
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bispecific circular aptamer tethering a built-in universal molecular tag for functional protein delivery†
Xiaoshu
Pan‡
a,
Yu
Yang‡
a,
Long
Li
a,
Xiaowei
Li
a,
Qiang
Li
a,
Cheng
Cui
 *ab,
Bang
Wang
a,
Hailan
Kuai
b,
Jianhui
Jiang
b and
Weihong
Tan
*abc
*ab,
Bang
Wang
a,
Hailan
Kuai
b,
Jianhui
Jiang
b and
Weihong
Tan
*abc
aDepartment of Chemistry, Department of Physiology and Functional Genomics, Center for Research at the Bio/Nano Interface, Health Cancer Center, UF Genetics Institute, McKnight Brain Institute, University of Florida, Gainesville, FL 32611-7200, USA. E-mail: tan@hnu.edu.cn; cuichengcuicheng@gmail.com
bMolecular Science and Biomedicine Laboratory (MBL), State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology, Aptamer Engineering Center of Hunan Province, Hunan University, Changsha, Hunan 410082, P. R. China
cThe Cancer Hospital of the University of Chinese Academy of Sciences, Institute of Basic Medicine and Cancer (IBMC), Chinese Academy of Sciences, Hangzhou, Zhejiang 310022, China
First published on 21st August 2020
Abstract
Chemically engineering endogenous amino acids with a molecular tag is one of the most common routes of artificially functionalizing proteins for identification or cellular delivery. However, it is challenging to make conjugation efficient, facile and productive as well as avoiding a high chance of deactivation of the functional proteins. Here we present a new and straightforward design to specifically tether the distinct six polyhistidine tag, terminally expressed on protein cargoes and cellular membrane proteins by using bispecific circular aptamers (bc-apts). The anti-His tag aptamer on one end of the bc-apt can easily recognize the biorthogonal six polyhistidine tag (His tag) on functional proteins like EGFP or RNase A. Meanwhile, a cell-specific aptamer, sgc8, on the other end efficiently facilitates the targeted delivery of functional proteins, improving their overall bioactivity in the cellular milieu by around 4 fold. Therefore, the nuclease-resistant bc-apt is a promising molecular tethering reagent to enable the noncovalent crosslink between live diseased cells and His tag protein cargoes.
Introduction
Proteins are essential biomacromolecules able to perform cell transduction, cell–cell interactions and other biological activities.1–3 Protein therapy, harnessing highly specific proteins to play specific roles, is considered one of the safest and most powerful therapeutic strategies.4–7 Nevertheless, it is difficult for naked proteins to reach their intracellular targets. Electrostatic adsorption, chemical conjugation and encapsulation are common strategies for loading protein cargos into or onto nanocarriers.8,9 For instance, Rotello et al. engineered proteins with a terminally expressed polyglutamine tag to amend the charge of proteins for robust loading of protein cargoes onto nanocarriers and efficient intracellular delivery. Nonetheless, the surface charge and the roughness of the bio-nano interface is likely to result in structural changes in different individual proteins and potential negative biological outcomes.10–12 Besides, site-selective conjugation of endogenous amino acids in order to avoid deactivation of proteins is challenging and would produce heterogeneous bioconjugates with various levels of activity and batch-to-batch differences.13 Thus, a general, biocompatible method, which can target proteins with little modification is required to realize target-specific delivery of functional protein cargos.Aptamers are short single-stranded oligonucleotides with a specific three-dimensional structure which can target various ligands ranging from ions to proteins.14–19 Because of their desirable features such as easy synthesis, facile chemical modification, and low immunogenicity, aptamers are widely conjugated with drugs, RNAi and even proteins for application in biomedicine and bioanalysis.17,20–25,45 However, heterogeneity and the need for purification of functional bioconjugates have limited successful applications of chemically engineered aptameric conjugates, especially aptamer–protein conjugates.21,26,27 Thus, in order to realize quantitative and spatial control of chemical modification, aptamer–mediated conjugation methods could be promising solutions,28–30 but they require multiple, labor-intensive preparation and purification steps. This calls for a facile and robust strategy to chemically bond aptamers with therapeutic ligands.
Recently, our group reported that a circular bivalent aptamer (cb-apt) with two identical cell-specific aptamers showed enhanced nuclease resistance and higher binding ability in vivo.31 Owing to the enhanced stability and affinity of cb-apts by virtue of chemically engineering one cell-specific aptamer with supramolecular ligands, cb-apts also enhanced the intracellular delivery of small-molecule drugs and functional proteins that adhere to supramolecular anchors on aptamers.21 Nonetheless, cb-apts themselves actually can be considered molecular crosslinkers without further modification. For example, two aptamer sequences in one linear nanoconstruct could bind with targets individually, but they were found to be relatively unstable under biological conditions.32–34 Because circular aptamers exhibit higher structural integrity and stability compared to linear nucleic acid assemblies,31,35 they are logical candidates for molecular crosslinking of two targets in biological milieu.
Herein we exploited bispecific circular aptamers as a general platform for targeted delivery of functional proteins by specific and strong interaction between a polyhistidine tag (His tag) of proteins and an anti-His tag aptamer selected by bead-based SELEX (“Systematic Evolution of Ligand Exponential Enrichment”).36,37 More specifically, an anti-His tag aptamer, which binds with a 6× His tag on the terminus of proteins,38,39 and sgc8, which recognizes protein tyrosine kinase 7 (PTK7) on the cell surface, were chosen to construct the bispecific circular anti-His tag/cell-specific aptamer (bc-apt), as shown in Fig. 1A. Next, the bc-apt is hypothesized to selectively, efficiently bind with His tag protein and target cells through twin contact. For a proof-of-concept, His tag enhanced Green Fluorescent Protein, EGFP, was incubated with the bc-apt for cellular analysis. H-tagged RNase A, a potent antineoplastic ribonuclease cleaving cytosolic RNA and then inducing cytotoxic effects upon cellular uptake, was investigated for its cytotoxic function after delivery mediated by the bc-apt.40,41 We demonstrated the enhanced binding and intracellular delivery of EGFP and enhanced inhibition of tumor cell growth induced by RNase A. Thus, using a bispecific circular aptamer as a practical, direct molecular crosslinker for the targeted delivery of functional therapeutic proteins is facile and efficient.
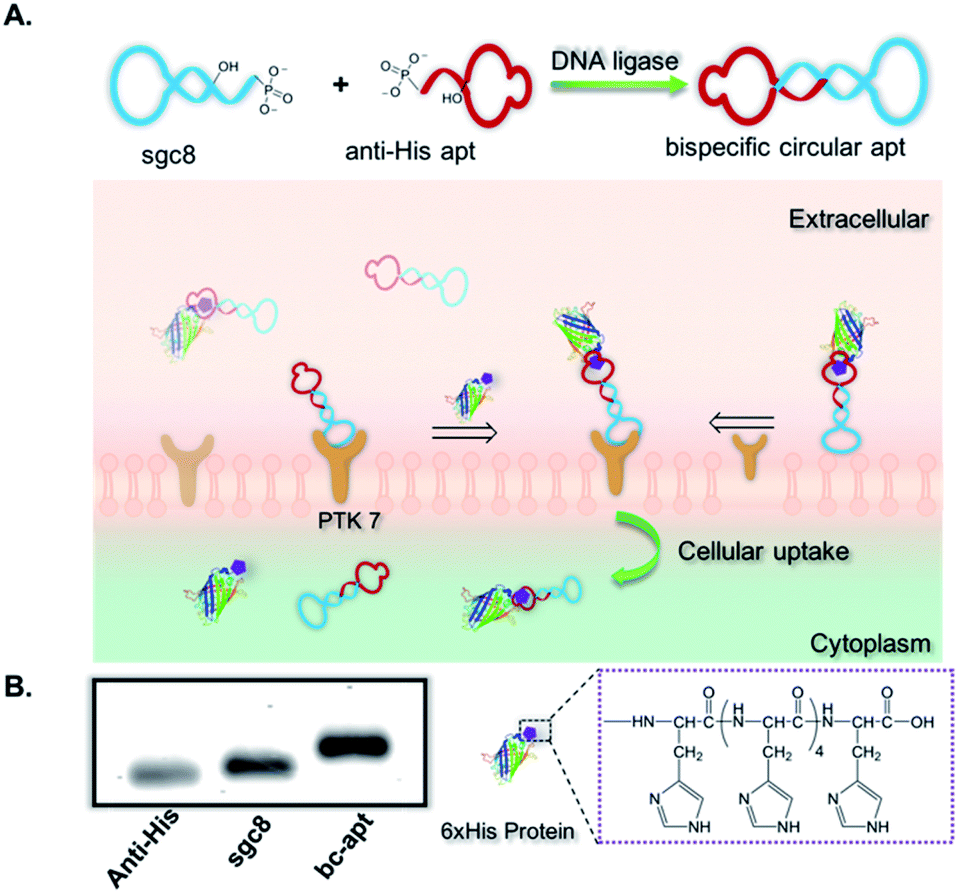 | ||
| Fig. 1 Design and characterization of the bispecific circular aptamer. (A) Scheme of construction of the bispecific circular aptamer (bc-apt) and artificial recognition process mediated by molecular linker bc-apt between His tagged protein cargoes and PTK7 on the targeted cells. (B) 3% agarose gel electrophoresis of mono aptamers, anti-His aptamer, sgc8, and the purified bc-apt. Agarose gel was stained by using ethidium bromide (EtBr). | ||
Results and discussion
To construct bc-apts, an anti-His tag aptamer and sgc8 with 13bp extended complementary sequences were efficiently ligated by T4 DNA ligase (Fig. 1A and detailed sequences shown in Table S1†). The resulting bc-apt was analyzed and purified by 8% 6M Urea-PAGE gel electrophoresis. The ready-to-use bc-apts were then characterized, along with the anti-His tag monomer and sgc8, by using 3% agarose gel. The anti-His tag aptamer and sgc8 exhibited similar mobility in the gel since they differ by only two nucleotides, while the mobility of the cyclized resulting aptamer was substantially retarded (Fig. 1B). Because of the presence of more hybridizing sites on nucleotides after cyclization of aptamer monomers, the bc-apt is more efficiently intercalated by using ethidium bromide (EtBr) compared to its components, showing the strongest band intensity. The anti-His tag aptamer intrinsically has 6 fewer base pairs than the sgc8 aptamer monomer (Table S1†); therefore, it exhibits the weakest band intensity, even with equal aptamer concentration. Anion exchange HPLC was also used to evaluate the purity of the bc-apt. The bc-apt eluted out at 14.5 min with a single peak showing satisfactory purity while sgc8 and the anti-His tag aptamer eluted at 15.0 and 13.5 min respectively (Fig. S1†). Although the bc-apt has the longest sequences, the structural rigidity of the bc-apt possibly weakened its interaction with the anionic resin which decreases the retention time on the column. Therefore, the bc-apt eluted a little bit earlier than sgc8 with a 55 mer and later than the anti-His tag aptamer with a 53 mer. HPLC and the agarose gel trace of the bc-apt together demonstrated that the purified bc-apt is sufficiently pure for further study.To evaluate the stability of the new constructs, we incubated the bc-apt in full culture medium containing 10% FBS and either exonuclease I or III. Exonuclease I (exo I) removes nucleotides from open-end DNA in the 3′ to 5′ direction, while exonuclease III (exo III) catalyses the removal of nucleotides from linear or nicked double-stranded DNA in the same direction. The fully cyclized bc-apt without free 3′ and 5′ ends exhibited greater physical stability in 1 U per μL exo I and III after 30 min of incubation, while linear aptamers were easily digested by either DNA exonucleases with no detectable band in agarose gel, as shown in Fig. 2A. We then asked whether the bc-apt is structurally stable under the biological culturing conditions where a broad variety of serum nucleases possibly cause more DNA attacks.42 Therefore, the bc-apt and the linear aptamer were treated with full culture medium for different time intervals. It is found that the fluorescently tagged bc-apt exhibited good binding properties against the targeted cells after exposure to serum nucleases for various hours while the linear aptamers showed the gradually weaker binding to the targeted cells (Fig. 2B). We then chose EtBr to illuminate the serum-treated bc-apt and linear aptamers in agarose gel for further stability analysis. Since EtBr inserted itself into stacking bases of DNA,48 it is easier for EtBr to illuminate the base pairing in DNA. Therefore, the bc-apt with 13 base pairings in the middle has the highest band intensity and then the band intensity of the bc-apt gradually decreased over hours which indicated that the stacking bases dissociating from each other over hours possibly resulted from serum nuclease. Consequently, the bc-apt showed fewer base pairings and weaker intercalation with EtBr indicated by the weaker band intensity after 36 hours (Fig. 2C and S2†). For linear aptamers with two open ends, the significant loss in band intensity of linear aptamers indicated that their stacking bases were more actively affected by serum nucleases and dissociated from each other in the first hour of incubation. Surprisingly, however, the binding ability of the aptamer is not greatly affected. We reasoned that there are still good amounts of surviving aptamers able to bind to the targeted CCRF-CEM cells considering the low Kd of sgc8, 0.78 nM. These results corresponded to the binding evaluation of the serum-treated sgc8 monomer and cb-apt at low concentration of 10 nM by Kuai et al.31 Therefore, compared to the linear aptamer, there are larger amounts of bc-apt maintaining good binding properties and structural integrity after serum treatment.
 | ||
| Fig. 2 Stability investigation of the bc-apt. (A) Bc-apt and linear aptamers were incubated with DNA exonuclease I and III respectively for 30 min, and then analyzed and imaged by using 3% agarose gel electrophoresis. (B) Flow cytometry analysis of a fluorescently labelled bc-apt and sgc8/His tag aptamer mixture targeting CCRF-CEM cells after treatment in full culture medium for various hours. (C) 3% agarose gel electrophoresis of the equivalent amount of the bc-apt and sgc8/His tag aptamer mixture after serum treatment for different hours. The gel was stained by using EtBr. | ||
After demonstrating the stability of the bc-apt, we next investigated the noncovalent complex formed between the bc-apt and His tagged proteins. The accessibility of His tag to the anti-His tag aptamer is reported to be individually different for different proteins due to size effects.39 Basically, His tag on larger proteins is less accessible to the aptamer than smaller ones. Therefore, given that the bc-apt investigated in the study has approximately 33.4 kDa, EGFP, as the model protein with an around 27 kDa C-terminally expressed 6× His tag, was first incubated with the bc-apt, as well as BSA, as the negative control protein without a 6× His tag, which is also used in the binding buffer for mitigating the non-specific binding. The protein–aptamer complex was then evaluated by band-shifting assay.43 As shown in Fig. 3A, after incubation with the monomeric anti-His tag aptamer and bc-apt, EGFP significantly retarded aptamer migration in native PAGE gel due to specific molecular recognition between the His tag on EGFP and the anti-His aptamer. However, BSA had little effect on the mobility of either the anti-His tag or bc-apts since BSA without the H tag did not complex with either of the aptamers. The results demonstrated that the anti-His tag aptamer on one end of the bc-apt exhibited good binding affinity and specificity against the His tag on EGFP compared to the anti-His tag aptamer only. A similar outcome was also exhibited in the electrophoretic mobility analysis of the recognition between the His tag on the C terminus of 16 kDa RNase A and the bc-apt, anti-His tag aptamer individually (Fig. 3B). Similarly, a slower mobility of the bc-apt/ and anti-His/His-RNase A was also observed suggesting their more intense interaction than the bc-apt/ and anti-His/BSA, which is highly consistent with the results of EGFP. Thus it is concluded that the bc-apt retains its strong binding specificity and affinity to the His tag on proteins like EGFP and RNase A. Even though the His tag with 1 kDa has a lower coverage ratio on larger proteins, it is exciting to see that His tagged Cas9 proteins also hinder the migration of the bc-apt on native PAGE gel after the incubation while there are amounts of free aptamers not targeting His-Cas9. The binding between the bc-apt and larger proteins like Cas9 is achievable but weaker than RNase A with the same DNA to protein ratio (Fig. S3†), agreeing with the size effects of proteins. Therefore, by direct incubation, we efficiently assembled protein–aptamer complexes based on the high specificity and affinity of the anti-His tag aptamer without further purification. The integrity of bc-apt-protein shows promise for the delivery of tethered proteins into specific cells.
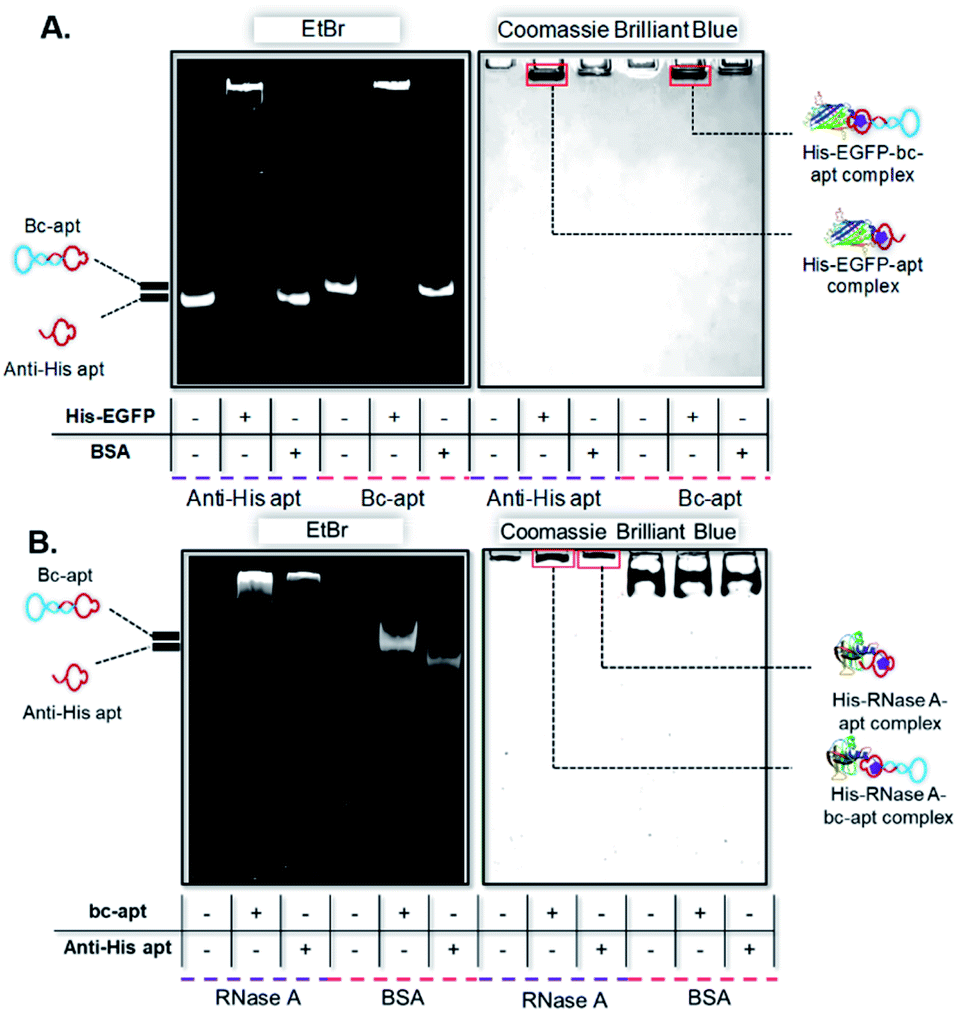 | ||
| Fig. 3 Electrophoretic mobility assay of targeted protein and aptamer complex. (A) Electrophoretic mobility analysis of the complex formed between aptamers and His-EGFP and non-His tag BSA protein in 1× Tris-Borate EDTA (1× TBE) buffer with addition of 50 mM potassium ions. From left to right: anti-His tag aptamer only; EGFP incubated with the His tag aptamer; BSA incubated with the His tag aptamer only; bc-apt only; EGFP incubated with the bc-apt; BSA incubated with the bc-apt. (B) Electrophoretic mobility analysis of complexes formed between aptamers and His-RNase A and non-His tag BSA protein in 1× native Tris-Glycine buffer. From left to right: RNase A only; RNase A incubated with the bc-apt; RNase A incubated with anti-His apt; BSA only; BSA incubated with the bc-apt; BSA incubated with anti-His apt. | ||
We then asked if sgc8 on the bc-apt could bind with biomarkers on the cell membrane, when the anti-His tag aptamer links with the HIS tag on the protein cargo. It is reported that the presence of 13 or more base pairs between two aptamers on each end led to no adverse effect on the binding affinity of aptamers against targeted cells.31 Therefore, to construct a suitable bc-apt, 13 base pairs were selected to link sgc8 and the anti-His tag aptamer (Fig. 1A and Table S1†). Meanwhile, the bc-apt, prepared fluorophore-free, was also purified with Urea PAGE gel, and DNA bands were visualized on the TLC plate illuminated by using a UV lamp with 260 nm. The fluorophore-free bc-apt was then analysed in 3% agarose gel corroborating the existence and purity of the bc-apt (Fig. 1B and 2A).
Next, EGFP as a model protein was firstly used to investigate whether the bc-apt can mediate the artificial recognition between protein cargoes and targeted cells. As shown in the scheme, two possible noncovalent crosslinkings of targeted cells and protein cargoes by the bc-apt are either cells as a solid support for recognizing the His tag on EGFP or the bc-apt/EGFP complex together with the targeted specific cell line respectively (Fig. 1A). It is reported that the binding affinity of the anti-His tag aptamer against different proteins is in the nM to μM range39 which is much higher than sgc8 so we assumed that the binding-limiting step is the recognition of the His tag on the proteins by the bc-apt. Meanwhile, the equilibrium state is attributed to not only the binding affinity but also the relative concentrations.47 With this in mind, we first investigated the binding between the preformed bc-apt/His-EGFP complex and CCRF-CEM cells. It is found that with the excessive bc-apt/His-EGFP complex, 15 μM bc-apt/3 μM His-EGFP shows good target binding ability, 3-fold higher compared to free EGFP (Fig. 4A, B and S4†). Though the excessive bc-apt was applied under the binding conditions, bc-apt did not completely saturate the His tag on EGFP in the binding buffer. Thus, we hypothesized that CCRF-CEM cells may function as a solid support for the cascade binding of bc-apt and the His tag on protein cargoes. Therefore, the bc-apt was first incubated with CCRF-CEM cells and then together with His-EGFP. Interestingly, 2.5 μM bc-apt shows similar target binding properties to the preformed bc-apt/His-EGFP, though with fewer percentages of EGFP-bound live cells with lower concentration of the bc-apt (Fig. S5†). By further increasing the concentration of bc-apt to 6 μM, bc-apt-mediated target binding is distinct and no significant binding shift is seen in EGFP intensity compared to the 2.5 μM group thereby suggesting an equilibrium state of binding with EGFP (Fig. S5 and S6†). Meanwhile, mono anti-His tag aptamers were added for outcompeting with the twin contact mediated bc-apt, but the results showed that the group had similar binding properties to the groups with addition of sgc8 and no addition of the anti-His aptamer (Fig. S6†). This suggested that CCRF-CEM cells targeted by the bc-apt can serve as a solid support to favour the specific binding of the bc-apt and His tag EGFP while free monomeric aptamers are generally less competitive than the bc-apt on the solid support for binding with the His tag. Besides, His-Cas9-GFP with 160 kDa, was also investigated for accessibility of the His tag to form larger proteins on biological membranes. It is found that, however, 5 μM bc-apt did not distinctly enhance the twin contact between 0.8 μM His-Cas9-GFP and CCRF-CEM cells possibly due to size effects and low concentration of Cas9 used in the study (Fig. S7†). Therefore, specific binding of His tagged proteins against CCRF-CEM cells by the bc-apt is good and feasible but limited by accessibility of the His tag on protein cargoes.
Due to the specific and stable binding property of the bc-apt, the intracellular delivery of EGFP was then investigated. It is reported that sgc8 binding with PTK7 on CCRF-CEM cells is taken by PTK7-mediated endocytosis into the cells and an sgc8-tethered nanotrain can also specifically deliver intercalated Dox into the targeted cells.28,44,46 Therefore, sgc8 as the tethering molecular agent is promising in the targeted delivery of molecular drug by noncovalent crosslinking. We reasoned that EGFP tethered to biological membranes by bc-apt can also be specifically taken by CCRF-CEM cells. The confocal results showed the good distribution of the higher EGFP signal inside CCRF-CEM cells after being coincubated with the bc-apt and lipofectamine 2000 and that some His-EGFP aggregates induced by lipofectamine 2000 are found to be tethered with the CCRF-CEM cell membrane by the bc-apt while EGFP only showed little EGFP signal inside the cells or on the cell membrane (Fig. 4C). This indicates the higher intracellular delivery of EGFP proteins into CCRF-CEM cells after being tethered on biological membranes by the bc-apt. In a PTK7-negative Ramos cell, however, the intracellular delivery of His-EGFP and EGFP only mediated by the bc-apt and lipofectamine 2000 is hardly detected since there is no binding between the bc-apt and nontargeted cells.44 (Fig. S9A†) Therefore, bc-apt enabled His-EGFP efficiently enter the targeted cells.
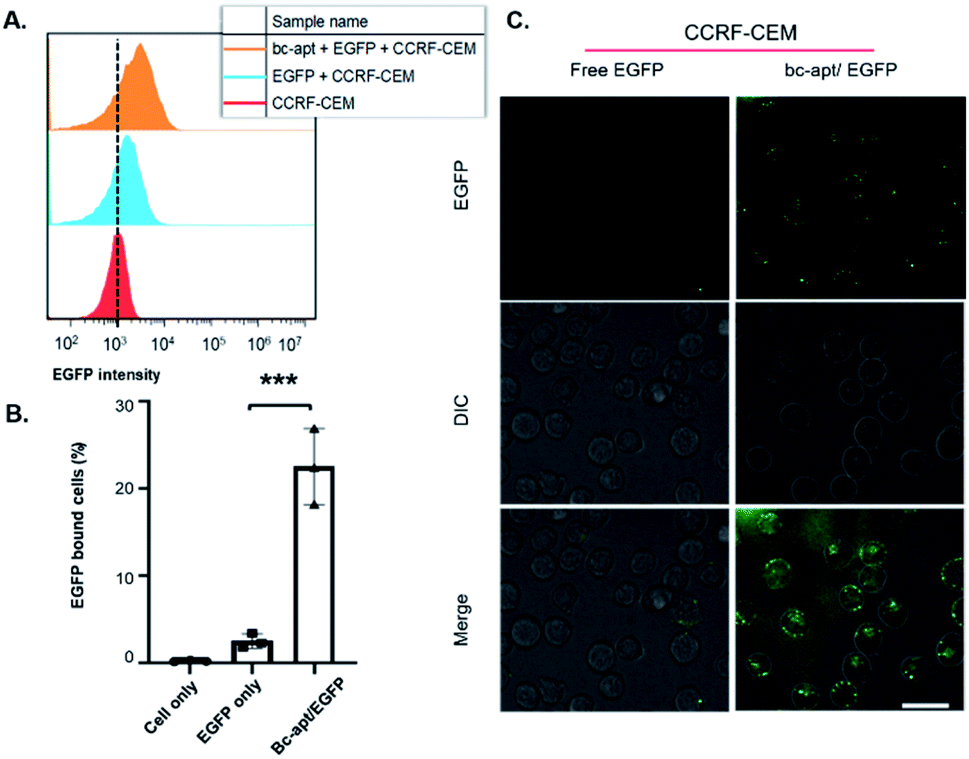 | ||
| Fig. 4 Investigation of specific binding and cellular uptake of His-EGFP mediated by the bc-apt. (A) Flow cytometry analysis of the bc-apt/EGFP complex binding with cells. CCRF-CEM cells were incubated with 15 μM bc-apt/3 μM EGFP. (B) Statistical analysis of EGFP bound CCRF-CEM cells after the incubation with 15 μM bc-apt/3 μM EGFP. The experiments were conducted in triplicate. ANOVA statistical analysis, with ***P < 0.001. (C) CCRF-CEM cell uptake of His-EGFP mediated by the bc-apt and lipofectamine 2000. The amount of EGFP used was equivalent to that of the bc-apt. Scale bar: 25 μm. | ||
Before investigating in vitro targeted therapeutic intervention, the cytotoxicity of bc-apts was first evaluated by using MTS cell proliferation colorimetric assay kit. CCRF-CEM cells were incubated with various concentrations of bc-apts under the desired conditions for 48 hours and then replaced with fresh culture medium with reagents for MTS analysis. As shown in Fig. S4,† the viability of CCRF-CEM cells treated with different concentrations of bc-apts is 90 percent, or even higher, meaning that bc-apts have excellent biocompatibility even at 10 μM concentration (Fig. 5 and S8A†). Encouraged by the dramatic biosafety of bc-apts, we then evaluated the potency of bc-apt-mediated therapeutic intervention by functional proteins. To accomplish this, His-RNase A, as a potent anticancer protein, was complexed with bc-apts characterized with Native PAGE (Fig. 2B). Also, we assessed whether the binding between the aptamer and His tag on proteins may influence the enzymatic activity of RNase A. It is found that noncovalent apt-RNase A conjugate does not affect the degradation of RNA by RNase A (Fig. 5A).
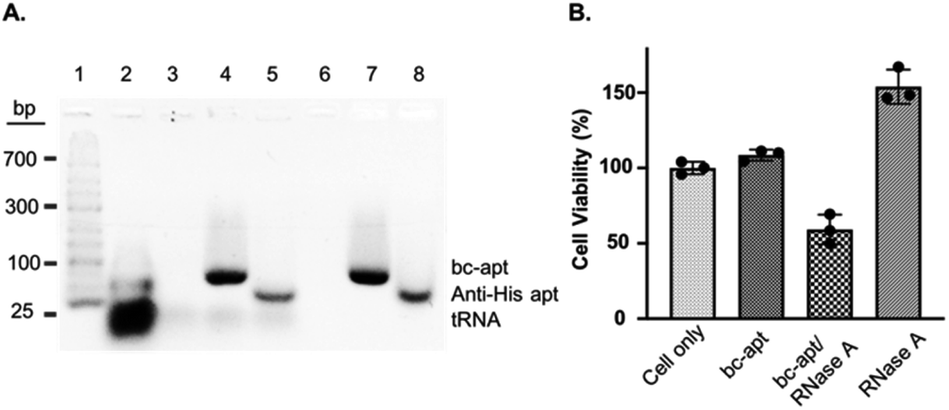 | ||
| Fig. 5 (A) Bioactivity analysis of RNase A noncovalently conjugated with bc-apt and anti-His apt in 3% agarose gel. Lane 1–8: DNA ladder; tRNA; tRNA and RNase A; tRNA and bc-apt/RNase A; tRNA and anti-His apt/RNase A; RNase A; bc-apt/RNase A; anti-His apt/RNase A. 120 μg tRNA was used in each incubation. The gel was stained with 1× Gelred nucleic acid staining reagent. (B) Cytotoxicity assay of the cytotoxic protein RNase A. The same number of CCRF-CEM cells was treated with PBS, bc-apt, bc-apt/RNase A and RNase A only. The cytotoxicity was normalized to cells treated with binding buffer only. | ||
We next investigated the targeted delivery of His-RNase A mediated by bc-apts. Since RNase A is a basic protein which can actively interact with anionic biopolymers, bc-apts after binding to the His-tag on RNase A may electrostatically adsorb onto RNase A and then block its binding to targeted cells.49 It is reasonable to envisage that bc-apts complexed with His-RNase A possibly required longer time to bind with the targeted cells. However, basic RNase A can also interact with anionic glycans on the membranes of cancer cells which would then enable the nonspecific intracellular trafficking of RNase A reported by Raines et al.49 Therefore, we hypothesized that if CCRF-CEM cells were first treated with bc-apts and then His-RNase A, His-RNase A may then be effectively delivered into targeted cells to enhance its therapeutic outcome. We found that, by using this strategy, 18 μM His-RNase A with bc-apts caused the robust killing efficacy to CCRF-CEM cells which is 2-fold higher than His-RNase A only (Fig. 5B). Surprisingly, however, CCRF-CEM cells had higher viability after being treated by free RNase A than those not treated in Fig. 5B. We reasoned that the protection effects possibly resulted from the non-toxic cryopreservation agent trehalose in RNase A sample which is found to help stressful cells retain biological membrane integrity under harsh conditions including low temperature.50 Meanwhile, we studied the cytotoxicity caused by preformed bc-apt/RNase A and free RNase A respectively. As expected, the similar killing efficacy of preformed bc-apt/RNase A to RNase A indicated that bc-apt/RNase A complexes exhibited no delivery advantage over the free RNase A over the longer incubating hours (Fig. S8B†) thereby showing that the bc-apt mediated delivery of His-RNase A promoted the therapeutic effects of His-RNase A in the targeted CCRF-CEM cells under the designated conditions. Since there is no reported protection effect of trehalose in physiological condition, higher viability in physiological temperature was not observed (Fig. S8B†). We then asked if His-RNase A with high concentration would also halt the growth of PTK7 negative Ramos cells. It is found that neither bc-apt/RNase A nor free RNase A effectively killed Ramos cells, but a similar higher percent of viable cells was also observed using the same incubation strategies (Fig. S9B†). Bc-apts, as molecular crosslinkers, hereby are shown to preferably recognize target cells and then specific His tag functional RNase A for higher specific intracellular protein delivery and induced cell death.
Conclusions
We herein constructed a fully cyclized bc-apt by hybridization between sgc8 and an anti-His tag aptamer and ligation by the T4 DNA ligase, as a compelling and promising bispecific molecular linker for the delivery of functional proteins. In this platform, bc-apt functions as a site- and ligand-specific molecular “glue” to tether the built-in His tag on functional proteins onto specific cells, which enables the specific delivery of functional proteins into CCRF-CEM cells. Basically, two unique advantages of bc-apts highlight their use in the delivery of functional proteins. Firstly, outstanding nuclease resistance endows bc-apts with high structural integrity and robust binding ability over time under physiological conditions. Next, for the efficient and specific delivery of His tagged functional proteins like EGFP and RNase A, a bc-apt-based platform demonstrated that protein cargoes intracellularly delivered can be completely free of chemical modification or physical adsorption to nanocarriers. This straightforward platform successfully enabled us to improve the specific killing of sgc8-targeted CCRF-CEM cells induced by RNase A without any disturbance in nontargeted Ramos cells. Furthermore, though we found a lower accessibility of the His tag on Cas9-GFP to bc-apts on the specific biological membrane, higher concentration of Cas9-GFP and the His tag expressed on both termini of Cas9-GFP, which is commercially available, are feasible solutions to this critical hurdle. To the best of our knowledge, this is the first time that a bc-apt tethering a built-in His tag on functional proteins of specific biological membranes for functional protein delivery has been reported. Finally, the bc-apt not only provides a general molecular recognition strategy to build efficient, bispecific protein–aptamer assemblies on the cell membrane for functional protein delivery but also can be rationally adapted to other built-in molecular tags like GST and Myc tag to solve biological and physiological challenges in the future protein therapy and gene editing field.Author contributions
X. Pan and Dr Y. Yang designed and performed all experiments and analyzed the experimental results. X. Pan wrote the manuscript. Prof. W. Tan supervised and led the discussion and analysis of the work in which all authors were involved.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSFC grants (NSFC 21827811 and 61527806), the Science and Technology Project of Hunan Province (2017XK2103), and Hunan Provincial Key Area R&D Program (2019SK2201).Notes and references
- W. E. Balch, R. I. Morimoto, A. Dillin and J. W. Kelly, Science, 2008, 319, 916–919 CrossRef CAS PubMed.
- S. Angers and R. T. Moon, Nat. Rev. Mol. Cell Biol., 2009, 10(7), 468–477 CrossRef CAS PubMed.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Angew. Chem., Int. Ed., 2006, 45, 2348–2368 CrossRef CAS PubMed.
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7(1), 21–39 CrossRef CAS PubMed.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53(46), 12320–12364 CAS.
- Y. Wu, S. M. Mikulski, W. Ardelt, S. M. Rybak and R. J. J. Youle, Biol. Chem., 1993, 268, 10686–10693 CAS.
- G. Walsh, Nat. Biotechnol., 2018, 36, 1136–1145 CrossRef CAS PubMed.
- Z. Gu, A. Biswas, M. X. Zhao and Y. Tang, Chem. Soc. Rev., 2011, 40, 3638–3655 RSC.
- (a) A. Fu, R. Tang, J. Hardie, M. E. Farkas and V. M. Rotello, Bioconjugate Chem., 2014, 25(9), 1602–1608 CrossRef CAS PubMed; (b) Y. W. Lee, D. C. Luther, R. Goswami, T. Jeon, V. Clark, J. Elia, S. Gopalakrishnan and V. M. Rotello, J. Am. Chem. Soc., 2020, 142(9), 4349–4355 CrossRef CAS PubMed.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8(7), 543–547 CrossRef CAS PubMed.
- Z. J. Deng, M. Liang, M. Monteiro, I. Toth and R. F. Minchin, Nat. Nanotechnol., 2011, 6, 39–44 CrossRef CAS PubMed.
- Y. Duan, Y. Liu, R. Coreas and W. Zhong, Anal. Chem., 2019, 91(6), 4204–4212 CrossRef CAS PubMed.
- (a) X. Liu, P. Zhang, W. Rödl, K. Maier, U. Lächelt and E. Wagner, Mol. Pharmaceutics, 2017, 14(5), 1439–1449 CrossRef CAS PubMed; (b) O. Boutureira and G. J. Bernardes, Chem. Rev., 2015, 115(5), 2174–2195 CrossRef CAS PubMed; (c) Y. Zhang, K. Y. Park, K. F. Suazo and M. D. Distefano, Chem. Soc. Rev., 2018, 47, 9106–9136 RSC.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1992, 355, 850–852 CrossRef CAS PubMed.
- K. Sefah, D. Shangguan, X. L. Xiong, M. B. O'Donoghue and W. Tan, Nat. Protoc., 2010, 5, 1169 CrossRef CAS PubMed.
- W. Tan, M. J. Donovan and J. Jiang, Chem. Rev., 2013, 113, 2842–2862 CrossRef CAS PubMed.
- M. R. Dunn, R. M. Jimenez and J. C. Chaput, Nat. Rev. Chem., 2017, 1(10), 0076 CrossRef CAS.
- X. Wu, J. Chen, M. Wu and J. X. Zhao, Theranostics, 2015, 5(4), 322 CrossRef PubMed.
- D. Castanotto and J. J. Rossi, Nature, 2009, 457(7228), 426 CrossRef CAS PubMed.
- Y. Jiang, X. Pan, J. Chang, W. Niu, W. Hou, H. Kuai, Z. Zhao, J. Liu, M. Wang and W. Tan, J. Am. Chem. Soc., 2018, 140, 6780–6784 CrossRef CAS PubMed.
- W. J. Niu, X. G. Chen, W. Tan and A. S. Veige, Angew. Chem., Int. Ed., 2016, 55, 8889 CrossRef CAS PubMed.
- S. Wan, L. Zhang, S. Wang, Y. Liu, C. Wu, C. Cui, H. Sun, M. Shi, Y. Jiang, L. Li, L. Qiu and W. Tan, J. Am. Chem. Soc., 2017, 139, 5289 CrossRef CAS PubMed.
- T. Chu, K. Twu, A. Ellington and M. Levy, Nucleic Acids Res., 2006, 34, e73 CrossRef PubMed.
- R. Wang, G. Zhu, L. Mei, Y. Xie, H. Ma, M. Ye, F. Qing and W. Tan, J. Am. Chem. Soc., 2014, 136, 2731 CrossRef CAS PubMed.
- N. Krall, F. P. Da Cruz, O. Boutureira and G. J. Bernardes, Nat. Chem., 2016, 8, 103–113 CrossRef CAS PubMed.
- Z. Zhou, Y. Xiang, A. Tong and Y. Lu, Anal. Chem., 2014, 86(8), 3869–3875 CrossRef CAS PubMed.
- G. Zhu, G. Niu and X. Chen, Bioconjugate Chem., 2015, 26, 2186–2197 CrossRef CAS PubMed.
- C. Cui, H. Zhang, R. Wang, S. Cansiz, X. Pan, S. Wan, W. Hou, L. Li, M. Chen and Y. Liu, Angew. Chem., 2017, 129, 12116–12119 CrossRef.
- R. Wang, D. Lu, H. Bai, C. Jin, G. Yan, M. Ye, L. Qiu, R. Chang, C. Cui, H. Liang and W. Tan, Chem. Sci., 2016, 7, 2157 RSC.
- H. Kuai, Z. Zhao, L. Mo, H. Liu, X. Hu, T. Fu, X. Zhang and W. Tan, J. Am. Chem. Soc., 2017, 139(27), 9128–9131 CrossRef CAS PubMed.
- R. Ueki, S. Atsuta, A. Ueki and S. Sando, J. Am. Chem. Soc., 2017, 139(19), 6554–6557 CrossRef CAS PubMed.
- X. Yu, S. Ghamande, H. Liu, L. Xue, S. Zhao, W. Tan, L. Zhao, S. C. Tang, D. Wu, H. Korkaya and N. J. Maihle, Nucleic Acid Ther., 2018, 10, 317–330 CrossRef CAS PubMed.
- J. Zheng, S. Zhao, X. Yu, S. Huang and H. Y. Liu, Theranostics, 2017, 7(5), 1373 CrossRef CAS PubMed.
- J. Li, J. Zhou, T. Liu, S. Chen, J. Li and H. Yang, Chem. Commun., 2018, 54, 896–899 RSC.
- M. B. M. Doyle, SA 2005/0142582 A1, Patent Application Publication, US, 2005.
- S. Tsuji, T. Tanaka, N. Hirabayashi, S. Kato, J. Akitomi, H. Egashira, I. Waga and T. Ohtsu, Biochem. Biophys. Res. Commun., 2009, 386, 227–231 CrossRef CAS PubMed.
- X. Tan, W. Chen, S. Lu, Z. Zhu, T. Chen, G. Zhu, M. You and W. Tan, Anal. Chem., 2012, 84, 8272–8276 CrossRef CAS PubMed.
- (a) Ö. Kökpinar, J. G. Walter, Y. Shoham, F. Stahl and T. Scheper, Biotechnol. Bioeng., 2011, 108(10), 2371–2379 CrossRef PubMed; (b) S. Tsuji, T. Tanaka, N. Hirabayashi, S. Kato, J. Akitomi, H. Egashira, I. Waga and T. Ohtsu, Biochem. Biophys. Res. Commun., 2009, 386(1), 227–231 CrossRef CAS PubMed.
- M. Wang, S. Sun, C. I. Neufeld, B. Perez-Ramirez and Q. Xu, Angew. Chem., 2014, 126, 13662 CrossRef.
- P. A. Leland and R. T. Raines, Chem. Biol., 2001, 8, 405–413 CrossRef CAS PubMed.
- (a) I. R. C. Hill, M. C. Garnett, F. Bignotti and S. S. Davis, Anal. Biochem., 2001, 291(1), 62–68 CrossRef CAS PubMed; (b) J. P. Shaw, K. Kent, J. Bird, J. Fishback and B. Froehler, Nucleic Acids Res., 1991, 19, 747 CrossRef CAS PubMed.
- L. M. Hellman and M. G. Fried, Nat. Protoc., 2007, 2(8), 1849 CrossRef CAS PubMed.
- (a) Z. Xiao, D. Shangguan, Z. Cao, X. Fang and W. Tan, Chem. – Eur. J., 2008, 14, 1769–1775 CrossRef CAS PubMed; (b) C. Lv, C. Yang, D. Ding, Y. Sun, R. Wang, D. Han and W. Tan, Anal. Chem., 2019, 91(21), 13818–13823 CrossRef CAS PubMed.
- Y. Yang, W. Zhu, L. Feng, Y. Chao, X. Yi, Z. Dong, K. Yang, W. Tan, Z. Liu and M. Chen, Nano Lett., 2018, 18(11), 6867–6875 CrossRef CAS PubMed.
- D. Shangguan, Z. Tang, P. Mallikaratchy, Z. Xiao and W. Tan, ChemBioChem, 2007, 8, 603–606 CrossRef CAS PubMed.
- (a) F. Spill, Z. B. Weinstein, A. Irani Shemirani, N. Ho, D. Desai and M. H. Zaman, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12076–12081 CrossRef CAS PubMed; (b) L. Long, X. Chen, C. Cui, X. Pan, X. Li, H. S. Yzad, Q. Wu, L. Qiu, J. Li and W. Tan, J. Am. Chem. Soc., 2019, 141(43), 17174–17179 CrossRef PubMed.
- A. Kellett, Z. Molphy, C. Slator, V. McKee and N. P. Farrell, Chem. Soc. Rev., 2019, 48, 971–988 RSC.
- T. Chao, L. D. Lavis and R. T. Raines, Biochemistry, 2010, 49(50), 10666–10673 CrossRef CAS PubMed.
- (a) A. Patist and H. Zoerb, Colloids Surf., B, 2005, 40, 107–113 CrossRef CAS PubMed; (b) N. K. Jain and I. Roy, Protein Sci., 2009, 18(1), 24–36 CAS; (c) H. Mussauer, V. L. Sukhorukov and U. Zimmermann, Cytometry, 2001, 45, 161–169 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: All experimental details are complete and written as a separate section in the SI materials. These include the ligation, purification and testing protocols. They are written in detail and are complete. See DOI: 10.1039/d0sc02279a |
| ‡ X. P. and Y. Y. contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2020 |