
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePosttranscriptional site-directed spin labeling of large RNAs with an unnatural base pair system under non-denaturing conditions†
Yan
Wang
 a,
Venkatesan
Kathiresan
b,
Yaoyi
Chen‡
a,
Yanping
Hu
a,
Wei
Jiang
b,
Guangcan
Bai
c,
Guoquan
Liu
c,
Peter Z.
Qin
*b and
Xianyang
Fang
*a
a,
Venkatesan
Kathiresan
b,
Yaoyi
Chen‡
a,
Yanping
Hu
a,
Wei
Jiang
b,
Guangcan
Bai
c,
Guoquan
Liu
c,
Peter Z.
Qin
*b and
Xianyang
Fang
*a
aBeijing Advanced Innovation Center for Structural Biology, School of Life Sciences, Tsinghua University, Beijing 100084, China. E-mail: fangxy@tsinghua.edu.cn
bDepartment of Chemistry, University of Southern California, Los Angeles, California 90089, USA. E-mail: pzq@usc.edu
cState Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China
First published on 20th August 2020
Abstract
Site-directed spin labeling (SDSL) of large RNAs for electron paramagnetic resonance (EPR) spectroscopy has remained challenging to date. We here demonstrate an efficient and generally applicable posttranscriptional SDSL method for large RNAs using an expanded genetic alphabet containing the NaM-TPT3 unnatural base pair (UBP). An alkyne-modified TPT3 ribonucleotide triphosphate (rTPT3COTP) is synthesized and site-specifically incorporated into large RNAs by in vitro transcription, which allows attachment of the azide-containing nitroxide through click chemistry. We validate this strategy by SDSL of a 419-nucleotide ribonuclease P (RNase P) RNA from Bacillus stearothermophilus under non-denaturing conditions. The effects of site-directed UBP incorporation and subsequent spin labeling on the global structure and function of RNase P are marginal as evaluated by Circular Dichroism spectroscopy, Small Angle X-ray Scattering, Sedimentation Velocity Analytical Ultracentrifugation and enzymatic assay. Continuous-Wave EPR analyses reveal that the labeling reaction is efficient and specific, and Pulsed Electron–Electron Double Resonance measurements yield an inter-spin distance distribution that agrees with the crystal structure. The labeling strategy as presented overcomes the size constraint of RNA labeling, opening new avenues of spin labeling and EPR spectroscopy for investigating the structure and dynamics of large RNAs.
Introduction
Many large RNAs, such as long non-coding RNAs that are arbitrarily defined as transcripts longer than 200 nucleotides (nts) but with no or little protein coding potentials, have been found to play widespread and crucial roles in a variety of biological processes, and have emerged as key players in the etiology of several disease states.1 Similar to proteins, RNAs are capable of forming complex secondary and high-order structures that dictate their functions. Knowledge about the structure, dynamics and interaction of these large RNAs is of critical importance to understand their functions, and lay the groundwork for the development of novel RNA-based and/or RNA-targeted therapeutics.2 Due to the increased flexibility of large RNAs, structural studies of large RNAs using techniques such as X-ray crystallography and cryo-EM remain challenging.3–5 While solution techniques, such as NMR and small angle X-ray and neutron scattering (SAXS/SANS), can provide unique structural information and address the inherent flexibility and dynamics of biomolecules, SAXS/SANS has relatively low resolution,6,7 and NMR is constrained by the size of the molecule, although the size-limit has been increased due to active developments, such as segment labelling.8,9 An exciting direction in investigating the structure, dynamics and interaction of large RNAs and RNA-protein complexes is via an integrative approach that combines multiple techniques, such as NMR, SAXS, and electron paramagnetic resonance (EPR).10–16 Among these techniques, EPR methods, for example pulsed electron–electron double resonance (PELDOR or DEER) spectroscopy, can provide pair-wise distance information on the 20–100 Å length scale under biologically relevant conditions with a small amount of materials, and the measurements are not restricted by the size of the biomolecule.17,18 A number of studies have demonstrated that EPR-measured distance constraints, coupled with NMR and computational modeling, are highly informative in investigating the global structure and conformational dynamics of RNA and RNA-protein complexes.12,17,19–24Given that the majority of biomolecules are diamagnetic, efficient and site-specific incorporation of spin labels such as nitroxide into biomolecules is a prerequisite for successful EPR (including PELDOR) measurements. However, site-directed spin labeling (SDSL) remains challenging for large RNAs.25 To date, RNAs have been prepared in sufficient amounts for structural studies by either solid-phase chemical synthesis or by in vitro transcription.26 The majority of established RNA spin-labeling methods rely on solid-phase chemical synthesis, with the labels either incorporated directly during synthesis or post-synthetically.12,25,27,28 The chemical synthesis method has great capability and flexibility in generating chemically diverse RNAs, but currently remains generally applicable to RNAs smaller than 100 nts. The size limits might be mitigated by combining chemical synthesis with splint-assisted enzymatic ligation,29,30 in which larger RNAs are assembled from smaller pieces or by complementary-addressed approaches, in which custom-designed DNA guides are used to direct a label to the desired RNA sites.31,32 However, these methods are generally laborious, resulting in low yield of the final products. In addition, they require denaturing purification steps to remove the DNA splints or guides, which may interfere with folding of RNAs, in particular large RNAs. As such, alternative efficient approaches for SDSL of large RNAs under non-denaturing conditions are highly desirable.
A promising approach has recently emerged for preparation of site-specifically labeled RNAs ranging from tens to thousands of nucleotides using a combination of the expanded genetic alphabets and in vitro transcription catalyzed by T7 RNA polymerase.33–35 Over the past two decades, Benner's, Hirao's and Romesberg's groups have reported a number of unnatural base pairs (UBPs) that can function as a third base pair in replication, transcription and/or translation, thus expanding the genetic alphabets.36 Through the formation of UBPs, new components with different physicochemical properties can be introduced site-specifically into DNAs or RNAs.34,37 For example, the UBPs of NaM-5SICS, developed by Romesberg's group, have been utilized for preparations of site-specifically labeled large RNAs with fluorophores Cy3 and Cy5 for the smFRET study.38
A TPT3-NaM UBP, which was originally developed by Romesberg's group, has been shown to maintain similar efficiency and fidelity to a natural base pair in in vitro replication, but its potential in site-specific labeling of large RNAs has not been fully explored.39,40 Very recently, a nitroxide-modified triphosphate TPT3 (TPT3No) was developed to achieve site-specific spin labeling of RNAs through in vitro transcription.41 Here, we present the synthesis of alkyne-derivatized TPT3 (rTPT3COTP) (Fig. 1) and demonstrate its application in posttranscriptional site-directed spin labeling of large RNAs. Using the TPT3-NaM UBP scaffold and click chemistry, we have succeeded in covalently attaching azide-modified nitroxide spin labels site-specifically to a 419 nt ribozyme, the ribonuclease P (RNase P) RNA from Bacillus stearothermophilus.42 Continuous-Wave (CW-) EPR analyses show that the nitroxide labels are attached to RNAs with high specificity and efficiency under non-denaturing conditions. Circular Dichroism (CD), small angle X-ray scattering (SAXS), sedimentation velocity analytical ultracentrifugation (SV-AUC) and enzymatic pre-tRNA processing assay demonstrate that the UBP modification and the subsequent spin labeling minimally interfere with the global folding and catalytic function of the RNAs. EPR measurements, in particular the inter-label distance, are shown to report the folded RNA structure. The data clearly demonstrate the applicability of this TPT3-based SDSL approach to large RNAs, thus opening new possibilities for application of spin labeling and EPR spectroscopy in investigating the structure and dynamics of large RNAs.
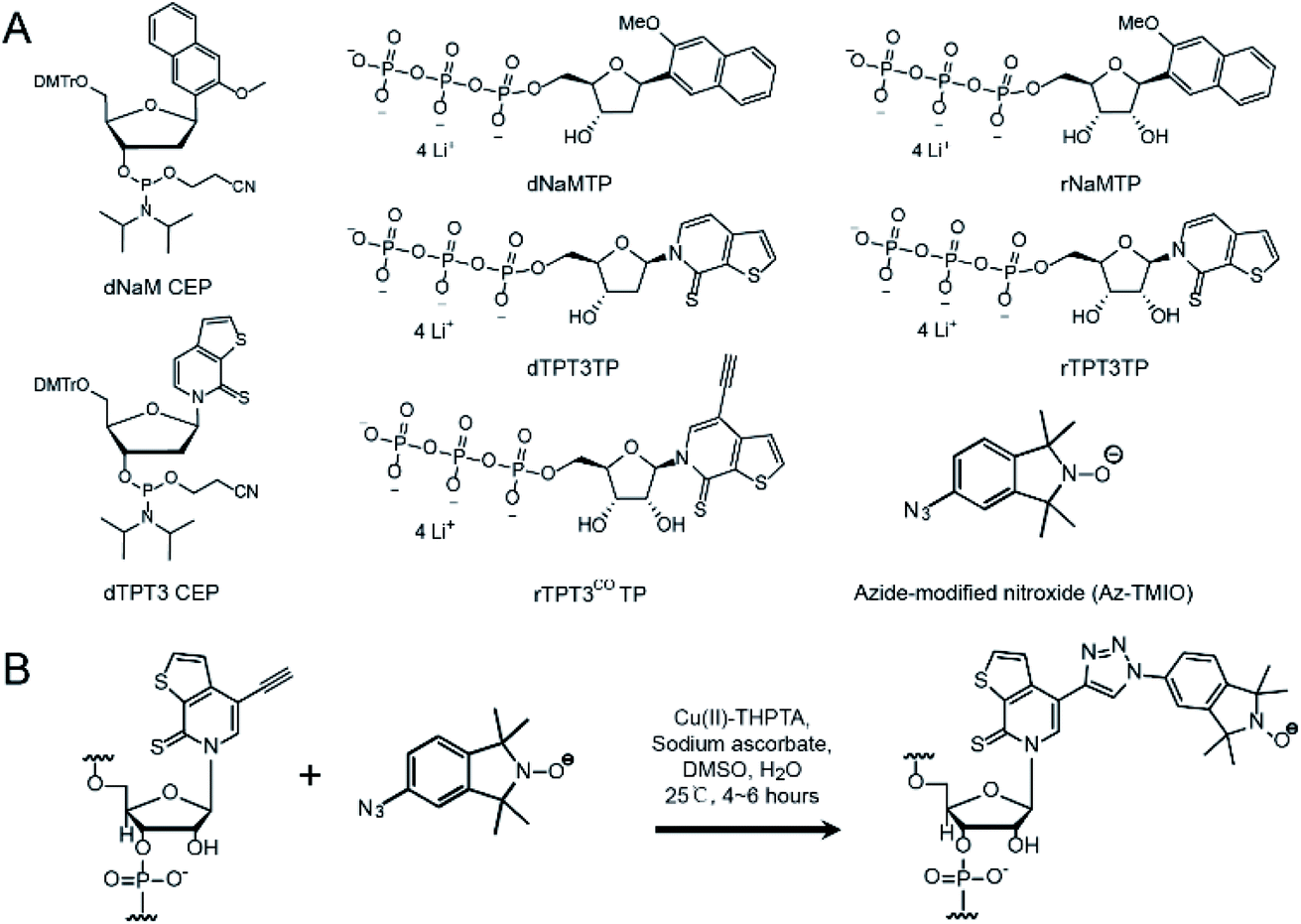 | ||
| Fig. 1 Compounds used in this work. (A) Chemical structure of TPT3 and NaM variants and the azide-modified nitroxide (AZ-TMIO). (B) Cu(I) catalyzed click chemistry reaction between alkyne containing TPT3 and azide modified nitroxide. | ||
Results and discussion
Synthesis of unnatural nucleotides and nitroxide
Three variants of TPT3 and NaM (Fig. 1A), including deoxyribonucleotide phosphoramidites (dTPT3-CEP and dNaM-CEP, for DNA primer synthesis), triphosphorylated deoxyribonucleotides (dTPT3TP and dNaMTP, for PCR) and ribonucleotides (rTPT3TP and rNaMTP, for transcription), were custom synthesized according to procedures described in the literature.34,40,43 In addition, a TMIO nitroxide functionalized with an azide (Az-TMIO) (Fig. 1A) was custom synthesized according to literature procedures,28,44 and an alkyne-modified ribonucleotide of TPT3 in the triphosphorylated form (rTPT3COTP) (Fig. 1A) was custom synthesized as described in detail in ESI S1.† Using these reagents, alkyne-modified TPT3 is stably incorporated at specific site(s) during RNA in vitro transcription, allowing subsequent covalent attachment of azide-containing nitroxide via click chemistry (Fig. 1B). This mitigates degradation of azide and nitroxide due to the reducing conditions used in in vitro transcription.UBP modification and spin labeling of RNase P RNAs
To demonstrate the applicability of the TPT3-NaM UBP in site-directed spin labeling of large RNAs, we have chosen the 419 nt RNase P RNA from Bacillus stearothermophilus as a model, for which a crystal structure is available (PDB ID: 2A64).45 Two nucleotides, U67 located at P5.1 and U86 located at the internal loop between P7, P5, and P5.1, are selected for nitroxide labeling (Fig. S1A†). The crystal structure shows that N 1s of these two nucleotides is separated by 3.32 nm (Fig. S1B†), and UBP modification and subsequent labeling at these sites are not expected to cause drastic perturbation to the global folding of the RNA.It has been previously reported that dNaM in the template strand of DNA is more efficient in directing the incorporation of its partner UBP into RNA.46 Therefore, dsDNA templates containing dNaM at the template strand are prepared to direct the incorporation of TPT3 or TPT3CO into RNase P transcripts at specific sites. To produce dsDNA templates containing UBP, phosphoramidites of dNaM or dTPT3 are used to first introduce UBPs into the DNA primers by solid-phase chemical synthesis (Fig. 2A). For single-site labeling, each DNA primer contains one dNaM or dTPT3 at the desired modification site. For double labeling, as the two modified sites (U67 and U86) in the RNase P RNA are close in the primary sequence, longer primers are designed to include either two dNaMs or two dTPT3s simultaneously. With the expanded six-letter genetic alphabet including the natural A–T and G–C base pairs and the unnatural NaM-TPT3 base pair,38,47 a two-step overlap extension PCR is employed to amplify and assemble the DNA templates containing one or two dNaMs at the template strand (Fig. 2B). Using the U67/86 double-modified RNase P as an example, four primers are initially synthesized: the pMVF (forward) primer targeting a sequence 624 bp upstream of the T7 promoter, the 67/86R (reverse) and 67/86F (forward) primers containing two dNaMs and dTPT3s, respectively, at sites corresponding to U67 and U86, and the RPR (reverse) primer targeting the 3′ end sequence of RNase P (Fig. 2A). The DNA template for transcription is then produced in two steps. In Step I, directed by the two pairs of DNA primers (pMVF, 67/86R, 67/86F and RPR), two intermediate DNA fragments (F1: 745bp, F2: 367 bp) with each containing two dNaMs in the template strand are amplified by PCR (Fig. 2C). In Step II, the F1 and F2 fragments are mixed and annealed as the PCR template, and then directed by the pMVF and RPR primers to assemble into the full length DNA template (1060 bp) by overlap extension PCR (Fig. 2C). Note that with the pMVF primer targeting 624 bp upstream of the T7 promoter, the DNA template (1060 bp) is significantly larger than the RNase P RNA transcript (419 nts). This enables subsequent direct RNA purification by SEC without the need of prior enzymatic digestion of the DNA template (see below, Fig. S2†). The TPT3-NaM UBP has been reported to show the best performance in replication efficiency and fidelity among the UBPs available, and no mutagenesis is expected to result from the PCR amplification.39,40
 | ||
| Fig. 2 A UBP-based strategy for posttranscriptional site-directed spin labeling of large RNAs using the 419 nt RNase P RNA as a model. (A) UBPs are incorporated into the ssDNA PCR primers by solid-phase chemical synthesis with the phosphoramidites of dNaM or dTPT3. (B) The full length double-stranded UBP-containing DNA templates for transcription are generated by a two-step overlap extension PCR using the UBP-containing primers. (C) 1% agarose gel analysis of dsDNA fragments obtained from the two-step overlap extension PCR reactions. F1 and F2 indicate fragments 1 and 2 generated from Step I, and FL indicates the full-length templates from Step II. (D) Site-specific incorporation of TPT3 or TPT3CO into RNAs by in vitro transcription. (E) 6% native PAGE analysis of in vitro transcription products from native or UBP-containing DNA templates. NaM is at the UBP-containing template strand. Transcription of the native DNA template using rNTP mix results in one single band. Efficient incorporation of rTPT3 or rTPT3CO into RNase P RNA transcripts is observed when either rTPT3TP or rTPT3COTP is provided with rNTP mix during in vitro transcription using UBP-containing DNA templates. If neither rTPT3TP nor rTPT3COTP is added to the in vitro transcription reaction, no full length RNA but only short abortive transcripts are observed. (F) Purified TPT3CO-containing RNAs are labeled with azide-modified nitroxide (Az-TMIO) via click chemistry. | ||
The resulting dsDNA templates containing dNaMs in the template strand are used to explore site-specific modifications of RNAs with UBPs (Fig. 2D). Although the TPT3-NaM UBP has been successfully utilized in creating an Escherichia coli and an eukaryotic semisynthetic organisms,48,49 the ribonucleotides of TPT3CO have not been tested in in vitro transcription. We therefore first characterize the ability of the T7 RNA polymerase to incorporate rTPT3 or rTPT3CO into the RNase P RNA (Fig. 2D). Under the conditions employed (Table S4†), no full-length product but only the abortive transcript is observed in the absence of UBP (Fig. 2E); this demonstrates that rTPT3 or rTPT3CO is essential for the correct transcription of dNaM-containing DNA templates. Furthermore, addition of cognate unnatural triphosphates, either rTPT3 or rTPT3CO, resulted in efficient production of the full-length transcription product (Fig. 2E). These data indicate that enzymatic incorporation of both rTPT3 and rTPT3CO into RNA is possible via in vitro transcription using T7 RNA polymerase. The RNase P RNA transcript with TPT3 or TPT3CO modification shows a major band with a purity of >94%, and is well-resolved from the significantly larger DNA template on native PAGE gel (Fig. 2E). This allows direct purification by SEC under non-denaturing conditions (Fig. S2†). Such non-denaturing purification preserves the co-transcriptionally derived structure of large RNAs50 and at the same time removes reducing agents (i.e. DTT) used in in vitro transcription that may interfere with the subsequent nitroxide labeling. Purified RNase P RNAs with single or double TPT3CO-modification are subjected to spin labeling via click chemistry (Fig. 2F). Upon conclusion of the reactions, non-denaturing SEC is again used to purify the labeled RNA from other undesired species, including the excess free nitroxide.
To confirm the successful coupling of spin labels to RNase P RNA, we measure the X-band CW-EPR spectra of the spin-labeled RNAs and compare them with those of free Az-TMIO (Fig. 3). The line-shape of a CW-EPR spectrum is dictated by the reorientation dynamics (rotational motions) of the nitroxide. As nitroxide motions reduce, averaging of its g- and hyperfine tensors becomes incomplete, resulting in line broadening and the appearance of extra features in the low-field and high-field regions.51 As shown in Fig. 3, the free Az-TMIO exhibits the typical three sharp lines of nearly equal amplitudes (upper spectrum), which is indicative of a low-molecular species undergoing isotropic tumbling with a rotational correlation time <1 ns. Spectra are clearly different for the two single spin-labeled (SSL) (U67TPT3CO- and U86TPT3CO-) and double spin-labeled (DSL) (U67/U86TPT3CO-) RNase P samples (lower spectra). Each RNA spectrum shows broad lines, indicating a large reduction of nitroxide rotational motions that is consistent with what is expected from attachment to the high molecular-weight RNAs. This clearly indicates successful covalent attachment of nitroxide spin labels to the RNase P RNAs.
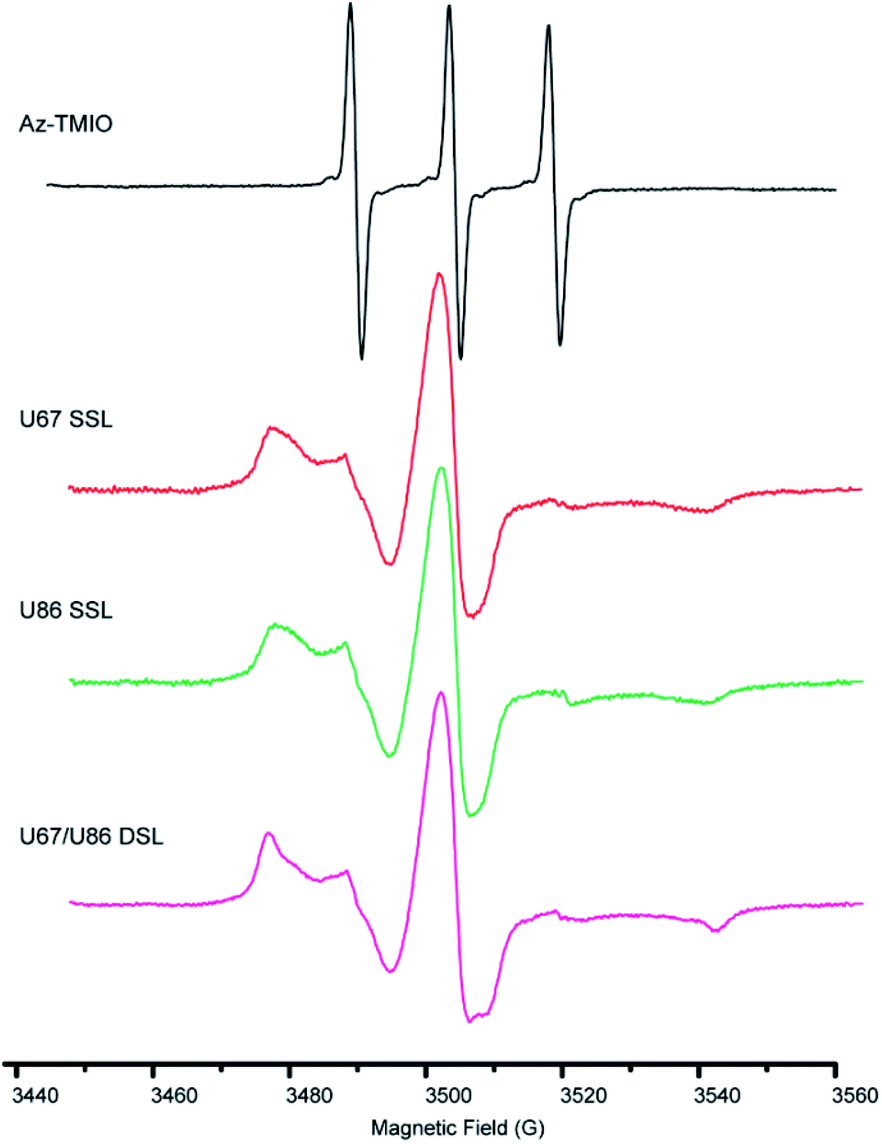 | ||
| Fig. 3 CW-EPR spectra of the free Az-TMIO (black), the U67 singly spin-labeled (red), U86 singly spin-labeled (green) and U67/U86 doubly spin-labeled (magenta) RNase P samples. All spectra are processed as described in Methods and as shown scaled to the same center-line amplitude. For the RNA spectra, a trace amount of free nitroxide signals (≤0.5%) has been subtracted. | ||
Using the measured EPR spectra of the RNAs, spin counting is carried out (Fig. S5 and Table S5†). The analyses show that the labeling efficiencies of the two SSL- (at sites of U67 or U86) and DSL- (at sites of both U67 and U86) RNase P samples are 84%, 82% and 85%, respectively (Table S5†). In a control experiment, nitroxide labeling of a wild type RNase P without TPT3 modification gives no observable CW-EPR signal (Fig. S6†). Together the data clearly demonstrate that the TPT3-based spin labeling scheme is efficient and specific. Furthermore, we note that a trace amount of free nitroxide can be detected in the measured EPR spectra of the labeled RNAs (Fig. S7,† indicated by “*”). Following a spectral decomposition procedure described previously,52 the amount of the free nitroxide is estimated to be ≤0.5% of the total spin population in all three samples reported. This indicates that the non-denaturing purification scheme is sufficient for producing pure spin-labeled large RNAs.
In addition, although urea is not required for the click reaction between Az-TMIO and rTPT3CO-containing RNAs, for RNase P RNA the presence of 0.5 M urea, which retains the RNA in the non-denaturing state,53 enhances the nitroxide labeling efficiency (Fig S7c and Table S5†). This is consistent with the notion that the small amount of urea enhances RNA conformational fluctuations, thus increasing accessibility of labeling reagents to give higher reaction yield.
The effects of spin labels on the RNase P structure and function
To study the effects of UBP modification and subsequent spin labeling on the structure of RNase P RNA, we measured CD spectra and SAXS profiles of wild type, UBP modified as well as spin-labeled RNase P RNAs under the same solution conditions. The CD spectrum provides information on the helical structure of DNA and RNA, and thus has been widely used to study secondary structure formation in nucleic acids.54 As shown in Fig. 4A and S4,† all CD spectra of RNase P RNAs (wild type, TPT3CO modified, and single- and double-spin labeled) display the same characteristic profile, indicating that UBP modification and spin labeling do not result in significant changes in the secondary structure of RNase P.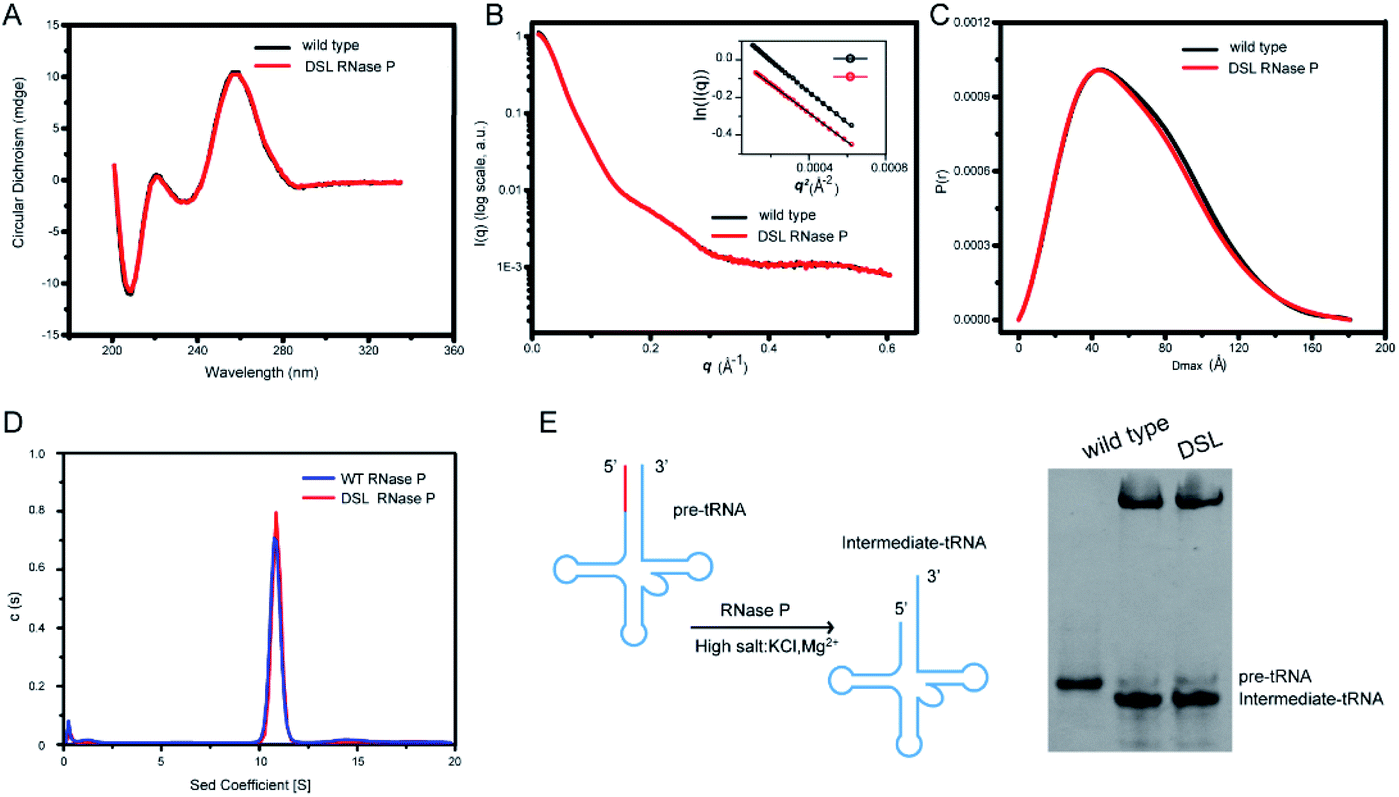 | ||
| Fig. 4 UBP modification and spin-labeling have marginal effects on the global folding and catalytic activity of RNase P RNA. (A) CD spectra of wild type and double spin labeled (DSL) RNase P RNAs, which exhibit very similar features. (B and C) The experimental scattering profiles (B) and PDDF (C) of wild type (black) and DSL (red) RNase P RNAs are superimposable, suggesting no significant overall structural changes of RNase P upon spin labeling. The inset in (B) shows good linear fitting in the Guinier regions of the respective scattering profiles, indicating monodispersity and homogeneity of the samples in solution. (D) SV-AUC profiles of wild type (blue) and DSL (red) RNase P RNAs. Both RNA profiles show a single and sharp peak with a similar sedimentation coefficient. (E) Pre-tRNA processing assay. Left: secondary structure of the yeast pre-tRNAPhe and the intermediate tRNA product. RNase P cleaves the 5′ leader sequence (colored in red) and generates an intermediate tRNA that is shorter than the pre-tRNAPhe. Right: DSL and wild type RNase P show a similar processing activity. | ||
We next characterize the RNAs using SAXS. The 1D SAXS profile of a biomolecule encodes structural information such as the size, shape, molecular weight, and the low-resolution 3D global structure, and therefore can be used to study structural changes.55 The scattering profiles, with scattering intensity I(q) plotted against momentum transfer q, along with the pair distance distribution function (PDDF) transformed from scattering profiles of the wild type and DSL RNase P RNAs are shown in Fig. 4B and C. The Guinier regions of both the scattering profiles are linear, indicating that both RNA samples are monodispersed and homogeneous in solution. The scattering profiles (Fig. 4B) and PDDFs (Fig. 4C) of the wild type and DSL RNase P RNAs are superimposable, suggesting that UBP modification and spin labeling have little effects on the 3D global structure of the RNase P RNA. The overall structural parameters, including the radius of gyration Rg calculated from Guinier slopes, Rg and the maximum diameter Dmax from PDDF functions, as well as molecular weights derived from volume-of-correlation (Vc),56 are summarized in Table S7.† The similar Rg and Dmax as well as the unchanged oligomerization state upon UBP modification and spin labeling (both are monomeric) are consistent with the absence of significant structural changes.
We further characterize the RNAs using sedimentation velocity analytical ultracentrifugation (SV-AUC), which is a sensitive technique for detecting the global conformational features of biological molecules in solution.57–59 As shown in Fig. 4D, both the wild type and DSL RNase P fold into a monodisperse species as indicated by the single sharp peak of their SV-AUC profiles. The homogeneity of wild type and DSL RNase P is 93.8% and 96.4%, respectively. This is comparable to the 95% purity/homogeneity requirement for X-ray crystallography and NMR.60 The calculated MWs are 136.4 kDa and 137.5 kDa for wild type and DSL RNase P, respectively, which nicely match the expected values (135.6 kDa for wild type and 137.8 kDa for DSL RNase P, respectively). The SV-AUC data reveal that the DSL RNase P shares a very similar solution conformation to the wild type, and UBP modification and spin labeling do not perturb the overall structure of RNase P.
We next assess whether the UBP-based spin labeling affects the catalytic activity of RNase P. RNase P is a ribozyme that catalyzes hydrolysis of a phosphodiester bond in precursors of transfer RNA (pre-tRNA), resulting in the formation of the 5′-phosphorylated intermediate tRNA and the release of a 5′-precursor fragment (Fig. 4E). Using a yeast pre-tRNAPhe as a substrate, we observe the formations of intermediate tRNAPhe products in the presence of both wild type and DSL RNase P, and the percentage of cleaved pre-tRNAPhe of three independent experiments for wild type and DSL RNase P is 91% ± 3% and 89% ± 5%, respectively (Fig. 4E). Given the high degree of labeling of the DSL RNase P (Fig. 3 and Table S5†), the pre-tRNA processing assay suggests that the UBP-based spin labeling does not impair the catalytic activity of the RNase P, indicating that the spin labels do not alter RNA folding. Overall, CD spectroscopy, SAXS, SV-AUC and activity assays show that the TPT3CO-modification and subsequent spin labeling at the sites of U67 and U87 minimally perturb the global folding and function of the RNase P RNA.
Furthermore, we analyze the X-band CW-EPR spectral lineshape of the spin labeled RNAs in order to assess the local RNA environment at the nitroxide labeling site(s). In our study, because of the high-molecular weight of the RNase P RNA (138 kDa) and the presence of glycerol, the global tumbling of the labeled RNA is slow and contributes very little to the magnetic tensor averaging reported in an X-band spectrum.51 As such, the measured spectrum shows primarily the local RNA environment at the nitroxide labeling site. Compared to the well-characterized R1 nitroxide attached to a cysteine of proteins,61 our observed RNA spectra show similar characteristics to “tertiary contact” sites of R1, but are broader than those at the “loop” and “helical surface” sites.61 This indicates that at these RNA labeling sites, the TPT3-based nitroxide label has a lower mobility than that of the R1 nitroxide at protein loops and contact-free helical surfaces. Such differences likely arise in part from the different linkers (i.e., bonds connecting the nitroxide pyrroline ring to the macromolecule): R1 is connected to a cysteine side-chain by an “–S–S–” bond, while the TPT3-nitroxide is linked via a triazole moiety (Fig. 1B) that has been shown to restrict motions between the nucleo-base and the nitroxide pyrroline ring.44
In the reported crystal structure, U67 locates in an internal bulge region and its base points outwards, and thus is expected to be dynamic. U86 resides in a junction region and its base stacks with the base of G85, and thus is expected to be less mobile. However, the single-labeled U67 and U86 spectra show very little difference (Fig. 3, S7A†), suggesting that the TPT3-nitroxides have similar mobility at these two sites. Furthermore, the double-labeled spectrum is slightly broader than the average of the two single-labeled RNA spectra (Fig. 3, S7B†). It should be noted that substituting a uridine with TPT3, which has a larger base, likely gives rise to some degree of changes in the parent RNA, although such changes seem rather confined and are sufficiently accommodated at the chosen sites, as little perturbation is detected by CD, SAXS, SV-AUC, and the activity assay. The observed CW-EPR spectra overall indicate that the TPT3-nitroxide at loop sites may come into contact with the parent RNA, either via the nitroxide pyrroline ring or the TPT3 base. This gives rise to the observed “tertiary contact” like spectra with low label mobility. This is also consistent with a previous study that the incorporation of UBP could partly disturb the local structure.62,63 In the future, it would be interesting to explore how the TPT3-nitroxide interacts with its local RNA environment, as well as strategies for selecting optimal sites for a particular RNA study.
Distance measurements of dual spin-labeled RNA by PELDOR
The DSL RNase P sample was used for distance measurements by PELDOR (Fig. 5, S8 and S9†). Fig. 5A shows the background-corrected X-band PELDOR trace, which clearly demonstrates the occurrence of dipolar oscillation. The absolute modulation depth is ∼30%, consistent with the measured high efficiency of labeling. The distance distributions obtained using the Tikhonov regularization approach are broad, with the mean distance <rDEER> = 4.02 nm and the width of distribution σ = 0.78 nm (Fig. 5B). In addition, in repeat PELDOR measurements (including lengthening the dipolar evolution time), <rDEER> are found to vary less than 7% of the reported value. Furthermore, no dipolar decay or oscillation is observed from the SSL RNase P samples (Fig. S8B and C†), indicating that our measurements are free of interference from either aggregations of the labeled RNA or non-specifically nitroxide attachment at RNase P. | ||
| Fig. 5 Distance measurements on U67/U86 doubly spin-labeled RNase P RNA by X-band PELDOR. (A) Background-corrected dipolar evolution time trace (black) and DeerAnalysis fitting (red). (B) Inter-spin distance distribution obtained using Tikhonov regularization with a regulation parameter of 100. | ||
According to the crystal structure, the distance between the respective N1 atoms of U67 and U86 labeling sites is 3.32 nm (Fig. S1†). Taking into account the inherent flexibility of the large RNase P RNA, the local dynamics of the base at the labeling sites, and the size of Az-TMIO nitroxide including the linker, the measured <rDEER> of 4.02 nm are reasonable. Furthermore, the “tertiary-contact” like CW-EPR spectra (Fig. 3) indicate potential contacts between the nitroxide label and the RNA. This can give rise to heterogeneous rotamer conformations at each labeling site and subsequently a broad multiple-population distance distribution profile (Fig. 5B). It should be noted that using the PELDOR setup reported in this work, the mean inter-spin distance is determined to a high degree of confidence, but the shape of the distance distribution profile is under-determined.64 As such, more detailed analyses of the distance distribution profile are not pursued.
Conclusions
In this work, we have demonstrated an efficient and generally applicable site-directed spin labeling strategy for large RNAs based on an expanded genetic alphabet containing the NaM-TPT3 unnatural base pair. Phosphoramidite and tri-phosphate variants of TPT3 and NaM have been synthesized, together with an alkyne-modified triphosphorylated ribonucleotide TPT3. These analogues allow site-specific incorporation of alkyne-modified TPT3 into the 419 nt RNase P RNA from Bacillus stearothermophilus by in vitro transcription, as well as posttranscriptional covalent attachment of an azide-modified nitroxide via click chemistry. Characterization by EPR spectroscopy as well as a number of biophysical and biochemical assays clearly demonstrates that the nitroxide labels are attached to the desired sites in a specific and efficient manner, and present minimal perturbation to the global folding and activity of RNase P. The measured inter-spin distance distribution is in good agreement with the crystal structure. Overall, the data validate the labeling strategy presented.Furthermore, by the judicial design of DNA templates and RNA constructs, we demonstrate in this work non-denaturing labeling and purification of the RNase P RNA. This preserves the co-transcriptional folding of the target RNA, and is advantageous for RNA structure–function relationship studies. We note that the rTPT3CO labeling scheme is compatible with denaturing methods, as either rTPT3/rTPT3CO incorporated RNAs41,65 or nitroxide-labeled RNAs20,66 have been purified with denaturing gels.
During our manuscript preparation, a similar UBP-based spin labeling approach for RNAs was reported by Domnick and co-workers.41 In that report, Domnick and co-workers synthesize a nitroxide-modified triphosphate ribonucleotide TPT3NO that is incorporated into RNAs during in vitro transcription. The direct use of TPT3NO in in vitro transcription incurs potential risks of damaging the nitroxide, for example, exposure to reducing agents during in vitro transcription, and may account for the slightly lower labeling efficiency reported by Domnick and co-workers41 (see Table S5†). In addition, constrained by the requirement that the RNA polymerase must accept the nitroxide-modified unnatural base (i.e., TPT3NO) as a substrate, a relatively long and flexible linker is utilized to connect the nitroxide to TPT3. In contrast, in work reported here, TPT3CO, which contains a rather small alkyne moiety, is efficiently incorporated into long RNA transcripts by T7 RNA polymerase, and the nitroxide is coupled to the transcribed RNAs. This posttranscriptional labeling strategy avoids the possibility of nitroxide (or any spin label) destruction by reducing agents, and would allow a much greater option for linker chemistry. These features could be beneficial to EPR studies.
In summary, work reported here clearly demonstrates the use of an unnatural base pair system to achieve specific and efficient posttranscriptional site-directed spin labeling of large RNAs, and the method can be applied under non-denaturing conditions. Studies are underway to further develop this labeling approach as well as to explore its applications in investigating biological functions of large RNAs, for example optimizing labeling site selections, computationally evaluating the inter-nitroxide distances, and detecting RNA conformational changes via CW- and pulsed-EPR. Combined with other biophysical techniques, such as SAXS and NMR, the method reported here will advance our understanding of the relationship of the structure, dynamics, and function of RNAs.
Experimental
Materials
The phosphoramidites of dNaM and dTPT3, triphosphorylated nucleotides of dNaM, dTPT3, rNaM, rTPT3, and azide modified nitroxide (5-azide-1,1,3,3-tetramethylisoindolin-2-yloxyl) (Az-TMIO) (Fig. 1) were custom synthesized according to the literature procedures by WuXi AppTec, China within 6–8 weeks.28,34,40,43 2 × Golden mix of Taq DNA polymerase (supplied with buffer and natural dNTPs mix) was purchased from TSINGKE Biological Technology Co., Ltd. (Beijing, China). The T7 RNA polymerase was homemade. Plasmids encoding the RNase P RNA from Bacillus stearothermophilus and the pre-tRNAPhe from yeast were generated by total-gene synthesis and confirmed by sequencing (Wuxi Qinglan Biotechnology Inc, Wuxi, China). All DNA oligonucleotide primers (containing natural and/or unnatural nucleotides) were synthesized by solid-phase chemical synthesis and purified by OPC purification by TSINGKE within one week. The DNA sequences of the synthetic plasmids and the native and unnatural primers can be found in Tables S1 and S2,† respectively. All polyacrylamide gels were stained with Gelsafe (YPR-Bio) and imaged with a Tanon 2500 Gel Imaging System. The gel images were quantified with ImageJ.Preparation of native or UBP modified DNA templates
The double-stranded (ds) DNA templates for in vitro transcription of wild type (unmodified) RNase P RNA and the yeast pre-tRNAPhe were generated by PCR using a forward primer (pMVF) targeting a common upstream sequence in the plasmids and a reverse primer specific to the respective cDNAs (RPR and tRNAR, respectively).The unnatural dsDNA templates containing one or two NaMs at the template strand for in vitro transcription of UBP-modified RNase P RNAs were generated by a two-step overlap extension PCR (Fig. 2B) within 2 days. In Step I, two intermediate DNA fragments were generated, and the PCR steps included initial denaturation at 95 °C for 5 minutes, 32 cycles of denaturation at 95 °C for 30 s, primer annealing at 62 °C for 30 s, extension at 72 °C for 20 s, and a final extension at 72 °C for 5 minutes. The two fragments were recovered with an EasyPure PCR Purification Kit from TransGen Biotech, and were mixed to assemble the full-length DNA templates in Step II. The PCR steps in Step II included initial denaturation at 95 °C for 5 minutes, 18 cycles of denaturation at 95 °C for 30 s, primer annealing at 62 °C for 30 s, extension at 72 °C for 20 s, and a final extension at 72 °C for 5 minutes. The full-length DNAs obtained from Step II were purified by size-exclusion chromatography, and then PCR-amplified to produce the respective templates for subsequent large-scale in vitro transcriptions. The final PCR-amplified dsDNA templates were directly used for in vitro transcription without further purification. The primers used in each step and the final concentrations for each component in PCR reactions are given in Table S3.†
Preparation of wild-type or UBP-containing RNAs
Prior to large scale RNA sample preparation, the optimal conditions for in vitro transcription of wild type or unnatural RNAs were screened against various Mg2+ concentrations for each transcript. The native dsDNA templates were used for preparation of wild type RNase P RNA and yeast pre-tRNAPhe RNA. The unnatural dsDNA templates were used for site-specific incorporation of rTPT3TP or rTPT3COTP. Details on in vitro transcription systems can be found in Table S4.†The transcription products were found to migrate as a single band at native PAGE gels (Fig. 2E), and were directly purified with SEC by passing through either a Hiload 16/600 Superdex 200 (for wild type or unnatural RNase P RNAs) or a Superdex 75 (for wild type pre-tRNAPhe) column with a SEC buffer containing 20 mM HEPES (pH 7.40), 100 mM KCl, and 5 mM MgCl2. Fractions containing the target RNAs were collected and concentrated with Amicon Centrifugal Filter Units, and then stored at −80 °C until use. The concentrations of RNAs were determined by UV-Vis absorption at 260 nm using a NanoDrop 2000 (Thermo Scientific). The molar extinction coefficients of RNAs were calculated from the primary RNA sequences using an OligoAnalyzer Tool (https://sg.idtdna.com/pages/tools/oligoanalyzer).
Site-directed spin labeling of TPT3CO-modified RNase P
Purified RNase P RNAs containing one or two rTPT3CO were first precipitated with ethanol, and then dissolved with appropriate RNase-free water to a final concentration of 200 μM. The reactive Az-TMIO nitroxide (dissolved in 100% DMSO) was added with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 or 1:100, for single-site or double labeling of RNase P RNAs, respectively. The click chemistry reactions were carried out under nearly physiological conditions with 10 mM THPTA (tris-hydroxypropyltriazolylmethylamine) at pH 7.0 (Sigma Aldrich), 0.5 mM copper(II)-TBTA (Lumiprobe Corporation, Maryland, USA), and 1 mM freshly prepared D-isoascorbic acid in 50% (v/v) DMSO/RNase-free water. 0.5 M urea was added to the reaction solutions to improve the labeling efficiency. The reaction mixtures were incubated at 25 °C for 4–6 hours to allow coupling of Az-TMIO nitroxide to RNAs. EDTA was then added to a final concentration of 5 mM to quench the click reaction and remove free copper. The mixtures were buffer-exchanged to the SEC buffer using Amicon Ultra Centrifugal Filter Devices (MWCO 10K, Millipore), and the spin-labeled RNAs were purified with SEC by passing the mixtures through a Superose 6 Increase 10/300 GL column. Fractions containing the spin-labeled RNAs were collected and concentrated for further use. The spin labeling and purification generally take one day.
:50 or 1:100, for single-site or double labeling of RNase P RNAs, respectively. The click chemistry reactions were carried out under nearly physiological conditions with 10 mM THPTA (tris-hydroxypropyltriazolylmethylamine) at pH 7.0 (Sigma Aldrich), 0.5 mM copper(II)-TBTA (Lumiprobe Corporation, Maryland, USA), and 1 mM freshly prepared D-isoascorbic acid in 50% (v/v) DMSO/RNase-free water. 0.5 M urea was added to the reaction solutions to improve the labeling efficiency. The reaction mixtures were incubated at 25 °C for 4–6 hours to allow coupling of Az-TMIO nitroxide to RNAs. EDTA was then added to a final concentration of 5 mM to quench the click reaction and remove free copper. The mixtures were buffer-exchanged to the SEC buffer using Amicon Ultra Centrifugal Filter Devices (MWCO 10K, Millipore), and the spin-labeled RNAs were purified with SEC by passing the mixtures through a Superose 6 Increase 10/300 GL column. Fractions containing the spin-labeled RNAs were collected and concentrated for further use. The spin labeling and purification generally take one day.
Circular dichroism spectroscopy
CD spectra of wild type and unnatural RNase P RNAs were measured on an Applied Photophysics Chirascan-plus spectrophotometer (Leatherhead) controlled by Pro-Data Chirascan v4 software. Spectra were recorded between 200 and 360 nm with a step-resolution of 1 nm, a slit-width of 0.6 nm, and an integration time of 5 s. Acquisition was performed at 25 °C using a 0.2 mm path length cuvette with RNA concentrations being 1 μM. The spectra were averaged over three scans and corrected by subtraction of the buffer signal.Small angle X-ray scattering
SAXS experiments for all RNA samples were carried out in a buffer containing 20 mM HEPES (pH 7.40), 100 mM KCl, 5 mM Mg2+, and 3% (v/v) glycerol. The data collection and processing procedures are similar to those described before.67 All parameters for data collection and software employed for data analysis are summarized in Table S6.† Briefly, SAXS measurements were performed at room temperature at the beamline 12 ID-B of the Advanced Photon Source, Argonne National Laboratory. The 2D images were reduced to one-dimensional scattering profiles using Matlab scripts on-site. The scattering profiles of the RNAs were calculated by subtracting the background buffer contribution from the sample-buffer profile using the program PRIMUS following standard procedures.68 The forward scattering intensity I(0), and the radius of gyration (Rg) were calculated from the Guinier analysis and the indirect Fourier transform method implemented in the program GNOM,69 along with the pair distance distribution function (PDDF), p(r), and the maximum dimension of the protein, Dmax. The Volume-of-correlation (Vc) was calculated using the program Scatter, and the molecular weights of solutes were calculated on a relative scale using the Rg/Vc power law developed by Rambo et al.,56 independent of the RNA concentration and with minimal user bias.Sedimentation velocity analytical ultracentrifugation
The SV-AUC experiment was performed with a Proteomelab XL-1 centrifuge with an An-60 Ti rotor (Beckman Coulter). Before centrifugation, RNAs were prepared with the SEC buffer, and the concentration was adjusted to obtain an initial absorption value of 0.7 at 260 nm. All experiments were performed at 20 °C at 50000 rpm. Data were analysed with the continuous c(s) distribution model as implemented in Sedfit.70
Pre-tRNA processing assay
The enzymatic activity of the wild-type and labeled (U67/U86 double spin-labeled) RNase P RNAs was tested by performing the pre-tRNA processing assay, in which the yeast pre-tRNAPhe substrate (40 nM) was mixed with 25 nM wild type or labeled RNase P RNAs. The reactions were carried out in the SEC buffer with the addition of 15 mM MgCl2 and 400 mM KCl. The mixtures were incubated at 50 °C for 30 minutes, and subsequently the reactions were quenched by adding 8 M urea and 5 mM EDTA. All reaction mixtures were denatured at 95 °C for 10 minutes and then analysed using 15% denaturing polyacrylamide gels.Continuous-wave EPR spectroscopy and spin counting
Prior to EPR measurements, the folded labeled RNAs were first lyophilized then re-dissolved in 50/50 (v/v) glycerol/water to obtain a stock solution with the same salt concentrations as that in the SEC buffer. Control studies have shown that lyophilization does not affect global folding of the RNase P RNAs (Fig. S3†).For CW-EPR measurements, each EPR sample was approximately 10 μL with the spin-labeled RNA concentrations being 50–120 μM, and was loaded into a borosilicate glass capillary (1.0 mm ID × 1.2 mm OD, Fiber Optic Center, Inc.) sealed at one end. CW-EPR spectra were obtained on a Bruker EMX X-band spectrometer equipped with an ER-041X microwave Bridge and a high sensitivity cavity (ER-4119HS, Bruker Biospin, Inc). All CW-EPR spectra were acquired at room temperature with an incident microwave power of 2 mW, a modulation frequency of 100 kHz, and a modulation amplitude of 1 G. For each sample, 10 scans were collected and accumulated. The spectra were background corrected and normalized following a previously reported procedure.51 Spin counting was performed based on the 2nd integral values of the baseline-corrected but not normalized CW-EPR spectra,51 and the details are described in Fig. S5 and Table S5.†
Pulsed electron–electron double resonance spectroscopy
To prepare PELDOR samples, an appropriate amount of folded spin-labeled RNase P RNAs was lyophilized and then re-dissolved to obtain a solution containing 20 mM HEPES (pH 7.0), 100 mM KCl, 10 mM MgCl2, and 50% (v/v) glycerol/water. The samples were loaded into a round quartz capillary (2.0 mm ID × 2.4 mm OD, Vitrocom, Inc., Mountain Lakes, NJ) sealed at one end, and were flash-frozen in liquid nitrogen. Each PELDOR sample was approximately 30 μL with the spin-labeled RNA concentrations being approximately 70–80 μM.PELDOR measurements were carried out at 78 K on a Bruker ELEXSYS E580 X-band spectrometer equipped with an ER4118-MS3-EN resonator. Previously reported procedures71 were used. Specifically, a dead-time free four-pulse scheme was used, with a pump set at the center of the nitroxide spectrum and the observer at the low-field shoulder. The observer π pulse was 32 ns. The pump π pulse was optimized using a nutation experiment and was at 16 or 18 ns. The video bandwidth was 20 MHz. The Shot Repetition Time was set at 1020 μs. The accumulation time was set to 17–22 hours with 100 shots per point.
The inter-spin distance distributions were computed from the resulting dipolar evolution data using the DeerAnalysis2013 program.72 In the analyses, background in the dipolar evolution data was corrected by fitting an exponential decay corresponding to a homogeneous 3-dimentional distribution of the electron spins. The Tikhonov regularization approach was used to obtain the inter-spin distance distribution, from which the mean distance <rDEER> and width of the distance distribution (σ, represented by the standard deviation of the distribution profile) were computed as previously described.73
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. U1832215), the Beijing Advanced Innovation Center for Structural Biology, the Tsinghua-Peking Joint Center for Life Sciences, and the China Youth 1000-Talent Program of the State Council of China (X. F.); and the US National Science Foundation (MCB-1818107) and National Institute of Health (S10 RR028992(P. Z. Q)). We thank Dr Xiaobing Zuo at the beamline 12-ID-B, Advanced Photon Source, Argonne National Laboratory, USA for assistance during SAXS data collection. We acknowledge the assistance of the Protein Preparation and Identification Facility at Technology Center for Protein Science, Tsinghua University.Notes and references
- J. M. Engreitz, N. Ollikainen and M. Guttman, Nat. Rev. Mol. Cell Biol., 2016, 17, 756–770 CrossRef CAS PubMed.
- A. Zampetaki, A. Albrecht and K. Steinhofel, Front. Physiol., 2018, 9, 1201 CrossRef PubMed.
- A. J. Blythe, A. H. Fox and C. S. Bond, Biochim. Biophys. Acta, 2016, 1859, 46–58 CrossRef CAS PubMed.
- I. V. Novikova, S. P. Hennelly, C. S. Tung and K. Y. Sanbonmatsu, J. Mol. Biol., 2013, 425, 3731–3746 CrossRef CAS PubMed.
- I. V. Novikova, S. P. Hennelly and K. Y. Sanbonmatsu, Int. J. Mol. Sci., 2013, 14, 23672–23684 CrossRef PubMed.
- X. Fang, J. R. Stagno, Y. R. Bhandari, X. Zuo and Y. X. Wang, Curr. Opin. Struct. Biol., 2015, 30, 147–160 CrossRef CAS PubMed.
- A. T. Tuukkanen, A. Spilotros and D. I. Svergun, IUCrJ, 2017, 4, 518–528 CrossRef CAS PubMed.
- J. L. J Xu and D. M. Crothers, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 44–48 CrossRef PubMed.
- Y. Liu, E. Holmstrom, P. Yu, K. Tan, X. Zuo, D. J. Nesbitt, R. Sousa, J. R. Stagno and Y. X. Wang, Nat. Protoc., 2018, 13, 987–1005 CrossRef CAS PubMed.
- A. Lapinaite, B. Simon, L. Skjaerven, M. Rakwalska-Bange, F. Gabel and T. Carlomagno, Nature, 2013, 502, 519–523 CrossRef CAS PubMed.
- M. Dimura, T. O. Peulen, C. A. Hanke, A. Prakash, H. Gohlke and C. A. Seidel, Curr. Opin. Struct. Biol., 2016, 40, 163–185 CrossRef CAS PubMed.
- O. Duss, M. Yulikov, G. Jeschke and F. H. Allain, Nat. Commun., 2014, 5, 3669 CrossRef CAS.
- G. Cornilescu, A. L. Didychuk, M. L. Rodgers, L. A. Michael, J. E. Burke, E. J. Montemayor, A. A. Hoskins and S. E. Butcher, J. Mol. Biol., 2016, 428, 777–789 CrossRef CAS PubMed.
- J. Wang, X. Zuo, P. Yu, H. Xu, M. R. Starich, D. M. Tiede, B. A. Shapiro, C. D. Schwieters and Y. X. Wang, J. Mol. Biol., 2009, 393, 717–734 CrossRef CAS PubMed.
- M. Falb, I. Amata, F. Gabel, B. Simon and T. Carlomagno, Nucleic Acids Res., 2010, 38, 6274–6285 CrossRef CAS PubMed.
- C. P. Jones, W. A. Cantara, E. D. Olson and K. Musier-Forsyth, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3395–3400 CrossRef CAS PubMed.
- I. Krstic, B. Endeward, D. Margraf, A. Marko and T. F. Prisner, Top. Curr. Chem., 2012, 321, 159–198 CrossRef CAS PubMed.
- K. Halbmair, J. Seikowski, I. Tkach, C. Höbartner, D. Sezer and M. J. C. Bennati, Chem. Sci., 2016, 7, 3172–3180 RSC.
- A. Ponce-Salvatierra, Astha, K. Merdas, C. Nithin, P. Ghosh, S. Mukherjee and J. M. Bujnicki, Biosci. Rep., 2019, 39, BSR20180430 CrossRef PubMed.
- X. Zhang, C. S. Tung, G. Z. Sowa, M. M. Hatmal, I. S. Haworth and P. Z. Qin, J. Am. Chem. Soc., 2012, 134, 2644–2652 CrossRef CAS PubMed.
- J. M. Esquiaqui, E. M. Sherman, J. D. Ye and G. E. Fanucci, Biochemistry, 2016, 55, 4295–4305 CrossRef CAS PubMed.
- O. Duss, M. Yulikov, F. H. T. Allain and G. Jeschke, Methods Enzymol., 2015, 558, 279–331 CAS.
- P. Z. Qin and T. Dieckmann, Curr. Opin. Struct. Biol., 2004, 14, 350–359 CrossRef CAS PubMed.
- D. K. Yadav and P. J. Lukavsky, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 97, 57–81 CrossRef CAS.
- G. Z. Sowa and P. Z. Qin, Prog. Nucleic Acid Res. Mol. Biol., 2008, 82, 147–197 CAS.
- L. Baronti, H. Karlsson, M. Marusic and K. Petzold, Anal. Bioanal. Chem., 2018, 410, 3239–3252 CrossRef CAS PubMed.
- S. Saha, A. P. Jagtap and S. T. Sigurdsson, Methods Enzymol., 2015, 563, 397–414 CAS.
- U. Jakobsen, S. A. Shelke, S. Vogel and S. T. Sigurdsson, J. Am. Chem. Soc., 2010, 132, 10424–10428 CrossRef CAS PubMed.
- I. Lebars, B. Vileno, S. Bourbigot, P. Turek, P. Wolff and B. Kieffer, Nucleic Acids Res., 2014, 42, e117 CrossRef PubMed.
- G. Hanspach, S. Trucks and M. Hengesbach, RNA Biol., 2019, 16, 1119–1132 CrossRef PubMed.
- E. S. Babaylova, A. A. Malygin, A. A. Lomzov, D. V. Pyshnyi, M. Yulikov, G. Jeschke, O. A. Krumkacheva, M. V. Fedin, G. G. Karpova and E. G. Bagryanskaya, Nucleic Acids Res., 2016, 44, 7935–7943 CrossRef CAS PubMed.
- M. Zhao, F. D. Steffen, R. Borner, M. F. Schaffer, R. K. O. Sigel and E. Freisinger, Nucleic Acids Res., 2018, 46, e13 CrossRef PubMed.
- T. Someya, A. Ando, M. Kimoto and I. Hirao, Nucleic Acids Res., 2015, 43, 6665–6676 CrossRef CAS PubMed.
- Y. J. Seo, D. A. Malyshev, T. Lavergne, P. Ordoukhanian and F. E. Romesberg, J. Am. Chem. Soc., 2011, 133, 19878–19888 CrossRef CAS PubMed.
- F. Eggert, K. Kulikov, C. Domnick, P. Leifels and S. Kath-Schorr, Methods, 2017, 120, 17–27 CrossRef CAS PubMed.
- D. A. Malyshev and F. E. Romesberg, Angew. Chem., Int. Ed., 2015, 54, 11930–11944 CrossRef CAS PubMed.
- R. Kawai, M. Kimoto, S. Ikeda, T. Mitsui, M. Endo, S. Yokoyama and I. Hirao, J. Am. Chem. Soc., 2005, 127, 17286–17295 CrossRef CAS PubMed.
- T. Lavergne, R. Lamichhane, D. A. Malyshev, Z. Li, L. Li, E. Sperling, J. R. Williamson, D. P. Millar and F. E. Romesberg, ACS Chem. Biol., 2016, 11, 1347–1353 CrossRef CAS PubMed.
- K. Dhami, D. A. Malyshev, P. Ordoukhanian, T. Kubelka, M. Hocek and F. E. Romesberg, Nucleic Acids Res., 2014, 42, 10235–10244 CrossRef CAS PubMed.
- L. Li, M. Degardin, T. Lavergne, D. A. Malyshev, K. Dhami, P. Ordoukhanian and F. E. Romesberg, J. Am. Chem. Soc., 2014, 136, 826–829 CrossRef CAS PubMed.
- C. Domnick, F. Eggert, C. Wuebben, L. Bornewasser, G. Hagelueken, O. Schiemann and S. Kath-Schorr, Angew. Chem., Int. Ed., 2020, 59, 7891–7896 CrossRef CAS.
- A. Mondragon, Annu. Rev. Biophys., 2013, 42, 537–557 CrossRef CAS.
- Y. J. Seo, G. T. Hwang, P. Ordoukhanian and F. E. Romesberg, J. Am. Chem. Soc., 2009, 131, 3246–3252 CrossRef CAS.
- M. Kerzhner, D. Abdullin, J. Wiecek, H. Matsuoka, G. Hagelueken, O. Schiemann and M. Famulok, Chemistry, 2016, 22, 12113–12121 CrossRef CAS.
- A. V. Kazantsev, A. A. Krivenko, D. J. Harrington, S. R. Holbrook, P. D. Adams and N. R. Pace, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13392–13397 CrossRef CAS PubMed.
- Y. J. Seo, S. Matsuda and F. E. Romesberg, J. Am. Chem. Soc., 2009, 131, 5046–5047 CrossRef CAS PubMed.
- M. P. Ledbetter, D. A. Malyshev and F. E. Romesberg, Methods Mol. Biol., 2019, 1973, 193–212 CrossRef CAS.
- Y. Zhang, J. L. Ptacin, E. C. Fischer, H. R. Aerni, C. E. Caffaro, K. San Jose, A. W. Feldman, C. R. Turner and F. E. Romesberg, Nature, 2017, 551, 644–647 CrossRef CAS PubMed.
- A. X. Zhou, K. Sheng, A. W. Feldman and F. E. Romesberg, J. Am. Chem. Soc., 2019, 141, 20166–20170 CrossRef CAS PubMed.
- E. P. Booy, H. Meng and S. A. McKenna, Methods Mol. Biol., 2012, 941, 69–81 CrossRef CAS PubMed.
- X. Zhang, P. Cekan, S. T. Sigurdsson and P. Z. Qin, Methods Enzymol., 2009, 469, 303–328 CAS.
- P. Nguyen, X. Shi, S. T. Sigurdsson, D. Herschlag and P. Z. Qin, Chembiochem, 2013, 14, 1720–1723 CrossRef CAS PubMed.
- V. M. Shelton, T. R. Sosnick and T. Pan, Biochemistry, 1999, 38, 16831–16839 CrossRef CAS PubMed.
- J. Kypr, I. Kejnovska, D. Renciuk and M. Vorlickova, Nucleic Acids Res., 2009, 37, 1713–1725 CrossRef CAS PubMed.
- X. Fang, J. R. Stagno, Y. R. Bhandari, X. Zuo and Y. X. Wang, Curr. Opin. Struct. Biol., 2015, 30, 147–160 CrossRef CAS PubMed.
- R. P. Rambo and J. A. Tainer, Nature, 2013, 496, 477–481 CrossRef CAS PubMed.
- S. Mitra, Methods Mol. Biol., 2014, 1086, 265–288 CrossRef CAS.
- F. Liu, S. Somarowthu and A. M. Pyle, Nat. Chem. Biol., 2017, 13, 282–289 CrossRef CAS PubMed.
- S. Somarowthu, M. Legiewicz, I. Chillon, M. Marcia, F. Liu and A. M. Pyle, Mol. Cell, 2015, 58, 353–361 CrossRef CAS PubMed.
- Y. Kim, L. Bigelow, M. Borovilos, I. Dementieva, E. Duggan, W. eschenfeldt, C. Hatzos, G. Joachimiak, H. Li, N. Maltseva, R. Mulligan, P. Quartey, A. Sather, L. Stols, L. Volkart, R. Wu, M. Zhou and A. Joachimiak, Structural Genomics, Part A, 2008, 85–105, DOI:10.1016/s0065-3233(07)75003-9.
- H. S. McHaourab, M. A. Lietzow, K. Hideg and W. L. Hubbell, Biochemistry, 1996, 35, 7692–7704 CrossRef CAS PubMed.
- R. Galindo-Murillo and J. Barroso-Flores, Phys. Chem. Chem. Phys., 2017, 19, 10571–10580 RSC.
- I. Negi, P. Kathuria, P. Sharma and S. D. Wetmore, Phys. Chem. Chem. Phys., 2017, 19, 16365–16374 RSC.
- G. Jeschke and Y. Polyhach, Phys. Chem. Chem. Phys., 2007, 9, 1895–1910 RSC.
- M. Kerzhner, H. Matsuoka, C. Wuebben, M. Famulok and O. Schiemann, Biochemistry, 2018, 57, 2923–2931 CrossRef CAS PubMed.
- N. S. Tangprasertchai, X. Zhang, Y. Ding, K. Tham, R. Rohs, I. S. Haworth and P. Z. Qin, Methods Enzymol., 2015, 564, 427–453 CAS.
- Y. Zhang, Y. Zhang, Z. Y. Liu, M. L. Cheng, J. Ma, Y. Wang, C. F. Qin and X. Fang, EMBO Rep., 2019, 20, e47016 CAS.
- P. V. Konarev, V. V. Volkov, A. V. Sokolova, M. H. J. Koch and D. I. Svergun, J. Appl. Crystallogr., 2003, 36, 1277–1282 CrossRef CAS.
- D. I. Svergun, J. Appl. Crystallogr., 1992, 25, 495–503 CrossRef CAS.
- P. H. Brown and P. Schuck, Biophys. J., 2006, 90, 4651–4661 CrossRef CAS PubMed.
- X. Zhang, C. X. Xu, R. Di Felice, J. Sponer, B. Islam, P. Stadlbauer, Y. Ding, L. Mao, Z. W. Mao and P. Z. Qin, Biochemistry, 2016, 55, 360–372 CrossRef CAS PubMed.
- G. Jeschke, V. Chechik, P. Ionita, A. Godt, H. Zimmermann, J. Banham, C. R. Timmel, D. Hilger and H. Jung, Appl. Magn. Reson., 2006, 30, 473–498 CrossRef CAS.
- Q. Cai, A. K. Kusnetzow, W. L. Hubbell, I. S. Haworth, G. P. Gacho, N. Van Eps, K. Hideg, E. J. Chambers and P. Z. Qin, Nucleic Acids Res., 2006, 34, 4722–4730 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01717e |
| ‡ Present address: Department of Mathematics and Computer Science, Freie Universität Berlin, Berlin 14195, Germany. |
| This journal is © The Royal Society of Chemistry 2020 |