
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of secondary and tertiary amides without coupling agents from amines and potassium acyltrifluoroborates (KATs)†
Anne
Schuhmacher
a,
Tomoya
Shiro‡
a,
Sarah J.
Ryan
b and
Jeffrey W.
Bode
 *a
*a
aLaboratory of Organic Chemistry, Eidgenössische Technische Hochschule (ETH) Zurich, Vladimir-Prelog-Weg 3, 8093 Zurich, Switzerland. E-mail: bode@org.chem.ethz.ch
bSmall Molecule Design and Development, Eli Lilly and Company Indianapolis, IN 46285, USA
First published on 10th March 2020
Abstract
Although highly effective for most amide syntheses, the activation of carboxylic acids requires the use of problematic coupling reagents and is often poorly suited for challenging cases such as N-methyl amino acids. As an alternative to both secondary and tertiary amides, we report their convenient synthesis by the rapid oxidation of trifluoroborate iminiums (TIMs). TIMs are easily prepared by acid-promoted condensation of potassium acyltrifluoroborates (KATs) and amines and are cleanly and rapidly oxidized to amides with hydrogen peroxide. The overall transformation can be conducted either as a one-pot procedure or via isolation of the TIM. The unique nature of the neutral, zwitterionic TIMs makes possible the preparation of tertiary amides via an iminium species that would not be accessible from other carbonyl derivatives and can be conducted in the presence of unprotected functional groups including acids, alcohols and thioethers. In preliminary studies, this approach was applied to the late-stage modifications of long peptides and the iterative synthesis of short, N-methylated peptides without the need for coupling agents.
Introduction
Amide bonds are the key connections between amino acids in peptides and are present in many marketed drug molecules.1 The synthesis of amides is typically achieved by the reaction of an activated carboxylic acid with an amine,2 a process that employs stoichiometric amounts of coupling reagents and environmentally concerning solvents, e.g. N,N-dimethylformamide or dichloromethane (Fig. 1a).3 While recycling of solvents on large scale can reduce the environmental impact of peptide synthesis, the use and handling of coupling agents results in acute safety hazards due to their explosive and allergenic nature.4 Current approaches are therefore suitable for making most amide-based structures, but sustainable approaches to peptides and other amides requires new reactions that do not rely on these problematic reagents.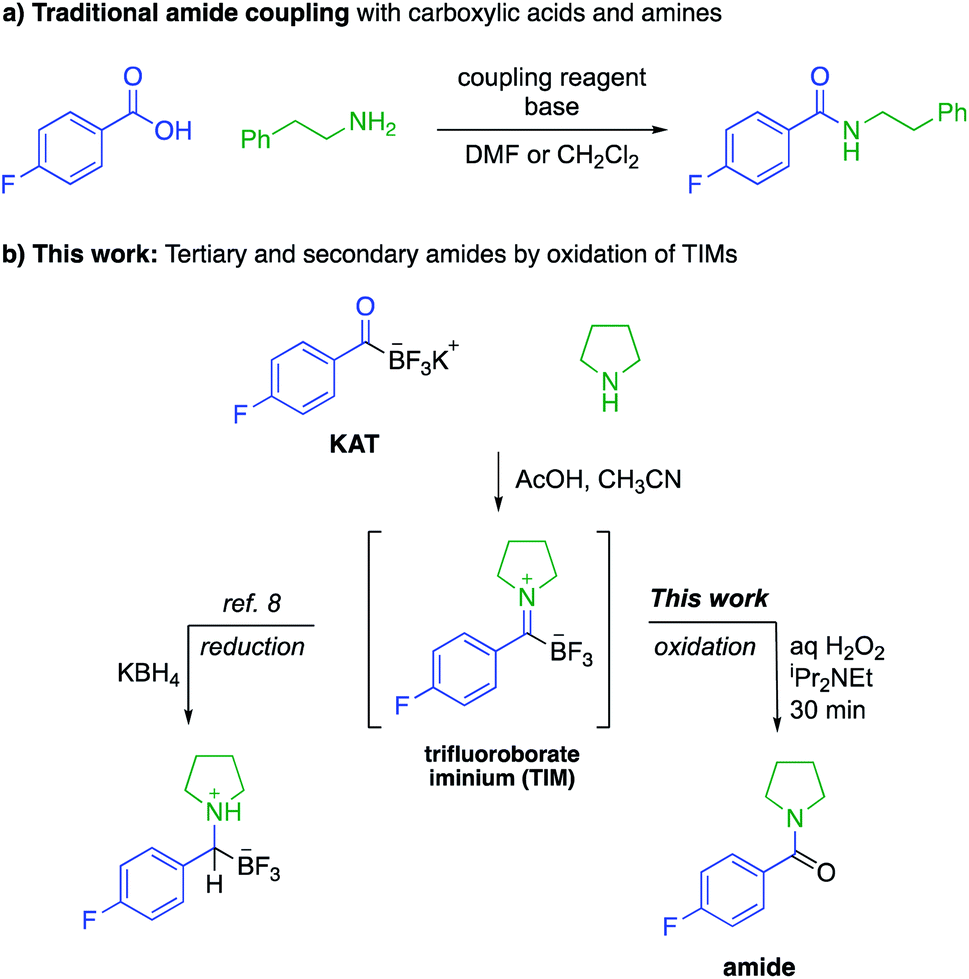 | ||
| Fig. 1 Amide-forming reaction with potassium acyltrifluoroborates (KATs) and amines via trifluoroborate iminiums (TIMs). (a) Traditional amide coupling methods using coupling reagents. (b) Formation of TIMs and oxidative conversion to amides. | ||
As part of a program aimed at new amide-forming methods, our group has identified potassium acyltrifluoroborates (KATs) as functional groups that form amide bonds with azides,5 hydroxylamines6 or in situ generated N-chloroamines.7 These methods, however, are mechanistically limited to the synthesis of secondary amides; the preparation of tertiary amides is not possible. In this report we document the oxidative amidation of KATs to form tertiary and secondary amides from amines via trifluoroborate iminiums (TIMs) (Fig. 1b). The TIMs are formed under simple conditions by mixing KATs and amine salts and their oxidation occurs rapidly and chemoselectively.8 Although KATs are far less common starting materials than carboxylic acids, they are increasingly available from commercial sources or a plethora of recently reported methods for conversion of common starting materials to KATs. In the past few years, their syntheses from aryl halides,9 Grignard reagents,10 aldehydes,11 vinyl boronic acids,12 alkynes,13 and acid chlorides14 have been reported. The important class of amino acid-derived KATs is expanding and has been shown to be configurationally stable.9b,11b Anticipating that these amino acid derivatives will become widely available in the near future, we sought to identify protocols for peptide synthesis under conditions where no activating agents are required.
Results and discussion
Amines and KATs condense to form TIMs – chromatographically stable, zwitterionic species that can be reduced with hydride sources or alkylated with organometallic reagents to give α-aminoboronates (Fig. 1).8 Given their ease of formation, we hypothesized that TIMs could also serve as intermediates to secondary and tertiary amides via oxidation. Using TIM 1a as a model substrate, we screened oxidation conditions and found successful formation of the desired tertiary amide with several oxidants and basic additives under a variety of conditions (detailed screening data can be found in the ESI†). As TIMs can hydrolyze to give the corresponding KATs under basic aqueous conditions, we were pleased to find that the oxidation is fast and occurs without competing hydrolysis of the TIM.We selected aqueous H2O2 and N,N-diisopropylethylamine in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3CN/H2O for 30 min as the standard conditions for further experiments. This method allows for the fast preparation of secondary and tertiary amides, including those derived from aliphatic or aromatic KATs with different primary and secondary amines. All underwent smooth oxidation to give the amide in good yields (Scheme 1). Unprotected functional groups including alcohols, indoles, and alkynes are not affected by the oxidation step. The reaction between a KAT and cysteine yielded the S,N-acetal 1b instead of a TIM. We found that the oxidation of 1b is possible under slightly modified conditions using 2 equiv. of hydrogen peroxide to achieve full conversion and adding TCEP before work-up to reduce the disulfide formed during the reaction.
:1 CH3CN/H2O for 30 min as the standard conditions for further experiments. This method allows for the fast preparation of secondary and tertiary amides, including those derived from aliphatic or aromatic KATs with different primary and secondary amines. All underwent smooth oxidation to give the amide in good yields (Scheme 1). Unprotected functional groups including alcohols, indoles, and alkynes are not affected by the oxidation step. The reaction between a KAT and cysteine yielded the S,N-acetal 1b instead of a TIM. We found that the oxidation of 1b is possible under slightly modified conditions using 2 equiv. of hydrogen peroxide to achieve full conversion and adding TCEP before work-up to reduce the disulfide formed during the reaction.
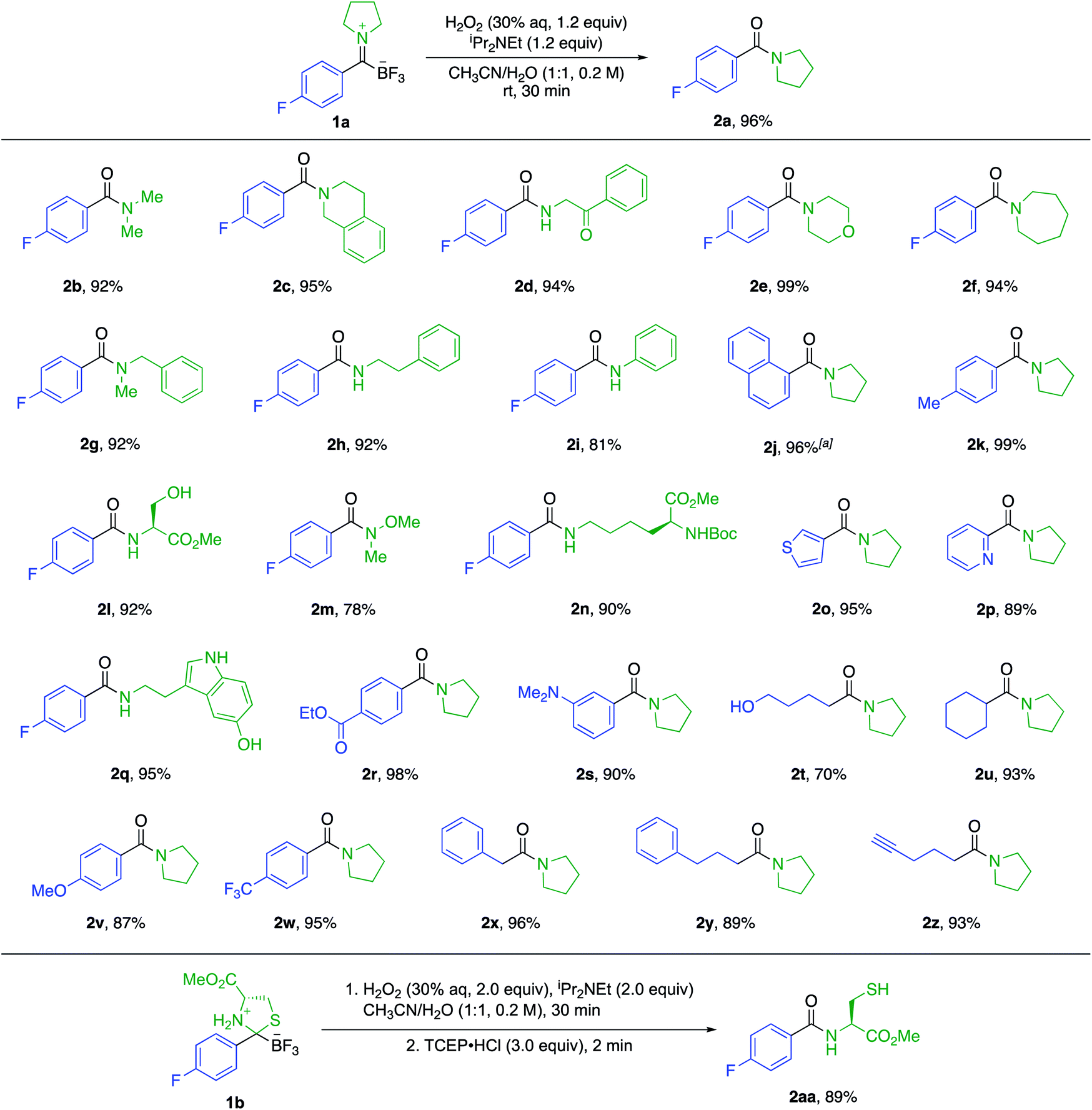 | ||
| Scheme 1 Substrate scope for the amide formation from trifluoroborate iminiums (TIMs). The yields are isolated yields. a Reaction time of 2 h. | ||
We also established a protocol for the one-pot preparation of amides from KATs and amines by TIM formation and subsequent oxidation (Scheme 2). This one-pot procedure offers both operational simplicity and slightly higher overall yields by obviating the isolation of the TIM intermediate. Since KATs are commercially available or easily prepared by several routes from commercial starting materials, this one step protocol offers a practical alternative to the formation of many simple amides. For substrate 2a, we demonstrated the applicability of the reaction on a larger scale (3.0 mmol) without a significant decrease in yield.
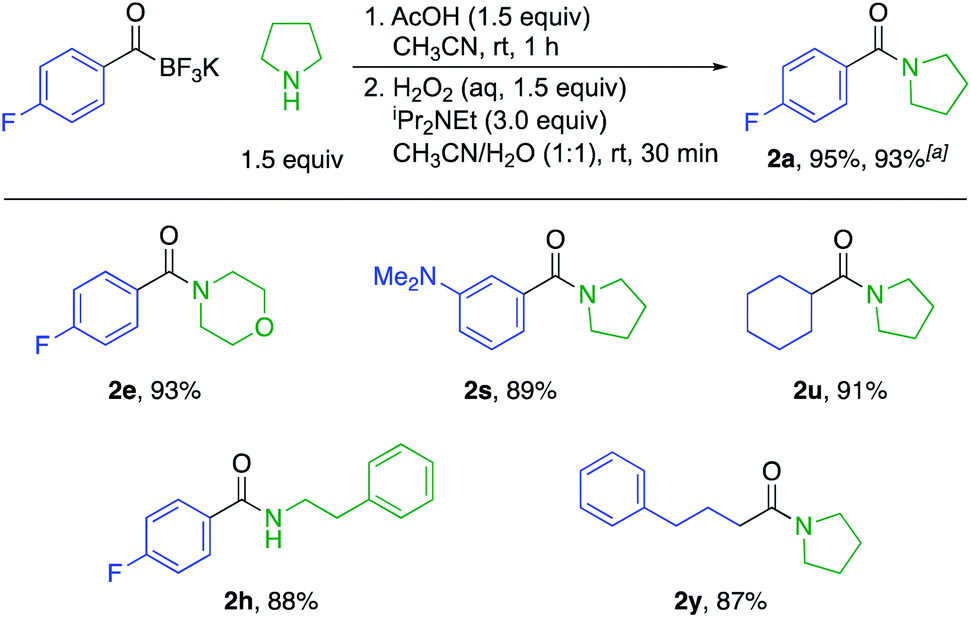 | ||
| Scheme 2 Substrate scope for the one-pot amide formation from potassium acyltrifluoroborates (KATs) and scale-up experiment. The yields are isolated yields. a Yield for the reaction at 3.0 mmol scale instead of 0.2 mmol scale. | ||
The success of the one-pot amide formation from KATs and amines encouraged us to make preliminary studies towards a solid-phase peptide synthesis (SPPS) method based on this chemistry. In standard SPPS, the carboxylic acid is used in excess and must be activated with a coupling reagent. This activation step prevents it from being recovered after the coupling reaction. This both contributes to the large volume of waste produced during standard SPPS15 and limits the number of equivalents that can be used to drive the reaction. The amide-forming reaction between KATs and amines does not require any activating agent and the KAT can be recovered and reused if used in excess. We hypothesized that SPPS could be performed using KAT amino acids as monomers by TIM formation and subsequent oxidation, allowing the excess KAT to be recycled (Scheme 3a).
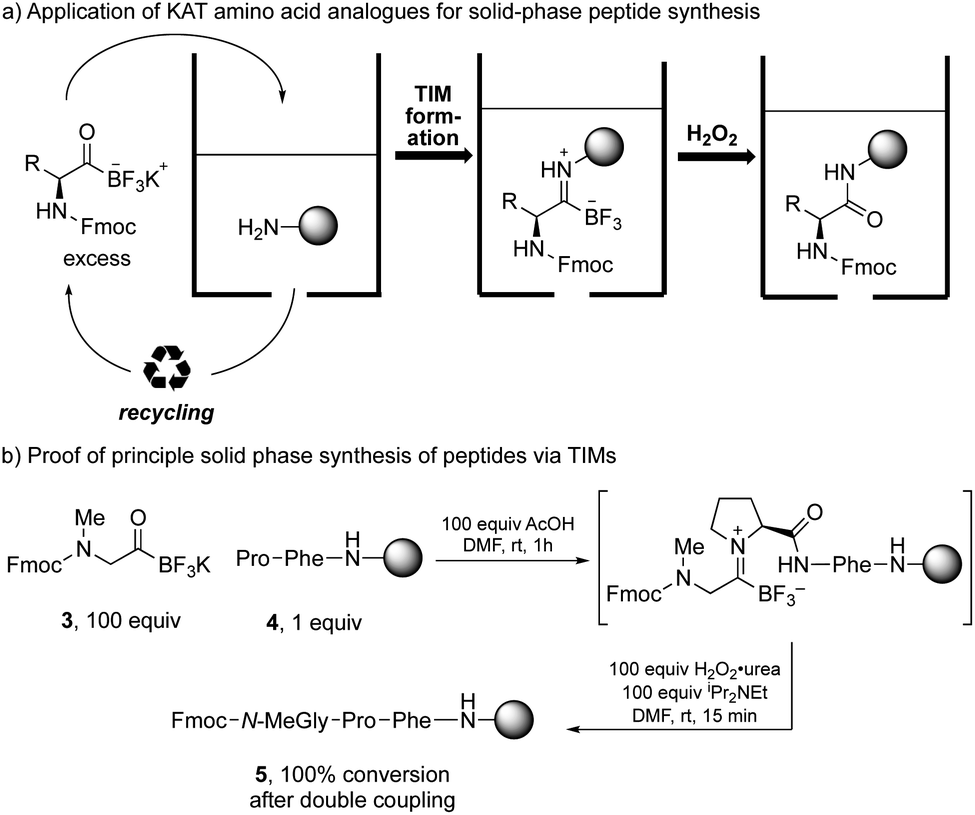 | ||
| Scheme 3 Preliminary studies toward solid-phase peptide synthesis (SPPS) using KAT amino acids as monomers. (a) Schematic representation of the application of KATs in SPPS. (b) Model synthesis of a peptide on solid support via TIMs. Conversion was determined by Fmoc loading test and HPLC analysis after cleavage from the resin. | ||
In preliminary studies, we achieved coupling between Fmoc-protected N-methylglycine KAT and solid-supported proline with full conversion. The conversion was determined after coupling of the N-methylglycine KAT analogue by Fmoc loading test of the resin and HPLC analysis after cleavage from the resin (Scheme 3b).
The acidic reaction conditions for TIM formation provides a unique environment for amide formation on unprotected substrates, such as peptides. The selective modification of peptides is important for the development of new peptide drugs, as most modern peptide therapeutics are modified with lipids or PEGs to increase their half-life and stability.16 Our method allows for the direct derivatization of an amine residue (lysine or N terminus), even when all other side chain functional groups are unprotected. We chose to investigate the modification of a lysine residue of an anti-diabetic GLP-1 analogue. Several GLP-1 analogues, e.g. Liraglutide or Semaglutide, are approved drugs for the treatment of diabetes.17 In both cases, the peptide is modified with a lipid side chain for binding to serum albumin.18 We were able to achieve the introduction of different KATs on Lys12 of peptide 6 with good conversions and yields, including KATs bearing a PEG chain (7c), an oleyl side chain (7a) and a diazo dye (7b) (Scheme 4a). Although it was necessary to preserve the N-terminal Fmoc group, this could be easily removed with Et2NH in DMSO to give target peptide 8.
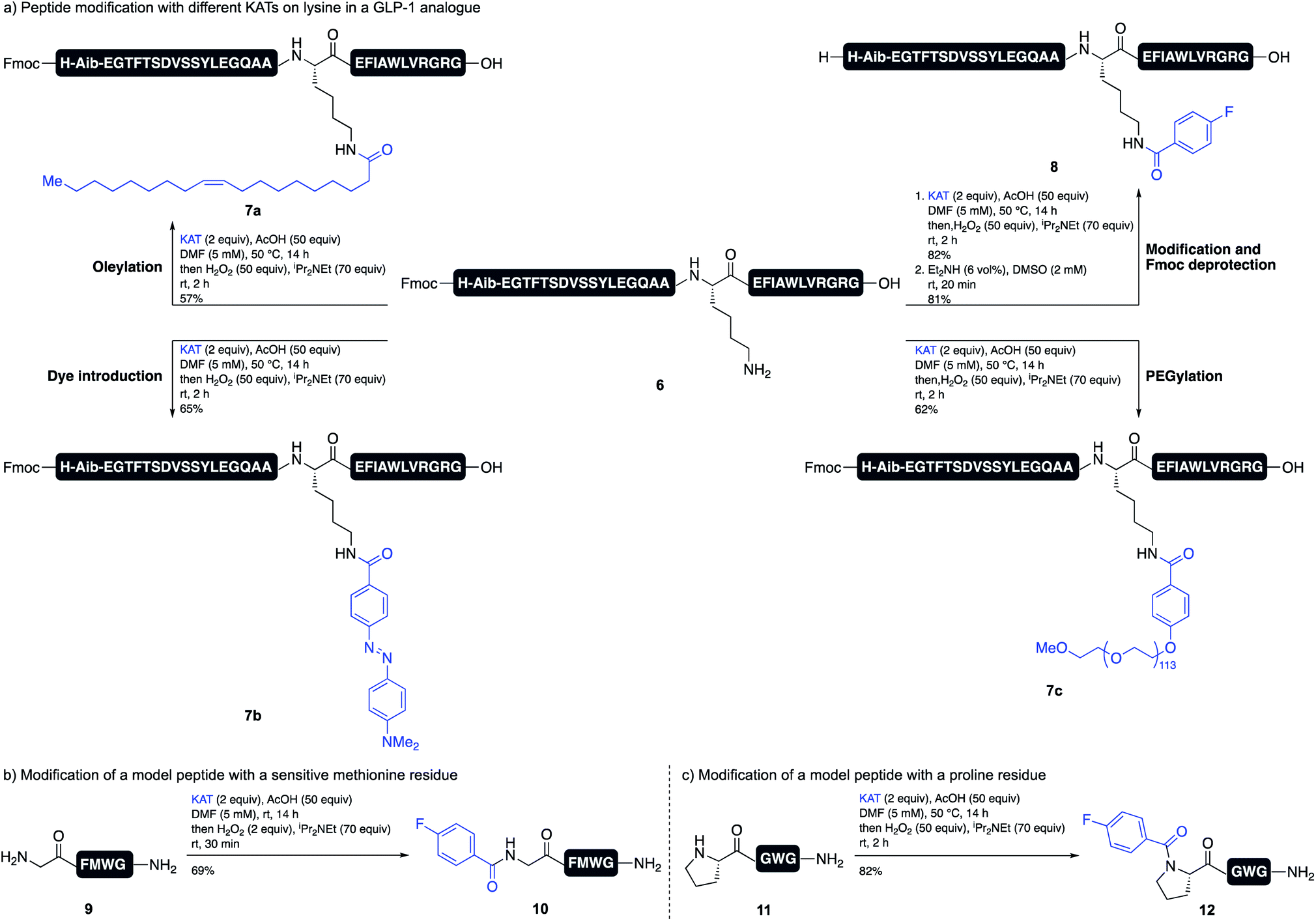 | ||
| Scheme 4 Application of the trifluoroborate iminium (TIM) formation and subsequent oxidation for the modification of different peptides. (a) Modification of a GLP-1 analogue with different KATs. (b) Modification of a peptide containing methionine. (c) Modification of a proline residue in a peptide. | ||
Amino acid side chains sensitive to oxidizing conditions in peptide 6, i.e. tryptophan, tyrosine, or histidine, tolerated the conditions and we did not observe any oxidation side products. We also showed that peptide 9, which contains methionine, tolerates slightly modified oxidation conditions. Using only 2 equiv. of hydrogen peroxide and quenching the reaction with Na2SO3 after 30 min gave the desired product 10 in good yield. The oxidized methionine residue was only observed as a minor side product (Scheme 4b). Since this method allows for the facile formation of tertiary amides, we furthermore showed that the modification of an N-terminal proline residue is possible and peptide 12 was obtained in high yield (Scheme 4c).
We considered several possible mechanisms for the formation of amides from TIMs with basic hydrogen peroxide. We showed with 18O-labelled hydrogen peroxide that the oxygen in the amide is incorporated from the hydrogen peroxide exclusively and no exchange with water is observed. With this in mind, we focused our preliminary studies on the two most likely pathways (Scheme 5). Hydrogen peroxide as well as other oxidants are well known to oxidize organotrifluoroborates and organoboronates to hydroxyl groups, making Path A a viable route. While there are many reports concerning the oxidation of organoboronates,19 only a few examples of the direct oxidation of organotrifluoroborates are known. Hu reported a copper-catalyzed oxidation of phenyltrifluoroborate,20 Fensterbank a TEMPO-promoted procedure,21 and Ochiai employed a hypervalent iodonium complex.22 Of greatest relevance are Molander's oxidation conditions using Oxone23 and Kandasamy's report using hydrogen peroxide in lactic acid for the oxidation of phenyltrifluoroborate.24 These conditions, however, are relatively harsh and not consistent with the rapid, clean oxidations we observed.
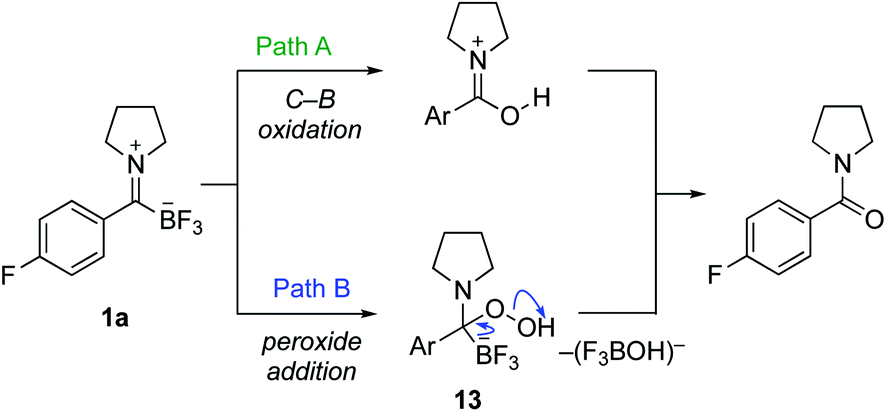 | ||
| Scheme 5 Possible mechanisms for the oxidative amide formation from trifluoroborate iminiums (TIMs). | ||
As an alternative, we considered nucleophilic attack of peroxide anion to the TIM (Path B). We have already established that TIMs are good electrophiles, which would lead to tetrahedral intermediate 13.7 Loss of BF3OH−, in a manner similar to the postulated mechanism of KAT ligations with hydroxylamines, would lead directly to the amide.6 In order to test this, we attempted the oxidation with NaClO2, which is used in the Pinnick oxidation via a similar mechanism.25 Upon exposure of TIMs to standard Pinnick conditions, the amide product was cleanly formed. We also conducted a competition experiment between potassium phenyltrifluoroborate and TIM 1a under our standard oxidation conditions. TIM 1a was oxidized to give the amide while potassium phenyltrifluoroborate was recovered unchanged (see ESI† for details). Takemoto and co-workers recently published oxidative amide formation from α-ketoacids and amines. They propose a similar mechanism via imine formation and subsequent oxidation via a tetrahedral intermediate.26 Together, these observations support Path B as the likely route for amide formation.
Conclusions
In summary, we have reported the oxidative synthesis of secondary and tertiary amides from KATs or TIMs under aqueous conditions. It offers a unique reactivity and chemoselectivity profile, features that should make it a viable alternative to classical methods for amide bond formation and a promising approach to chemoselective modification of peptides and proteins. The unique ability to use two stable precursors – KATs and amines – without any additional reagents to form the TIM intermediate provides a promising approach to a new form of iterative peptide synthesis that avoids problematic coupling reagents.Experimental section
General procedure for the oxidation of TIMs to amides
TIM (1.0 equiv.) was dissolved in CH3CN/H2O (1:1, 0.2 M), H2O2 (30% aq., 1.2 equiv.) and N,N-diisopropylethylamine (1.2 equiv.) were added and the reaction was allowed to stir for 30 min. The reaction was diluted with H2O and EtOAc and the aqueous layer was extracted three times with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. If necessary, the crude product was purified with flash column chromatography.
General procedure for the one-pot TIM formation and oxidation
KAT (1.0 equiv.) was dissolved in CH3CN (0.5 M), AcOH (1.5 equiv.) and the amine (1.5 equiv.) were added and the reaction was stirred for 1 hour. H2O (0.5 M), H2O2 (aq, 30%, 1.5 equiv.), and N,N-diisopropylethylamine (3.0 equiv.) were added and the reaction was allowed to stir for an additional 30 min. The reaction was diluted with H2O and EtOAc and the aqueous layer was extracted three times with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. If necessary, the crude product was purified with flash column chromatography.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by Eli Lilly through the Lilly Research Award Program (LRAP). A. Schuhmacher acknowledges the Scholarship Fund of the Swiss Chemical Industry (SSCI) for a fellowship. Support for T. Shiro was provided by Sumitomo Dainippon Pharma. Co., Ltd. We thank the Molecular and Biomolecular Analytical Services (MoBiAS) and the LOC NMR service for analyses. Alessandro Fracassi (ETH Zurich) is acknowledged for providing oleyl KAT. Dino Wu (ETH Zurich) is acknowledged for developing the synthesis of potassium (N-(tert-butoxycarbonyl)-N-methylglycyl)trifluoroborate.Notes and references
- (a) The Amide Linkage: Selected Structural Aspects in Chemistry, Biochemistry and Material Science, ed. A. Greenburg, C. M. Breneman and J. S. Liebman, Wiley, New York, 2000 Search PubMed; (b) J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS.
- (a) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC; (b) D. S. MacMillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. B. Watson, Green Chem., 2013, 15, 596–600 RSC; (c) M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS.
- K. J. McKnelly, W. Sokol and J. S. Nowick, J. Org. Chem., 2020, 85, 1764–1768 CrossRef CAS PubMed.
- G. A. Molander, J. Raushel and N. M. Ellis, J. Org. Chem., 2010, 75, 4304–4306 CrossRef CAS PubMed.
- (a) A. M. Dumas, G. A. Molander and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 5683–5686 CrossRef CAS PubMed; (b) H. Noda, G. Erős and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 5611–5614 CrossRef CAS PubMed.
- A. Osuna Gálvez, C. P. Schaack, H. Noda and J. W. Bode, J. Am. Chem. Soc., 2017, 139, 1826–1829 CrossRef PubMed.
- T. Shiro, A. Schuhmacher, M. K. Jackl and J. W. Bode, Chem. Sci., 2018, 9, 5191–5196 RSC.
- (a) G. Erős, Y. Kushida and J. W. Bode, Angew. Chem., Int. Ed., 2014, 53, 7604–7607 CrossRef PubMed; (b) D. Wu, N. A. Fohn and J. W. Bode, Angew. Chem., Int. Ed., 2019, 58, 11058–11062 CrossRef CAS PubMed.
- S. M. Liu, D. Wu and J. W. Bode, Org. Lett., 2018, 20, 2378–2381 CrossRef CAS PubMed.
- (a) A. M. Dumas and J. W. Bode, Org. Lett., 2012, 14, 2138–2141 CrossRef CAS PubMed; (b) J. Taguchi, T. Takeuchi, R. Takahashi, F. Masero and H. Ito, Angew. Chem., Int. Ed., 2019, 58, 7299–7303 CrossRef CAS PubMed.
- (a) Z. He, P. Trinchera, S. Adachi, J. D. S. Denis and A. K. Yudin, Angew. Chem., Int. Ed., 2012, 51, 11092–11096 CrossRef CAS PubMed; (b) S. Adachi, S. K. Liew, C. F. Lee, A. Lough, Z. He, J. D. S. Denis, G. Poda and A. K. Yudin, Org. Lett., 2015, 17, 5594–5597 CrossRef CAS PubMed; (c) D. Tan, Y. Cai, Y. Zeng, W. Lv, L. Yang, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 13784–13788 CrossRef CAS PubMed; (d) S. Lin, L. Wang, N. Aminoleslami, Y. Lao, C. Yagel and A. Sharma, Chem. Sci., 2019, 10, 4684–4691 RSC.
- (a) M. L. Lepage, S. Lai, N. Peressin, R. Hadjerci, B. O. Patrick and D. M. Perrin, Angew. Chem., Int. Ed., 2017, 56, 15257–15261 CrossRef CAS PubMed; (b) J. Taguchi, T. Ikeda, R. Takahashi, I. Sasaki, Y. Ogasawara, T. Dairi, N. Kato, Y. Yamamoto, J. W. Bode and H. Ito, Angew. Chem., Int. Ed., 2017, 56, 13847–13851 CrossRef CAS PubMed.
- (a) Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS PubMed; (b) J. Campos and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 14159–14163 CrossRef CAS PubMed.
- (a) R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS; (b) M. Amblard, J. Fehrentz, J. Martinez and G. Subra, Mol. Biotechnol., 2006, 33, 239–254 CrossRef CAS PubMed; (c) K. G. Varnava and V. Sarojini, Chem.–Asian J., 2019, 14, 1088–1097 CrossRef CAS PubMed.
- (a) Q. Hu, F. Berti and R. Adamo, Chem. Soc. Rev., 2016, 45, 1691–1719 RSC; (b) N. Yin, M. A. Brimble, P. W. R. Harris and J. Wen, Med. Chem., 2014, 4, 763–769 Search PubMed; (c) O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- (a) A. Andersen, A. Lund, F. K. Knop and T. Vilsbøll, Nat. Rev. Endocrinol., 2018, 14, 390–403 CrossRef CAS PubMed; (b) D. Sharma, S. Verma, S. Vaidya, K. Kalia and V. Tiwari, Biomed. Pharmacother., 2018, 108, 952–962 CrossRef CAS PubMed; (c) S. Dhillon, Drugs, 2018, 78, 275–284 CrossRef CAS PubMed.
- (a) D. Irby, C. Du and F. Li, Mol. Pharmaceutics, 2017, 14, 1325–1338 CrossRef CAS PubMed; (b) M. T. Larsen, M. Kuhlmann, M. L. Hvam and K. A. Howard, Mol. Cell. Ther., 2016, 4, 3 CrossRef PubMed.
- (a) J. Simon, S. Salzbrunn, G. K. S. Prakash, N. A. Petasis and G. A. Olah, J. Org. Chem., 2001, 66, 633–634 CrossRef CAS PubMed; (b) G. K. S. Prakash, S. Chacko, C. Panja, T. E. Thomas, L. Gurung, G. Rasul, T. Mathew and G. A. Olah, Adv. Synth. Catal., 2009, 351, 1567–1574 CrossRef CAS; (c) K. S. Webb and D. Levy, Tetrahedron Lett., 1995, 36, 5117–5118 CrossRef CAS; (d) R. E. Maleczka, F. Shi, D. Holmes and M. R. Smith III, J. Am. Chem. Soc., 2003, 125, 7792–7793 CrossRef CAS PubMed; (e) E. Kianmehr, M. Yahyaee and K. Tabatabai, Tetrahedron Lett., 2007, 48, 2713–2715 CrossRef CAS; (f) E. Shin, H. Kim, S. Joo, U. S. Shin and S. Kim, Catal. Lett., 2019, 149, 1560–1564 CrossRef CAS; (g) G. Cheng, X. Zeng and X. Cui, Synthesis, 2014, 46, 295–300 Search PubMed.
- J. Xu, X. Wang, C. Shao, D. Su, G. Cheng and Y. Hu, Org. Lett., 2010, 12, 1964–1967 CrossRef CAS PubMed.
- G. Sorin, R. Martinez Mallorquin, Y. Contie, A. Baralle, M. Malacria, J. Goddard and L. Fensterbank, Angew. Chem., Int. Ed., 2010, 49, 8721–8723 CrossRef CAS PubMed.
- M. Ochiai, K. Miyamoto, Y. Yokota, T. Suefuji and M. Shiro, Angew. Chem., Int. Ed., 2005, 44, 75–78 CrossRef CAS PubMed.
- (a) G. A. Molander and L. N. Cavalcanti, J. Org. Chem., 2011, 76, 623–630 CrossRef CAS PubMed; (b) G. A. Molander, S. Z. Siddiqui and N. Fleury-Brégeot, Org. Synth., 2013, 90, 153–163 CrossRef CAS.
- (a) S. Gupta, P. Chaudhary, L. Seva, S. Sabiah and J. Kandasamy, RSC Adv., 2015, 5, 89133–89138 RSC; (b) S. Gupta, P. Chaudry, V. Srivastava and J. Kandasamy, Tetrahedron Lett., 2016, 57, 2506–2510 CrossRef CAS.
- (a) B. O. Lindgren and T. Nilsson, Acta Chem. Scand., 1973, 27, 888–890 CrossRef CAS; (b) G. A. Kraus and B. Roth, J. Org. Chem., 1980, 45, 4825–4830 CrossRef CAS; (c) B. S. Bal, W. E. Childers Jr and H. W. Pinnick, Tetrahedron, 1981, 37, 2091–2096 CrossRef CAS; (d) V. P. Kudesia, Bull. Soc. Chim. Belg., 1972, 81, 623–628 CrossRef CAS.
- T. Nanjo, N. Kato, X. Zhang and Y. Takemoto, Chem.–Eur. J., 2019, 25, 15504–15507 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01330g |
| ‡ Current address: Sumitomo Dainippon Pharma Co., Ltd., 3-1-98 Kasugade-naka, Konohana-ku, Osaka 554-0022, Japan. |
| This journal is © The Royal Society of Chemistry 2020 |