
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogen-directed chemical sialylation: pseudo-stereodivergent access to marine ganglioside epitopes†‡
Taiki
Hayashi
,
Alexander
Axer
,
Gerald
Kehr
,
Klaus
Bergander
and
Ryan
Gilmour
 *
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstrasse 40, Münster, Germany. E-mail: ryan.gilmour@uni-muenster.de
First published on 26th March 2020
Abstract
Sialic acids are conspicuous structural components of the complex gangliosides that regulate cellular processes. Their importance in molecular recognition manifests itself in drug design (e.g. Tamiflu®) and continues to stimulate the development of effective chemical sialylation strategies to complement chemoenzymatic technologies. Stereodivergent approaches that enable the α- or β-anomer to be generated at will are particularly powerful to attenuate hydrogen bond networks and interrogate function. Herein, we demonstrate that site-selective halogenation (F and Br) at C3 of the N-glycolyl units common to marine Neu2,6Glu epitopes enables pseudo-stereodivergent sialylation. α-Selective sialylation results from fluorination, whereas traceless bromine-guided sialylation generates the β-adduct. This concept is validated in the synthesis of HLG-1 and Hp-s1 analogues.
Gangliosides are pervasive in terrestrial and marine organisms where their structural heterogeneity manifests itself in the regulation of a broad spectrum of cellular processes.1 These intriguing natural products are conspicuous on account of their amphiphilic nature, where the simplicity of the lipid anchor contrasts sharply with the increasing stereochemical complexity of the carbohydrate epitope. Whilst the core of the epitope consists of common monosaccharides, the periphery is functionalised with sialic acids units that may vary in their N-substitution.2 The topology of the epitope encodes for a precise hydrogen bond network that underpins function,3 and thus the stereocontrolled construction of gangliosides is pivotal in delineating structure–function interplay across the glycosciences.4 The ubiquity and exterior position of sialic acid units in bioactive gangliosides therefore requires that efficient and selective (α or β) chemical sialylation strategies be devised to complement chemoenzymatic approaches.5
Gangliosides derived from marine echinoderms are particularly attractive targets on account of their complexity and importance in chemical neurology.6,7 Our interest in modulating non-neuronal glial cell behaviour with fluorinated carbohydrates8 led us to identify the Neu2,6Glu fragment as a promising target for neuropathy.9 This structural feature is common to a plenum of bioactive gangliosides including HLG-1, isolated from the sea cucumber Holothuria leucospilota,6 and Hp-s1, isolated from sea urchin Hemicentrotus pulcherrimus7a,b and Diadema setosum7c (Fig. 1, top).
 | ||
| Fig. 1 Top: Naturally occurring gangliosides HLG-1 and Hp-s1. Bottom: Regulating the stereochemical outcome of sialylation by halogen introduction at the C3 position. | ||
In developing a pseudo-stereodivergent platform to access the Neu2,6Glu epitope, the concept of halogen-dependent sialylation was investigated (Fig. 1, bottom). It was envisaged that halogen installation at C3 (ref. 10) would provide a steering group to direct the stereochemical course of sialylation (α or β) and offer the possibility to simultaneously modulate the physicochemical profile of the product. This strategy was motivated by the observation that fluorination of the GM4 epitope upregulates oligodendrocyte differentiation,8 and that this process of molecular editing can direct sialylation in the N-acetyl derivatives common to terrestrial gangliosides. As a working hypothesis, it was reasoned that sialylation with the fluorinated N-Gc derivative would proceed via a contact ion pair with the counterion distal from the Fδ− to minimize electrostatic repulsion.11,12 This strategy to access the α-anomer with the axial C(sp3)–F bond would also circumvent competing elimination that is often observed in the parent scaffolds. In contrast, C3 bromination would allow anchimeric assistance to be invoked to favour formation of the β-anomer.13 Moreover, the axial-bromo substituent would mitigate elimination but also provide the opportunity to devise a traceless protocol by virtue of a subsequent reduction step. To validate this pseudo-stereodivergent synthesis of the Neu2,6Glu unit, the fluorinated and brominated analogues of the HLG-1 epitope were prepared from the N–Ac glycal (Scheme 1). Mild and efficient conversion of the commercially available N–Ac derivative to the requisite to N-Gc (Gc = glycolyl) derivative was achieved through conversion of the amide to the carbamate as reported by Burk et al.14 and applied to sialic acid chemistry by Izumi et al.15 Initial Boc protection of glycal 1 (Boc2O, DMAP, in THF, reflux) followed by methanolysis of the acetyl group afforded N-Boc methyl ester 3 (24%) along with recovered carboxylic acid 2 (73%), which could be reprocessed to 3 by methylation with MeI and K2CO3. Exposure of 3 to a TMSCl–phenol combination16 effectively cleaved the N-Boc group to generate the free amine: this was processed further with treatment with benzyloxyacetyl chloride {(Bn)GcCl} in the presence of NEt3 to afford N-Gc glycal 4. Hydroxyfluorination of 4 was carried out with Selectfluor® in wet DMF. The diastereoisomers were easily separated by column chromatography to give the axial-F 5 (61%) and equatorial-F 6 (19%) donors. Installation of the C3 bromo group proved facile by exposing 4 to NBS in wet DMF. The diastereomers axial-Br 7 and equatorial-Br 8 were isolated in 58 and 32% yield, respectively. Given the lower levels of selectivity observed with equatorial-F donors in N–Ac systems,11d compound 6 was not investigated further and the remainder of the synthesis was completed with the axial derivative 5.
 | ||
| Scheme 1 Site selective halogenation of the sialyl donors. | ||
Prior to imidate formation, the anomeric configuration of the sialoside donors was defined based on the 3J(13C1–C2–C3–19Fax/eq) and/or 3J(13C1–C2–C3–1Hax/eq) coupling constant from the 13C NMR. Given the coupling constant (3JCF(H)) dependence on torsion angle, values of ca. 3.7 and ca. 5.5 Hz were expected for the α-configuration, reflecting the antiperiplanar arrangements of 13C1 and 19F or 1H, respectively, at C3. Small values around 1 Hz (perhaps not even resolved in the NMR spectrum) should result from the syn- and anti-clinal relationships of 13C and 19F/1H in both the α- and in the β-configuration.10a,c,17 To that end, selective decoupling experiments were carried out in order to determine the requisite coupling constants. Fig. 2 demonstrates two representative examples, one for compound 6 and the other for compound 8 thereby establishing the β-configuration indicated. In both cases the carbonyl C1(sp2)-atom of the ester substituent was investigated. Standard broadband 1H decoupled (CPD) 13C experiments directly revealed the 3JFC (1.1 Hz) coupling constant of compound 6. Determination of the respective 3JCH coupling constant (1.5 Hz) was achieved by simultaneous selective decoupling of 19F and the 1H resonance of the OMe group. For compound 8, the 3JCH coupling constant was not resolved by a selective 1H decoupling 13C NMR experiment, but its value could be estimated to be less than 1 Hz due to the line width. These data indicate that C3 halogenated donors favour the β-configuration, likely due to an enhanced anomeric effect.
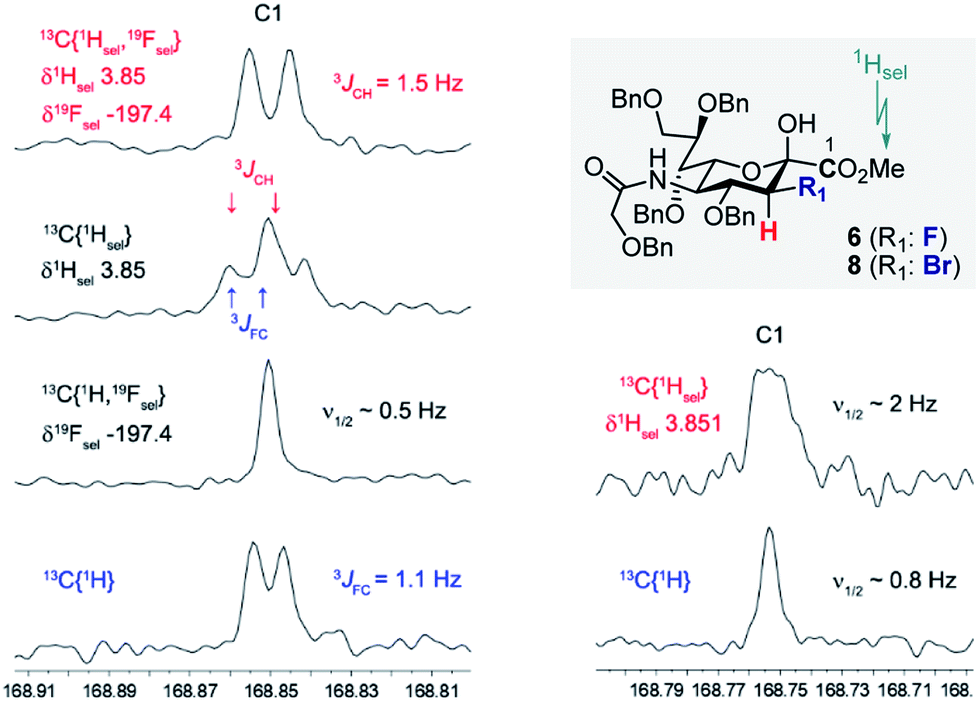 | ||
| Fig. 2 Right: Decoupling experiments carried out on compound 6 to determine 3JCH and 3JFC. Left: Decoupling experiments carried out on compound 8 to determine 3JCH. In both examples, the OMe group of the ester was selectively decoupled (1Hsel). | ||
Attempted generation of the imidate derivative of 7 (ax-Br) under standard conditions (2,2,2-trifloroacetimidoyl chloride, K2CO3, acetone, rt),18 proved to be problematic, resulting in the formation of epoxide 12 as the major product (Scheme 2). This is consistent with intramolecular cyclisation via the anomeric alkoxide generated by exposure to K2CO3 (A). However, through a process of optimisation it was possible to generate donor 9 exclusively (86%) and these conditions were extended to generate 10 (71%, from 8). In the case of the fluorinated substrate 5, imidate 11 was generated under standard conditions described above (93%). In all cases, β-anomers were generated as established by NMR analyses (see ESI‡).
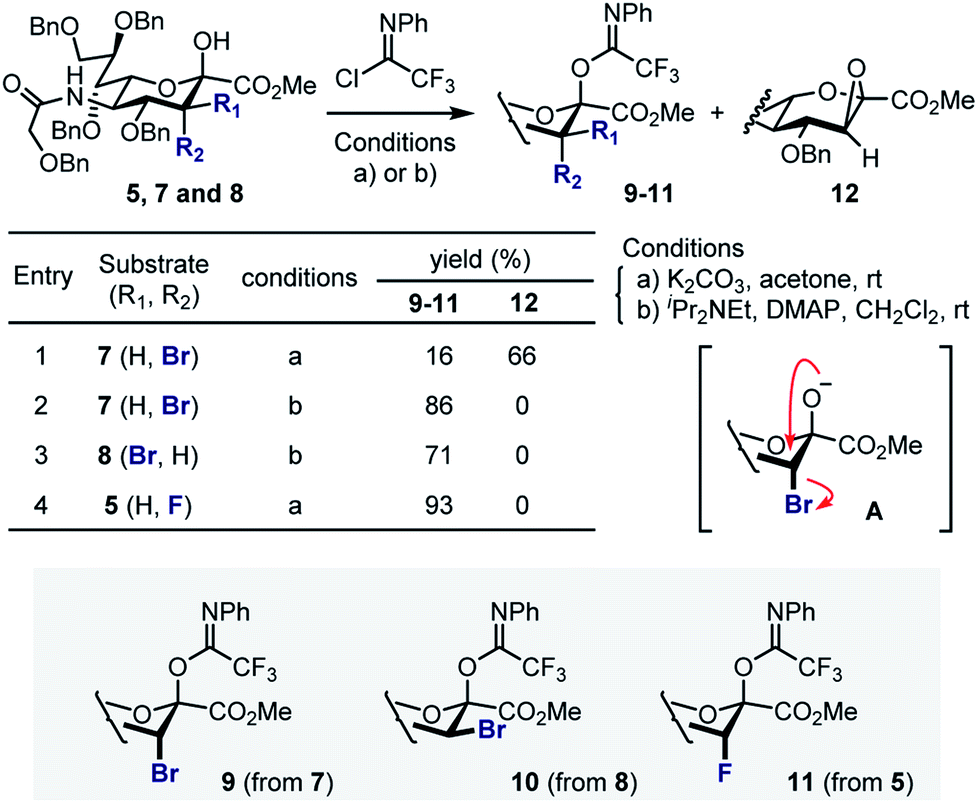 | ||
| Scheme 2 Preparation of the sialyl donors 9–11. | ||
With the halogenated donors 9–11 in hand, formation of the protected Neu2,6Glu fragment was attempted via coupling with 13, which was prepared from D-glucose (Scheme 3). Initially the fluorinated donor 11 and acceptor 13 were treated with TMSOTf in CH2Cl2 at 0 °C. Under these conditions, the sialylation reaction proved to be completely stereospecific, affording the disaccharide 14 61% exclusively as the α-anomer. The stereochemical course is consistent with the induction model described in Fig. 1.11d
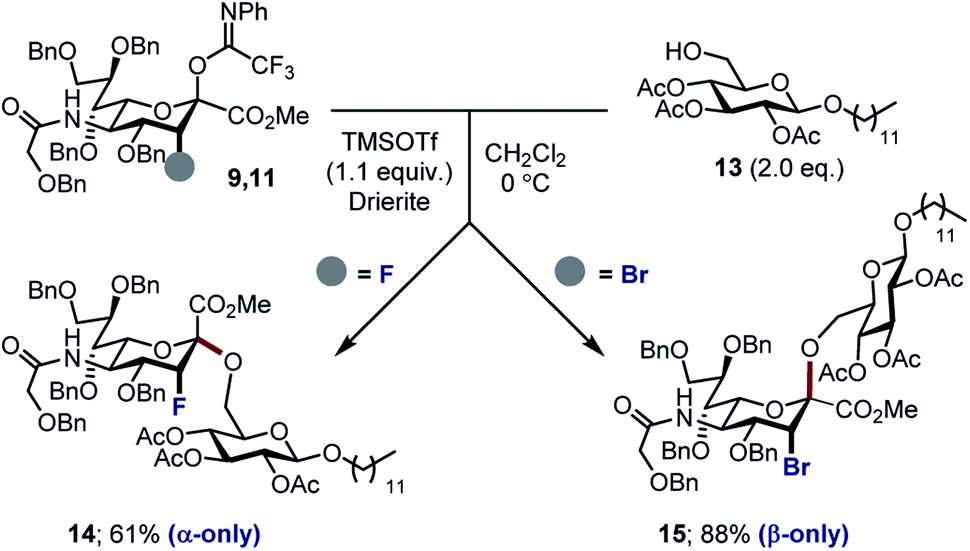 | ||
| Scheme 3 Sialylation of 3-Brax donor 9 and 3-Fax donor 11. | ||
By comparison, sialylation using the brominated donor 9 under identical conditions generated the β-anomer 15 (88%). This can be rationalized by invoking anchimeric assistance via the bromonium ion intermediate with concomitant ring opening. These data indicate that C3 halogen-directed pseudo-stereodivergence is efficient and stereospecific in steering chemical sialylation. As a control experiment, donor 10 was subjected to the sialylation conditions. This system, in which the C(sp3)–Br bond is pseudo-equatorial, proved to be challenging and gave an inseparable mixture of disaccharides. However, a subsequent radical reduction (Bu3SnH, AIBN, toluene, reflux) enabled the separation of both anomers. The sialylation is clearly sensitive to the configuration at C3, with an overall drop in selectivity and efficiency being observed. Nonetheless, it was possible to isolate the α-anomer 16 in 30% yield, and the β-anomer 17 in 4% (Scheme 4). The predominant generation of the α-anomer is also consistent with neighbouring group participation, but the pseudo-equatorial orientation no longer mitigates potential elimination following activation and this may be reflected in yield. The reduction does, however, validate the use of the C3 bromo substituent as a traceless directing group.
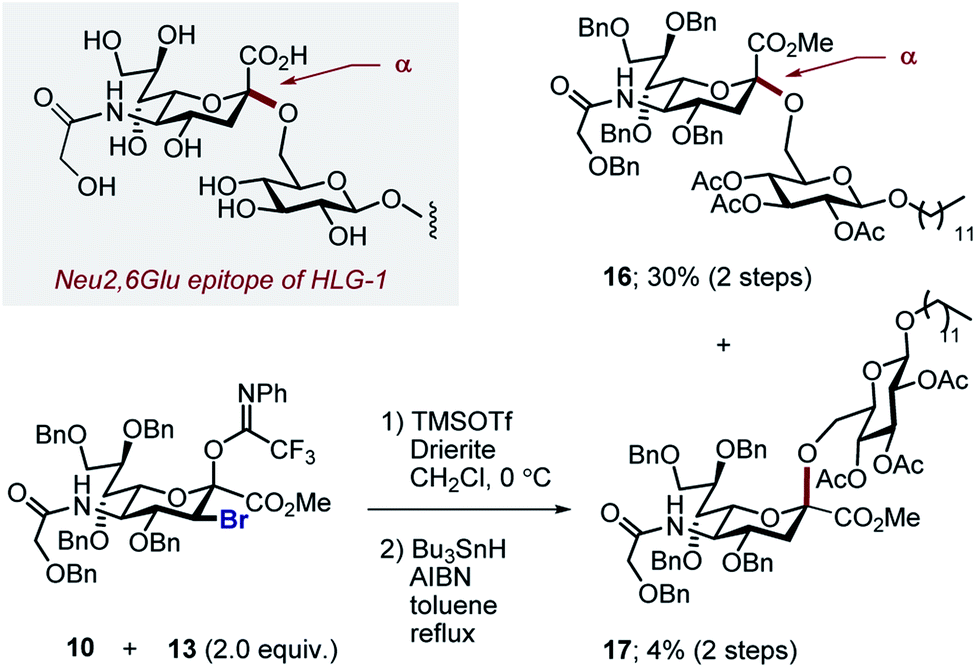 | ||
| Scheme 4 Comparative sialylation of 3-Breq donor 10. | ||
With this preliminary validation of halogen-dependent pseudo-stereodivergence, it was necessary to demonstrate (a) that the fluorinated epitope is compatible with standard deprotection conditions, and (b) that the selectivity observed represents a methodological advance over the non-fluorinated case.
To that end, the tolerance of 14 towards standard global deprotection conditions was investigated (Scheme 5). Gratifyingly, hydrogenolysis yielded the target disaccharide 18 (81% yield), and this could be further hydrolysed to 19 in 94% yield.
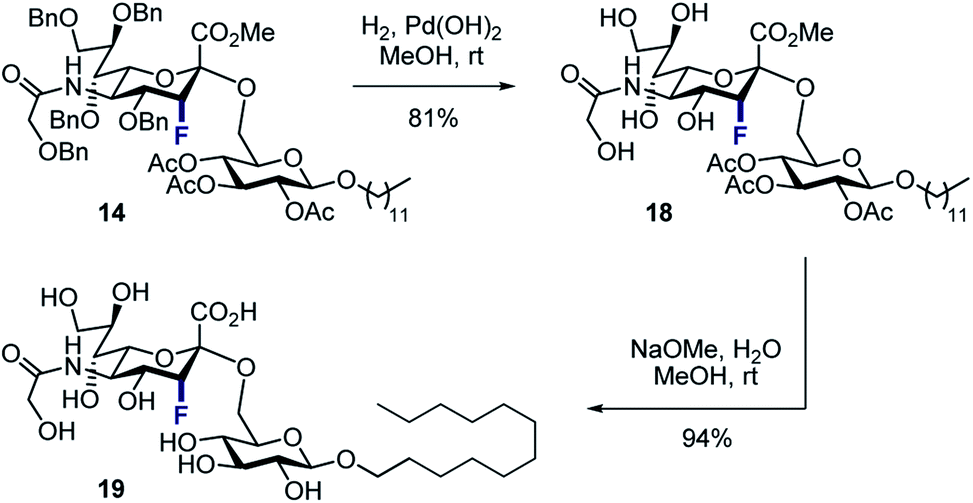 | ||
| Scheme 5 Deprotection of disaccharide 14 to generate 19. | ||
Finally, to ensure a direct comparison of the fluorinated versus non-fluorinated donors, the commonly employed phosphite donor 20 was employed for simplicity (Scheme 6). Moreover, performing this with the N–Ac derivatives allowed the fluorinated and non-fluorinated epitopes of Hp-s1 to be accessed. Glycosylation of N–Ac fluorinated phosphite donor 20 with 13, followed by hydrogenolysis of benzyl groups afforded disaccharide 21 exclusively as the α-anomer in 58% (2 steps). Subsequent hydrolysis liberated the target scaffold 22 in 85% yield.
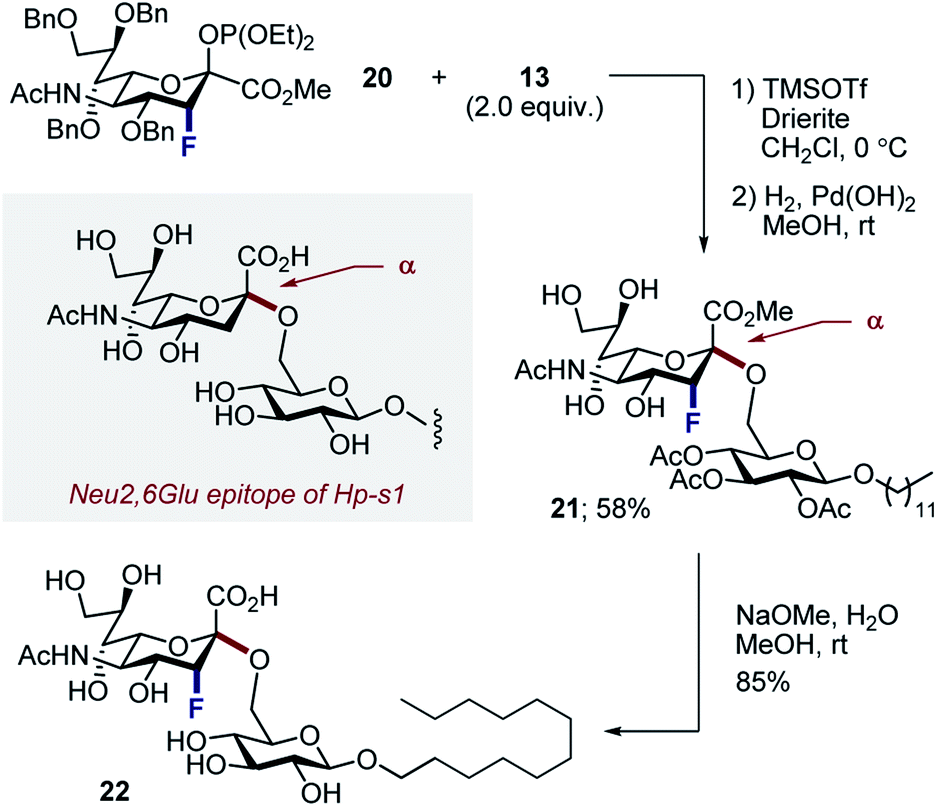 | ||
| Scheme 6 Synthesis of the fluorinated Hp-s1 analogue 22. | ||
For comparison, the non-fluorinated Hp-s1 was prepared by exposure of phosphite 23 and the benzyl-protected donor 24 to TMSOTf in a mixed solvent (MeCN, CH2Cl2, v/v = 2/1). The reaction proceeded non-selectively to give an approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of anomers (α-anomer 25, 25%; β-anomer 26, 21%. Scheme 7). For completeness, global deprotection by sequential hydrogenolysis/hydrolysis furnished the α- and β-anomers 27 and 28, respectively.
:1 mixture of anomers (α-anomer 25, 25%; β-anomer 26, 21%. Scheme 7). For completeness, global deprotection by sequential hydrogenolysis/hydrolysis furnished the α- and β-anomers 27 and 28, respectively.
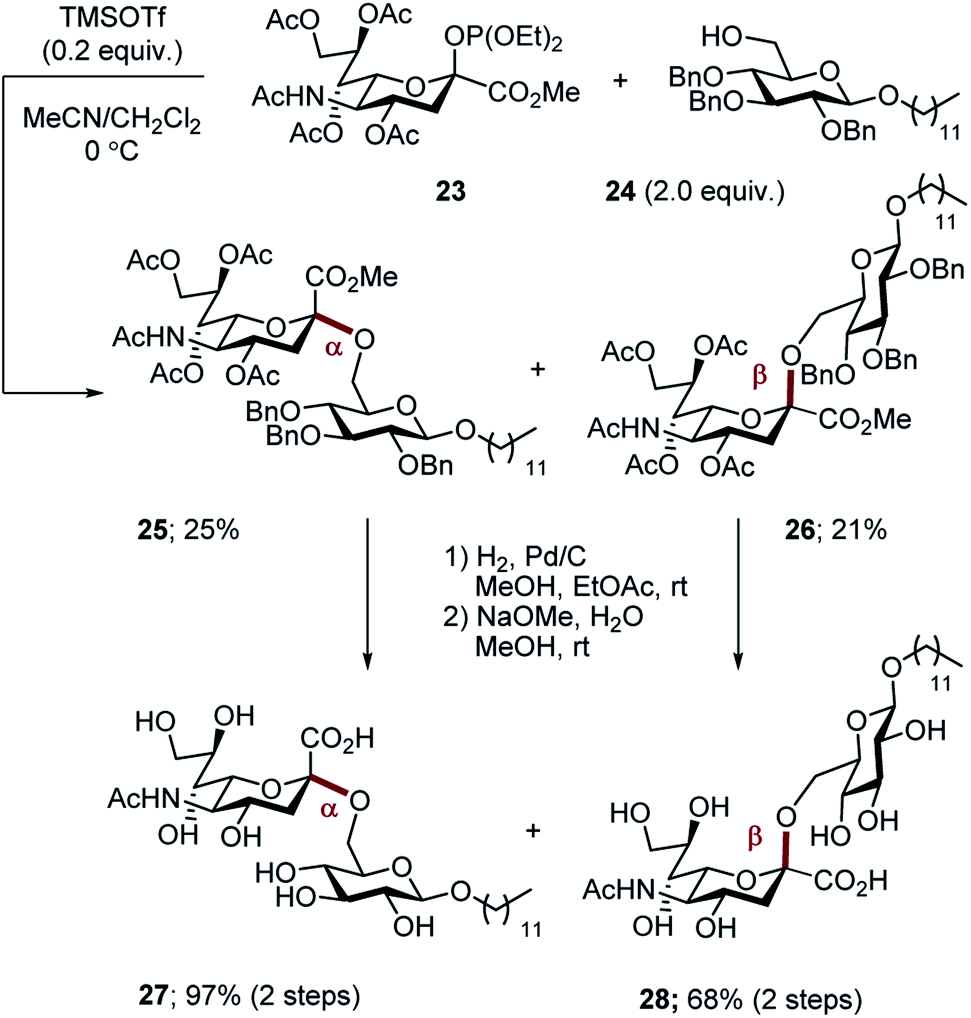 | ||
| Scheme 7 Synthesis of non-fluorinated Hp-s1 analogues 27 and 28. | ||
Conclusions
The concept of pseudo-stereodivergent sialylation is disclosed and applied to the synthesis of the Neu2,6Glu core of the marine gangliosides HLG-1, and Hp-s1. In the case of N-Gc sialic acid donors, the introduction of axial halogen substituents supresses competing elimination, and facilitates stereospecific sialylation (Scheme 8). It is postulated that in the case of the C-3 fluoro substituent, the close contact ion pair (CIP) is highly pre-organised with the triflate on the opposite face to mitigate electrostatic repulsion. Nucleophilic attack thus favours formation of the α-anomer in a stereospecific manner. Whilst an SN2-like process cannot be completely discounted, the steric demand of the D-glucose-derived acceptor renders it unlikely. In contrast, the C-3 bromo substituent can generate a bromonium ion intermediate upon imidate activation. The stereoelectronic requirements for productive bond formation ensure that the β-anomer is generated. Facile reduction of the C(sp3)–Br bond renders this process traceless, whereas the fluorine substituent provides a useful NMR probe for structural analyses.19 Exploring the biological effects on these halogenated epitopes on non-neuronal glial cell behaviour will be the subject of future research endeavours in our laboratory.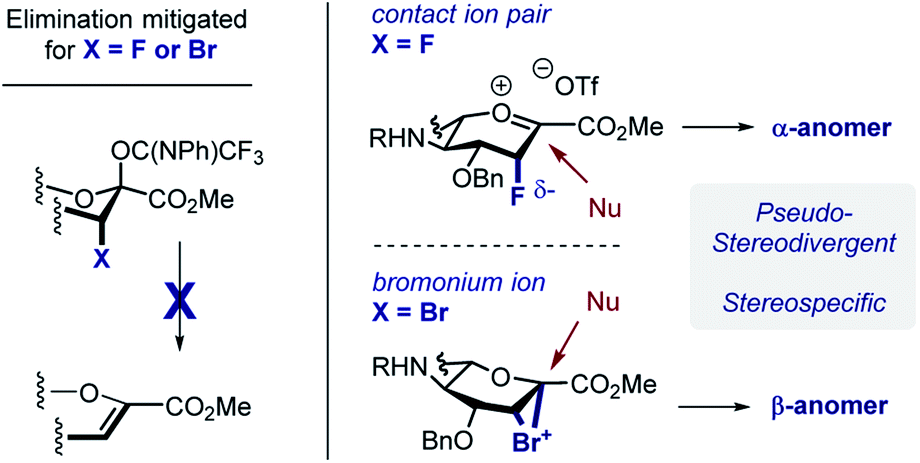 | ||
| Scheme 8 Pseudo-stereodivergent sialylation. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge generous financial support from the WWU Münster.Notes and references
- (a) A. Varki, Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 3rd edn, 2017 Search PubMed; (b) D. B. Werz, R. Ranzinger, S. Herget, A. Adibekian, C.-W. Von der Lieth and P. H. Seeberger, ACS Chem. Biol., 2007, 2, 685 CrossRef PubMed; (c) A. Adibekian, P. Stallforth, M.-L. Hecht, D. B. Werz, P. Gagneux and P. H. Seeberger, Chem. Sci., 2011, 2, 337 RSC; (d) I. Cheng-Sánchez and F. Sarabia, Mar. Drugs, 2018, 16, 294 CrossRef PubMed.
- (a) T. Kolter, ISRN Biochem., 2012, 1 Search PubMed; (b) L. Wang, Y. Liu, L. Wu and X.-L. Sun, Biochim. Biophys. Acta, 2016, 1864, 143 CrossRef PubMed.
- Y. Yu, T. Tyrikos-Ergas, Y. Zhu, G. Fittolani, V. Bordoni, A. Singhal, R. J. Fair, A. Grafmüller, P. H. Seeberger and M. Delbianco, Angew. Chem., Int. Ed., 2019, 58, 13127 CrossRef CAS PubMed.
- (a) J. Finkelstein, Nature Insight: Glycochemistry and Glycobiology, 2007, vol. 446, p. 999 Search PubMed; (b) N. S. Sampson, M. Mrksich and C. R. Bertozzi, Proc. Natl. Acad. Sci. U.S.A, 2001, 98, 12870 CrossRef CAS PubMed; (c) C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357 CrossRef CAS PubMed; (d) P. H. Seeberger, Nat. Chem. Biol., 2009, 5, 368 CrossRef CAS.
- (a) Selective Glycosylation: Synthetic Methods and Catalysts, ed. C. S. Bennet, Wiley-VCH Verlag GmbH & Co. KGaA, 2017, pp. 155–172 Search PubMed; (b) G.-J. Boons and A. V. Demchenko, Chem. Rev., 2000, 100, 4539 CrossRef CAS.
- (a) K. Yamada, R. Matsubara, M. Kaneko, T. Miyamoto and R. Higuchi, Chem. Pharm. Bull., 2001, 49, 447 CrossRef CAS; (b) M. Kaneko, K. Yamada, T. Miyamoto, M. Inagaki and R. Higuchi, Chem. Pharm. Bull., 2007, 55, 462 CrossRef CAS.
- (a) T. Ijuin, K. Kitajima, Y. Song, S. Kitazume, S. Inoue, S. M. Haslam, H. R. Morris, A. Dell and Y. Inoue, Glycoconjugate J., 1996, 13, 401 CrossRef CAS; (b) K. Yamada, K. Tanabe, T. Miyamoto, T. Kusumoto, M. Inagaki and R. Higuchi, Chem. Pharm. Bull., 2008, 56, 734 CrossRef; (c) J.-T. Hung, C.-H. Yeh, S.-A. Yang, C.-Y. Lin, H. J. Tai, G. B. Shelke, D. M. Reddy, A. L. Yu and S.-Y. Luo, ACS Chem. Neurosci., 2016, 7, 1107 CrossRef PubMed.
- T. J. Kieser, N. Santschi, L. Nowack, G. Kehr, T. Kuhlmann, S. Albrecht and R. Gilmour, ACS Chem. Neurosci., 2018, 9, 1159 CrossRef PubMed.
- R. Higuchi, M. Inagaki, K. Yamada and T. Miyamoto, J. Nat. Med., 2007, 61, 367 CrossRef.
- (a) T. Nakajima, H. Hori, H. Ohrui, H. Meguro and T. Ido, Agric. Biol. Chem., 1988, 52, 1209 Search PubMed; (b) M. D. Burkart, Z. Zhang, S.-C. Hung and C.-H. Wong, J. Am. Chem. Soc., 1997, 119, 11743 CrossRef CAS; (c) K. Suzuki, S. Daikoku, S.-H. Son, Y. Ito and O. Kanie, Carbohydr. Res., 2015, 406, 1 CrossRef CAS; (d) H. A. Chokhawala, H. Cao, H. Yu and X. Chen, J. Am. Chem. Soc., 2007, 129, 10630 CrossRef CAS PubMed; (e) Z. Gao, M. Niikura and S. G. Withers, Angew. Chem., Int. Ed., 2017, 56, 6112–6116 CrossRef CAS PubMed.
- (a) C. Bucher and R. Gilmour, Angew. Chem., Int. Ed., 2010, 49, 8724 CrossRef CAS PubMed; (b) E. Durantie, C. Bucher and R. Gilmour, Chem.–Eur. J., 2012, 18, 8208 CrossRef CAS PubMed; (c) N. Santschi and R. Gilmour, Eur. J. Org. Chem., 2015, 32, 6983 CrossRef; (d) T. Hayashi, G. Kehr, K. Bergander and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 3814 CrossRef CAS PubMed; (e) H.-J. Lo, L. Krasnova, S. Dey, T. Cheng, H. Liu, T.-I. Tsai, K. B. Wu, C.-Y. Wu and C.-H. Wong, J. Am. Chem. Soc., 2019, 141, 6484 CrossRef CAS PubMed.
- (a) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (c) L. E. Zimmer, C. Sparr and R. Gilmour, Angew. Chem., Int. Ed., 2011, 50, 11860 CrossRef CAS.
- (a) J. C. Castro-Palomino, Y. E. Tsvetkov and R. R. Schmidt, J. Am. Chem. Soc., 1998, 120, 5434 CrossRef CAS; (b) K. Okamoto and T. Goto, Tetrahedron, 1990, 46, 5835 CrossRef CAS.
- M. J. Burk and J. G. Allen, J. Org. Chem., 1997, 62, 7054 CrossRef CAS.
- M. Izumi, G.-J. Shen, S. Wacowich-Sgarbi, T. Nakatani, O. Plettenburg and C.-H. Wong, J. Am. Chem. Soc., 2001, 123, 10909 CrossRef CAS.
- E. Kaiser, J. P. Tam, T. M. Kubiak and R. B. Merrifield, Tetrahedron Lett., 1988, 29, 303 CrossRef CAS.
- (a) K. Bock and C. Pedersen, Acta Chem. Scand., Ser. B, 1975, 29, 682 CrossRef; (b) H. Hori, T. Nakajima, Y. Nishida, H. Ohrui and H. Meguro, Tetrahedron Lett., 1988, 29, 6317 CrossRef CAS.
- S. Cai and B. Yu, Org. Lett., 2003, 5, 3827 CrossRef CAS.
- (a) H. Chen, S. Viel, F. Ziarelli and L. Peng, Chem. Soc. Rev., 2013, 42, 7971 RSC; (b) C. P. Rosenau, B. J. Jelier, A. D. Gossert and A. Togni, Angew. Chem., Int. Ed., 2018, 57, 9528 CrossRef CAS.
Footnotes |
| † This manuscript is dedicated to Prof. Dr Jack David Dunitz FRS “the Professor's Professor” on the occasion of his 97th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01219j |
| This journal is © The Royal Society of Chemistry 2020 |