
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnravelling the effect of N(ε)-(carboxyethyl)lysine on the conformation, dynamics and aggregation propensity of α-synuclein†
Laura
Mariño
 ,
Rafael
Ramis
,
Rodrigo
Casasnovas
,
Joaquín
Ortega-Castro
,
Bartolomé
Vilanova
,
Juan
Frau
and
Miquel
Adrover
*
,
Rafael
Ramis
,
Rodrigo
Casasnovas
,
Joaquín
Ortega-Castro
,
Bartolomé
Vilanova
,
Juan
Frau
and
Miquel
Adrover
*
Institut Universitari d'Investigació en Ciències de la Salut (IUNICS), Institut d’Investigació Sanitària Illes Balears (IdISBa), Departament de Química, Universitat de les Illes Balears, Ctra. Valldemossa km 7.5, E-07122 Palma de Mallorca, Spain. E-mail: miquel.adrover@uib.es; Fax: +34 971 173426; Tel: +34 971 173491
First published on 10th March 2020
Abstract
α-Synuclein (αS) aggregation is a hallmark in several neurodegenerative diseases. Among them, Parkinson's disease is highlighted, characterized by the intraneuronal deposition of Lewy bodies (LBs) which causes the loss of dopaminergic neurons. αS is the main component of LBs and in them, it usually contains post-translational modifications. One of them is the formation of advanced glycation end-products (mainly CEL and MOLD) arising from its reaction with methylglyoxal. Despite its biological relevance, there are no data available proving the effect of glycation on the conformation of αS, nor on its aggregation mechanism. This has been hampered by the formation of a heterogeneous set of compounds that precluded conformational studies. To overcome this issue, we have here produced αS homogeneously glycated with CEL. Its use, together with different biophysical techniques and molecular dynamics simulations, allowed us to study for the first time the effect of glycation on the conformation of a protein. CEL extended the conformation of the N-terminal domain as a result of the loss of transient N-/C-terminal long-range contacts while increasing the heterogeneity of the conformational population. CEL also inhibited the αS aggregation, but it was not able to disassemble preexisting amyloid fibrils, thus proving that CEL found on LBs must be formed in a later event after aggregation.
Introduction
Parkinson's disease (PD) is a neurodegenerative movement disorder characterized by the loss of dopamine-producing neurons as a result of the accumulation of intraneuronal protein deposits (known as Lewy bodies (LBs)).1 LBs interfere with the trafficking in neurons, disrupt membranes and sequester proteins.2 Their main component is α-synuclein (αS), a small monomeric and intrinsically disordered protein (IDP).3 Its sequence contains three domains: (i) an N-terminal lipid-binding domain (M1-K60); (ii) a non-amyloid-β central domain (NAC; E61-V95) with a highly hydrophobic motif indispensable for αS aggregation; and (iii) a C-terminal acidic domain (K96-A140) involved in the biological binding of αS (Fig. 1A). In vivo, αS can either display an α-helical structure when it is bound to vesicles, or an unfolded conformation. Hence, this dynamical conformation suggests specific roles in different cellular locations that must be involved in the maintenance of the function of dopaminergic neurons. In fact, αS acts as a chaperon of synaptic SNARE proteins,4 regulates the neuronal redox balance,5 inhibits apoptosis, participates in the regulation of glucose levels, and modulates the calmodulin activity, among others.6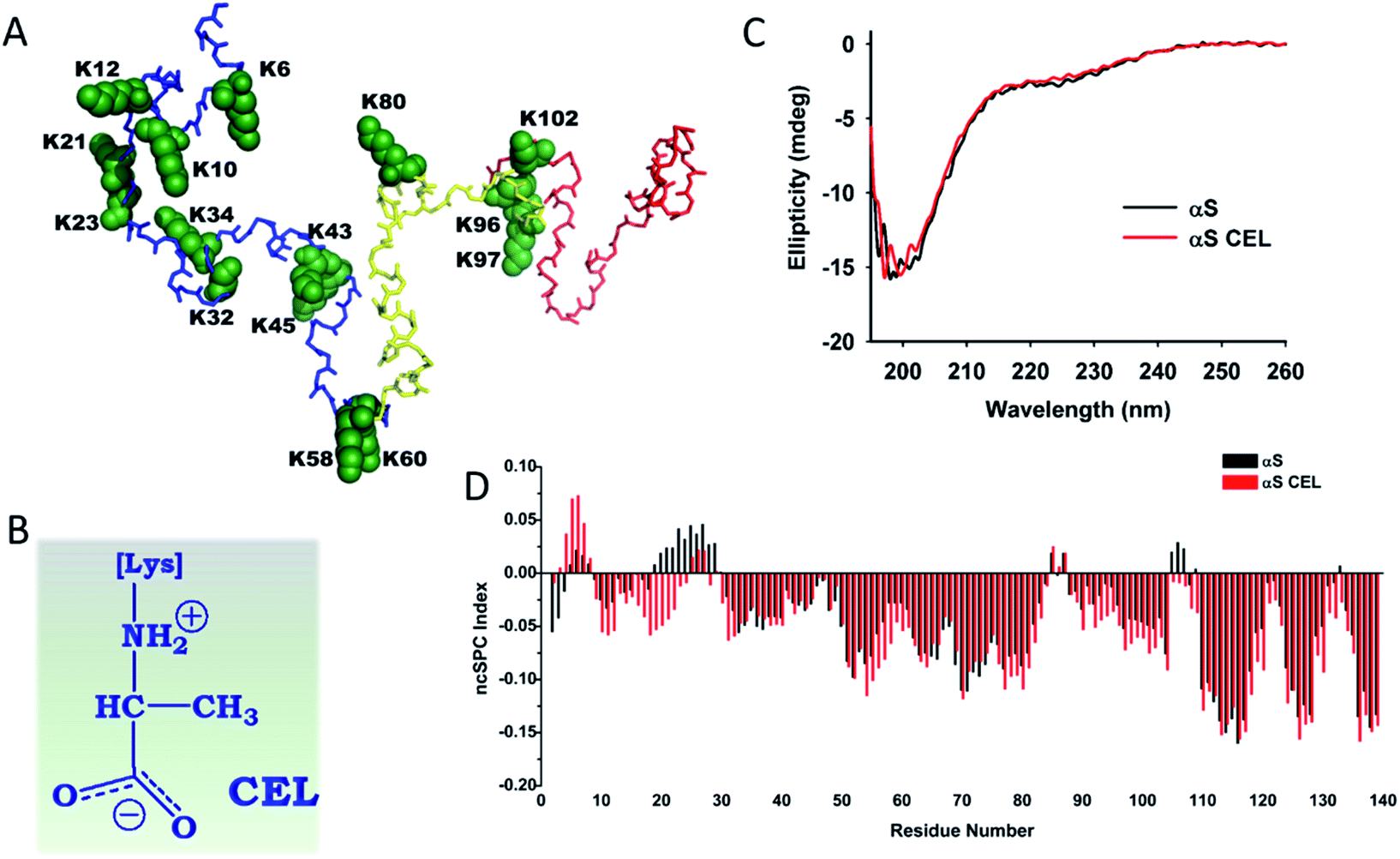 | ||
| Fig. 1 Effect of CEL formation on the secondary structure of αS. (A) Structural conformation corresponding to the averaged ensemble of native αS, which was obtained from the Protein Ensemble Database (PED9AAC).59 The backbone atoms corresponding to the N-terminal domain (M1-K60) are colored in blue. Those corresponding to the NAC domain (E61-V95) are colored in yellow, whereas the backbone atoms corresponding to the C-terminal domain (K96-A140) are colored in red. The backbone and the side chain atoms of the fifteen Lys residues included in the αS sequence are shown as spheres and colored in green. Each Lys has been labelled according to their residue number. (B) The chemical structure of CEL. It is shown under its zwitterionic form, which must be predominant form at physiological pH. (C) Overlapping of the far-UV CD spectrum of αS with that corresponding to αS-CEL. The spectra were recorded in 200 mM phosphate buffer (pH 7.4) at 25 °C. (D) Residue-specific ncSPC scores obtained for αS (black) and for αS-CEL (red) calculated from the HN, N, Hα, Cα, Cβ, and CO chemical shifts at 12.5 °C and at pH 6.5. “+1” indicates the maximum propensity to form a full α-helix, “−1” indicates a fully formed β-sheet, and “0” indicates disorder. | ||
Regardless of its biological role, αS is a highly aggregation-prone protein. It initially forms soluble oligomers, which might bind to the neuronal membrane and induce the formation of reactive oxygen species (ROS),7 thus facilitating PD.8 Later, these oligomers further evolve into amyloid fibrils, which consist of two protofilaments (involving the V37-Q99 stretch) that intertwine by forming a left-handed helix.9 αS fibrils finally clump into LBs.1
Many molecular mechanisms contribute to stimulate the αS aggregation. An increased αS expression is sufficient to trigger its aggregation and neurodegeneration.10 This occurs as a result of the duplication11 or triplication12 of the αS encoding gene (SCNA). Genetic mutations (e.g. A53T, A30P, E46K or G51D)13 are also able to promote αS aggregation and cause early-onset forms of PD. The formation of metal-αS complexes enhances the αS aggregation rate; meanwhile they also exert neuronal toxicity through the formation of ROS.14
In addition to all these factors, most of the αS found in vivo includes post-translational modifications (PTMs) such as acetylation, phosphorylation, ubiquitination, nitration, sumoylation, truncation, oxidation or glycosylation.15 These PTMs can have either positive or negative effects on αS aggregation. While S129 phosphorylation promotes its aggregation, that occurring on Y39, S87, Y125 or Y133 diminishes its aggregation propensity.15b,16 Ubiquitination and sumoylation display a site-dependent effect, although they mainly delay or even inhibit αS aggregation.15b Tyr nitration (on Y39, Y125, Y133 and/or Y136) stabilizes αS oligomers, but inhibits fibril formation.17aNα-acetylation does not affect the aggregation of αS,15b whereas C-terminal truncated αS displays a higher aggregation propensity than full-length αS.18 Met oxidation (detected in LBs) notably stabilizes neurotoxic αS oligomers.19 Recently, αS has been found O-GlcNAcylated in vivo, which completely inhibits its aggregation, and thus it could constitute a plausible cellular strategy to protect neurons.20
αS within LBs can also be non-enzymatically glycosylated. This random process, also known as glycation, occurs on Lys side chains as a result of their reaction with reducing sugars, or with the oxidative by-products of intraneuronal glycolysis. These reactions yield a heterogeneous set of compounds, known as advanced glycation end-products (AGEs), which change the chemical nature of Lys and therefore alter the biophysical features of proteins (Fig. S1†). Accumulation of AGEs on LBs becomes more relevant to people suffering from diabetes mellitus (DM),21 which could explain the increased prevalence of PD in DM patients.22
AGEs such as Nε-(carboxymethyl)lysine (CML)23 or pyrraline21b have been detected on αS. However, the most prevalent AGEs found on LBs are MOLD and Nε-(carboxyethyl)lysine (CEL)23,24 (Fig. 1B and S1†). Both arise from the reaction of αS with methylglyoxal (MG), a prominent product of intraneuronal glycolysis25 with notorious glycation potential on αS.24 Glycation of αS mediated by MG enhances its neurotoxicity through different mechanisms, such as reducing its membrane binding ability or facilitating the accumulation of toxic oligomers.24 Moreover, we recently proved that MG also diminishes the metal-binding ability of recombinant αS (Fig. S2†), while reducing its protective role against oxidative stress.26
All these initial insights are still far from providing a full molecular-level comprehension of the glycation effect on the biophysical properties of αS. This has been hampered by the formation of a heterogeneous set of AGEs, as well as by the formation of a heterogeneous mixture of protein molecules with different glycation degrees.26,27 Therefore, it has been nearly impossible to assign each glycation-induced effect to the formation of a specific AGE and therefore unveil the glycation effect on αS conformation or aggregation propensity. This information would become crucial to be able to design effective therapies to diminish the predisposition of DM patients to suffer from synucleinopathies.
Being aware of the importance of obtaining homogeneously glycated αS to enable the understanding of its biophysical and biochemical properties in the context of PD, we have synthesized a homogeneously glycated αS through the attachment of CEL moieties on each of its fifteen Lys residues (αS-CEL) (Fig. S3A–D and S4†). The effect of CEL on the structural descriptors of αS has been analyzed at the residue level by using NMR in combination with a coarse-grained molecular dynamics approach (CG-MD), which we have recently adapted to specifically study αS and αS-CEL.28 Besides studying the structural effects, we have used fluorescence spectroscopy, molecular dynamics simulations and steered molecular dynamics (SMD) to investigate the effect of CEL not only on the aggregation process of αS, but also on the inter- and intra-molecular interactions between the different NAC domains assembling the architecture of αS amyloid fibrils. To our knowledge, this is the first study that describes at the residue level (using high-resolution techniques) the specific effect of a single AGE (i.e. CEL) on the biophysical properties of a protein.
Results
Obtaining homogeneously glycated αS with Nε-(carboxyethyl)lysine (CEL)
Glycation of αS with MG results in the formation of a heterogeneous set of molecules that preclude structural studies (Fig. S5†). To overcome this drawback, we chemically synthesized CEL (Fig. 1B) on αS. The modified αS (αS-CEL) was isolated using SEC (Fig. S3B†), and its monomeric state was confirmed by SDS-PAGE (Fig. S3C†) and MALDI-TOF/TOF analysis. Its mass spectrum displayed a narrow and unique peak with an increase in its Δm/z ∼1162 Da (Fig. S3D†), which proves that CEL moieties (72 Da) have been added on the 15 Lys of αS and on its N-terminal amino group. NMR spectroscopy additionally confirmed the Lys carboxyethylation, since the Lys-Cε signals downfield shifted by ∼6.5 ppm in αS-CEL (Fig. S3E†). This synthetic approach yielded a homogeneous sample, which allowed us to study the specific effect of CEL on the conformation, dynamics and aggregating features of αS.The effect of CEL on the secondary structure content of αS
Native αS is a disordered protein3 especially in its C-terminal region (Fig. S6†). Its overall unfolding degree was not severely altered as a result of CEL formation (Fig. 1C). However, CD spectroscopy and CG-MD simulations suggested a slight increase of its disordered percentage upon CEL formation (Table S1†). These subtle differences were deeply analysed at the residue level using NMR spectroscopy. The chemical shifts of N, HN, Cα, Cβ, Hα and CO were achieved for all residues between D2 and A140 in αS (BMRB code 27796) and αS-CEL (BMRB code 27797), and they were used to estimate the secondary structure content using the ncSPC index,29 the SSP scores,30 the TALOS + program31a and the NOE intensity ratios. The CEL-induced modifications can be ascribed to a CEL-induced conformational change. Controls carried out on Nα-Ac-Lys proved that CEL does not have any inductive effect on Hα, Cα, Hβ, Cβ, Hγ or Cγ chemical shifts, and it only slightly affected the Cδ chemical shift (∼0.1 ppm) and largely that of Cε (Table S2 and Fig. S7A†). This trend was also observed when comparing the chemical shifts of the Lys side chains between αS and αS-CEL (Figs. S3E and S7B†).CEL formation on the N-terminal amino group and/or K6 seems to slightly increase the transient α-helicity of the F4-L8 region, while CEL21 and/or CEL23 reduced the α-helicity of the E20-A29 stretch (Fig. 1D and S8†). This was additionally confirmed by the increase in the dαN(i,i)/dαN(i − 1,i) NOE intensity ratios of the F4-L8 stretch, whereas those corresponding to the E20-A29 stretch decreased (Fig. S9A†).
CEL formation on K10 and K12, on K58 and K60, and on K96, K97 and K102 results in a conformational extension of the S9-G25, T54-E61 and A89-Q109 regions, respectively (Fig. 1D and S8†). This elongation cannot be attributed to the partial acquisition of a polyproline II helix (PPII) conformation, since CEL did not increase the dαN(i,i)/dαN(i − 1,i) NOE intensity ratios in these stretches (Fig. S9A†), nor the ellipticity at 217 nm (Fig. 1C).32 A CEL-induced PPII conformation is also discarded by the disappearance of some dNN(i − 1,i) NOEs (Fig. S10†) and the negligible change in the dNN(i − 1,i)/dNN(i,i) NOE intensity ratios32 (Fig. S9B†).
The secondary structure content of the G31-A53 stretch was not affected by the formation of CEL on K32, K34, K43 and K45 (Fig. 1D and S8†). In addition, CEL formation did not affect the transient β-hairpin loops typically formed within the NAC domain or between this and the N-terminus of the C-terminal domain (Fig. S11†).
Hence, CEL does not induce the acquisition of any secondary structure on αS, but likely transiently extends the conformation of most of the regions holding CEL-modified Lys.
CEL formation changes the chemical environment of most of the N-terminal residues
NMR assignments were also used to evaluate how CEL affects the local chemical environments of the population-weighted average over all αS conformations. In IDPs, the chemical shift dispersion of N, HN and CO is much greater than others because of their higher sensitivity to the molecular environment.33 Indeed, the Hα, Cα or Cβ chemical shifts of Lys were not affected by the formation of CEL (Fig. S12 and S13†). Furthermore, the 1H,15N-HSQC spectrum of αS-CEL was nearly identical to that of αS (Fig. 2A), thus pointing out that CEL does not significantly modify the weighted average conformation. However, many residues displayed subtle chemical shift variations, which were mainly mapped on the N-terminal domain (Fig. 2B). These perturbations became greater when comparing the HNi–COi−1 cross-peaks (Fig. 2C), but higher ΔδCO still occurred on the N-terminal residues (Fig. 2D). In fact, CEL formation on K80 (NAC domain), K96, K97 and K102 (C-terminal domain) (Fig. 1A) had a negligible effect on ΔδCO and thus on the chemical environment of their neighboring regions (Fig. 2C, D).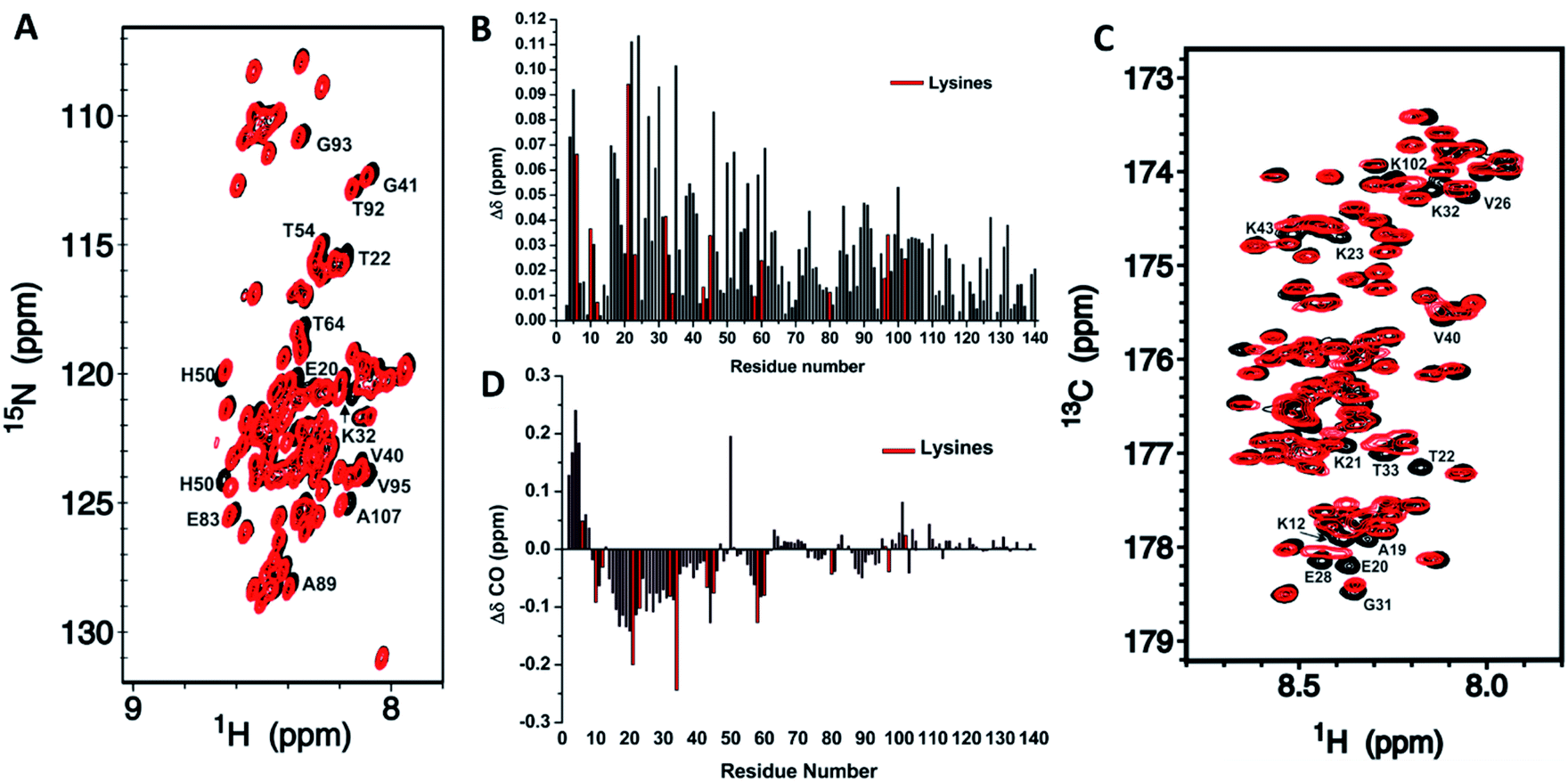 | ||
Fig. 2 Mapping of the residue-specific effect of CEL on αS. (A) Overlapping of the 15N-HSQC spectrum of αS-CEL (red) with that corresponding to native αS (black). Cross-peaks of residues displaying a high chemical shift perturbation are labelled with their residue number. (B) Amide chemical shift perturbations (Δδ) of the HN and N backbone resonances of αS as a result of the formation of CEL on its Lys. For each residue, 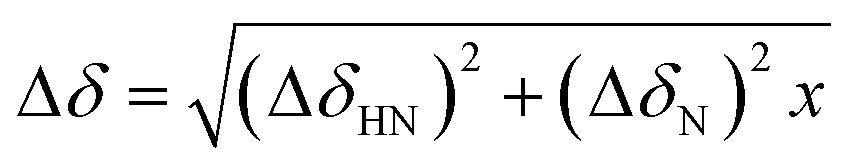 , where x is 0.2 for Gly and 0.14 for the other residues.60 ΔδHN and ΔδN are the amide proton and the amide nitrogen chemical shift differences, respectively. Experimental data corresponding to the Lys are colored in red. (C) Overlapping of the projections of the HN–CO plane in the HNCO spectra of αS (black) and αS-CEL (red). Cross-peaks of residues displaying a high chemical shift perturbation are labelled with their residue number. (D) Chemical shift perturbations (ΔδCO = ΔδαS-CEL − ΔδαS) occurring on the CO chemical shifts of αS as a result of the formation of CEL on its Lys. Experimental data corresponding to the Lys residues are colored in red. , where x is 0.2 for Gly and 0.14 for the other residues.60 ΔδHN and ΔδN are the amide proton and the amide nitrogen chemical shift differences, respectively. Experimental data corresponding to the Lys are colored in red. (C) Overlapping of the projections of the HN–CO plane in the HNCO spectra of αS (black) and αS-CEL (red). Cross-peaks of residues displaying a high chemical shift perturbation are labelled with their residue number. (D) Chemical shift perturbations (ΔδCO = ΔδαS-CEL − ΔδαS) occurring on the CO chemical shifts of αS as a result of the formation of CEL on its Lys. Experimental data corresponding to the Lys residues are colored in red. | ||
Consequently, CEL formation over the entire sequence of αS must display a sequence-related effect, since it only affects the environments of those residues within the N-terminal Lys-rich domain.
CEL formation induces a slight increase of the αS hydrodynamic radius
NMR data point towards an increase in the populations displaying a more extended N-terminal domain. This should result in a slightly higher hydrodynamic radius (Rh), which is suggested by the lower elution volume of the SEC peak of αS-CEL than that of αS (Fig. S3B†). However, this could also be due to a change in the column affinity without the need for a structural alteration, as we reported for lysozyme glycated with ribose27d and with glycolaldehyde.27eTo clarify this issue, we carried out DLS, SAXS and DOSY measurements, as well as CG-MD simulations. DLS evidenced that the most populated conformations of αS displayed a Rh of ∼2.06 nm, whereas for αS-CEL it shifted up to ∼2.80 nm (Fig. 3A). Accordingly, diffusion-ordered spectroscopy also evidenced a greater Rh for αS-CEL than for αS (3.15 ± 0.03 nm vs. 2.89 ± 0.04 nm). This trend was also confirmed by SAXS measurements, which were used to calculate the radius of gyration (Rg) from Guinier plots. The obtained data indicated that αS has a Rg of 3.33 ± 0.05 nm, whereas the Rg of αS-CEL is 3.73 ± 0.13 nm (Fig. S14†). CG-MD simulations also pointed towards a higher Rg for αS-CEL (Fig. 3B). In addition, DLS and CG-MD simulations revealed a more heterogeneous size distribution for αS-CEL than for αS, which implied an increase in the populations of the ensembles with a Rg >3.5 nm. This greater population heterogeneity was also confirmed by the HSQC peak intensities, which were mostly lower and wider in αS-CEL than in αS (Fig. S15†), thus reflecting a broader heterogeneous ensemble of transiently interacting states.
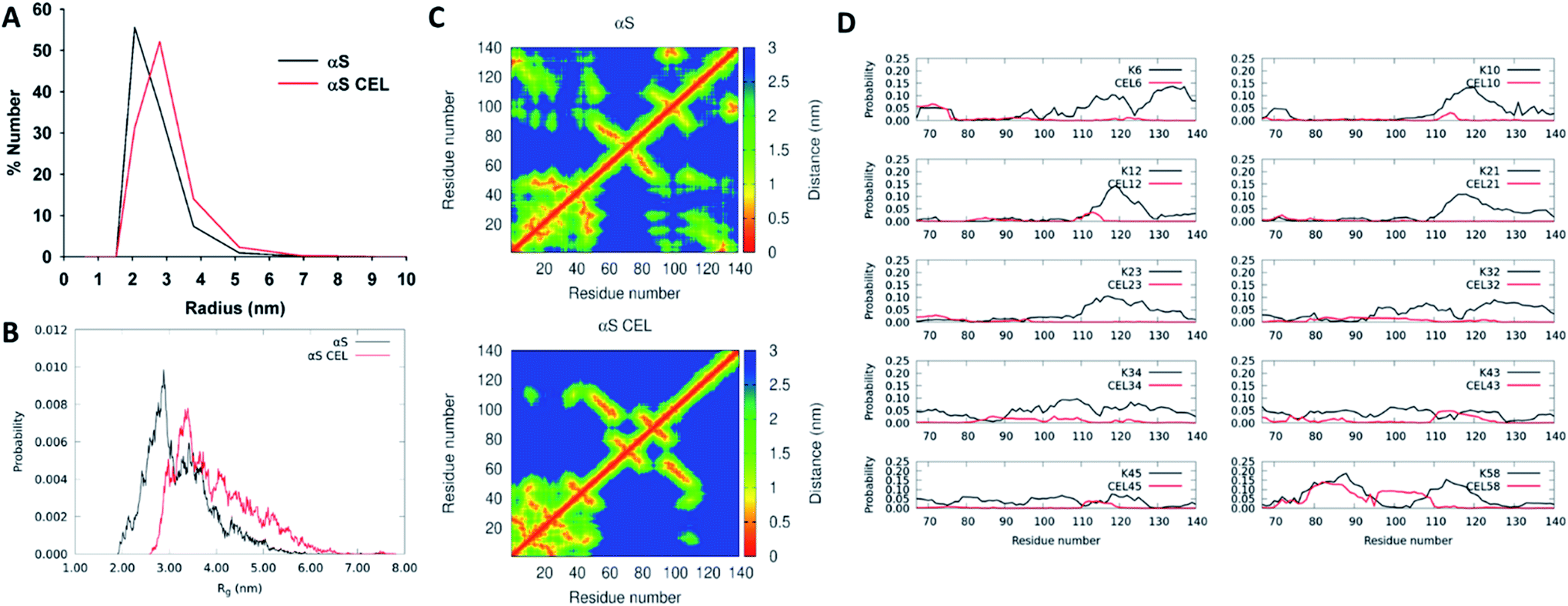 | ||
| Fig. 3 Effect of CEL on the radius of αS and on its intramolecular contacts. (A) Number-weighted dynamic light scattering (DLS) size distributions obtained for monomeric αS (black) and monomeric αS-CEL (red). (B) Overlapping of the Rg histograms obtained for αS (black) and αS-CEL (red). These histograms were calculated from the ensembles computed by using CG-MD simulations and a factor f of 1.3.28 (C) Contact maps corresponding to the central structure of the most populated cluster in αS (top) and in αS-CEL (bottom) obtained from CG-MD simulations (f = 1.3). (D) Propensity of each Lys (in αS; black) or of each CEL moiety (in αS-CEL; red) in the N-terminal domain (M1-K60) to stablish transient contacts with residues at the NAC or at the C-terminal domains. A contact is defined as a distance ≤1.5 nm. The residue–residue propensity contacts were determined from the CG-MD structures obtained during 1400 ns simulations (f = 1.3). | ||
These results indicate that the conformational extension of the N-terminal domain in αS-CEL might be responsible for the increase in the protein radius, as well as for the enhanced heterogeneity of the different conformational populations.
CEL formation truncates the transient long-range contacts in αS
To better understand the effect of CEL on the radius of αS we analysed the most populated clusters of αS and αS-CEL obtained by CG-MD simulations. CEL increased the average distances between the geometry center of the C-terminal domain and that of the N-terminal domain (4.5 ± 2.2 nm in αS and 6.7 ± 2.5 nm in αS-CEL) (Fig. S16A†) and that of the NAC domain (3.3 ± 1.3 nm in αS and 4.6 ± 1.7 nm in αS-CEL) (Fig. S16B†). These increase in the separations occurred due to a CEL-induced reduction of the transient N-terminal/C-terminal contacts (Fig. 3C). In fact, the propensity of the N-terminal Lys to be in close contact with other residues of the C-terminal domain significantly decreases when Lys are replaced by CEL (Fig. 3D).Regardless of the domain separation, the average solvent-exposed surface area (SASA) per residue was not drastically affected (Fig. S17A†). However, the mapping of the ΔSASA evidenced subtle differences. CEL reduced the SASA of the residues within the M1-V40 stretch, while increasing that of most of the residues of NAC and C-terminal domains (Fig. S17B†). This increase might be related to the breakage of the transient ion pair contacts between the amphipathic N-terminal domain and the acidic C-terminal domain, which should be linked to the replacement of positively charged Lys by zwitterionic CEL. In addition, the reduction of the SASA of the N-terminal residues might respond to an increase in the CEL-induced local interactions within the N-terminal domain, as suggested by the contact maps (Fig. 3C).
Although the conformational extension of the N-terminal domain might be involved in the increase of Rg/Rh, it seems that the breakage of the transient N-terminal/C-terminal contacts as a result of CEL formation is the driving force that structurally unpacks αS and increases the heterogeneity of its conformational ensemble.
Cis/trans Pro isomerization in αS is not affected by CEL formation
To further understand the effect of CEL on αS, we also studied whether it affected the cis/trans Pro isomerization. The C-terminal domain of αS contains five Pro (P108, P117, P120, P128 and P138) with a cis population less than 5%.34 To determine how CEL affected this percentage, we used the N, HN, Cα, Cβ, Hα and CO chemical shifts as input for Promega.31b Independently, we also evaluated the intensity ratios of the 15N-HSQC resonances affected by the cis- and trans-Pro states (Fig. S18†).34 In both cases, we found that cis-Pro bonds varied between 2 and 8%, but these percentages were not altered by CEL formation (Table S3†). Hence, CEL does not change the cis-Pro fraction in αS. However, this cannot be taken as a general rule in all proteins, since all Lys residues in αS are sequentially far away from Pro.J couplings also point towards a CEL-induced extension of the N-terminal domain and suggest a change in the N- and C-terminal side chain topology
J couplings are independent reporters for the backbone and side chain conformations in folded proteins35 and in IDPs.36 Hence, we used the HNHA experiment to determine the 3JHNHα coupling constants37 for αS and αS-CEL (Fig. S19A†). Their values ranged between 4.5 and 7 Hz, thus indicating random coil conformations.38 In addition, their variations within each residue type (Δ3JHNHα ∼1–3 Hz) (Fig. S19B†) must correspond to sequence/structure-dependent effects31c since 3JHNHα is quite insensitive to the residue type.39a CEL formation did not directly affect the 3JHNHα values, as the 3JHNHα of Nα-Ac-Lys was identical to that of CEL-modified Nα-Ac-Lys (i.e. 7.65 Hz). CEL formation mainly increased the 3JHNHα values of Lys neighboring residues within the N-terminal domain (Δ3JHN-Hα ∼0.6 ± 1.1 Hz), whereas NAC and C-terminal domains were less affected (Fig. 4A). This increase can be interpreted as a gain in the conformational populations with more extended backbone geometry.36a Nonetheless, the lack of 3JHNHα >8 Hz also proves that these extensions do not involve the formation of β-sheets.38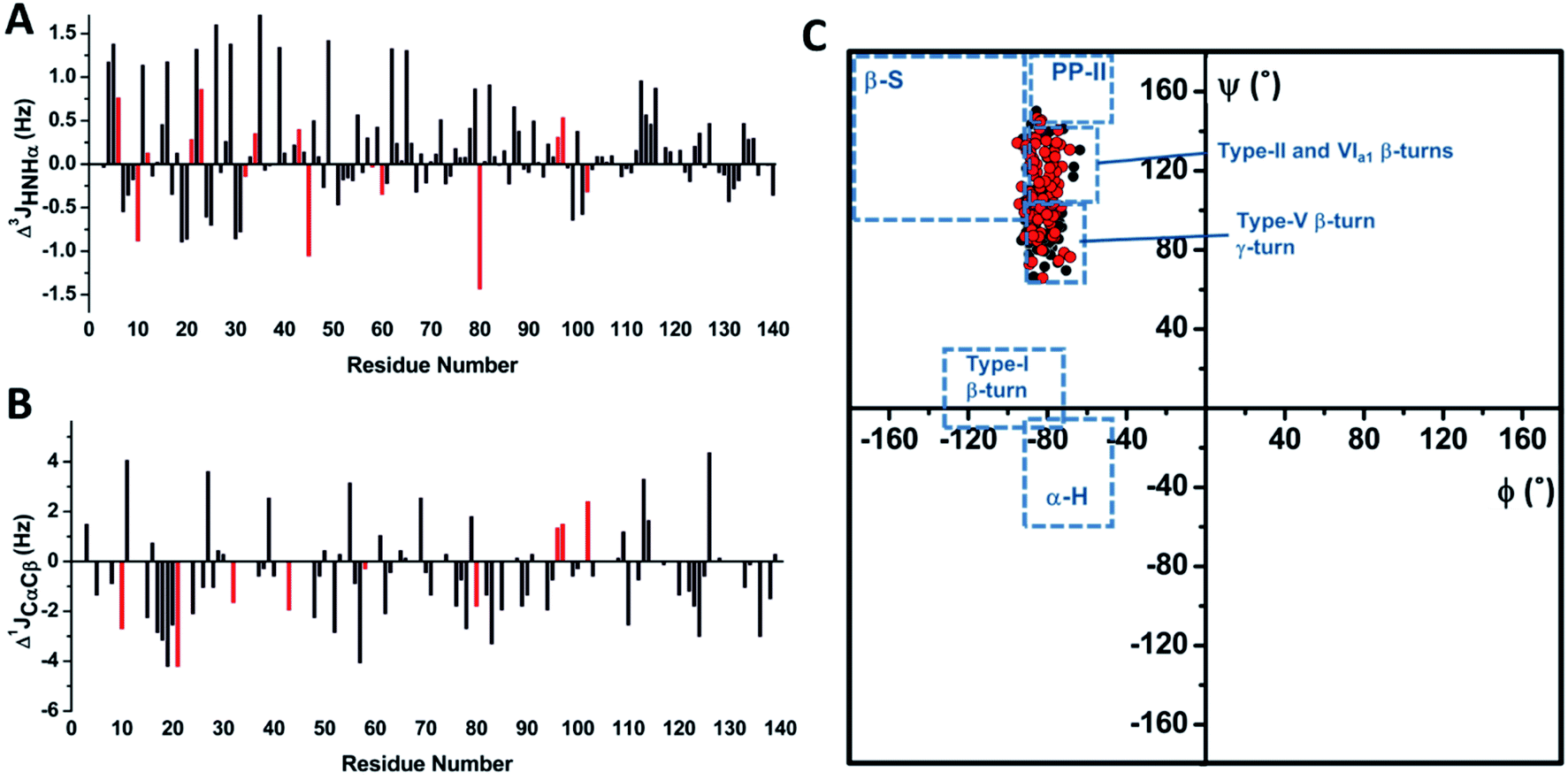 | ||
| Fig. 4 Determining the J coupling constants and the dihedral angles for αS and αS-CEL. (A) Sequence-dependent variations in the 3JHN-Hα coupling constants of αS as a result of CEL formation (Δ3JHN-Hα = 3JHN-Hα_αS-CEL − 3JHN-Hα_αS). Data corresponding to the Lys residues are colored in red. (B) Sequence-dependent variations in the 1JCαCβ coupling constants of αS as a result of CEL formation (Δ1JCαCβ = 1JCαCβ-αS-CEL − 1JCαCβ-αS). Gly 1JCαCβ is non-existent, whereas the Δ1JCαCβ values of Asn, Asp, Ser and Thr are not plotted since these residues exhibit slightly high 1JCαCβ values (Fig. S20A†) that could involve the misinterpretation of the structural data derived from 1JCαCβ. Data corresponding to the Lys residues are colored in red. (C) Ramachandran plot obtained for monomeric αS (black) and monomeric αS-CEL (red). The ϕ and ψ dihedral angles were obtained from the 3JHN-Hα and 1JCαCβ coupling constants using the corresponding Karplus equations. The different conformational regions in the Ramachandran plot are classified as follows: α-helix, −90°<ϕ<−45° and −60°<ψ<−15°; β-sheet, −180°<ϕ<−90° and 90°<ψ<180°; PPII, −90°<ϕ<−45° and 105°<ψ<180°; type I β-turn, −135°<ϕ<−75° and −15°<ψ<30°;31c type II β-turn, −50<ϕ<−80° and 120°<ψ<150°; type V β-turn, −75<ϕ<−85° and 75°<ψ<85°; type VIa1 β-turn, −60<ϕ<−70° and 130°<ψ<140°; and γ-turn, −80<ϕ<−90° and 60°<ψ<75°.44 | ||
The one-bond 1JCαCβ values were also measured for αS and αS-CEL from their HN(CO)CA spectra, as they exhibit substantial conformational dependences.40 However, 1JCαCβ values seem to also display certain amino acid type dependence. For instance, Asp, Asn, Ser and Thr had slightly high 1JCαCβ values (Fig. S20A†), which might mask the structural effects.35 The averaged values for αS and αS-CEL were 37.2 ± 1.4 and 36.6 ± 1.5 Hz respectively (Fig. S20B†), which are greater than the averaged values found in globular proteins (∼34.9 ± 2.5 Hz).35 In any case, CEL reduced the overall 1JCαCβ values (Δ1JCαCβ ∼−0.5 ± 1.7 Hz). This decrease mainly occurred for the V15-Q24 stretch (Δ1JCαCβ ∼−2.3 ± 1.7 Hz), and it could also confirm the CEL-induced decrease in the local turns as a result of a conformational extension.35 However, this local decrease could also be related to the loss of the N-terminal/C-terminal contacts, since the 1JCαCβ values are affected by the side chain topology and therefore, they are dependent on the side chain torsion angles.35 This idea is supported by the decrease in the 1JCαCβ values of the A76-F94 (Δ1JCαCβ ∼−1.2 ± 1.2 Hz), P120-A124 (Δ1JCαCβ ∼−1.6 ± 0.9 Hz) and Y133–P138 (Δ1JCαCβ ∼−1.1 ± 1.2 Hz) stretches, which was larger than the average (Fig. 4B).
CEL formation enhances the conformational space explored by the N-terminal domain
3 J HNHα couplings are sensitive to ϕ dihedrals,36a,39a whereas 1JCαCβ values vary as a function of ϕ, ψ,40χ1(Cα–Cβ) and χ2 (Cβ–Cγ) dihedrals.35 Hence, Karplus equations were applied to estimate the backbone torsion angles (ϕ and ψ) of αS and αS-CEL. In both cases, most of the ϕ angles were between −70° and −90°, whereas ψ values ranged from 65° to 140° (Fig. S21†), thus indicating that the averaged conformations of αS and αS-CEL explore the same conformational space. The positive ψ values are additionally confirmed by the dαN(i,i)/dαN(i − 1,i) NOE intensity ratios <1 (Fig. S9A†),41 which are typical of extended conformations,42 whereas the lack of large 1JCαCβ values (∼40 Hz) also discards positive ϕ angles.35The Ramachandran plots (Fig. 4C) reveal that the averaged conformations of αS and αS-CEL do not display structural motifs resembling β-sheet, α-helix, PP-II or type-I β-turns. As expected, they are similar to those observed in random coil peptides,43 and their ϕ/ψ pair angles are located in regions characteristic of type-II, -V, and -VIa1 β-turns, as well as in regions typical of γ-turns.44
Although the ϕ/ψ pair angles of the averaged conformations of αS and αS-CEL explore the same conformational space, CEL induced a slight decrease and increase in the averaged ϕ (Δϕavg ∼ −0.97 ± 3.8°) and ψ values (Δψavg ∼ 5.1 ± 20°), respectively (Fig. S22A–C†). The main differences between the ϕαS and ϕαS-CEL values map on most of the residues at the N-terminal domain, although the ϕ angle of K80 and those of their neighboring residues were also affected (Fig. 5A and S22A†). In contrast, the residues with the greatest differences between ψαS and ψαS-CEL angles were found along the entire N-terminal and NAC domains, as well as in the N-terminal region of the C-terminal domain (Fig. 5B and S22B†). Consequently, these results suggest that CEL formation may induce a shift between the different populations of turns, which would mostly occur at the N-terminal domain.
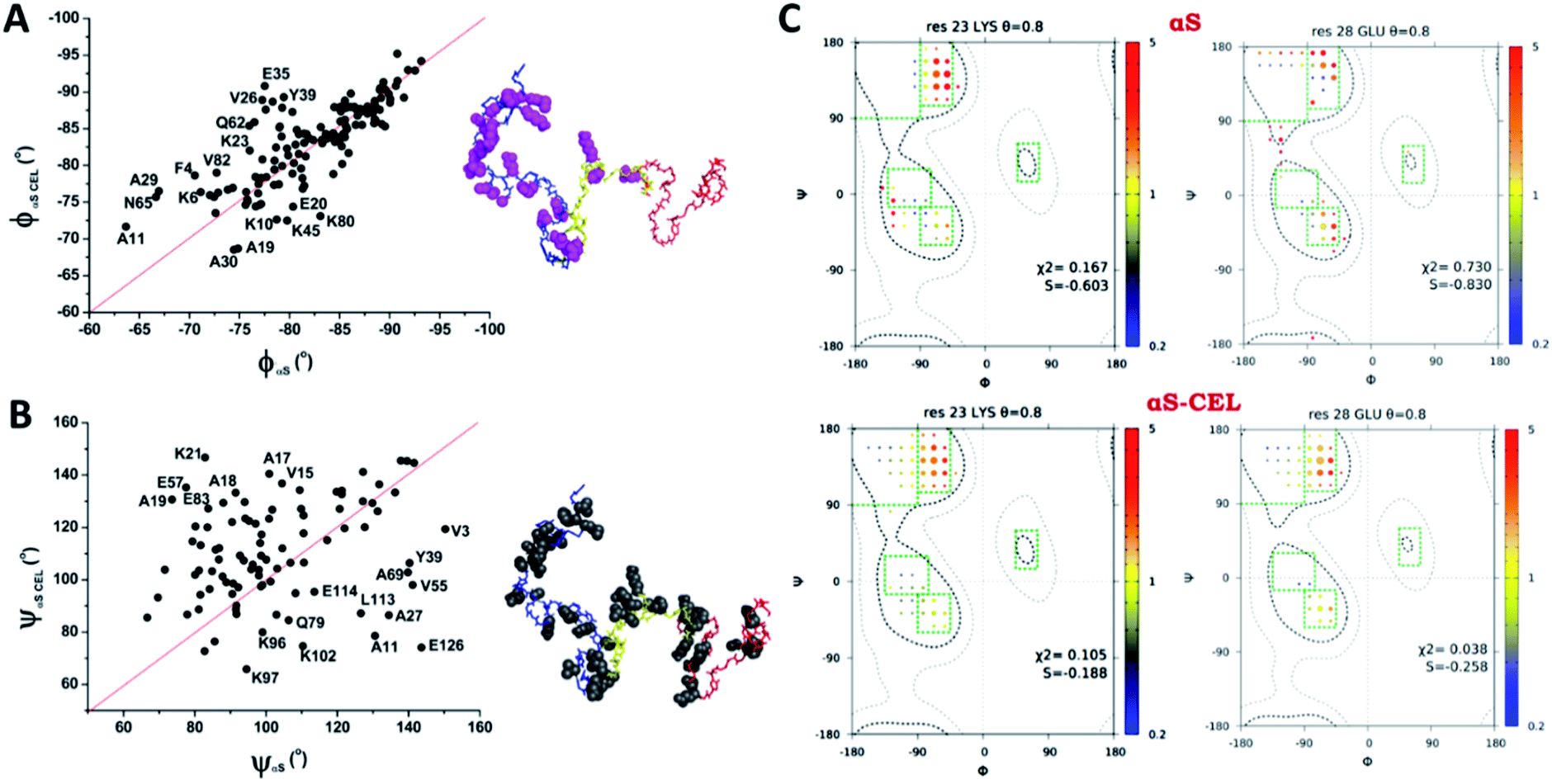 | ||
Fig. 5 Effect of CEL on the ϕ/ψ dihedral angles of αS. (A, B) Plots of the ϕ (A) or ψ (B) dihedral angles obtained for each residue of αS-CEL vs. the corresponding values obtained for αS (black dots). The red lines simulate the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 relationship between the dihedral angles of αS and αS-CEL. The experimental points corresponding to those residues with a high divergence between its ϕ/ψαS values and its ϕ/ψαS-CEL values have been labelled with its one letter amino acid code followed by its sequence number. In addition, these residues are highlighted [pink space fill (A) or grey space fill (B)] on the averaged structural ensemble obtained for native αS (PED9AAC)59 (right). The color code of each domain is given in the legend of Fig. 1A. (C) Examples of ϕ/ψ distributions derived from MERA calculations for K23 (left) and E28 (right) in αS (top) and αS-CEL (bottom). The surface area of each circle is proportional to the population of its 15° × 15° voxel, and its color represents the ratio relative to that of the population seen in the coil database for that residue type, from 0.2 (blue) to 5 (red). An entropy weight factor of 0.8 was used. Each Ramachandran plot includes its χ2 and S values. :1 relationship between the dihedral angles of αS and αS-CEL. The experimental points corresponding to those residues with a high divergence between its ϕ/ψαS values and its ϕ/ψαS-CEL values have been labelled with its one letter amino acid code followed by its sequence number. In addition, these residues are highlighted [pink space fill (A) or grey space fill (B)] on the averaged structural ensemble obtained for native αS (PED9AAC)59 (right). The color code of each domain is given in the legend of Fig. 1A. (C) Examples of ϕ/ψ distributions derived from MERA calculations for K23 (left) and E28 (right) in αS (top) and αS-CEL (bottom). The surface area of each circle is proportional to the population of its 15° × 15° voxel, and its color represents the ratio relative to that of the population seen in the coil database for that residue type, from 0.2 (blue) to 5 (red). An entropy weight factor of 0.8 was used. Each Ramachandran plot includes its χ2 and S values. | ||
Nonetheless, the estimation of ϕ/ψ dihedrals from J couplings only gave information about the weighted-average conformations of αS and αS-CEL. Consequently, we used several restraints (i.e.3JHNHα coupling constants; N, Cα, and CO chemical shifts; and dNN(i,i+1), dαN(i,i) and dαN(i,i+1) NOEs) and the MERA program to generate the Ramachandran map distribution for each residue. We obtained a wide distribution of the ϕ/ψ pairs for all residues, which is typical of disordered conformations.39b Yet, CEL formation expanded, even more, the number of conformational states around regions characteristic of β- and γ-turns, which occurred concomitant with a change in the voxel populations. However, this was mainly observed for the Lys located at the N-terminal domain and for their neighboring residues, as the Ramachandran map distributions for the residues at the NAC and the C-terminal domains were always nearly the same (Fig. 5C and S23†). Accordingly, CEL increased the conformational entropy of most of the N-terminal residues, which might occur as a consequence of the loss of transient N-terminal/C-terminal contacts.
Our data reveal that the average conformations of αS and αS-CEL mostly explore regions typical of β- and γ-turns. However, CEL expanded the conformational space explored by the N-terminal domain, although all the new populations were placed near to Ramachandran regions characteristic of turns.
The comparison of the αS and αS-CEL ensembles evidences a tiny but preferred CEL-induced structural rearrangement at the N-terminal domain
NMR chemical shifts (N, HN, Cα, Cβ, Hα and CO) were also used to generate ensembles characteristic of the average conformations of αS and αS-CEL. The initial random structures were grouped into clusters with cut-offs of 1.7, 1.8, 1.9 and 2.0 nm (Table S4†), and then used to generate weighted ensembles of αS and αS-CEL that best reproduced the chemical shifts (Tables S5–S7†). The weight of each structure in the final linear combination was used to calculate the weighted ensemble averages of the Rg and the ensemble RMSD of different sections of αS-CEL with respect to αS (Table 1). The αS-CEL ensembles calculated at each cutoff always displayed greater Rg values than those obtained for the ensembles of αS, which constitutes additional evidence (besides the DLS, SAXS, DOSY and the CG-MD simulation data) that CEL slightly enhances the radius of αS. In addition, the structural comparison between the ensembles of αS-CEL and αS proves that CEL-induced conformational perturbations are larger at the N-terminal domain than at the NAC and C-terminal domains (Table 1).CEL induces changes in the motional properties of the Lys-containing domains
To study whether CEL modifies the dynamic features of αS, we acquired the R2 values (sensitive to slow [ns] motions, and to very slow [μs to ms] conformational exchange processes) and performed HET-NOE (sensitive to fast motions [from ps to sub-ns]) backbone relaxation experiments. The averaged R2 and HET-NOE values are lower than those found in folded proteins of similar size (i.e. ∼10 s−1 and 0.8 respectively) (Fig. 6A, B), thus confirming that αS and αS-CEL are mainly unfolded. However, CEL slightly increased the R2 values of Lys, and in some cases, those of their neighboring residues (e.g. G31-G47 stretch) (Fig. 6A). This did not occur as a result of a CEL-induced conformational slow exchange process, as the ΔR2eff values determined for αS and αS-CEL using Carr-Purcell Meiboom-Gill relaxation dispersion (CPMG RD) experiments were almost negligible (Fig. S24†). This indicates that CEL-modified Lys has a greater propensity for slower motions than Lys in αS, which could be related to the increase in the CEL-induced intra-domain interactions suggested by the contact maps (Fig. 3C).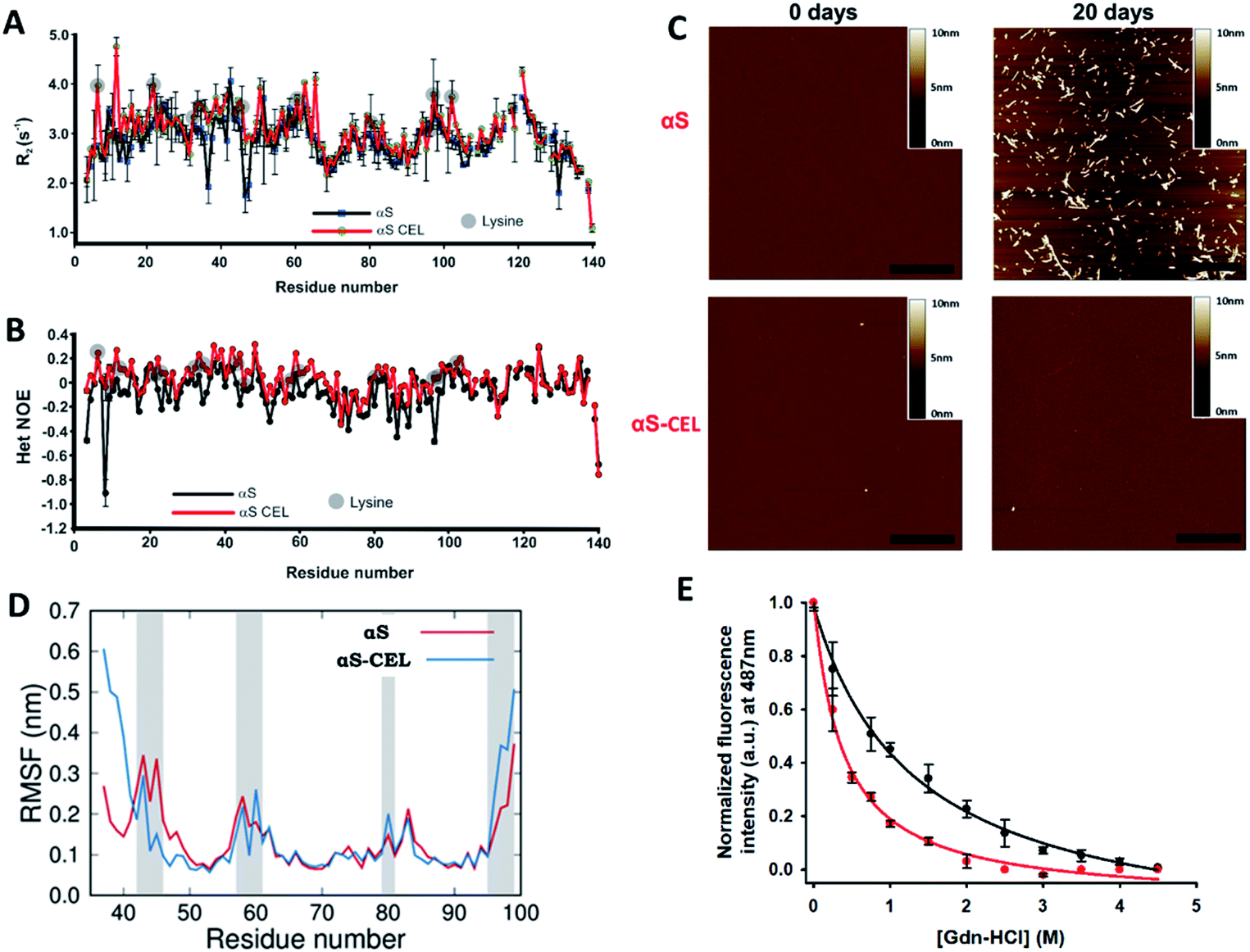 | ||
| Fig. 6 Effect of CEL formation on the dynamics and aggregation propensity of αS. (A, B) Plots of the R2 (s−1) (A) and HET NOE (B) relaxation data obtained for αS (black) and αS-CEL (red). The relaxation measurements were carried out at 12.5 °C in 20 mM phosphate buffer (pH 6.5) in the presence of 150 mM NaCl. Data corresponding to the different Lys residues are labelled with grey circles. (C) AFM micrographs of αS (top) and αS-CEL (bottom) solutions previously incubated for 0 (left) and 20 days (right) at pH 7.4 and at 37 °C in phosphate buffer in the presence of NaCl (150 mM), while shaking at 1000 rpm. The scale bar represents 0.5 μm. (D) Root mean square fluctuation (RMSF) per residue of αS and αS-CEL monomers when assembled in the cryo-EM structure of the αS amyloid fibrils (PDB code 6A6B).9 The RMSF values were computed for the monomers located at the ending extreme of the fibril by using MD simulations. Grey areas indicate the location of Lys/CEL side chains. (E) ThT fluorescence study (λexc 440 nm) of Gdn-HCl-assisted denaturation of full-length αS amyloid fibrils (black) and full-length αS amyloid fibrils previously modified with CEL (red). | ||
CEL also increased the HET-NOE values of the N-terminal and NAC domains (Fig. 6B), which additionally confirms a preference for slower over faster motions of these domains in αS-CEL. In contrast, it seems that the flexibility of the C-terminal domain was not affected by the formation of CEL. These results prove that CEL reduces the propensity of fast motions of those αS stretches containing Lys.
CEL formation inhibits the αS amyloid fibril formation
Glycation has been pointed out as an inducer of amyloid fibril formation.45 However, more recent data suggested the opposite, since AGEs seem to inhibit the amyloid formation through the stabilization of preformed oligomers.24,27d,46 To clarify this controversy we also studied the specific effect of CEL on the aggregation of αS.The incubation of αS involved the time-dependent formation of ThT-active aggregates (Fig. S25A, C†) through the typical profile of a nucleation-dependent pathway. These aggregates displayed a linear and unbranched morphology, typical of amyloid fibrils (Fig. 6C). In contrast, the incubation of αS-CEL under the same conditions did not induce the formation of ThT-active aggregates (Figs. S25B, C†). In fact, its AFM micrographs did not show any kind of aggregate bound to the mica surface (Fig. 6C). To further confirm this finding, we acquired the DLS autocorrelation functions for αS and αS-CEL (Fig. S26A, B†). At increasing incubation times, αS displayed higher correlations for longer delay times, thus indicating the formation of large aggregates. This did not occur for αS-CEL, as its correlation functions did not increase with the delay times. In fact, the average particle size of αS-CEL scarcely changed during incubation, whereas that of αS notably increased (Fig. S26C, D†). These results reveal that CEL completely inhibits the αS fibrillization and likely diminishes the oligomer formation process.
αS chelates a wide number of metal cations, and most of the resulting complexes display an enhanced fibrillation propensity.17b
Hence, we studied whether metals could induce fibril formation on αS-CEL, which possesses a higher chelating ability than αS.26 The presence of Fe3+, Al3+ or Cu2+ was not sufficient to induce the formation of ThT-active aggregates in αS-CEL (Fig. S27†), thus proving that CEL is able to inhibit αS fibrillation even when metal cations are present.
Our results conclusively prove that CEL completely inhibits the fibrillation of αS.
CEL formation is able to destabilize preformed αS fibrils
To better understand the mechanism through which CEL inhibits fibril formation, we carried out all-atom MD and SMD simulations on the native and the CEL-modified cryo-EM structure of αS fibrils (Fig. S28A†).9 We initially studied if CEL was able to destabilize the amyloid assembly. The RMSD value of the backbone of the αS monomer at the ending of the fibril (0.4 nm) was smaller than that obtained for the αS-CEL monomer (0.5 nm) (Fig. S28B†). Hence, αS-CEL relaxes to a more different conformation than αS, thus their incorporation into the fibril should be less favorable.In addition, the RMSF per residue of the two monomers at the end of the fibril evidenced larger fluctuations for αS-CEL than for αS. The mobility of the V37-S42 stretch was enhanced, since the protonated N-terminal amino group interacted with the carboxylate groups of CEL43 and CEL80. However, this might occur to a lesser extent on fibrils formed by the full-length protein. The large RMSF of the C-terminal region was caused by the repulsion of K96 and K97 with their counterparts in the monomer below, although the side chain of D98 of the adjacent monomers sees to form salt bridges that might retain the integrity of the fibril. The formation of CEL on K96 and K97 hinders their salt bridges with D98, and thus the mobility of the C-terminal region is increased (Fig. 6D).
The CEL-induced formation of new salt bridges between CEL43 and CEL45 in αS-CEL – which do not occur between K43 and K45 in αS – explains the decrease in their local mobility. In addition, the K45-H50-E57’ (of the opposite monomer) H-bond observed in the αS fibrils is maintained in the αS-CEL fibrils (Fig. S29A†).
CEL58 did not evidence larger mobility than K58, and thus the interaction between K58 and E61 is maintained in the αS-CEL fibrils (Fig. S29B†). CEL60 became more dynamic than K60 because it is able to exchange interactions with other CEL60 moieties, E62 and water molecules (Fig. 6D). Moreover, the interaction between K80 and E46 in the αS fibrils was still present when K80 was replaced by CEL80 (Fig. S29C†), and thus the mobility of K80 did not notably change (Fig. 6D).
The mechanical work performed in the SMD simulations to detach a monomer from the ending of the fibril is, on average, smaller for αS-CEL than for αS (Fig. S28C†). The application of the Jarzynski equation (eqn (8) in the ESI†) allowed us to estimate that the free energy cost to detach the αS-CEL monomers from the CEL-modified protofibril is ∼18 kcal mol−1 smaller than to detach αS monomers from the native protofibril.
We then studied if the synthesis of CEL on preformed amyloid fibrils could trigger their disassembly. The incubation of αS fibrils with NaBH3CN and pyruvic acid did not change their ThT fluorescence profile (Fig. S30†), thus indicating that CEL formation inhibits their formation but it might not be sufficient to induce their disaggregation. Accordingly, Gdn-HCl-assisted (de)polymerization studies further confirmed that CEL weakens the inter-monomer interactions tying the αS fibrils (as already suggested by MD and SMD simulations). In fact, the ThT fluorescence intensity decrease upon increasing the Gdn-HCl concentration was greater in the CEL-modified fibrils than in the native fibrils (Fig. 6E).
Our results indicate that the low tendency of αS-CEL to form amyloid fibrils might be due to the disruption of the monomer–monomer interaction at the N- and C-terminal domains of the protofilament. This, together with the estimated ΔG for the monomer detachment from the two types of fibrils, indicates that αS-CEL is less prone to form amyloid fibrils than αS.
Discussion
Since 1980, when Monnier and Cerami postulated the “glycation hypothesis of aging”, which linked non-enzymatic protein browning with aging47, most of the efforts in this field have been focused on understanding the molecular mechanism underlying protein glycation. In spite of that, the effect of glycation on the protein conformation is not yet understood because spontaneous glycation always results in the formation of a heterogeneous set of protein molecules with different glycation degrees, which hampered structural studies at the residue level.27 To date, most of the structural data arising from the use of low-resolution techniques (e.g. CD or fluorescence spectroscopy) might be easily misinterpreted as a result of concomitant aggregation or due to AGE-induced fluorescence quenching effects.27dTo overcome this drawback, we have synthesized a homogeneously glycated protein, which enables conformational studies at the residue level. Here, we have studied αS since LBs isolated from PD patients with DM display abnormally high levels of AGEs,21 which might explain the higher prevalence of PD in DM patients (∼38%).22 We have synthesized CEL, one of the main AGEs found in vivo on LBs,23,24 on all fifteen Lys residues of αS. The combined use of different biophysical techniques, together with different molecular dynamics approaches, has allowed us to describe, for the first time, the precise effect of a single AGE on the biophysical properties of an IDP.
CEL formation involved the replacement of fifteen positively charged Lys residues by fifteen zwitterionic CEL moieties (Fig. 1B), and thus the decrease in the protein pI and the increase in its negative electrostatic potential at the N-terminal and NAC domains.26 These changes did not induce a remarkable structuration on αS, differently from the increase of its β-sheet content detected when it was modified with 4-hydroxy-2-nonenal.48 However, the obtained data (i.e. ncSPC index in Fig. 1D; ΔδCO in Fig. 2D; SSP index, TALOS + data and CG-MD simulation data in Fig. S8†) unequivocally indicate that CEL slightly increases the transient α-helicity of the F4-L8 region while decreasing it in the E20-A29 stretch. These data, together with the Δ3JHNHα and Δ1JCαCβ values (Fig. 4A,B), also indicate a CEL-induced fluctuating extended character in most of the N-terminal stretches (e.g. S9-G25 or T54-E61), which is not related to the acquisition of any transient β-sheet or PPII conformation.
NMR was used to map those residues whose chemical environment was mostly affected by the formation of CEL, which might differ from those changing their transient structure. Residues located at the N-terminal domain were those displaying higher chemical shift perturbations (Fig. 2B,D), which agrees with the data reported by Miranda et al., who proved that glycation of αS mediated by MG essentially affected the N-terminal domain.24 Although CEL was also formed on K80, K96, K97 and K102 (Fig. 1A), we could neither detect a significant perturbation of their chemical shifts, nor those of their neighboring residues (Fig. 2D). This let us hypothesize that in an IDP, the sole formation of CEL on Lys would not be sufficient to change its chemical environment, but it could certainly be modified due to the CEL-induced disruption of the medium and long-range intramolecular interactions.
In fact, αS displays lasting long-range interactions between the N-terminal and the C-terminal domains,49 which are lost upon CEL formation (Fig. 3C, D). This would occur as a result of the loss of the salt-bridges tying the N-terminal positively charged Lys with the C-terminal negatively charged Asp/Glu, through a mechanism resembling the conformational process occurring on αS upon lowering the pH.50 This distancing was suggested not only by CG-MD simulations, but also by the 1JCαCβ values, which are dependent on the side chain topology,35 on the steric effects and on interactions with lone-electron pairs.40 In fact, the Δ1JCαCβ values of the N-terminal and C-terminal stretches decreased more than the average (Fig. 4B). Additionally, CEL increases the propensity of local interactions within the N-terminal domain because its zwitterionic character enhances its ability to stablish additional hydrogen bonds. This idea is also supported by the ΔSASA data (Fig. S17B†) and by NMR relaxation data. The CEL-induced increase in the R2 and HET-NOE relaxation values (which were similar to those reported for αS51) of the residues located at the N-terminal domain indicates their preference for slower over faster motions, which might arise from the CEL-induced intradomain interactions.
The breakage of these long-range interactions implies an enlargement of the averaged distances between the geometric centres of the N-terminal and the C-terminal domains, which results in an increase of the radius of the protein by ∼1 nm (Fig. 3A, B and S14†).28 The Rg values obtained for αS and αS-CEL are similar to those reported for αS,50,52 and differences must arise from its high dependence on the environment; in TRIS it is 4.27 nm, whereas in acetic acid buffer it is 2.72 nm.52 In addition, the breakage of the N-/C-terminal contacts induced a more heterogeneous size distribution, as proved by DLS (Fig. 3A) and CG-MD simulation (Fig. 3B) data. In fact, the greater heterogeneity in the HSQC peak intensities and peak shapes (Fig. S15†) indicates that αS-CEL displays a larger interconverting and transiently interacting heterogeneous ensemble of states than αS.
NMR spectroscopy also enabled us to obtain the J coupling constants, which arise from the hyperfine interaction between two different nuclei. While the 1JCαCβ couplings for αS have not yet been reported, the 3JHNHα values correlate with those already published (Pearson's r was 0.67; rmsd ∼0.50 Hz),39a although our experimental conditions were slightly different – we added 150 mM NaCl. CEL mainly increased the 3JHNHα values of the Lys neighboring residues within the N-terminal domain (in ∼0.6 ± 1.1 Hz), and these changes were larger than those arising from the conformational change induced by pressure.36a In any case, the 3JHNHα and 1JCαCβ couplings were used together with the Karplus equations to derive the ϕ/ψ angles. The obtained values suggest that the averaged ensembles of αS and αS-CEL explored the same conformational regions, which are those typically occupied by random coil peptides – mainly β- and γ-turns. However, CEL induced a remarkable decrease in the ϕ values of the N-terminal residues, and an increase in the ψ values along the entire N-terminal and NAC domains, as well as in the N-terminus of the C-terminal domain (Fig. S21†). Therefore, it seems that CEL induces a shift between the different populations of turns. The Ramachandran map distributions obtained for each residue proved that Lys located at the N-terminal domain and its neighboring residues expanded the number of conformational states around regions characteristic of β- and γ-turns. This proves that CEL increases the conformational entropy of most of the N-terminal residues, which might occur as a consequence of the loss of transient N-terminal/C-terminal contacts. Besides, it confirms that CEL enhances the heterogeneity of the conformational populations and proves that this change directly arises from the structural perturbation occurring at the N-terminal domain demonstrated by comparing the ensembles of αS with those of αS-CEL (Table 1).
After the detailed description of the effect of CEL on the conformation of αS, we wanted to further study whether CEL is able to influence its aggregation mechanism. Differently from what we observed when using αS, the incubation of αS-CEL did not involve the formation of amyloid fibrils or any other aggregate (e.g. soluble oligomers). The presence of metal cations, well known to induce αS aggregation,17b was also not sufficient to induce αS-CEL fibrillation. Hence, we proved that CEL completely inhibits αS aggregation. The inhibition of αS fibrillation due to its glycation was already reported when using MG24,54 or ribose.55 However, both were able to stabilize cytotoxic soluble oligomers, which proves that this must occur due to the formation of other AGEs different from CEL. These findings, together with our conformational studies comparing αS and αS-CEL, would reinforce the idea that the release of the long-range N-terminal/C-terminal contacts is not a triggering factor for aggregation,56 differently of what was thought before.49
To date, the scientific community had been still debating whether AGEs found on LBs are already formed on the αS monomer, before its aggregation, or on preexisting amyloid fibrils. αS fibrils are made of a left-handed helix composed of two protofilaments assembled from the V37-Q99 stretch. This region folds into a β-strand-rich architecture with a Greek key-like topology, facilitated by the formation of intramolecular K58-E61 and E46-K58 salt bridges. The dimer interface involves the interaction between antiparallel V37-Q99 stretches, which is stabilized by hydrophobic (e.g. A53-V55′) and electrostatic (e.g. E57-H50-K45) interactions9 (Fig. S28A and S29†). Our data prove that, at least in the case of CEL detected on LBs,23 its formation must occur on preformed LBs since αS-CEL monomers are not able to fibrillate. On the other hand, CEL formation on preexisting αS amyloid fibrils did not induce their disassembly (Fig. S30†), although it seems that it might weaken the driving forces intertwining consecutive monomers. This is supported by the CEL-induced increased dynamics of K60 and that of the N- and C-terminal regions of the amyloidogenic stretch. In addition, the decrease in the free energy cost to detach the monomers from the CEL-modified fibrils and their higher sensitivity against the chemically induced disassembly (Fig. 6E) also support this idea. Hence, the lack of a completely CEL-induced disassembly could occur because some of the Lys residues are buried in the inner part of the fibril (e.g. K58), thus being inaccessible to glycation.
Besides the effect of CEL on the conformation and aggregation propensity of αS, our data enable us to go further and argue about the possible effect of CEL on the biological function of αS. The N-terminal domain of αS57 and specially its Lys58 are essential to bind biological vesicles. Hence, the replacement of these cationic Lys residues by zwitterionic (e.g. CEL) or neutral AGEs must have a disrupting effect on αS-vesicle binding. In fact, MG impairs the α-helical folding of αS typically occurring in the presence of SUVs.24 Hence, it is likely that AGEs formed from MG (among them CEL) inhibit vesicle binding. However, further structural studies need to be performed to validate this hypothesis.
Conclusion
Here we prove that CEL formation on αS did not induce its folding process, but increased the population of transient conformations displaying a more extended N-terminal domain. This seems to result from the breakage of the long-range transient contacts between the N- and C-terminal domains, which induced a slight increase in the radius of the protein, and an increase in the population heterogeneity. In addition, CEL completely inhibited αS aggregation, but the destabilizing effect caused by its formation on preexisting αS fibrils is not sufficient to induce their disaggregation.Conflicts of interest
There are no conflicts to declare.Author contributions
L.M. produced αS; synthetized αS-CEL; performed SEC, electrophoretic, mass spectrometry, NMR, CD and fluorescence studies. In addition, she and M.A carried out the data analysis and interpretation. R.R carried out most of the CG-MD simulations. R.C. performed the MD and SMD simulations, and he also calculated the αS and αS-CEL ensembles. J.O.-C. carried out the DLS studies and analyzed the CG-MD simulation results. B.V. performed the AFM and ThT studies. J.F. and M.A. conceived and designed the experiments. M.A. and L.M. wrote the manuscript.Acknowledgements
The authors are grateful for the excellent technical assistance from the Serveis Cientificotècnics at the UIB, especially to Dr Gabriel Martorell for his generous help with NMR measurements and to Dr Rosa Gomila for her aid with the MALDI-TOF set-up and analysis. We thank Dr Kris Pauwels for helping in the acquisition of the CD data and analysis. L.M. wants to thank MINECO for the FPU PhD grant FPU14/01131. R.R. acknowledges his PhD scholarship granted by the FPU program (FPU16/00785). R.C. acknowledges a Margalida Comas-CAIB postdoctoral fellowship granted by the “Govern de les Illes Balears, Conselleria d'Innovació, Recerca i Turisme” (PD/11/2016). The authors are grateful to “Consorci de Serveis Universitaris de Catalunya (CSUC)”, the “Centro de Cálculo de Supercomputación de Galicia (CESGA)”, and the “Centre de Tecnologies de la Informació (CTI) de la UIB” for providing access to their computational facilities. This work was funded by the Spanish Government in the framework of the Project CTQ2014-55835-R.Notes and references
- G. M. Spillantini, M. L. Schmidt, V. M.-Y. Lee, J. Q. Trojanowski, R. Jakes and M. Goedert, Nature, 1997, 839–840 CrossRef PubMed.
- C. W. Shults, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1661–1668 CrossRef CAS PubMed.
- F.-X. Theillet, A. Binolfi, B. Bekei, A. Martorana, H. M. Rose, M. Stuiver, S. Verzini, D. Lorenz, M. van Rossum, D. Goldfarb and P. Selenko, Nature, 2016, 530, 45–50 CrossRef CAS PubMed.
- N. M. Bonini and B. I. Giasson, Cell, 2005, 123, 359–361 CrossRef CAS PubMed.
- D. Béraud, H. A. Hathaway, J. Trecki, S. Chasovskikh, D. A. Johnson, J. A. Johnson, H. J. Federoff, M. Shimoji, T. R. Mhyre and K. A. Maguire-Zeiss, Journal of Neuroimmune Pharmacology, 2013, 8, 94–117 CrossRef PubMed.
- F. N. Emamzadeh, J. Res. Med. Sci., 2016, 21, 29 CrossRef PubMed.
- M. S. Parihar, A. Parihar, M. Fujita, M. Hashimoto and P. Ghafourifar, Cell. Mol. Life Sci., 2008, 65, 1272–1284 CrossRef CAS PubMed.
- L. V. Kalia, S. K. Kalia, P. J. McLean, A. M. Lozano and A. E. Lang, Ann. Neurol., 2013, 73, 155–169 CrossRef CAS PubMed.
- Y. Li, C. Zhao, F. Luo, Z. Liu, X. Gui, Z. Luo, X. Zhang, D. Li, C. Liu and X. Li, Cell Res., 2018, 28, 897–903 CrossRef CAS PubMed.
- (a) E. Junn and M. M. Mouradian, Neurosci. Lett., 2002, 320, 146–150 CrossRef CAS; (b) D. Kirik, C. Rosenblad, C. Burger, C. Lundberg, T. E. Johansen, N. Muzyczka, R. J. Mandel and A. Björklund, J. Neurosci., 2002, 22, 2780–2791 CrossRef CAS.
- M.-C. Chartier-Harlin, J. Kachergus, C. Roumier, V. Mouroux, X. Douay, S. Lincoln, C. Levecque, L. Larvor, J. Andrieux, M. Hulihan, N. Waucquier, L. Defebvre, P. Amouyel, M. Farrer and A. Destée, Lancet, 2004, 364, 1167–1169 CrossRef CAS.
- (a) A. B. Singleton, M. Farrer, J. Johnson, A. Singleton, S. Hague, J. Kachergus, M. Hulihan, T. Peuralinna, A. Dutra, R. Nussbaum, S. Lincoln, A. Crawley, M. Hanson, D. Maraganore, C. Adler, M. R. Cookson, M. Muenter, M. Baptista, D. Miller, J. Blancato, J. Hardy and K. Gwinn-Hardy, Science, 2003, 302, 841 CrossRef CAS PubMed; (b) F. Zafar, R. A. Valappil, S. Kim, K. K. Johansen, A. L. S. Chang, J. W. Tetrud, P. S. Eis, E. Hatchwell, J. W. Langston, D. W. Dickson and B. Schüle, npj Parkinson's Dis., 2018, 4, 18 CrossRef PubMed.
- P. Flagmeier, G. Meisl, M. Vendruscolo, T. P. J. Knowles, C. M. Dobson, A. K. Buell and C. Galvagnion, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10328–10333 CrossRef CAS PubMed.
- E. Carboni and P. Lingor, Metallomics, 2015, 7, 395–404 RSC.
- (a) A. W. Schmid, B. Fauvet, M. Moniatte and H. A. Lashuel, Mol. Cell. Proteomics, 2013, 12, 3543–3558 CrossRef CAS PubMed; (b) H. Chen, Y.-F. Zhao, Y.-X. Chen and Y.-M. Li, ACS Chem. Neurosci., 2019, 10, 910–921 CrossRef CAS PubMed.
- (a) K. E. Paleologou, A. Oueslati, G. Shakked, C. C. Rospigliosi, H.-Y. Kim, G. R. Lamberto, C. O. Fernandez, A. Schmid, F. Chegini, W. P. Gai, D. Chiappe, M. Moniatte, B. L. Schneider, P. Aebischer, D. Eliezer, M. Zweckstetter, E. Masliah and H. A. Lashuel, J. Neurosci., 2010, 30, 3184–3198 CrossRef CAS PubMed; (b) A. Kleinknecht, B. Popova, D. F. Lázaro, R. Pinho, O. Valerius, T. F. Outeiro and G. H. Braus, PLoS Genet., 2016, 12, e1006098 CrossRef PubMed.
- (a) V. N. Uversky, G. Yamin, L. A. Munishkina, M. A. Karymov, I. S. Millett, S. Doniach, Y. L. Lyubchenko and A. L. Fink, Mol. Brain Res., 2005, 134, 84–102 CrossRef CAS PubMed; (b) V. N. Uversky, J. Li and A. L. Fink, J. Biol. Chem., 2001, 276, 44284–44296 CrossRef CAS PubMed.
- W. Li, N. West, E. Colla, O. Pletnikova, J. C. Troncoso, L. Marsh, T. M. Dawson, P. Jäkälä, T. Hartmann, D. L. Price and M. K. Lee, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2162–2167 CrossRef CAS PubMed.
- C. B. Glaser, G. Yamin, V. N. Uversky and A. L. Fink, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1703, 157–169 CrossRef CAS PubMed.
- P. M. Levine, A. Galesic, A. T. Balana, A.-L. Mahul-Mellier, M. X. Navarro, C. A. De Leon, H. A. Lashuel and M. R. Pratt, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1511–1519 CrossRef CAS PubMed.
- (a) A. Oueslati, M. Fournier and H. A. Lashuel, Prog. Brain Res., 2010, 183, 115–145 CAS; (b) R. Castellani, M. A. Smith, G. L. Richey and G. Perry, Brain Res., 1996, 737, 195–200 CrossRef CAS; (c) G. Münch, H.-J. Lüth, A. Wong, T. Arendt, E. Hirsch, R. Ravid and P. Riederer, J. Chem. Neuroanat., 2000, 20, 253–257 CrossRef.
- X. Yue, H. Li, H. Yan, P. Zhang, L. Chang and T. Li, Medicine, 2016, 95, e3549 CrossRef PubMed.
- Y. G. Choi and S. Lim, Biochimie, 2010, 92, 1379–1386 CrossRef CAS PubMed.
- H. Vicente Miranda, É. M. Szegő, L. M. A. Oliveira, C. Breda, E. Darendelioglu, R. M. de Oliveira, D. G. Ferreira, M. A. Gomes, R. Rott, M. Oliveira, F. Munari, F. J. Enguita, T. Simões, E. F. Rodrigues, M. Heinrich, I. C. Martins, I. Zamolo, O. Riess, C. Cordeiro, A. Ponces-Freire, H. A. Lashuel, N. C. Santos, L. V. Lopes, W. Xiang, T. M. Jovin, D. Penque, S. Engelender, M. Zweckstetter, J. Klucken, F. Giorgini, A. Quintas and T. F. Outeiro, Brain, 2017, 140, 1399–1419 CrossRef PubMed.
- I. Allaman, M. Bélanger and P. J. Magistretti, Front. Neurosci., 2015, 9, 23 Search PubMed.
- H. Martínez-Orozco, L. Mariño, A. B. Uceda, J. Ortega-Castro, B. Vilanova, J. Frau and M. Adrover, ACS Chem. Neurosci., 2019, 10, 2919–2930 CrossRef PubMed.
- (a) H. F. Bunn, R. Shapiro, M. McManus, L. Garrick, M. J. McDonald, P. M. Gallop and K. H. Gabbay, J. Biol. Chem., 1979, 254, 3892–3898 CAS; (b) R. A. Gomes, L. M. A. Oliveira, M. Silva, C. Ascenso, A. Quintas, G. Costa, A. V Coelho, M. Sousa Silva, A. E. N. Ferreira, A. Ponces Freire and C. Cordeiro, Biochem. J., 2008, 416, 317–326 CrossRef CAS PubMed; (c) Y. Zhang, E. P. Go and H. Desaire, Anal. Chem., 2008, 80, 3144–3158 CrossRef CAS PubMed; (d) M. Adrover, L. Marino, P. Sanchis, K. Pauwels, Y. Kraan, P. Lebrun, B. Vilanova, F. Munoz, K. Broersen and J. Donoso, Biomacromolecules, 2014, 15, 3449–3462 CrossRef CAS PubMed; (e) L. Mariño, C. A. Maya-Aguirre, K. Pauwels, B. Vilanova, J. Ortega-Castro, J. Frau, J. Donoso and M. Adrover, ACS Chem. Biol., 2017, 12, 1152–1162 CrossRef PubMed; (f) S. Leone, J. Fonderico, C. Melchiorre, A. Carpentieri and D. Picone, Mol. Cell. Biochem., 2019, 451, 165–171 CrossRef CAS PubMed.
- R. Ramis, J. Ortega-Castro, R. Casasnovas, L. Mariño, B. Vilanova, M. Adrover and J. Frau, J. Chem. Inf. Model., 2019, 59, 1458–1471 CrossRef CAS PubMed.
- K. Tamiola and F. A. A. Mulder, Biochem. Soc. Trans., 2012, 40, 1014–1020 CrossRef CAS PubMed.
- J. A. Marsh, V. K. Singh, Z. Jia and J. D. Forman-Kay, Protein Sci., 2006, 15, 2795–2804 CrossRef CAS PubMed.
- (a) Y. Shen, F. Delaglio, G. Cornilescu and A. Bax, J. Biomol. NMR, 2009, 44, 213 CrossRef CAS PubMed; (b) Y. Shen and A. Bax, J. Biomol. NMR, 2010, 46, 199–204 CrossRef CAS PubMed; (c) Y. Shen, J. Roche, A. Grishaev and A. Bax, Protein Sci., 2018, 27, 146–158 CrossRef CAS PubMed.
- J. Makowska, S. Rodziewicz-Motowidlo, K. Baginska, M. Makowski, J. A. Vila, A. Liwo, L. Chmurzynski and H. A. Scheraga, Biophys. J., 2007, 92, 2904–2917 CrossRef CAS PubMed.
- J. Yao, H. J. Dyson and P. E. Wright, FEBS Lett., 1997, 419, 285–289 CrossRef PubMed.
- T. R. Alderson, J. H. Lee, C. Charlier, J. Ying and A. Bax, ChemBioChem, 2018, 19, 37–42 CrossRef PubMed.
- J. M. Schmidt, M. J. Howard, M. Maestre-Martínez, C. S. Pérez and F. Löhr, Magn. Reson. Chem., 2009, 47, 16–30 CrossRef CAS PubMed.
- (a) J. Roche, J. Ying, A. S. Maltsev and A. Bax, ChemBioChem, 2013, 14, 1754–1761 CrossRef CAS PubMed; (b) R. Schweitzer-Stenner and S. E. Toal, Mol. BioSyst., 2016, 12, 3294–3306 RSC.
- G. W. Vuister and A. Bax, J. Am. Chem. Soc., 1993, 115, 7772–7777 CrossRef CAS.
- A. Pardi, M. Billeter and K. Wüthrich, J. Mol. Biol., 1984, 180, 741–751 CrossRef CAS PubMed.
- (a) A. B. Mantsyzov, A. S. Maltsev, J. Ying, Y. Shen, G. Hummer and A. Bax, Protein Sci., 2014, 23, 1275–1290 CrossRef CAS PubMed; (b) A. B. Mantsyzov, Y. Shen, J. H. Lee, G. Hummer and A. Bax, J. Biomol. NMR, 2015, 63, 85–95 CrossRef CAS PubMed.
- G. Cornilescu, A. Bax and D. A. Case, J. Am. Chem. Soc., 2000, 122, 2168–2171 CrossRef CAS.
- S. M. Gagné, S. Tsuda, M. X. Li, M. Chandra, L. B. Smillie and B. D. Sykes, Protein Sci., 1994, 3, 1961–1974 CrossRef PubMed.
- A. S. Maltsev, J. Ying and A. Bax, Biochemistry, 2012, 51, 5004–5013 CrossRef CAS PubMed.
- B. Uluca, T. Viennet, D. Petrović, H. Shaykhalishahi, F. Weirich, A. Gönülalan, B. Strodel, M. Etzkorn, W. Hoyer and H. Heise, Biophys. J., 2018, 114, 1614–1623 CrossRef CAS PubMed.
- (a) M. Shapovalov, S. Vucetic and R. L. Dunbrack, PLoS Comput. Biol., 2019, 15, e1006844 CrossRef PubMed; (b) A. G. de Brevern, Sci. Rep., 2016, 6, 33191 CrossRef CAS PubMed.
- (a) Y.-H. Hsu, Y.-W. Chen, M.-H. Wu and L.-H. Tu, Biophys. J., 2019, 116, 2304–2313 CrossRef CAS PubMed; (b) M. S. Khan, N. Rabbani, S. Tabrez, B. Ul Islam, A. Malik, A. Ahmed, M. A. Alsenaidy and A. M. Alsenaidy, Protein Pept. Lett., 2016, 23, 892–897 CrossRef CAS PubMed.
- A. Emendato, G. Milordini, E. Zacco, A. Sicorello, F. Dal Piaz, R. Guerrini, R. Thorogate, D. Picone and A. Pastore, J. Biol. Chem., 2018, 293, 13100–13111 CrossRef CAS PubMed.
- V. M. Monnier and A. Cerami, Science, 1981, 211, 491–493 CrossRef CAS PubMed.
- Z. Qin, D. Hu, S. Han, S. H. Reaney, D. a. Di Monte and A. L. Fink, J. Biol. Chem., 2007, 282, 5862–5870 CrossRef CAS PubMed.
- C. W. Bertoncini, Y.-S. Jung, C. O. Fernandez, W. Hoyer, C. Griesinger, T. M. Jovin and M. Zweckstetter, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1430–1435 CrossRef CAS PubMed.
- M.-K. Cho, G. Nodet, H.-Y. Kim, M. R. Jensen, P. Bernado, C. O. Fernandez, S. Becker, M. Blackledge and M. Zweckstetter, Protein Sci., 2009, 18, 1840–1846 CrossRef CAS PubMed.
- R. Bussell and D. Eliezer, J. Biol. Chem., 2001, 276, 45996–46003 CrossRef CAS PubMed.
- K. Araki, N. Yagi, R. Nakatani, H. Sekiguchi, M. So, H. Yagi, N. Ohta, Y. Nagai, Y. Goto and H. Mochizuki, Sci. Rep., 2016, 6, 30473 CrossRef CAS PubMed.
- D. Lee, C. W. Park, S. R. Paik and K. Y. Choi, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 421–430 CrossRef CAS PubMed.
- L. Chen, Y. Wei, X. Wang and R. He, PLoS One, 2010, 5, e9052 CrossRef PubMed.
- S. McClendon, C. C. Rospigliosi and D. Eliezer, Protein Sci., 2009, 18, 1531–1540 CrossRef CAS PubMed.
- T. Bartels, L. S. Ahlstrom, A. Leftin, F. Kamp, C. Haass, M. F. Brown and K. Beyer, Biophys. J., 2010, 99, 2116–2124 CrossRef CAS PubMed.
- Y. Zarbiv, D. Simhi-Haham, E. Israeli, S. A. Elhadi, J. Grigoletto and R. Sharon, Neurobiol. Dis., 2014, 70, 90–98 CrossRef CAS PubMed.
- J. R. Allison, P. Varnai, C. M. Dobson and M. Vendruscolo, J. Am. Chem. Soc., 2009, 131, 18314–18326 CrossRef CAS PubMed.
- M. P. Williamson, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 73, 1–16 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational methodology, supplementary figures, and supplementary tables. See DOI: 10.1039/d0sc00906g |
| This journal is © The Royal Society of Chemistry 2020 |