH), 114.5 (CCN), 64.1 (NC(CH3)2), 58.1 (CH2 pyrrolidine), 49.1 (NCH3), 43.8 (NCC(CH3)2), 31.3 (CH3 pyrrolidine), 30.0 (CH3 pyrrolidine), 29.0 (CH(CH3)2iPr), 26.0 (CH(CH3)2iPr), 25.2 (CH(CH3)2iPr) ppm. Elemental analysis for C28H38N2S·0.05KI: calcd: C 76.26; H 8.68; N 6.35, S 7.26. Found: C 76.22; H 8.37; N 6.25, S 6.84%.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
D. M. and M. M. H. thank the Fonds der Chemischen Industrie for Liebig fellowships. Financial Support by the Boehringer-Ingelheim Fonds and the German-American Fulbright commission are gratefully acknowledged. We thank the RRZE Erlangen for computational resources. Continuous support by K. Meyer and M. Alcarazo is gratefully acknowledged.
Notes and references
- R. W. Hoffmann, Angew. Chem., Int. Ed., 1968, 7, 754 CrossRef CAS.
- J. Hocker and R. Merten, Angew. Chem., Int. Ed., 1972, 11, 964 CrossRef CAS.
- M. F. Lappert, J. Organomet. Chem., 1988, 358, 185 CrossRef CAS.
- J. Broggi, T. Terme and P. Vanelle, Angew. Chem., Int. Ed., 2014, 53, 384 CrossRef CAS PubMed.
- J. A. Murphy, J. Org. Chem., 2014, 79, 3731 CrossRef CAS PubMed.
- E. Doni and J. A. Murphy, Chem. Commun., 2014, 50, 6073 RSC.
- J. A. Murphy, J. Garnier, S. R. Park, F. Schoenebeck, S. Z. Zhou and A. T. Turner, Org. Lett., 2008, 10, 1227 CrossRef CAS PubMed.
- S. Z. Zhou, G. M. Anderson, B. Mondal, E. Doni, V. Ironmonger, M. Kranz, T. Tuttle and J. A. Murphy, Chem. Sci., 2014, 5, 476 RSC.
- S. Rohrbach, R. S. Shah, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2019, 58, 11454 CrossRef CAS PubMed.
- F. Wudl, D. Wobschall and E. J. Hufnagel, J. Am. Chem. Soc., 1972, 94, 670 CrossRef CAS.
- S. Hünig, G. Kießlich, H. Quast and D. Scheutzow, Liebigs Ann., 1973, 310 CrossRef.
- N. Martin, Chem. Commun., 2013, 49, 7025 RSC.
- D. Canevet, M. Salle, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245 RSC.
- M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891 CrossRef CAS PubMed.
- J. Garnier, A. R. Kennedy, L. E. A. Berlouis, A. T. Turner and J. A. Murphy, Beilstein J. Org. Chem., 2010, 6, 73 Search PubMed.
- S. Hünig, J. Groβ and W. Schenk, Liebigs Ann., 1973, 324 CrossRef.
- E. Weitz, Angew. Chem., 1954, 66, 658 CrossRef CAS.
- S. Hünig, B. J. Garner, G. Ruider and W. Schenk, Liebigs Ann., 1973, 1036 CrossRef.
- R. J. Mortimer, Chem. Soc. Rev., 1997, 26, 147 RSC.
- C. G. Claessens and J. F. Stoddart, J. Phys. Org. Chem., 1997, 10, 254 CrossRef CAS.
- S. Hünig, H. Schlaf, G. Kieβlich and D. Scheutzow, Tetrahedron Lett., 1969, 10, 2271 CrossRef.
- For concise and recent reviews on carbene ligands, see:
(a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed;
(b) R. S. Ghadwal, Dalton Trans., 2016, 45, 16081 RSC;
(c) D. Munz, Organometallics, 2018, 37, 275 CrossRef CAS. For thematic issues and books on NHCs, see:
(d) A. J. Arduengo and G. Bertrand, Chem. Rev., 2009, 109, 3209 CrossRef CAS PubMed;
(e)
S. Díez González, N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, Royal Society of Chemistry, Cambridge, UK, 2010 RSC;
(f) T. Rovis and S. P. Nolan, Synlett, 2013, 24, 1188 CrossRef CAS;
(g)
S. P. Nolan, N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis; Wiley-VCH, Weinheim, Germany, 2014 Search PubMed;
(h)
H. V. Huynh, The Organometallic Chemistry of N-heterocyclic Carbenes, John Wiley & Sons, 2017 CrossRef;
(i) F. E. Hahn, Chem. Rev., 2018, 118, 9455 CrossRef CAS PubMed ; For a review with a focus on cyclic singlet carbenes, see:;
(j) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810 CrossRef CAS PubMed. For a visual tutorial on isolating free carbenes and heterodimers, see:
(k) A. Grünwald, S. J. Goodner and D. Munz, J. Visualized Exp., 2019, e59389 Search PubMed.
- H. W. Wanzlick and E. Schikora, Chem. Ber./Recl., 1961, 94, 2389 CrossRef CAS.
- H. W. Wanzlick and E. Schikora, Angew. Chem., Int. Ed., 1960, 72, 494 CrossRef CAS.
- Y. Liu and D. M. Lemal, Tetrahedron Lett., 2000, 41, 599 CrossRef CAS.
- M. K. Denk, K. Hatano and M. Ma, Tetrahedron Lett., 1999, 40, 2057 CrossRef CAS.
- D. M. Lemal, R. A. Lovald and K. I. Kawano, J. Am. Chem. Soc., 1964, 86, 2518 CrossRef CAS.
- R. W. Alder, L. Chaker and F. P. V. Paolini, Chem. Commun., 2004, 2172 RSC.
- R. W. Alder, M. E. Blake, L. Chaker, J. N. Harvey, F. Paolini and J. Schütz, Angew. Chem., Int. Ed., 2004, 43, 5896 CrossRef CAS PubMed.
- V. P. W. Böhm and W. A. Herrmann, Angew. Chem., Int. Ed., 2000, 39, 4036 CrossRef.
- V. Gierz, J. Melomedov, C. Förster, C. Deißler, F. Rominger, D. Kunz and K. Heinze, Chem.–Eur. J., 2012, 18, 10677 CrossRef CAS PubMed.
- P. I. Jolly, S. Zhou, D. W. Thomson, J. Garnier, J. A. Parkinson, T. Tuttle and J. A. Murphy, Chem. Sci., 2012, 3, 1675 RSC.
- T. A. Taton and P. Chen, Angew. Chem., Int. Ed., 1996, 35, 1011 CrossRef CAS.
- Y. Liu, P. E. Lindner and D. M. Lemal, J. Am. Chem. Soc., 1999, 121, 10626 CrossRef CAS.
- F. E. Hahn, L. Wittenbecher, D. Le Van and R. Fröhlich, Angew. Chem., Int. Ed., 2000, 39, 541 CrossRef CAS PubMed.
- D. Munz, J. Chu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2016, 55, 12886 CrossRef CAS PubMed.
- For the first report of a CAAC, see:
(a) V. Lavallo, Y. Canac, C. Prasang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 CrossRef CAS PubMed. For reviews on CAACs, see:
(b) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef CAS PubMed;
(c) S. Roy, K. C. Mondal and H. W Roesky, Acc. Chem. Res., 2016, 49, 357 CrossRef CAS PubMed;
(d) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046 CrossRef CAS PubMed;
(e) U. S. D. Paul and U. Radius, Eur. J. Inorg. Chem., 2017, 3362 CrossRef CAS ; For the synthesis of donor substituted CAACs, see:;
(f) J. Chu, D. Munz, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 7884 CrossRef CAS PubMed.
- J. Messelberger, A. Grünwald, P. Pinter, M. M. Hansmann and D. Munz, Chem. Sci., 2018, 9, 6107 RSC.
- T. Ullrich, P. Pinter, J. Messelberger, P. Haines, R. Kaur, M. M. Hansmann, D. Munz and D. M. Guldi, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202001286.
- M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2206 CrossRef CAS PubMed.
- M. M. Hansmann, M. Melaimi, D. Munz and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2546 CrossRef CAS PubMed.
- D. Mandal, S. Sobottka, R. Dolai, A. Maiti, D. Dhara, P. Kalita, R. S. Narayanan, V. Chandrasekhar, B. Sarkar and A. Jana, Chem. Sci., 2019, 10, 4077 RSC.
- D. Mandal, R. Dolai, N. Chrysochos, P. Kalita, R. Kumar, D. Dhara, A. Maiti, R. S. Narayanan, G. Rajaraman, C. Schulzke, V. Chandrasekhar and A. Jana, Org. Lett., 2017, 19, 5605 CrossRef CAS PubMed.
- G. P. McGlacken and T. A. Khan, Angew. Chem., Int. Ed., 2008, 47, 1819 CrossRef CAS PubMed.
- A. J. Arduengo, F. Davidson, H. V. R. Dias, J. R. Goerlich, D. Khasnis, W. J. Marshall and T. K. Prakasha, J. Am. Chem. Soc., 1997, 197, 12742 CrossRef.
- P. W. Antoni, T. Bruckhoff and M. M. Hansmann, J. Am. Chem. Soc., 2019, 141, 9701 CrossRef CAS PubMed.
- P. W. Antoni and M. M. Hansmann, J. Am. Chem. Soc., 2018, 140, 14823 CrossRef PubMed.
- S. S. Hanson, E. Doni, K. T. Traboulsee, G. Coulthard, J. A. Murphy and C. A. Dyker, Angew. Chem., Int. Ed., 2015, 54, 11236 CrossRef CAS PubMed.
- S. V. Klementyeva, P. A. Abramov, N. V. Somov, Y. B. Dudkina, Y. H. Budnikova and A. I. Poddel’sky, Org. Lett., 2019, 21, 946 CrossRef CAS PubMed.
- C. M. Weinstein, C. D. Martin, L. Liu and G. Bertrand, Angew. Chem., Int. Ed., 2014, 126, 6550 CrossRef PubMed.
- L. Y. M. Eymann, P. Varava, A. M. Shved, B. F. E. Curchod, Y. Liu, O. M. Planes, A. Sienkiewicz, R. Scopelliti, F. Fadaei Tirani and K. Severin, J. Am. Chem. Soc., 2019, 141, 17112 CrossRef CAS PubMed.
- B. Eberle, E. Kaifer and H. J. Himmel, Angew. Chem., Int. Ed., 2017, 56, 3360 CrossRef CAS PubMed.
- The Z configuration was assigned based on calculations (B3LYP/def2-SVP: ΔG = 4.0 kcal mol−1) and 2D-NMR (NOESY/ROESY) spectra (Fig. S10†).
- Reinvestigation of the CV data of 14 revealed that the two halfwave potentials are located at E1,2 = +0.01 V and E2,3 = −0.33 V vs. SCE.
- Electrochemical data for 13 has been reported (E1,2 = −0.02 V; E2,3 = −0.17 V), but was repeated in order to reference vs. Fc/Fc+. S. Hünig, H. Schlaf, G. Kieβlich and D. Scheutzow, Tetrahedron Lett., 1969, 10, 2271 CrossRef.
- CVs in THF showed an irreversible redox wave.
- CVs in THF showed a very broad wave and did not resolve the two redox events well.
- For a detailed investigation of solvation effects, see: B. Eberle, O. Hubner, A. Ziesak, E. Kaifer and H. J. Himmel, Chem.–Eur. J., 2015, 21, 8578 CrossRef CAS PubMed.
- W. F. Cooper, N. C. Kenny, J. W. Edmonds, A. Nagel, F. Wudl and P. Coppens, Chem. Commun., 1971, 889 RSC.
- D. H. Evans, Chem. Rev., 2008, 108, 2113 CrossRef CAS PubMed.
- For leading references on the calculation of solvation free energies with a focus on electrochemistry, see:
(a) A. V. Marenich, J. M. Ho, M. L. Coote, C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2014, 16, 15068 RSC;
(b) J. M. Ho, Phys. Chem. Chem. Phys., 2015, 17, 2859 RSC;
(c) J. Cheng, X. D. Liu, J. VandeVondele, M. Sulpizi and M. Sprik, Acc. Chem. Res., 2014, 47, 3522 CrossRef CAS PubMed. For general references on modeling solvation effects, see:
(d) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS;
(e) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed;
(f) A. Klamt, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1338 Search PubMed;
(g) W. L. Jorgensen and J. Tirado-Rives, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6665 CrossRef CAS PubMed;
(h) J. Zhang, H. Y. Zhang, T. Wu, Q. Wang and D. van der Spoel, J. Chem. Theory Comput., 2017, 13, 1034 CrossRef CAS PubMed.
- S. Grimme and P. R. Schreiner, Angew. Chem., Int. Ed., 2018, 57, 4170 CrossRef CAS PubMed.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793 CrossRef CAS PubMed.
- J. Moens, P. Jaque, F. De Proft and P. Geerlings, J. Phys. Chem. A, 2008, 112, 6023 CrossRef CAS PubMed.
-
R. G. Parr and W. Yang, Density-Functional Theory of Atoms and Molecules, Oxford University Press, USA, 1989 Search PubMed.
- R. G. Pearson, J. Chem. Sci., 2005, 117, 369 CrossRef CAS.
- T. Koopmans, Physica, 1934, 1, 104 CrossRef.
- J. F. Janak, Phys. Rev. B: Solid State, 1978, 18, 7165 CrossRef CAS.
- R. G. Pearson, J. Mol. Struct., 1992, 255, 261 CrossRef.
- Even though
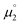 is seemingly also well in agreement with the experimental values for E2,3 when using eqn 2 (Fig. S22,†R2 = 0.95), a substantially worse fit (Fig. S23,†R2 = 0.61) is obtained for the difference of the halfwave potentials ΔE.
is seemingly also well in agreement with the experimental values for E2,3 when using eqn 2 (Fig. S22,†R2 = 0.95), a substantially worse fit (Fig. S23,†R2 = 0.61) is obtained for the difference of the halfwave potentials ΔE.
-
(a) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS PubMed;
(b) Z. F. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed;
(c) R. Gershoni-Poranne and A. Stanger, Chem. Soc. Rev., 2015, 44, 6597 RSC;
(d) A. Stanger, Eur. J. Org. Chem., 2020 DOI:10.1002/ejoc.201901829.
- Note that NICS values, especially NICS0, include σ-effects and hence can lead to erroneous results.
(a) C. Corminboeuf, T. Heine, G. Seifert, P. V. Schleyer and J. Weber, Phys. Chem. Chem. Phys., 2004, 6, 273 RSC;
(b) H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. Schleyer, Org. Lett., 2006, 8, 863 CrossRef CAS PubMed;
(c) A. Stanger, J. Org. Chem., 2010, 75, 2281 CrossRef CAS PubMed;
(d) A. Stanger, J. Phys. Chem. A, 2019, 123, 3922 CrossRef CAS PubMed.
- A. T. Balaban, D. C. Oniciu and A. R. Katritzky, Chem. Rev., 2004, 104, 2777 CrossRef CAS PubMed.
- A. R. Katritzky, V. Feygelman, G. Musumarra, P. Barczynski and M. Szafran, J. Prakt. Chem., 1990, 332 Search PubMed.
- K. E. Horner and P. B. Karadakov, J. Org. Chem., 2015, 80, 7150 CrossRef CAS PubMed.
-
T. M. Krygowski, M. K. Cyranski and M. A. R. Matos, Aromaticity in Heterocyclic Compounds, Springer, Berlin, Germany, 2009 Search PubMed.
- Equally note that antiaromaticity should be important for pyrylenes and pyridylidenes.
- V. Khodorkovsky, A. Edzifna and O. Neilands, J. Mol. Electron., 1989, 5, 33 Search PubMed.
- D. M. Lemal and K. I. Kawano, J. Am. Chem. Soc., 1962, 84, 1761 CrossRef CAS.
- R. C. Wheland, J. Am. Chem. Soc., 1976, 98, 3926 CrossRef CAS.
- P. B. Hitchcock, Dalton Trans., 1979, 1314 RSC.
- CVs in THF, dimethylformamide or acetonitrile showed either a very broad wave and did not resolve the two redox events (THF, DMF) well or showed an irreversible redox wave (MeCN).
- J. A. Murphy, S. Z. Zhou, D. W. Thomson, F. Schoenebeck, M. Mahesh, S. R. Park, T. Tuttle and L. E. Berlouis, Angew. Chem., Int. Ed., 2007, 46, 5178 CrossRef CAS PubMed.
- J. R. Ames, M. A. Houghtaling, D. L. Terrian and T. P. Mitchell, Can. J. Chem., 1997, 75, 28 CrossRef CAS.
- S. Hünig, D. Scheutzow, H. Schlaf and H. Pütter, Liebigs Ann., 1974, 1436 CrossRef.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
-
A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, US, 2000 Search PubMed.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97 CrossRef CAS.
- Y. Gothe, T. Marzo, L. Messori and N. Metzler-Nolte, Chem.–Eur. J., 2016, 22, 12487 CrossRef CAS PubMed.
- R. Jazzar, R. D. Dewhurst, J.-B. Bourg, B. Donnadieu, Y. Canac and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 2899 CrossRef CAS PubMed.
- P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360 CrossRef CAS PubMed.
- M. G. Capdevila, E. Emer, A. Gualandi, D. Petruzziello, S. Grilli and P. G. Cozzi, ChemCatChem, 2012, 4, 968 CrossRef CAS.
- F. Hanasaka, K. Fujita and R. Yamaguchi, Organometallics, 2005, 24, 3422 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98 CrossRef CAS.
- M. K. Denk, A. Thadani, K. Hatano and A. J. Lough, Angew. Chem., Int. Ed., 1997, 36, 2607 CrossRef CAS.
- H. W. Wanzlick, Org. Synth., 1967, 47, 14 CrossRef CAS.
- A. J. Arduengo, R. Krafczyk, R. Schmutzler, H. A. Craig, J. R. Goerlich, W. J. Marshall and M. Unverzagt, Tetrahedron, 1999, 55, 14523 CrossRef CAS.
- J. E. Thomson, C. D. Campbell, C. Concellón, N. Duguet, K. Rix, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2008, 73, 2784 CrossRef CAS PubMed.
- H. Quast and S. Hünig, Chem. Ber., 1966, 99, 2017 CrossRef CAS.
- A. J. Arduengo, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114, 5530 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1964614 and 1964615. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00699h |
| ‡ Both authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence bc and
Dominik
Munz
bc and
Dominik
Munz
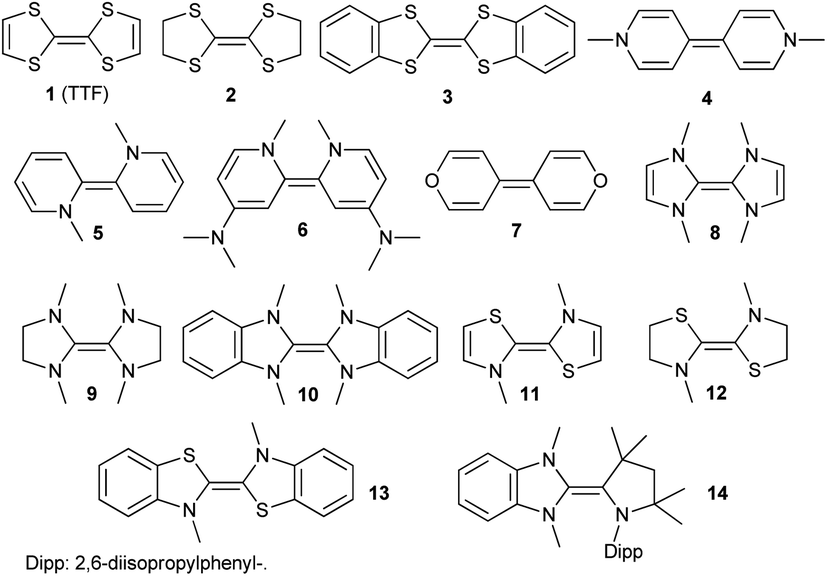
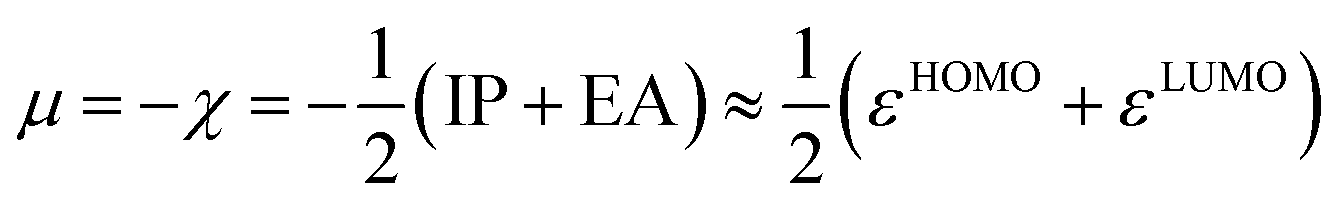

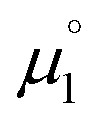 for the biscations, which are very difficult to be further oxidized, should be solely related with their electron affinity according to eqn (2). Differently, the chemical potential
for the biscations, which are very difficult to be further oxidized, should be solely related with their electron affinity according to eqn (2). Differently, the chemical potential 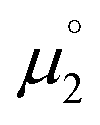 for the reduction of the cationic radicals should be approximated by eqn (1). Importantly, the cationic radicals will only be resistant towards disproportionation, if the reductive formation of the radical cation FROM the biscation
for the reduction of the cationic radicals should be approximated by eqn (1). Importantly, the cationic radicals will only be resistant towards disproportionation, if the reductive formation of the radical cation FROM the biscation 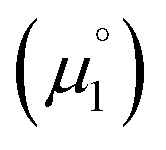 is more favorable than further reduction TO the olefin
is more favorable than further reduction TO the olefin 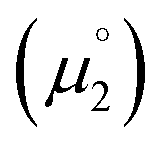 . Consequently, a persistent radical cation is formed (Δμ° > 0), if the first reduction to give the radical cation
. Consequently, a persistent radical cation is formed (Δμ° > 0), if the first reduction to give the radical cation 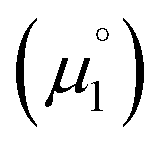 is more facile than the second reduction to the olefin
is more facile than the second reduction to the olefin 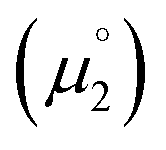 .
.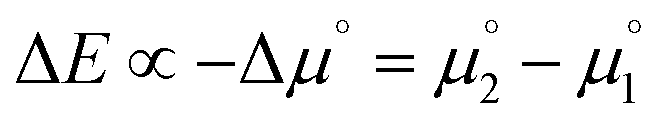
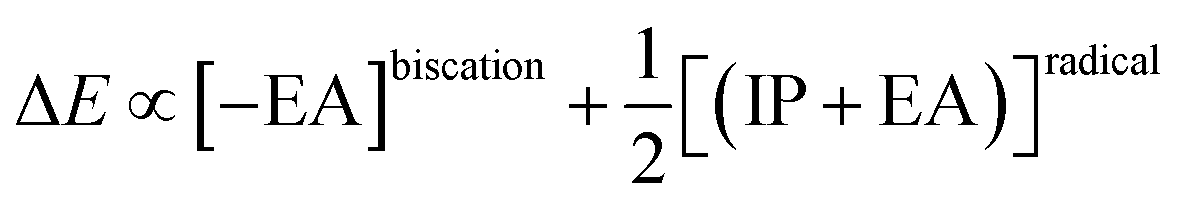
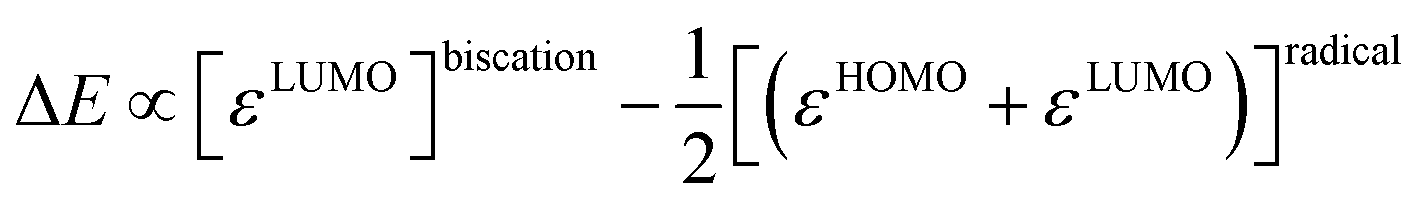
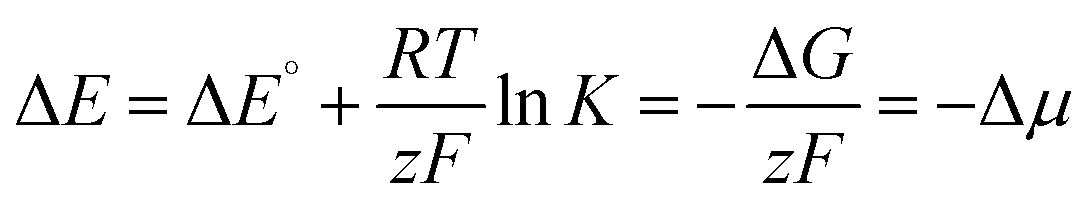
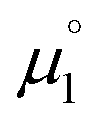 using eqn (2) (Fig. 7). A worse fit was obtained when approximating the electron affinity with the respective LUMO energies (Fig. S19;†R2 = 0.88). Accordingly, no satisfying correlation was obtained with eqn (1) (Fig. S20;†R2 = 0.60). The second redox step E2,3 was well (R2 = 0.93) reproduced with eqn (1) (Fig. 8), where the energies of the α-HOMO and β-LUMO were used for the ionization potential and the electron affinity, respectively.70 Modelling ΔE of the two halfwaves showed an equally good agreement with the experimental data (Fig. 9; R2 = 0.94). Also here, reasonable results were only (Fig. S22–S24†) obtained when using eqn (2) for
using eqn (2) (Fig. 7). A worse fit was obtained when approximating the electron affinity with the respective LUMO energies (Fig. S19;†R2 = 0.88). Accordingly, no satisfying correlation was obtained with eqn (1) (Fig. S20;†R2 = 0.60). The second redox step E2,3 was well (R2 = 0.93) reproduced with eqn (1) (Fig. 8), where the energies of the α-HOMO and β-LUMO were used for the ionization potential and the electron affinity, respectively.70 Modelling ΔE of the two halfwaves showed an equally good agreement with the experimental data (Fig. 9; R2 = 0.94). Also here, reasonable results were only (Fig. S22–S24†) obtained when using eqn (2) for 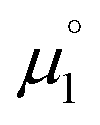 and eqn (1) for
and eqn (1) for 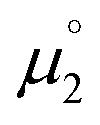 . Impressively, the accuracy (mean absolute deviation MAD) of the computational predictions is within 0.06 V deviation from the experiment (0.07 V if including the further examples discussed below; Table S2†). The calculations predict a large separation for TTF 1 (ΔE = 0.4 V; Δμ° = 0.36 eV) and saTTF 2 (ΔE = 0.5 V; Δμ° = 0.45 eV) and two-electron processes for the NHC- (8), saNHC- (9) and benzNHC (10) homodimers (ΔE = 0 V; Δμ°< 0). The same trend is obtained for the heterodimers 14, 18, 22, 25 (Fig. S26†).
. Impressively, the accuracy (mean absolute deviation MAD) of the computational predictions is within 0.06 V deviation from the experiment (0.07 V if including the further examples discussed below; Table S2†). The calculations predict a large separation for TTF 1 (ΔE = 0.4 V; Δμ° = 0.36 eV) and saTTF 2 (ΔE = 0.5 V; Δμ° = 0.45 eV) and two-electron processes for the NHC- (8), saNHC- (9) and benzNHC (10) homodimers (ΔE = 0 V; Δμ°< 0). The same trend is obtained for the heterodimers 14, 18, 22, 25 (Fig. S26†).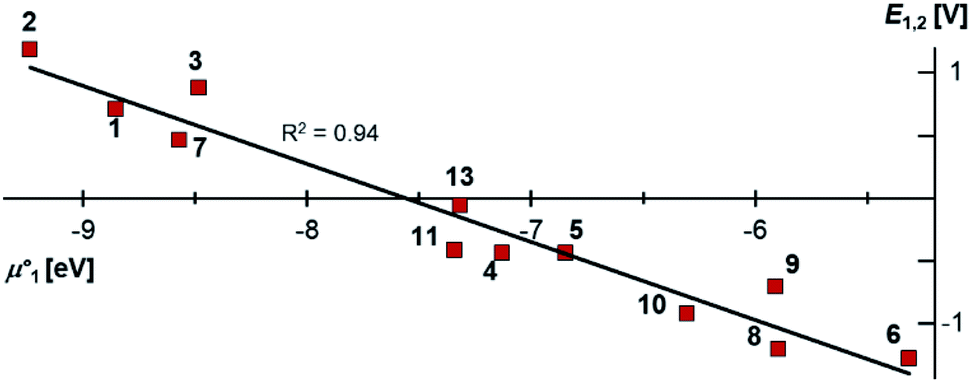
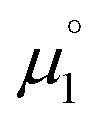 according to eqn (2). E1,2 is given vs. SCE.
according to eqn (2). E1,2 is given vs. SCE.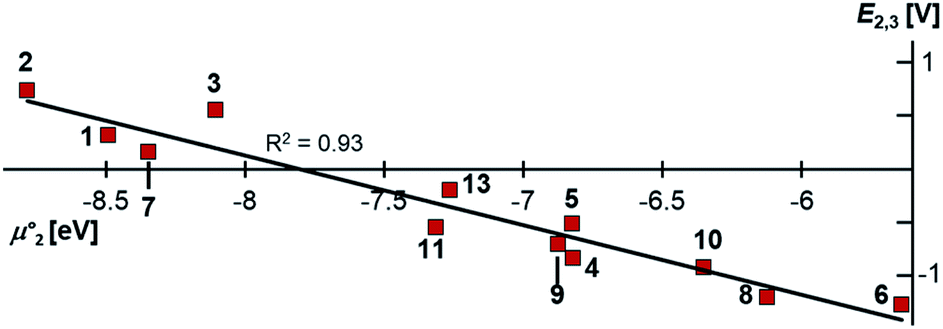
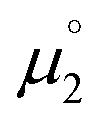 according to eqn (1). E2,3 is given vs. SCE.
according to eqn (1). E2,3 is given vs. SCE.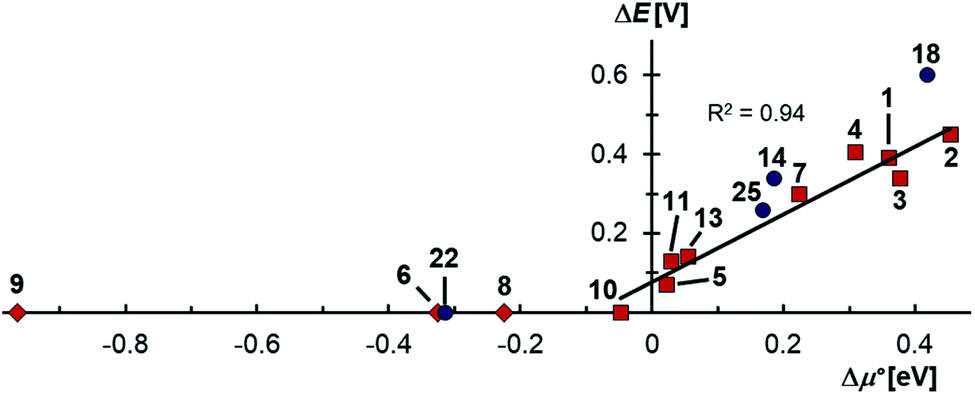
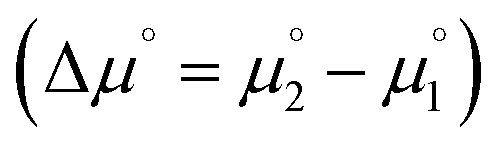 . Heterodimers are given in blue circles, homodimers are given in red squares and diamonds. Heterodimers and homodimers given in red diamonds are not included in the linear regression.
. Heterodimers are given in blue circles, homodimers are given in red squares and diamonds. Heterodimers and homodimers given in red diamonds are not included in the linear regression.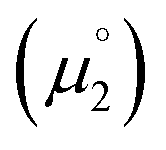 than in the first step from the biscations to the cationic radicals
than in the first step from the biscations to the cationic radicals 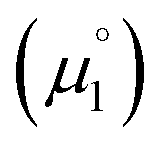 . Consequently, we had a look at the biscations' nucleus-independent chemical shift (NICS) of the NHC derived carbene rings.71 NICS is a convenient and straightforward method to approximately assess the aromaticity of planar π rings. The NICS0zz value relates to the calculated out-of-plane (“zz”) part of the isotropic negative chemical NMR shift of a dummy hydrogen atom in the center of an aromatic ring. This method seems well suitable for the molecules studied herein due to the two diastereotopic faces of the aromatic rings.72 The more negative the chemical shift of the dummy atom, the “more aromatic” is the carbene entity.
. Consequently, we had a look at the biscations' nucleus-independent chemical shift (NICS) of the NHC derived carbene rings.71 NICS is a convenient and straightforward method to approximately assess the aromaticity of planar π rings. The NICS0zz value relates to the calculated out-of-plane (“zz”) part of the isotropic negative chemical NMR shift of a dummy hydrogen atom in the center of an aromatic ring. This method seems well suitable for the molecules studied herein due to the two diastereotopic faces of the aromatic rings.72 The more negative the chemical shift of the dummy atom, the “more aromatic” is the carbene entity.
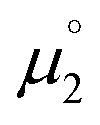 unfavorable and hence prevents overall the disproportionation to the olefin and biscation. For the homodimers,77 the aromaticity seems to be almost irrelevant to the stability of the radical cations as evidenced by the large separation ΔE obtained for both aromatic 1 (ΔE = 0.39 V)4 as well as aliphatic 2 (ΔE = 0.45 V).78 Accordingly, the aromatization energy, which originates in the oxidation of the olefin to its biscation, does not seem to have an influence on the (in)stability of the homodimeric radical cations towards disproportionation.
unfavorable and hence prevents overall the disproportionation to the olefin and biscation. For the homodimers,77 the aromaticity seems to be almost irrelevant to the stability of the radical cations as evidenced by the large separation ΔE obtained for both aromatic 1 (ΔE = 0.39 V)4 as well as aliphatic 2 (ΔE = 0.45 V).78 Accordingly, the aromatization energy, which originates in the oxidation of the olefin to its biscation, does not seem to have an influence on the (in)stability of the homodimeric radical cations towards disproportionation.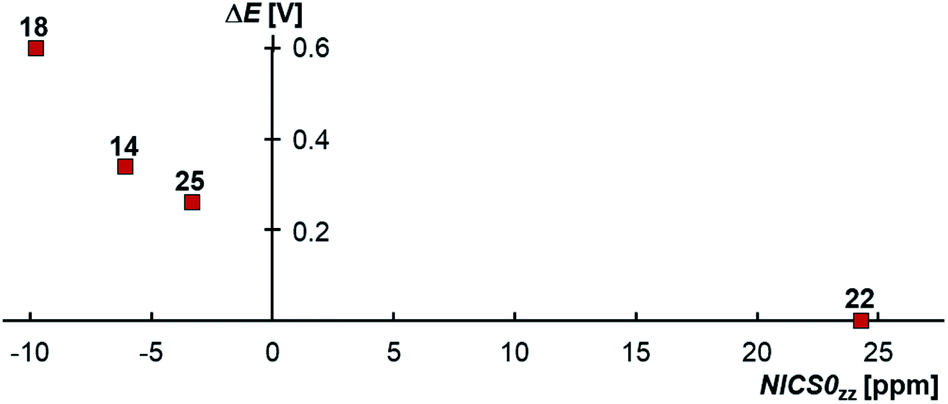
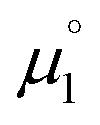 , the energies of the α-HOMO and the β-LUMO for the optimized structural parameters of the cationic radicals were used. For the calculation of
, the energies of the α-HOMO and the β-LUMO for the optimized structural parameters of the cationic radicals were used. For the calculation of 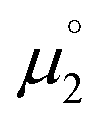 , the vertical electron affinity was used, i.e. the energy of the SOMO upon addition of one electron for the optimized structural parameters of the biscations.
, the vertical electron affinity was used, i.e. the energy of the SOMO upon addition of one electron for the optimized structural parameters of the biscations.