
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sequential C–O decarboxylative vinylation/C–H arylation of cyclic oxalates via a nickel-catalyzed multicomponent radical cascade†
Huan
Li‡
,
Lei
Guo‡
,
Xiaoliang
Feng
,
Liping
Huo
,
Shengqing
Zhu
and
Lingling
Chu
 *
*
State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Chemistry, Chemical Engineering and Biotechnology, Center for Advanced Low-Dimension Materials, Donghua University, Shanghai 201620, China. E-mail: Lingling.chu1@dhu.edu.cn
First published on 14th April 2020
Abstract
A selective, sequential C–O decarboxylative vinylation/C–H arylation of cyclic alcohol derivatives enabled by visible-light photoredox/nickel dual catalysis is described. This protocol utilizes a multicomponent radical cascade process, i.e. decarboxylative vinylation/1,5-HAT/aryl cross-coupling, to achieve efficient, site-selective dual-functionalization of saturated cyclic hydrocarbons in one single operation. This synergistic protocol provides straightforward access to sp3-enriched scaffolds and an alternative retrosynthetic disconnection to diversely functionalized saturated ring systems from the simple starting materials.
Introduction
In the last decade, the nickel-catalyzed cross-coupling reaction has emerged as a powerful technique to construct C–C bonds in chemical synthesis.1 Notably, nickel-catalyzed multicomponent reactions, that allow for the formation of multiple C–C and/or C–heteroatom bonds thus enabling the streamlined synthesis of complex molecular scaffolds in a single operation, are particularly attractive due to their good compatibility and unique selectivity.2 Significant progress has been achieved in the area of catalytic multicomponent difunctionalization of unsaturated systems, enabling the simultaneous, one-pot installation of two functionalities over double bonds or triple bonds via nickel catalysis (Fig. 1).3,4 In contrast, there has been a lack of reports in which saturated hydrocarbons have been manipulated for cross-coupling at more than one reaction site in one single operation.5 This catalytic strategy would simultaneously install two sp3 C–C bonds in saturated hydrocarbons, readily available and abundant building blocks in organic synthesis; nevertheless, the realization of such a strategy requires the ability to overcome the relative inertness of saturated hydrocarbons and to control the selectivity in the presence of multiple similar chemical bonds.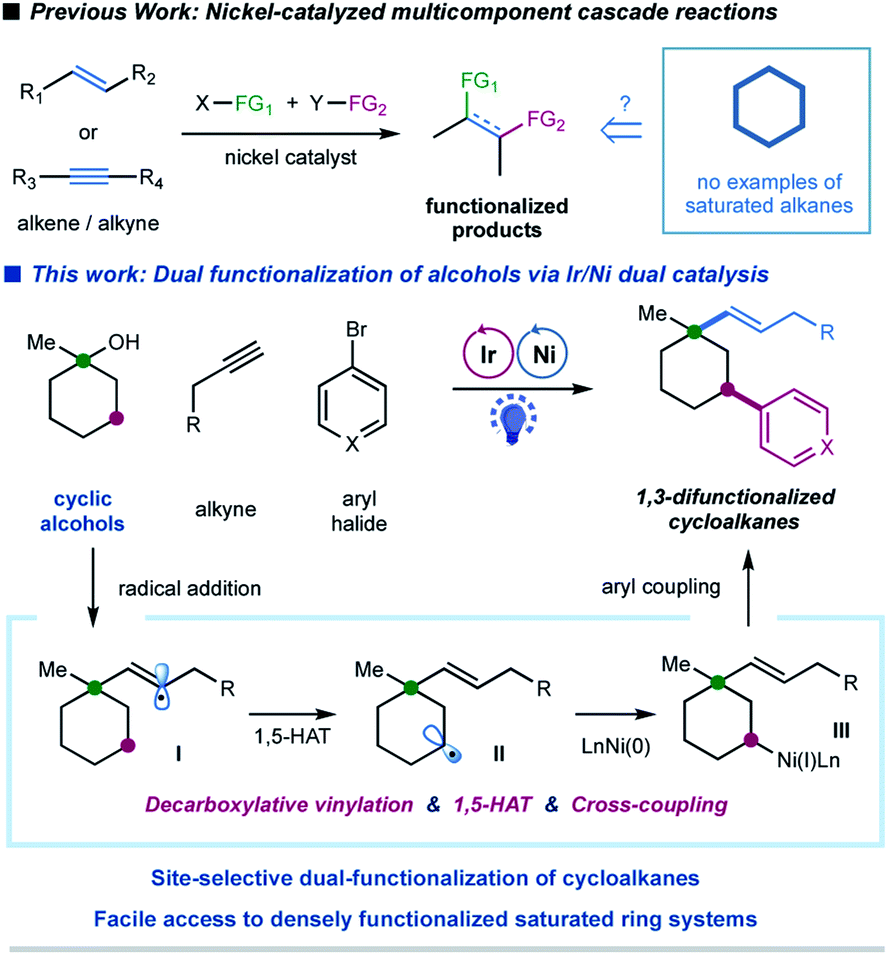 | ||
| Fig. 1 Multicomponent cascade reaction of cycloalkanes enabled by photoredox/nickel dual catalysis. | ||
Alcohols have been widely employed as salient synthetic building blocks in chemical synthesis. Notable advances have been made in selective functionalization of sp3 C–H bonds of alcohols through the hydrogen-atom-transfer (HAT) process.6,7 Moreover, alcohols can function as latent alkylating agents through transition-metal-catalyzed ipso-C–O activation8 or homolytic C–O cleavage.4j,k,9,10 These catalytic approaches enable the facile construction of diverse structural motifs from abundant alcohols. Nevertheless, one single transformation that combines ipso-C–O functionalization and remote C–H functionalization of alcohols, which would provide an attractive platform for the synthesis of highly functionalized sp3-rich scaffolds,11 has not been reported yet. We herein report a selective, cascade C–O bond vinylation/C–H bond arylation of alcohols that achieves site-selective dual carbofunctionalization of simple abundant aliphatic alcohols via a photoredox/nickel12-enabled multicomponent radical cascade process. This strategy relies on a synergistic combination of alkyl oxalate decarboxylation, 1,5-hydrogen atom transfer (1,5-HAT) of the vinyl radical, and C(sp3)–C(sp2) cross-coupling. Our original design for this radical cascade strategy is outlined in Fig. 1. Specifically, we envisioned that a radical addition of alkylalkyne would give rise to a σ-type vinyl radical I, which would prefer to undergo an intramolecular 1,5-HAT process to yield a nucleophilic alkyl radical species II.13 Subsequent nickel-mediated coupling of alkyl radial II with aryl halides would forge a C(sp3)–Ar bond,12 and finally lead to a sequential two-site functionalization of oxalates, i.e. C–O decarboxylative vinylation & C–H arylation (Fig. 1). We are particularly interested in exploring cyclic alkyl oxalates to construct highly functionalized cycloalkanes,14 due to two considerations: (i) this synergistic cascade process would undoubtedly expedite the synthesis of challenging aliphatic rings, and more importantly, would provide a novel retrosynthetic disconnection for complex saturated ring systems from simple starting materials; (ii) conformation of cyclic substrates might be beneficial for the intramolecular 1,5-HAT migration of vinyl radical species.15
Results and discussion
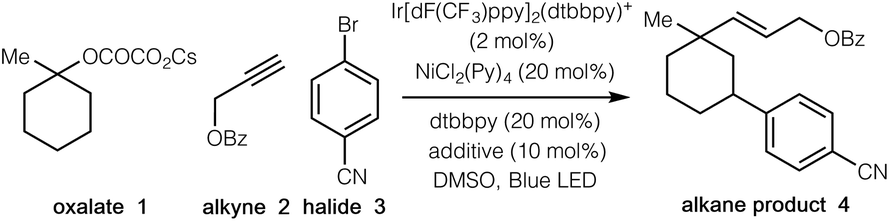
We began our investigations by employing tertiary cyclic oxalate 1 as a model substrate to test the possibilities (Table 1). Pleasingly, we found that irradiation of a DMSO solution of 1, aliphatic alkyne 2, and 4-bromobenzonitrile 3 did afford the desired 1,3-vinylarylation product 4 in 84% yield in the presence of catalytic Ir[dF(CF3)ppy]2(dtbbpy)PF6, NiCl2(Py)4, 4,4′-di-tert-butyl-2,2′-dipyridyl (dtbbpy), and bis(4-methoxyphenyl)methanone (Table 1, entry 1). The countercation of oxalates impacted the reaction efficiency to a small extent, while the use of cesium oxalates afforded higher yields than the corresponding Li, Na, K salts (entries 2–4). Further evaluation indicated that both Ni(II) and Ni(0) catalysts were able to promote the desired transformation, with the precatalyst NiCl2(Py)4 proving optimal (entries 5–9). Control experiments demonstrated that the photocatalyst, the nickel catalyst, and visible light were all essential to this synergistic cascade process, as the desired 1,3-vinylarylation products were not observed in the absence of any of these components (entries 10–12). Nevertheless, low conversion (27% yield) was still observed in the absence of the bipyridine ligand (entry 13). The use of bis(4-methoxyphenyl)methanone as an additive proved to be slightly beneficial to the reaction efficiency (entry 14).13|
|
||
|---|---|---|
| Entry | Variations from standard conditions | Yieldb |
| a Reaction conditions: Ir-1 (2 mol%), NiCl2(Py)4 (20 mol%), dtbbpy (20 mol%), alkene 2 (0.1 mmol), bromide 3 (2.0 equiv.), oxalate 1 (3.0 equiv.), bis(4-methoxyphenyl)methanone (additive) (10 mol%), DMSO [0.05 M], 37 °C, 90W blue LED. b Yields were determined by 1H NMR analysis of the crude reaction mixtures. | ||
| 1 | None | 84% |
| 2 | ROCOCO2Li | 55% |
| 3 | ROCOCO2Na | 71% |
| 4 | ROCOCO2K | 62% |
| 5 | NiCl2·DME | 70% |
| 6 | NiCl2·(PPh3)2 | 63% |
| 7 | NiBr2·dtbbpy | 52% |
| 8 | NiI2 | 40% |
| 9 | Ni(COD)2 | 34% |
| 10 | w/o photocatalyst | 0 |
| 11 | w/o nickel catalyst | 0 |
| 12 | w/o visible light | 0 |
| 13 | w/o ligand | 27% |
| 14 | w/o bis(4-methoxyphenyl)methanone | 78% |
With the optimal conditions in hand, we next turned our attention to evaluating the applicability of substrates as well as the potential limitations of this dual functionalization protocol. As shown in Scheme 1, a number of tertiary cyclic oxalates, readily prepared from the corresponding alcohols, could undergo the sequential C–O vinylation/C–H arylation with excellent site-selectivity, installing both vinyl and aryl functionalities onto the skeletons of alcohols under redox-neutral and mild conditions (products 4–14, 36–77% yields). A number of substituents including alkyl, ketal, ketone, and alkene on the cyclohexyl alcohols were tolerated in this dual catalytic cascade system, furnishing multi-substituted cyclic alkanes in a single operation under mild conditions (products 5–9, 36–77% yields). Nonetheless, steric hindrance of the substituents was found to have a considerable effect on the reaction efficiency: installing a methyl group at the α- or γ-position, or replacing methyl with ethyl at the ipso position of cyclohexyl oxalates resulted in decreased efficiency (products 6–8, 36–54% yields). Pleasingly, saturated O-, S-, and N-heterocyclic oxalates turned out to be viable substrates, yielding the vinyl/aryl-disubstituted saturated heterocycles with good efficiency (products 10–13, 50–76% yields). Moreover, cyclopentyl oxalates were also competent substrates, delivering the 1,3-difunctionalized cyclopentane products in synthetically useful yields (product 14, 40% yield).
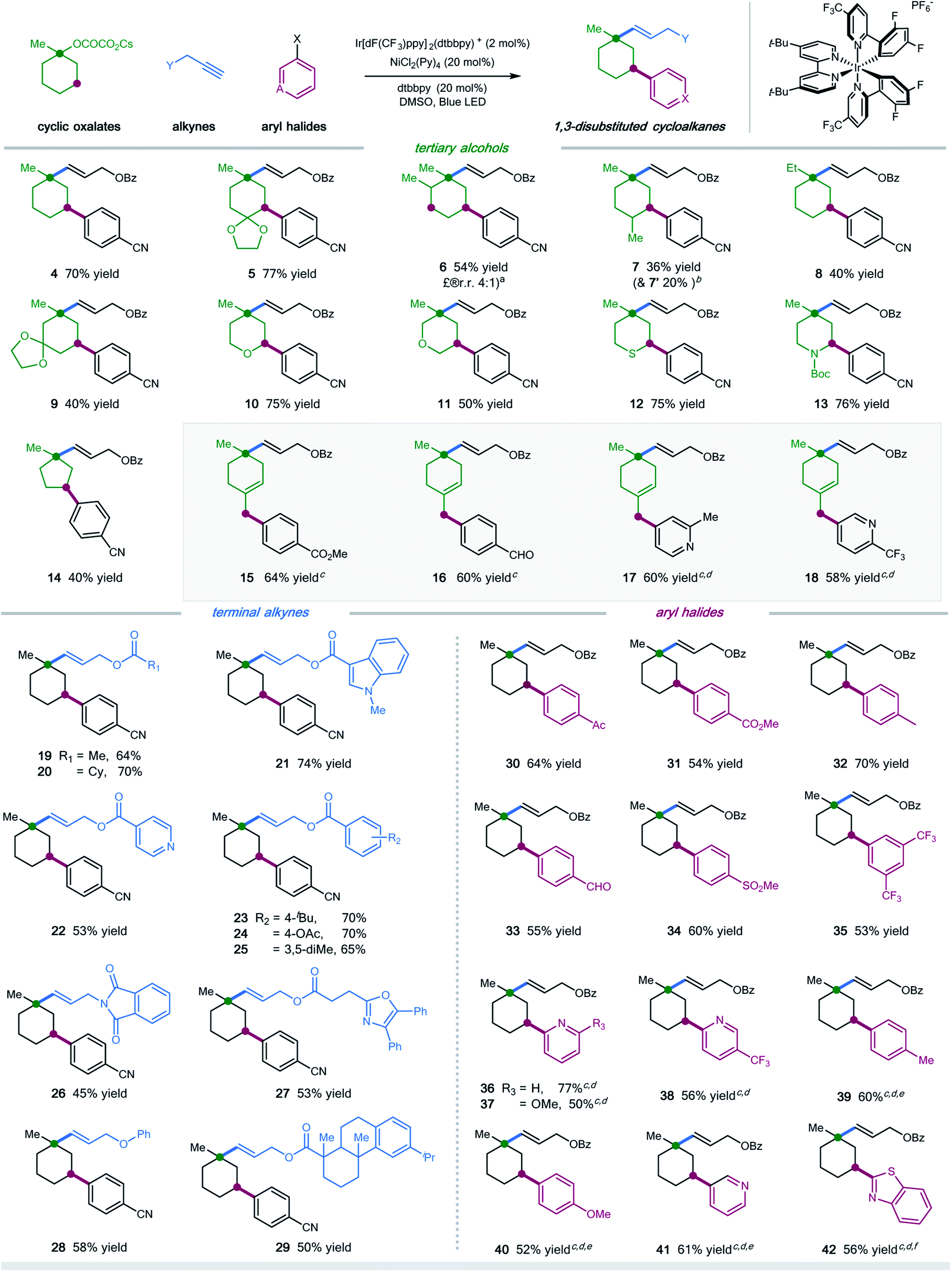 | ||
Scheme 1 Substrate scope. Ir[dF(CF3)ppy]2(dtbbpy)PF6 (2 mol%), NiCl2(Py)4 (20 mol%), dtbbpy (20 mol%), bis(4-methoxyphenyl)methanone (10 mol%), alkyne (0.1 mmol), oxalate (3.0 equiv.), aryl halide (2.0 equiv.), DMSO [0.05 M], 37 °C, 90W blue LED. Isolated yields, ratios of diastereoisomers determined by 1H NMR analysis are between 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:1.2. See the ESI† for experimental details. aRatio of regioisomers was determined via HPLC; bmolecular structure of 7′ is shown in the ESI;†cw/o bis(4-methoxyphenyl)methanone as additive; dDMSO/EA (4:1); eemploying aryl iodides; femploying aryl chlorides. :1 and 1:1.2. See the ESI† for experimental details. aRatio of regioisomers was determined via HPLC; bmolecular structure of 7′ is shown in the ESI;†cw/o bis(4-methoxyphenyl)methanone as additive; dDMSO/EA (4:1); eemploying aryl iodides; femploying aryl chlorides. | ||
Unfortunately, cyclic oxalates with larger or smaller ring sizes (e.g. 4- and 7-membered cyclic oxalates), bicyclic oxalates, and linear oxalates were unsuccessful substrates for this cascade protocol, probably due to the less favorable conformation (for unsuccessful oxalates, see Section 5, page S62 in the ESI†). Interestingly, the reaction of 4-methylenecyclohexyl oxalate with aryl bromides under optimal conditions afforded the 1,5-vinylarylation products in moderate yields (products 15–18, 58–64% yields). Excellent chemoselectivity was observed in this case, with no observations of 1,3-difunctionalized products. We reasoned that allyl-Ni species, generated via intramolecular 1,5-HAT followed by nickel trapping, underwent a selective coupling with aryl bromide at the terminal position, probably due to steric hindrance, to afford the cyclohexene product. This protocol represents a new and efficient platform to construct highly functionalized saturated heterocycles, important structural scaffolds for bioactive molecules, from simple starting materials.
Next, we examined the scope with respect to the alkyne component (Scheme 1). Pleasingly, a wide range of terminal alkylalkynes could be efficiently employed in this cascade protocol, yielding the corresponding trans-alkenes in moderate to good yields (products 19–29, 45–74% yields). Notably, aliphatic alkynes tethered with complex molecules, exemplified by oxaprozin and dehydroabietic acid, also worked with moderate efficiency, further demonstrating the amenability of this synergistic strategy for late-stage manipulations (products 27 and 29, 53% and 50% yield, respectively). Nevertheless, internal alkynes proved to be inefficient substrates, most of which remained intact under the standard conditions.
Finally, we explored the scope of aryl halides in this multicomponent transformation (Scheme 1). Aryl bromides containing electron-withdrawing substituents, including aldehydes, ketones, esters, nitriles, sulfones, and trifluoromethylates, are competent coupling partners under optimal conditions, delivering the desired products with good efficiency (products 30–35, 53–70% yields). The mild conditions allow for the good tolerance of these important functionalities. This reaction is amenable to heteroaryl halides, selectively installing pyridines and benzothiazoles into cyclohexanes with moderate yields (products 36–38 and 41–42, 50–77% yields). Additionally, (hetero)aryl iodides/chlorides also participated in this sequential C–O/C–H dual functionalization process smoothly (products 39–42, 52–61% yields). Electron-rich aryl halides were applicable coupling partners, albeit with decreased efficiency (products 39–40).
To further demonstrate the synthetic application of our cascade protocol, we have performed several transformations by utilizing the alkene functionality (Scheme 2). The double bond of compound 41 readily underwent selective hydrogenation with H2 in the presence of a Pd/C catalyst (product 43). Epoxidation of compound 30 with m-CPBA gave epoxide 44 in 65% yield. Furthermore, ozonolysis of 30 led to the formation of aldehyde 45, which could subsequently be oxidized to the corresponding carboxylic acid 46, or be reduced to the related alcohol 47 in good yield.
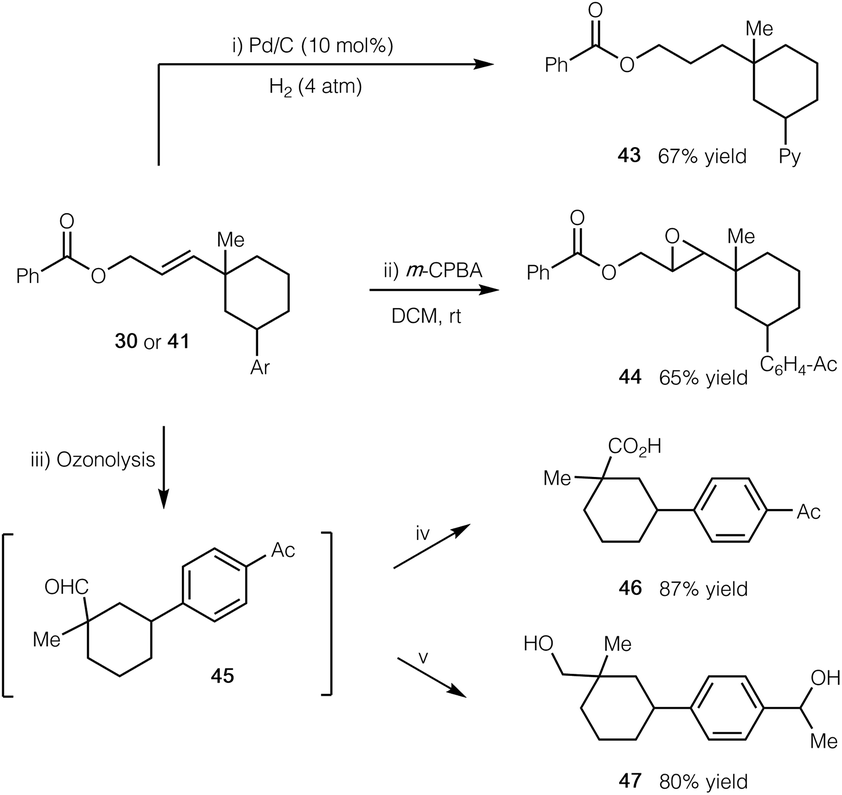 | ||
| Scheme 2 Synthetic manipulations of products. (i) Pd/C (10 mol%), H2 (4 atm), THF, rt; (ii) m-CPBA (2 equiv.), CH2Cl2, rt; (iii) O3, CH2Cl2, −78 °C; Me2S (10 equiv.), rt; (iv) NaClO2 (5 equiv.), H2O2 (4 equiv.), MeCN, 0 °C to rt; (v) NaBH4 (10 equiv.), MeOH, 0 °C to rt. See the ESI† for experimental details. | ||
To shed some light on the potential reaction pathway, we have conducted several mechanistic experiments (Scheme 3). Reaction of ethynylcyclopropane with oxalate 1 and aryl bromide 3 gave the allene product 48, presumably generated via a ring-opening/cross-coupling process, indicating the involvement of vinyl radical species (Scheme 3A). Initially, we assumed that the 1,5-HAT process could be related to the subtle conformation of oxalates. We also found that the expected 1,5-HAT process is reliant on the nature of the vinyl radicals (σ-type vs. π-type). For instance, competitive experiments between aryl and aliphatic alkynes showed that arylalkynes exhibited higher reactivity to afford the exclusive formation of 1,2-alkylarylation products,4j and the 1,5-HAT/coupling product was not observed in this case (Scheme 3B). Regarding the coupling step, we prepared the ligated Ar–Ni(II) complex 50,16 and found that the stoichiometric reaction of the Ar–Ni(II) complex with alkyne and oxalate didn't form the desired 1,3-disubstituted cycloalkane product 51, suggesting that Ar–Ni(II) might not be a reactive intermediate for this cascade transformation (Scheme 3C). Nevertheless, the Ar–Ni(II) complex was able to catalyze the synergistic cascade reaction, giving the 1,3-disubstituted cyclohexane 33 in 30% yield in acetone (Scheme 3D).
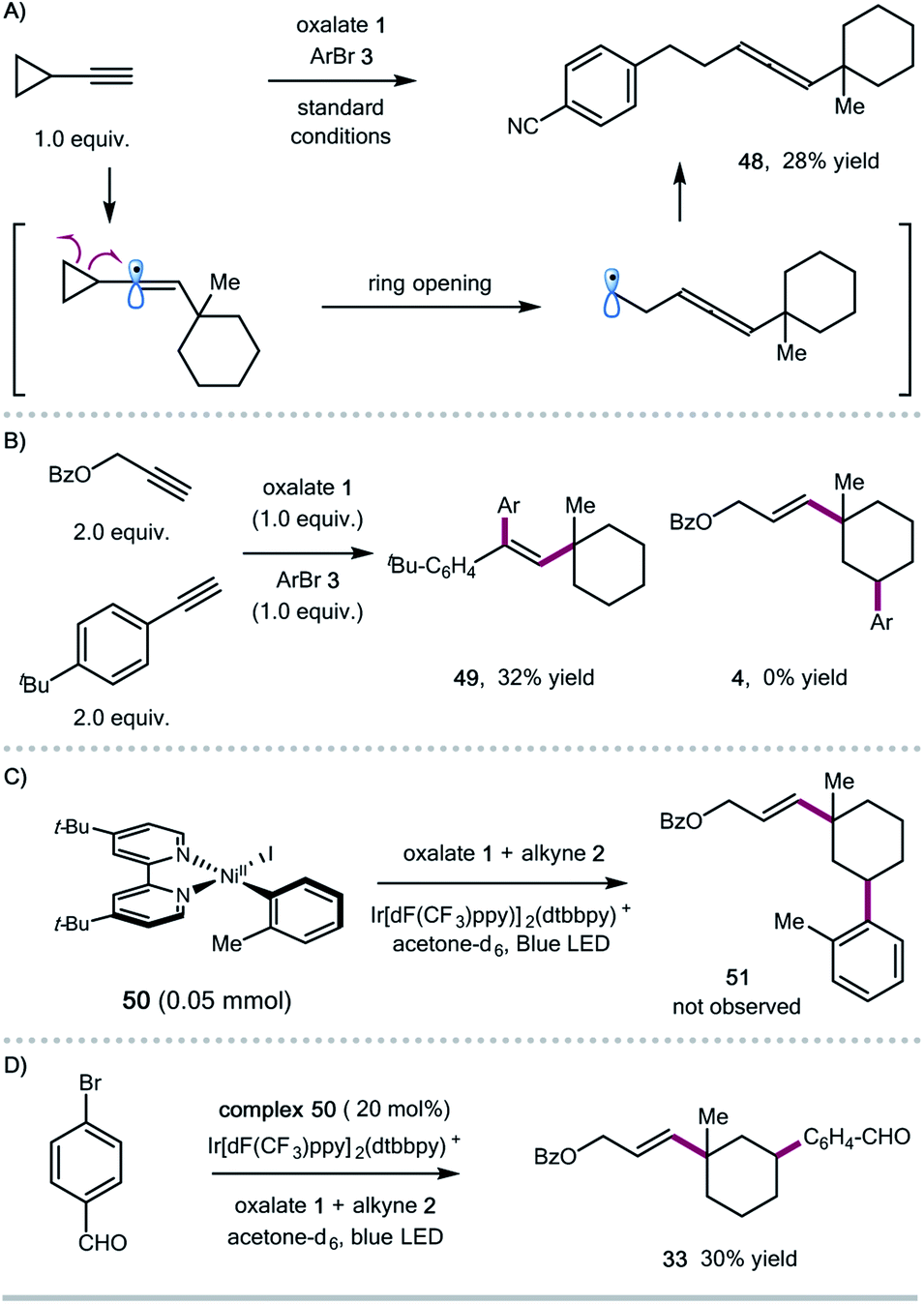 | ||
| Scheme 3 Mechanistic studies: (A) competitive experiments between arylalkyne and alkylalkyne; (B) radical clock experiment; (C) stoichiometric reaction of the Ar–Ni(II) complex; (D) reaction in the presence of a catalytic amount of the Ar–Ni(II) complex. | ||
On the basis of these experimental results as well as literature precedents,12,17 a plausible mechanism for this photoredox/nickel-catalyzed dual functionalization is depicted in Scheme 4. A thermodynamically feasible SET event between the photoexcited Ir-catalyst B and oxalate C would generate a tertiary alkyl radical Dvia decarboxylation, followed by a radical addition of alkyne to give rise to a σ-type vinyl radical E. The resulting vinyl radical E would go through an intramolecular 1,5-HAT to selectively activate the sp3 C–H of oxalates and to afford a nucleophilic, secondary alkyl radical species G. Subsequent interception of alkyl radical G by Ni(0) H would generate an alkyl-Ni(I) species I, which then undergoes an oxidative addition with aryl halide J to yield an (alkyl)(aryl)Ni(III) intermediate K.17 This high-valent Ni(III) complex K would undergo a feasible reductive elimination to forge the C(sp3)–Ar bond and furnish the final product L as well as Ni(I) species M. Finally, a SET event between Ni(I) M and the reduced Ir(II) F would regenerate Ni(0) H and the ground state Ir(III) A to close these two catalytic cycles. At this stage, we could not preclude another pathway that proceeds via oxidative addition of aryl bromide to Ni(0) followed by interception of Ar–Ni(II) N by alkyl radical species to afford the same (alkyl)(aryl)Ni(III) K.
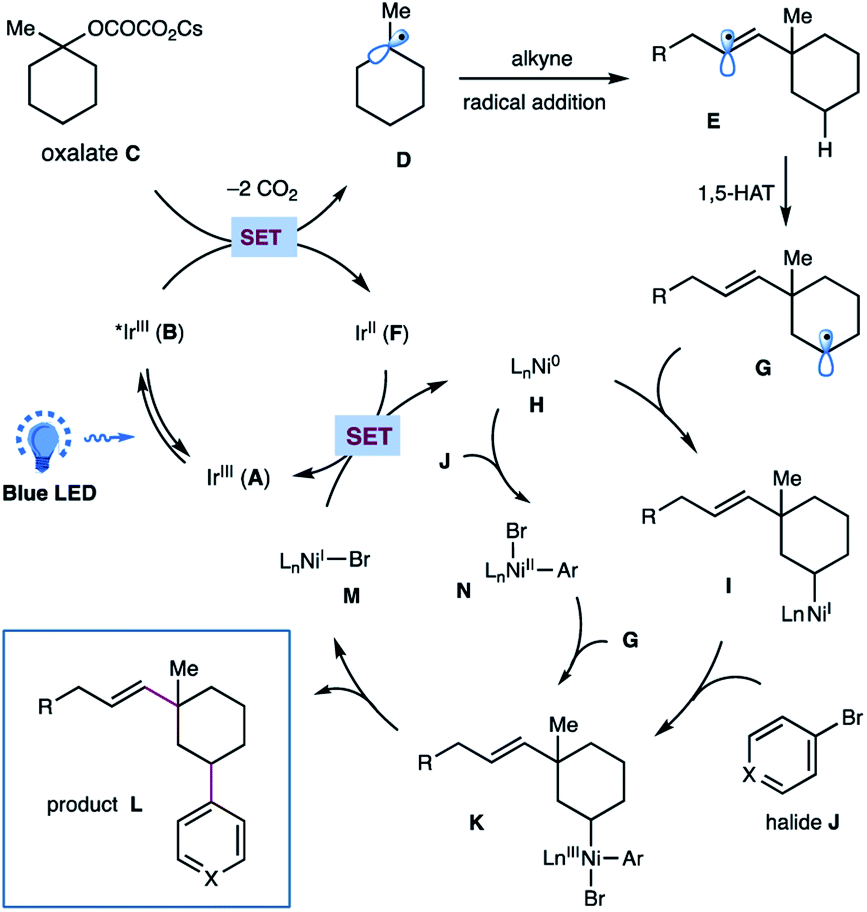 | ||
| Scheme 4 Proposed mechanism for this metallaphotoredox-catalyzed dual functionalization of cyclic oxalates. | ||
Conclusions
In summary, we have developed a sequential C–O decarboxylative vinylation/C–H arylation of cyclic oxalates via photoredox/nickel dual catalysis. This synergistic protocol enables efficient and selective assembly of both vinyl and aryl functionalities onto saturated cyclic hydrocarbons in one single operation under mild and redox-neutral conditions, providing a new and complementary retrosynthetic method for densely functionalized saturated cyclic hydrocarbons. The mild conditions allow for excellent compatibility of functional groups and substrate scope in the oxalates, alkynes, and (hetero)aryl halides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21971036, 21702029, and 21901036), the Shanghai Sailing Program (19YF1400300), and the Fundamental Research Funds for the Central Universities for financial support.Notes and references
- For selected reviews, see: (a) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed; (b) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed; (c) V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS; (d) J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed; (e) G. C. Fu, ACS Cent. Sci., 2017, 3, 692–700 CrossRef CAS PubMed; (f) A. Kaga and S. Chiba, ACS Catal., 2017, 7, 4697–4706 CrossRef CAS; (g) M. R. Kwiatkowski and E. J. Alexanian, Acc. Chem. Res., 2019, 52, 1134–1144 CrossRef CAS PubMed.
- For selected reviews, see: (a) H.-M. Huang, M. H. Garduño-Castro, C. Morrill and D. J. Procter, Chem. Soc. Rev., 2019, 48, 4626–4638 RSC; (b) J. Montgomery, Acc. Chem. Res., 2000, 33, 467–473 CrossRef CAS PubMed.
- For recent reviews, see: (a) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS PubMed; (b) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598–6608 RSC; (c) R. Giri and S. Kc, J. Org. Chem., 2018, 83, 3013–3022 CrossRef CAS PubMed; (d) R. K. Dhungana, S. Kc, P. Basnet and R. Giri, Chem. Rec., 2018, 18, 1314–1340 CrossRef CAS PubMed; (e) J.-S. Zhang, L. Liu, T. Chen and L.-B. Han, Chem.–Asian J., 2018, 13, 2277–2291 CrossRef CAS PubMed; (f) J. Derosa, V. T. Tran, V. A. van der Puyl and K. M. Engle, Aldrichimica Acta, 2018, 51, 21–32 CAS; (g) J. Lin, R.-J. Song, M. Hu and J.-H. Li, Chem. Rec., 2019, 19, 440–451 CrossRef CAS PubMed; (h) J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Chem. Sci., 2020 10.1039/c9sc06006e; (i) H.-Y. Tu, S. Zhu, F.-L. Qing and L. Chu, Synthesis, 2020, 52, 1346–1356 CrossRef CAS.
- For recent examples of Ni-catalyzed intermolecular difunctionalization of unsaturated bonds, see: (a) F. Xue, J. Zhao, T. S. A. Hor and T. Hayashi, J. Am. Chem. Soc., 2015, 137, 3189–3192 CrossRef CAS PubMed; (b) Z. Li, A. García-Domínguez and C. Nevado, Angew. Chem., Int. Ed., 2016, 55, 6938–6941 CrossRef CAS PubMed; (c) J. Derosa, V. T. Tran, M. N. Boulous, J. S. Chen and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 10657–10660 CrossRef CAS PubMed; (d) A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835–6838 CrossRef PubMed; (e) B. Shrestha, P. Basnet, R. K. Dhungana, S. Kc, S. Thapa, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2017, 139, 10653–10656 CrossRef CAS PubMed; (f) J. Derosa, R. Kleinmans, V. T. Tran, M. K. Karunananda, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 17878–17883 CrossRef CAS PubMed; (g) S. Thapa, R. K. Dhungana, R. T. Magar, B. Shrestha, S. Kc and R. Giri, Chem. Sci., 2018, 9, 904–909 RSC; (h) X. Zhao, H.-Y. Tu, L. Guo, S. Zhu, F.-L. Qing and L. Chu, Nat. Commun., 2018, 9, 3488 CrossRef PubMed; (i) D. Anthony, Q. Lin, J. Baudet and T. Diao, Angew. Chem., Int. Ed., 2019, 58, 3198–3202 CrossRef CAS PubMed; (j) L. Guo, F. Song, S. Zhu, H. Li and L. Chu, Nat. Commun., 2018, 9, 4543 CrossRef PubMed; (k) L. Guo, H. Y. Tu, S. Zhu and L. Chu, Org. Lett., 2019, 21, 4771–4776 CrossRef CAS PubMed; (l) K. F. Zhang, K. J. Bian, C. Li, J. Sheng, Y. Li and X. S. Wang, Angew. Chem., Int. Ed., 2019, 58, 5069–5074 CrossRef CAS PubMed; (m) C. Zhu, H. F. Yue, B. Maity, I. Atodiresei, L. Cavallo and M. Rueping, Nat. Catal., 2019, 2, 678–687 CrossRef CAS; (n) A. A. Kadam, T. L. Metz, Y. Qian and L. M. Stanley, ACS Catal., 2019, 5651–5656 CrossRef CAS; (o) A. Garcia-Dominguez, R. Mondal and C. Nevado, Angew. Chem., Int. Ed., 2019, 58, 12286–12290 CrossRef CAS PubMed; (p) C. Xu, Z.-F. Yang, L. An and X. Zhang, ACS Catal., 2019, 8224–8229 CrossRef CAS; (q) W. Shu, A. Garcia-Dominguez, M. T. Quiros, R. Mondal, D. J. Cardenas and C. Nevado, J. Am. Chem. Soc., 2019, 141, 13812–13821 CrossRef CAS PubMed; (r) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069–20078 CrossRef CAS PubMed; (s) L. Huang, C. Zhu, L. Yi, H. Yue, R. Kancherla and M. Rueping, Angew. Chem., Int. Ed., 2020, 59, 457–464 CrossRef CAS PubMed; (t) H. Yue, C. Zhu, R. Kancherla, F. Liu and M. Rueping, Angew. Chem., Int. Ed., 2020, 59, 5738–5746 CrossRef CAS PubMed.
- For selected reviews, see: (a) Q.-F. Wu, X.-B. Wang, P.-X. Shen and J.-Q. Yu, ACS Catal., 2018, 8, 2577–2581 CrossRef CAS PubMed; (b) M. Shang, K. S. Feu, J. C. Vantourout, L. M. Barton, H. L. Osswald, N. Kato, K. Gagaring, C. W. McNamara, G. Chen, L. Hu, S. Ni, P. Fernández-Canelas, M. Chen, R. R. Merchant, T. Qin, S. L. Schreiber, B. Melillo, J.-Q. Yu and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8721 CrossRef CAS PubMed.
- (a) S.-Y. Zhang, F.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2011, 40, 1937–1949 RSC; (b) E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70–73 CrossRef CAS PubMed; (c) Z. Ren, F. Mo and G. Dong, J. Am. Chem. Soc., 2012, 134, 16991–16994 CrossRef CAS PubMed; (d) M. Bender, B. W. H. Turnbull, B. R. Ambler and M. J. Krische, Science, 2017, 357, 779 CrossRef CAS PubMed; (e) S. W. Kim, W. Zhang and M. J. Krische, Acc. Chem. Res., 2017, 50, 2371–2380 CrossRef CAS PubMed; (f) D. Lichosyt, Y. Zhang, K. Hurej and P. Dydio, Nat. Catal., 2019, 2, 114–122 CrossRef CAS.
- (a) J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532–1536 CrossRef CAS PubMed; (b) E. A. Wappes, K. M. Nakafuku and D. A. Nagib, J. Am. Chem. Soc., 2017, 139, 10204–10207 CrossRef CAS PubMed; (c) J. Twilton, M. Christensen, D. A. DiRocco, R. T. Ruck, I. W. Davies and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 5369–5373 CrossRef CAS PubMed; (d) A. Hu, J.-J. Guo, H. Pan, H. Tang, Z. Gao and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 1612–1616 CrossRef CAS PubMed; (e) Y. Zhu, K. Huang, J. Pan, X. Qiu, X. Luo, Q. Qin, J. Wei, X. Wen, L. Zhang and N. Jiao, Nat. Commun., 2018, 9, 2625 CrossRef PubMed; (f) K. M. Nakafuku, S. C. Fosu and D. A. Nagib, J. Am. Chem. Soc., 2018, 140, 11202–11205 CrossRef CAS PubMed; (g) L. Niu, J. Liu, X.-A. Liang, S. Wang and A. Lei, Nat. Commun., 2019, 10, 467 CrossRef PubMed; (h) G.-X. Li, X. Hu, G. He and G. Chen, Chem. Sci., 2019, 10, 688–693 RSC.
- For selected reviews on TM-catalyzed C–O activations, see: (a) Y. Obora, ACS Catal., 2014, 4, 3972–3981 CrossRef CAS; (b) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed; (c) D. Shen, D. L. Poole, C. C. Shotton, A. F. Kornahrens, M. P. Healy and T. J. Donohoe, Angew. Chem., Int. Ed., 2015, 54, 1642–1645 CrossRef CAS PubMed; (d) W. M. Akhtar, C. B. Cheong, J. R. Frost, K. E. Christensen, N. G. Stevenson and T. J. Donohoe, J. Am. Chem. Soc., 2017, 139, 2577–2580 CrossRef CAS PubMed; (e) S. M. Pound and M. P. Watson, Chem. Commun., 2018, 54, 12286–12301 RSC; (f) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717–1726 CrossRef CAS PubMed; (g) H. Zeng, Z. Qiu, A. Domínguez-Huerta, Z. Hearne, Z. Chen and C.-J. Li, ACS Catal., 2017, 7, 510–519 CrossRef CAS; (h) A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- For recent examples of visible light photoredox-catalyzed homolytic C–O cleavage, see 4i–4j, as well as (a) C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270–11273 CrossRef CAS PubMed; (b) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87–90 CrossRef CAS PubMed; (c) C. R. Jamison and L. E. Overman, Acc. Chem. Res., 2016, 49, 1578–1586 CrossRef CAS PubMed; (d) X. Zhang and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 13862–13865 CrossRef CAS PubMed; (e) W. Liu, X. Yang, Z.-Z. Zhou and C.-J. Li, Chem, 2017, 2, 688–702 CrossRef CAS; (f) E. D. Nacsa and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 3322–3330 CrossRef CAS PubMed; (g) S. Y. Abbas, P. Zhao and L. E. Overman, Org. Lett., 2018, 20, 868–871 CrossRef CAS PubMed; (h) J. Y. Su, D. C. Grünenfelder, K. Takeuchi and S. E. Reisman, Org. Lett., 2018, 20, 4912–4916 CrossRef CAS PubMed; (i) S. P. Pitre, M. Muuronen, D. A. Fishman and L. E. Overman, ACS Catal., 2019, 9, 3413–3418 CrossRef CAS; (j) N. A. Weires, Y. Slutskyy and L. E. Overman, Angew. Chem., Int. Ed., 2019, 58, 8561–8565 CrossRef CAS PubMed.
- For selected examples on metal-mediated homolytic C–O cleavage, see: (a) H. R. Diéguez, A. López, V. Domingo, J. F. Arteaga, J. A. Dobado, M. M. Herrador, J. F. Quílez del Moral and A. F. Barrero, J. Am. Chem. Soc., 2010, 132, 254–259 CrossRef PubMed; (b) T. Suga and Y. Ukaji, Org. Lett., 2018, 20, 7846–7850 CrossRef CAS PubMed; (c) X.-G. Jia, P. Guo, J. Duan and X.-Z. Shu, Chem. Sci., 2018, 9, 640–645 RSC; (d) Y. Ye, H. Chen, J. L. Sessler and H. Gong, J. Am. Chem. Soc., 2019, 141, 820–824 CrossRef CAS PubMed; (e) Y. Ye, H. Chen, K. Yao and H. Gong, Org. Lett., 2020, 22, 2070–2075 CrossRef CAS PubMed; (f) T. Suga, S. Shimazu and Y. Ukaji, Org. Lett., 2018, 20, 5389–5392 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- For recent reviews on metallaphotoredox catalysis, see: (a) J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS PubMed; (b) Y.-Y. Gui, L. Sun, Z.-P. Lu and D.-G. Yu, Org. Chem. Front., 2016, 3, 522–526 RSC; (c) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (d) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS PubMed.
- (a) D. P. Curran and W. Shen, J. Am. Chem. Soc., 1993, 115, 6051–6059 CrossRef CAS; (b) U. Wille, Chem. Rev., 2013, 113, 813–853 CrossRef CAS PubMed.
- (a) J. C. C. Stephen and H. G. Cutler, Biologically Active Natural Products: Pharmaceuticals, CRC Press, Boca Raton, FL, 2000 Search PubMed; (b) L. N. Mander, Comprehensive Natural Products II: Chemistry and Biology, Elsevier, Amsterdam, 2010 Search PubMed.
- (a) A. Martín, I. Pérez-Martín, L. M. Quintanal and E. Suárez, Org. Lett., 2007, 9, 1785–1788 CrossRef PubMed; (b) L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 50, 1569–1586 CrossRef CAS PubMed.
- K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016–5019 CrossRef CAS PubMed.
- O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896–4899 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01471k |
| ‡ H. L. and L. G. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |