
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiastereoselective olefin amidoacylation via photoredox PCET/nickel-dual catalysis: reaction scope and mechanistic insights†
Shuai
Zheng
 a,
Shuo-Qing
Zhang
b,
Borna
Saeednia
a,
Jiawang
Zhou
a,
Jessica M.
Anna
a,
Xin
Hong
*b and
Gary A.
Molander
*a
a,
Shuo-Qing
Zhang
b,
Borna
Saeednia
a,
Jiawang
Zhou
a,
Jessica M.
Anna
a,
Xin
Hong
*b and
Gary A.
Molander
*a
aRoy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: gmolandr@sas.upenn.edu
bDepartment of Chemistry, Zhejiang University, Hangzhou, Zhejiang 310027, China
First published on 1st April 2020
Abstract
The selective 1,2-aminoacylation of olefins provides opportunities for the rapid construction of nitrogen-containing molecules. However, the lack of CO-free acylation reactions has limited their application. By using photoredox proton-coupled electron transfer (PCET)/Ni dual-catalysis, a highly regio- and diastereoselective amidoacylation of unactivated olefins has been developed. Various acyl electrophiles are compatible, including alkyl- and aryl acyl chlorides and anhydrides, as well as in situ activated carboxylic acids. Hammett studies and other mechanistic experiments to elucidate features of the diastereoselectivity, a transient absorption study of the PCET step, as well as computational evidence, provide an in-depth understanding of the disclosed transformation.
Introduction
Transition-metal catalyzed 1,2-difunctionalizations of unactivated olefins have emerged as powerful methods for the elaboration of commodity chemicals over the past decades, with attractive capabilities for rapidly building molecular complexity.1–5 Multiple strategies have been employed to achieve selectivity in the heterodifunctionalization, such as directed migratory insertion,6 nucleometalation,7 radical-polar crossover of activated olefins,8 utilization of bulky coupling partners and/or polarity reversal,9–13 as well as kinetically favorable radical cyclization approaches.14,15 Among these transformations, carboamination has received significant attention because of the prevalence of nitrogen-containing small molecules and heterocycles in bio-active and drug-like molecules.16 Progress has been achieved using amines as nucleophilic species for directed aminopalladation6,17 or trapping of cationic species after C–C bond formation.8,18 Efforts have also been made to utilize prefunctionalized, reductively generated amidyl- and iminyl radicals19–21 to improve efficiency and selectivity, while the oxidative proton-coupled electron transfer (PCET) approach has been utilized to generate N-centered radicals directly from N–H functional units.14,15,22–25Although β-amino ketone structures are reasonably prevalent among bioactive molecules (Fig. 1A), aminoacylation reactions and aminocarbonylative transformations of olefins appear relatively rare (Fig. 1B). Early efforts were focused on net-oxidative transformations, rendering β-amino esters as the final product,26,27 while the Lambert group accessed amidoacylation products by tethering a Pd-catalyzed carbonylation process with a Lewis acid-catalyzed Friedel–Crafts acylation of electron-rich (hetero)aromatic systems.28 Later, others utilized an Umpolung approach using a tethered oxime ester as an electrophilic nitrogen source and disclosed iminoacylation transformations using tetraaryl borates or organozinc reagents.29,30 In a more recent report,31 such transformations were achieved with aryl iodides using an 8-aminoquinoline directing group.
 | ||
| Fig. 1 Amidoacylation of olefin species. | ||
Together, these approaches provide a powerful suite of methods for the synthesis of N-heterocycles as well as β-amino ketone structures, yet many challenges remain to be addressed. One common feature of these transformations is the need for CO gas, which, although inexpensive and abundant, is toxic and flammable. Additionally, the majority of present transformations require aryl electrophiles or nucleophiles, while alkyl electrophiles remain challenging to incorporate. Because of their good commercial availability and convenient preparation, acyl (pseudo)halides are useful surrogates to consider in such transformations.32
As attractive as the utilization of N-centered radicals might be,20,33–35 the application of such intermediates in transition metal catalysis still remains rare (Fig. 1C). Apart from several reports facilitating direct C–N bond construction,36 transition metal-catalyzed cascade transformations, where C-centered radicals were generated via either 1,5-HAT37,38 or 5-exo-trig cyclization,15,20 to this point are scarce, possibly because of potential selectivity issues and the incompatibility between transition metal catalysts and N-radical generation. Therefore, inspired by our recent efforts to meld photoredox PCET with nickel catalysis for amidoarylation of olefins,15 we sought to address the challenges of amidoacylation by utilizing acyl electrophiles in photoredox PCET/Ni dual-catalyzed olefin difunctionalization (Fig. 1D).
Results and discussion
To test this strategy, an investigation was initiated using N-phenyl hex-4-enamide and benzoyl chloride as cross-coupling partners (Scheme 1). Previous reports pointed to 1,2-dichloroethane (DCE) as a potential solvent for photoredox PCET/Ni dual catalysis,15 which indeed rendered a 46% yield in the initial attempt (Scheme 1, entry 1). Although it is noteworthy that the less expensive organic photocatalyst 4CzIPN gave a comparable yield (entry 3), neither this nor a more oxidizing Ir photocatalyst ([Ir-2], entry 2) significantly improved the yield. Several ligands successfully promoted the reaction, with the electron-rich dMeObpy being most appropriate (entries 1, 4, 5). A careful examination of the reaction profile indicated a significant amount of hydroamidation byproduct, likely caused by hydrogen atom abstraction from the solvent,23 which led us to a further solvent optimization. Acetonitrile was eventually identified as the most suitable solvent, rendering a 75% yield of 1 (entry 7). Owing to the vast availability of carboxylic acid functional groups in a variety of commodity chemicals, we also examined the possibilities of employing in situ activation of carboxylic acids as the source of the acyl electrophile.39 After several attempts (entries 8–11), oxalyl chloride was identified as an appropriate activator, affording a 73% yield.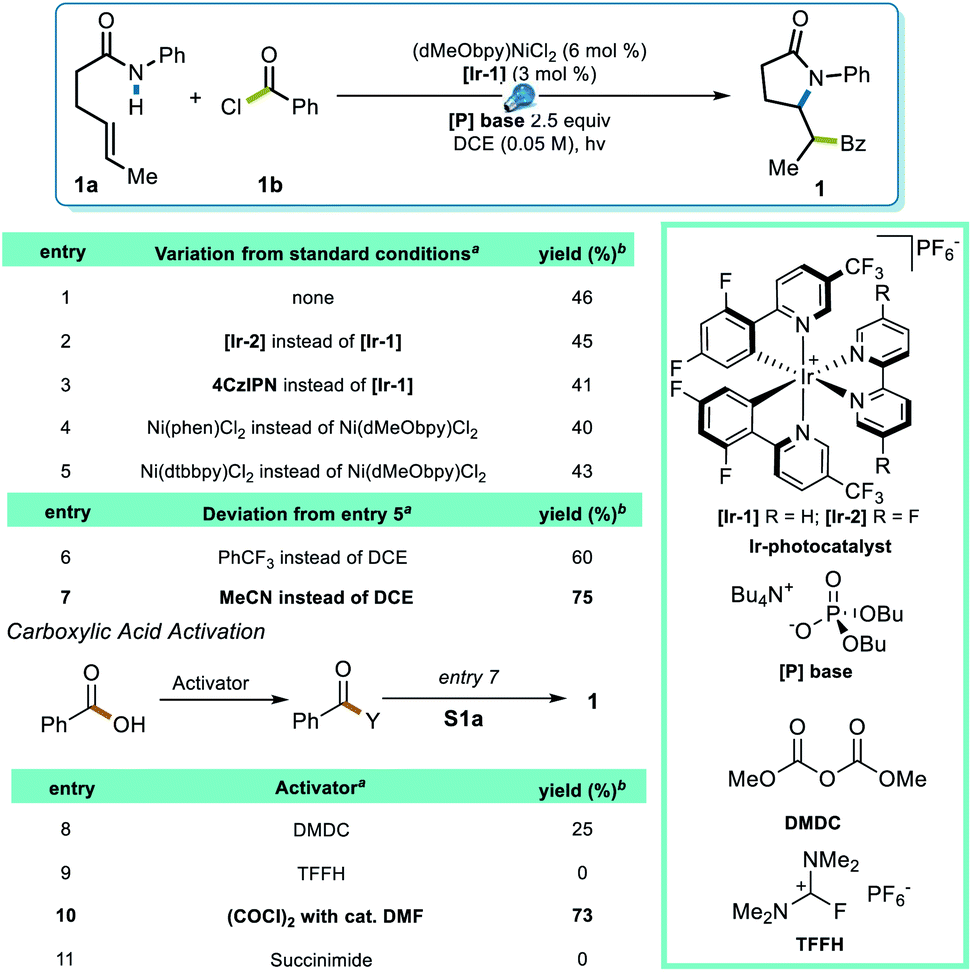 | ||
| Scheme 1 Reaction Optimization. aPerformed on 0.1 mmol scale. bIsolated yield. | ||
With proper conditions in hand, an exploration of the scope of the reaction was initiated. The developed reaction conditions allowed excellent functional group tolerance. For substituted benzoyl chlorides, electron-rich, electron-neutral, and electron-poor substrates all rendered reasonably good yields (Scheme 2, entries 1–7). Interestingly, even though the terminal methyl group on the alkene seemed rather sterically undemanding, exceptional diastereoselectivities were observed in all of these examples. Other types of electrophiles, such as acyl anhydrides (8) and alkyl acyl halides (9–11), are also compatible, despite leading to a lower diastereoselectivity in some cases. Perhaps most importantly, in situ activation of carboxylic acids did not lead to a compromise in either the yield or dr of the reactions (12, 13), and interesting substructures, such as substituted cyclopropyl (14) and difluorobenzodioxolyl (15), were installed with reasonable yields directly from the corresponding carboxylic acids.
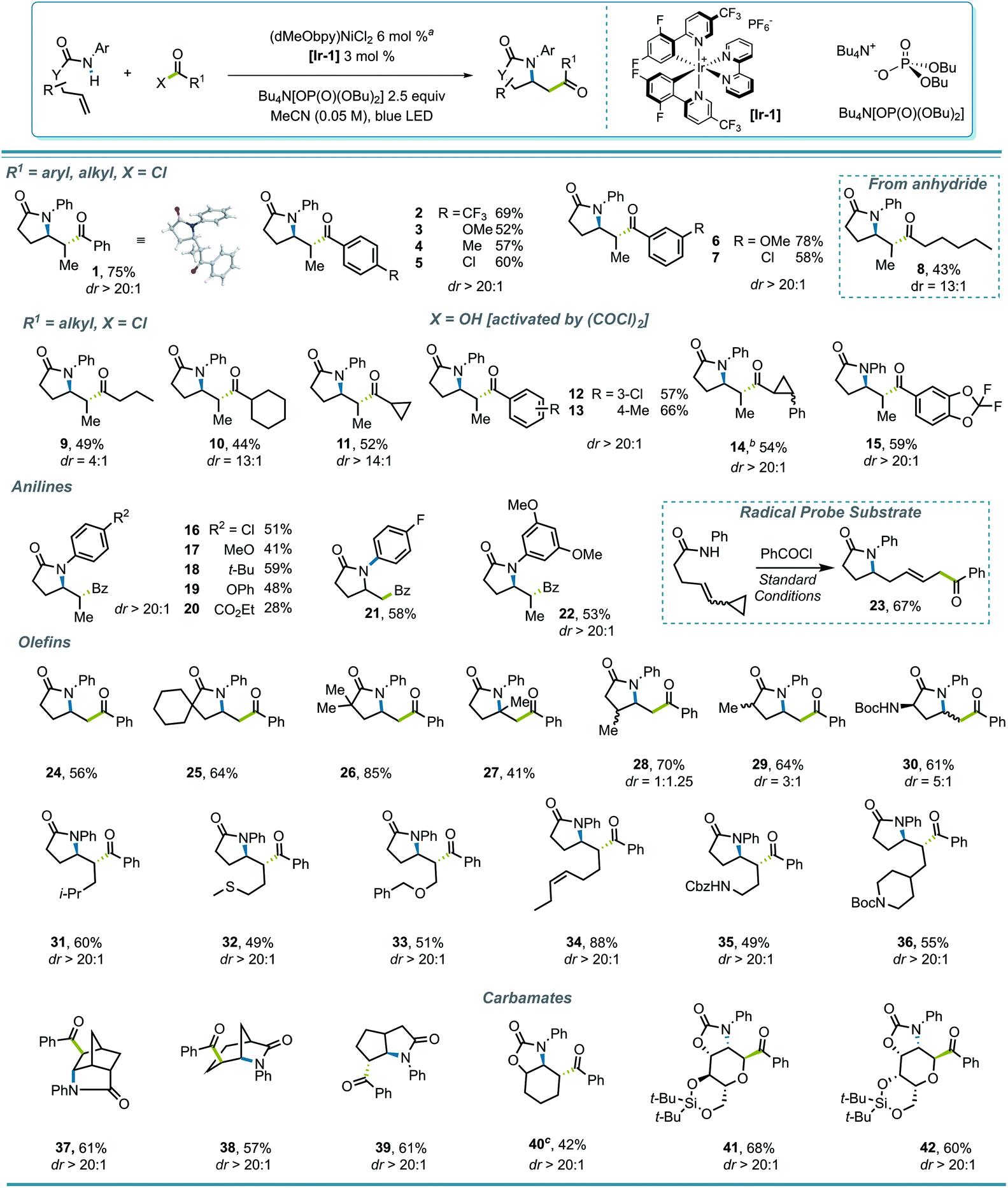 | ||
Scheme 2 Reaction scope. aReaction conditions: amide S1–S42 (0.36 mmol, 1.2 equiv.), acyl (pseudo)halides (0.3 mmol, 1.0 equiv.), Bu4N[OP(O)(OBu)2] (0.75 mmol, 2.5 equiv.), Ni(dMeObpy)Cl2 (0.018 mmol, 6 mol%), [Ir-1] (0.009 mmol, 3 mol%), blue LED, 6 mL MeCN. bA 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of two diastereoisomers originating from the acyl coupling partner. c0.006 mmol, 2 mol% of Ni precatalyst was used. :1 mixture of two diastereoisomers originating from the acyl coupling partner. c0.006 mmol, 2 mol% of Ni precatalyst was used. | ||
Various anilide- and alkenyl backbones were next examined, and their influence in this reaction was tested both to expand the scope and to shed light on the diastereoselectivity. For aniline derivatives bearing different substituents (16–22), although the yields varied, the diastereoselectivities, where relevant, remained consistent. Substrates leading to primary alkyl radicals were generally compatible in the reaction (21, 24–30), and although both methyl and a bulky Boc-protected amino group at the α-carbonyl position induced diastereoselectivity to some extent (29, 30), substitution β to the carbonyl seemed to have minimal influence (28). Therefore, it seems that substitution δ to the carbonyl, where the alkyl radical is generated after cyclization, is crucial for the high diastereoselectivity observed. Not surprisingly, polycyclic structures retain their high diastereoselectivity in the reaction (37–39).
Several functional groups were incorporated to test compatibility, including thioether (32), ether (33), and pendant olefin (34) groups, as well as protected primary- (35) and secondary (36) amines, all showing excellent dr and good to excellent yields. The applicability of the protocol for amidoacylation of alkenyl carbamates was also examined, which initially led to a diminished yield (20%) of 40, with 35% of N-phenyl benzamide byproduct. This byproduct likely results from an SN2′-type oxidative addition by the Ni(0) catalyst to the ene carbamate,40 with loss of CO2 and capture of the resulting aniline with the acid chloride. Lowering the loading of Ni precatalyst to 2% improved the yield of 40 to 42%. Interestingly, under standard reaction conditions, such a side reaction was not observed in monosaccharide-derived substrates (41, 42), rendering C-acyl aminoglycosides diastereoselectively in good yields.
Several experiments were performed to aid in deciphering the mechanism of the reaction. First, control experiments suggested that nickel, Ir photocatalyst, light, and base are all essential for the transformation (see ESI† for further details). Radical probe substrate 23 supported the proposed reaction pathway and indicated that the reaction indeed underwent a single electron transfer process. To rule out the possibility of energy transfer mechanism as seen in related systems,41 we performed a transient absorption spectroscopy study of the PCET step (Fig. 2). By comparing the well-studied triethanolamine (TEOA) system (black trace, Fig. 2), which is known to reduce the excited state of the Ir photocatalyst,42,43 with substrate 1a in the presence of base [Bu4N[OP(O)(OBu)2] (red trace, Fig. 2), the resulting spectrum suggested an evident generation of reduced [Ir-1] photocatalyst, supporting the proposed electron transfer mechanism, generating an N-centered radical. Then, considering the versatility of different electrophilic species, the nature of the actual electrophile in the reaction was sought. After stirring a 1:1 mixture of benzoyl chloride and Bu4N[OP(O)(OBu)2] in CH2Cl2 for 20 min, the mixed benzoic-phosphonic anhydride was isolated and subjected to the reaction conditions, rendering a 73% yield and a comparable diastereoselectivity to that observed under the normal reaction conditions (Scheme 3a). Further computational studies also provide support that, compared to benzoyl chloride, the benzoic-phosphonic anhydride appears to be a more stable species (see ESI† for further details). Interestingly, a diminished yield was observed when an alkyl acyl chloride was used under the same protocol.
 | ||
| Fig. 2 Transient absorption studies on PCET-mediated N-radical generation at 10 μs time delay. | ||
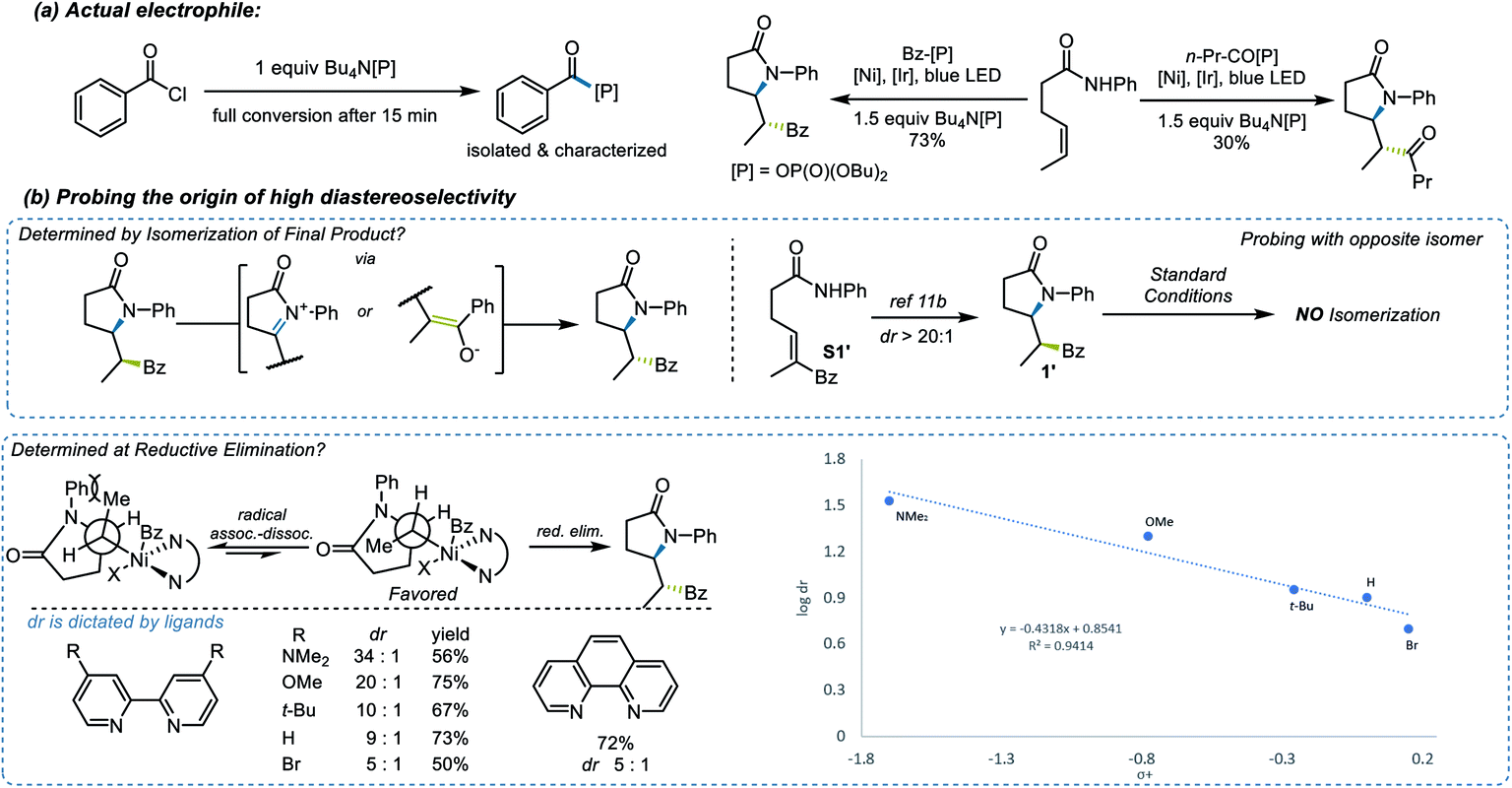 | ||
| Scheme 3 Experimental mechanistic insights. | ||
The origin of the high diastereoselectivity of this reaction was next explored. To investigate the stability of both stereoisomers, a synthesis of the opposite diastereoisomer was carried out. Thus, by applying the hydroamination reaction developed by Knowles et al.,23 the designed substrate S1′ led to diastereoisomer 1′. This isomer was subsequently subjected to the standard reaction conditions, and no isomerization to 1 was observed (Scheme 3b). Further investigation revealed a dependence between the electron density on the ligands and the dr values (Scheme 3b). When an analysis was performed relating logdr to σ+ parameters,44 a good linear correlation was found. The negative ρ value from this plot indicated that higher electron density on the ligand, which stabilizes the putative Ni(III) intermediates in the catalytic cycle, will lead to a higher diastereoselectivity in this transformation. Taken together, these results suggested that the high dr must originate from the catalytic process and not epimerization after product formation.45
To gain deeper insights on the diastereo-determining step, the investigation was continued using computational tools. First, the free energy calculation of 1 and 1′ indicated that 1′ is the thermodynamically more favorable isomer (Fig. 3a). Therefore, the diastereoselective formation of 1 is a kinetically controlled process. Although either a Ni(0)–Ni(I)–Ni(III) mechanism,46 where radical addition takes place before oxidative addition of acyl halide, or a Ni(0)–Ni(II)–Ni(III) mechanism appear viable, because Bz–X is in much higher concentration than the photogenerated radicals A and B, the major reaction pathway proposed is one in which oxidative addition of Bz–X to a catalytically active nickel complex that initiates the process. Regardless of the mechanistic pathway, the diastereo-determining step will be the addition of radical B to nickel complex D (vide infra). Thus, Ni(0) catalyst C first undergoes oxidative addition to Bz–X, forming Ni(II) species D, which subsequently binds the radical B generated through a kinetically favorable 5-exo-trig cyclization.46 The formed Ni(III) species E then undergoes reductive elimination, releasing product 1 and generating Ni(I) species F. Further SET between F and reduced photocatalyst [PC]˙− regenerates active Ni(0) catalyst C.
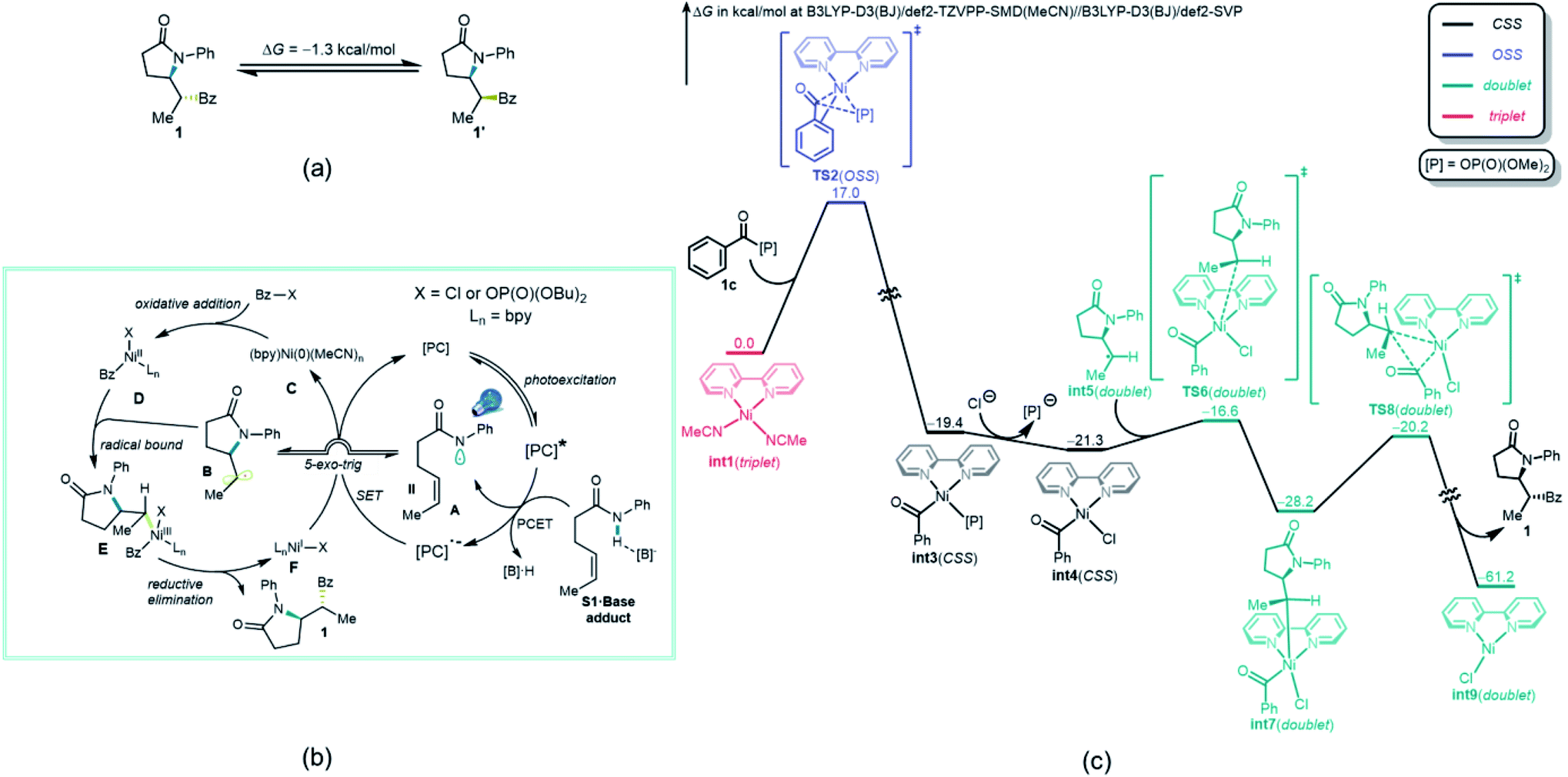 | ||
| Fig. 3 (a) Free energy difference between 1 and 1′. (b) Proposed reaction mechanism of the acylation between 1a and 1b. (c) DFT computed free energy diagram between 1a and 1c of the thermochemical part (from C to F) of the catalytic cycle calculated at B3LYP-D3(BJ)/def2-TZVPP-SMD(MeCN)//B3LYP-D3(BJ)/def2-SVP. | ||
Based on the proposed reaction mechanism in Fig. 3b, the most favorable computed free energy pathway for the thermochemical part (B to F) of the catalytic cycle calculated by density functional theory (DFT) is shown in Fig. 3c. Although both BzCl and Bz-[P] (1c) exhibited reasonable reactivity toward oxidative addition, the mixed anhydride Bz-[P] was applied as the initial point because of its greater stability and ease of formation (see ESI† for further details). The active triplet Ni(0) catalyst int1 is oxidized by Bz-[P] (1c) via an open-shell singlet (OSS) transition state TS2, which is quite facile, with a free energy barrier of only 17.0 kcal mol−1. This generates closed-shell Ni(II) species int3. This step is strongly exothermic and thus irreversible as proposed. Ligand exchange between the Cl− anion and the [P]− anion then generates a more stable Ni(II) species int4. The subsequent radical binding step between int4 and int5viaTS6 is also very fast, with a free energy barrier of only 4.7 kcal mol−1, generating doublet Ni(III) species int7. Further irreversible and highly exothermic reductive elimination viaTS8 possesses a free energy barrier of 8.0 kcal mol−1, generating product 1 and doublet Ni(I) species int9.
Somewhat surprisingly, TS6 is calculated to be 3.6 kcal mol−1 higher in energy than TS8, and conversion of int7 through TS8 is fast and irreversible. Thus, it is the radical binding step viaTS6 that determines the observed diastereoselectivity. As shown in Fig. 4, TS10, which leads to the diastereomeric product 1′, is 1.0 kcal mol−1 higher in energy than TS6, which is in good agreement with the observed dr of 9:1 using bpy as ligand. The highlighted steric issues between the Me and Ph groups elevate the free energy TS10 and is responsible for the observed diastereoselectivity.
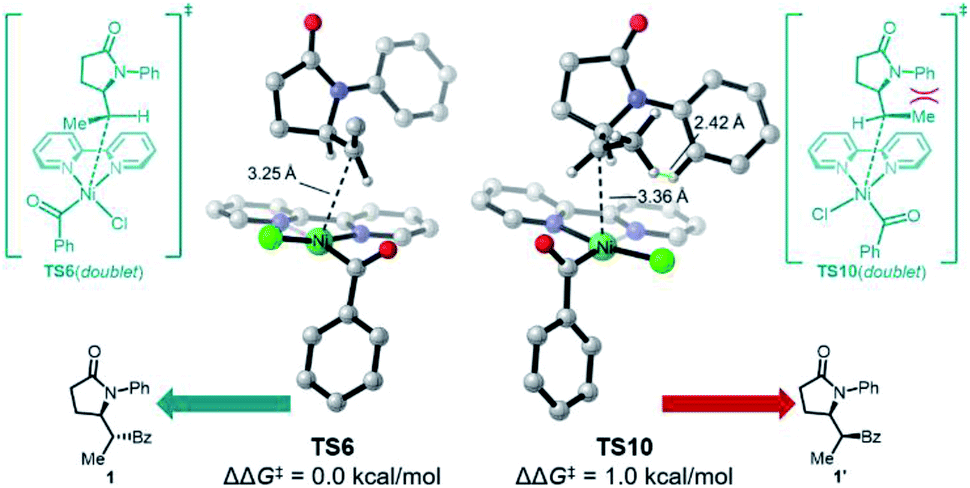 | ||
| Fig. 4 3D renderings of diastereoselectivity-determining radical bound transition states TS5 and TS9 calculated at B3LYP-D3(BJ)/def2-TZVPP-SMD(MeCN)//B3LYP-D3(BJ)/def2-SVP. Trivial H atoms are omitted for clarity. | ||
Conclusions
In conclusion, we have disclosed a photoredox PCET/Ni dual-catalyzed diastereoselective amidoacylation of unfunctionalized olefins. Various acyl electrophiles, including alkyl- and aryl acyl chlorides and anhydrides, as well as carboxylic acids activated in situ, are incorporated and retain excellent dr values in most of the examples. Thanks to the mild conditions, various functional groups, such as thioethers, protected amines, and saccharide derivatives are compatible. Transient spectroscopy provides strong support for electron transfer between the amide substrates and the Ir photocatalyst in the presence of phosphate base. Other mechanistic experiments indicate that the diastereoselectivity originates in the Ni-catalytic cycle, while the Hammett plot as well as a detailed computational study provide insight into the electronic and steric effects that lead to the diastereo-determining step toward the kinetic product. This study not only provides a powerful tool toward olefin heterodifunctionalization, but also sheds light on future reaction development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support provided by NIGMS (R35 GM 131680 to G. A. M.). We thank Dr Charles W. Ross, III (University of Pennsylvania) for obtaining HRMS data. We gratefully acknowledge Dr Mike Gau and Dr Pat Carroll (University of Pennsylvania) for acquiring X-ray crystal structures. We thank Dr Alvaro Gutierrez-Bonet (Merck & Co.) and Prof. Marisa C. Kozlowski (University of Pennsylvania) for helpful discussions. We acknowledge Johnson-Matthey for fine chemicals donations. Financial support from the NSFC (21702182 and 21873081 to X. H.), “Fundamental Research Funds for the Central Universities” (2019QNA3009 to X. H.) and China Postdoctoral Science Foundation (2018M640546 to S.-Q. Z.) is gratefully acknowledged. Calculations were performed on the high-performance computing system at the Department of Chemistry, Zhejiang University.Notes and references
- M.-Y. Cao, X. Ren and Z. Lu, Tetrahedron Lett., 2015, 56, 3732–3742 CrossRef CAS.
- X.-W. Lin, N.-X. Wang and Y. Xing, Eur. J. Org. Chem., 2017, 5821–5851 CrossRef.
- H.-M. Huang, M. H. Garduño-Castro, C. Morrill and D. J. Procter, Chem. Soc. Rev., 2019, 48, 4626–4638 RSC.
- J. Derosa, V. T. Tran, V. A. van der Puyl and K. M. Engle, Aldrichimica Acta, 2018, 51, 21–32 CAS.
- R. K. Dhungana, S. KC, P. Basnet and R. Giri, Chem. Rec., 2018, 18, 1314–1340 CrossRef CAS PubMed.
- J. E. Ney and J. P. Wolfe, Angew. Chem., Int. Ed., 2004, 43, 3605–3608 CrossRef PubMed.
- R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS PubMed.
- G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2013, 15, 4398–4401 CrossRef CAS PubMed.
- M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069–20078 CrossRef CAS PubMed.
- A. García-Domínguez, R. Mondal and C. Nevado, Angew. Chem., Int. Ed., 2019, 58, 12286–12290 CrossRef PubMed.
- W. Shu, A. García-Domínguez, M. T. Quirós, R. Mondal, D. J. Cárdenas and C. Nevado, J. Am. Chem. Soc., 2019, 141, 13812–13821 CrossRef CAS PubMed.
- S. W. Lardy and V. A. Schmidt, Eur. J. Org. Chem., 2019, 6796–6799 CrossRef CAS.
- J. M. Lear, J. Q. Buquoi, X. Gu, K. Pan, D. N. Mustafa and D. A. Nagib, Chem. Commun., 2019, 55, 8820–8823 RSC.
- G. J. Choi and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 9226–9229 CrossRef CAS PubMed.
- S. Zheng, Á. Gutiérrez-Bonet and G. A. Molander, Chem, 2019, 5, 339–352 CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- Z. Liu, Y. Wang, Z. Wang, T. Zeng, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 11261–11270 CrossRef CAS PubMed.
- B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505–5508 CrossRef CAS PubMed.
- J. Davies, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 13361–13365 CrossRef CAS PubMed.
- L. Angelini, J. Davies, M. Simonetti, L. M. Sanz, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2019, 58, 5003–5007 CrossRef CAS PubMed.
- K. M. Nakafuku, S. C. Fosu and D. A. Nagib, J. Am. Chem. Soc., 2018, 140, 11202–11205 CrossRef CAS PubMed.
- A. J. Musacchio, L. Q. Nguyen, G. H. Beard and R. R. Knowles, J. Am. Chem. Soc., 2014, 136, 12217–12220 CrossRef CAS PubMed.
- D. C. Miller, G. J. Choi, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 13492–13495 CrossRef CAS PubMed.
- G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268 CrossRef CAS PubMed.
- J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272 CrossRef PubMed.
- L. S. Hegedus, G. F. Allen and D. J. Olsen, J. Am. Chem. Soc., 1980, 102, 3583–3587 CrossRef CAS.
- L. S. Hegedus, P. M. Winton and S. Varaprath, J. Org. Chem., 1981, 46, 2215–2221 CrossRef CAS.
- T. A. Cernak and T. H. Lambert, J. Am. Chem. Soc., 2009, 131, 3124–3125 CrossRef CAS PubMed.
- A. Faulkner, J. S. Scott and J. F. Bower, J. Am. Chem. Soc., 2015, 137, 7224–7230 CrossRef CAS PubMed.
- L. M. Ambrosini, T. A. Cernak and T. H. Lambert, Synthesis, 2010, 2010, 870–881 CrossRef.
- J.-B. Peng, F.-P. Wu, D. Li, H.-Q. Geng, X. Qi, J. Ying and X.-F. Wu, ACS Catal., 2019, 9, 2977–2983 CrossRef CAS.
- L. Wang and C. Wang, Org. Chem. Front., 2018, 5, 3476–3482 RSC.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
- G. B. Boursalian, M.-Y. Ngai, K. N. Hojczyk and T. Ritter, J. Am. Chem. Soc., 2013, 135, 13278–13281 CrossRef CAS PubMed.
- T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069–3087 RSC.
- L. Zhou, S. Tang, X. Qi, C. Lin, K. Liu, C. Liu, Y. Lan and A. Lei, Org. Lett., 2014, 16, 3404–3407 CrossRef CAS PubMed.
- Z. Zhang, L. M. Stateman and D. A. Nagib, Chem. Sci., 2019, 10, 1207–1211 RSC.
- S. M. Thullen, S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2019, 141, 14062–14067 CrossRef CAS PubMed.
- J. Amani and G. A. Molander, Org. Lett., 2017, 19, 3612–3615 CrossRef CAS PubMed.
- A. Agarkov, E. W. Uffman and S. R. Gilbertson, Org. Lett., 2003, 5, 2091–2094 CrossRef CAS PubMed.
- T. Kim, S. J. McCarver, C. Lee and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 3488–3492 CrossRef CAS PubMed.
- A. Paul, M. D. Smith and A. K. Vannucci, J. Org. Chem., 2017, 82, 1996–2003 CrossRef CAS PubMed.
- C.-F. Leung, S.-M. Ng, C.-C. Ko, W.-L. Man, J. Wu, L. Chen and T.-C. Lau, Energy Environ. Sci., 2012, 5, 7903–7907 RSC.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- N. Y. Shin, J. M. Ryss, X. Zhang, S. J. Miller and R. R. Knowles, Science, 2019, 366, 364–369 CrossRef CAS PubMed.
- See ESI† for details. For references, see: (a) O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896–4899 CrossRef CAS PubMed; (b) E. Martinez and M. Newcomb, J. Org. Chem., 2006, 71, 557–561 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1981876. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01459a |
| This journal is © The Royal Society of Chemistry 2020 |