
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Introduction of a 7-aza-6-MeO-indoline auxiliary in Lewis-acid/photoredox cooperative catalysis: highly enantioselective aminomethylation of α,β-unsaturated amides†
Santosh K.
Pagire
 ,
Naoya
Kumagai
* and
Masakatsu
Shibasaki
*
,
Naoya
Kumagai
* and
Masakatsu
Shibasaki
*
Institute of Microbial Chemistry (BIKAKEN), 3-14-23, Kamiosaki, Shinagawa-ku, Tokyo 141-0021, Japan. E-mail: mshibasa@bikaken.or.jp; nkumagai@bikaken.or.jp
First published on 27th April 2020
Abstract
An efficient cooperative chiral Lewis acid/photoredox catalytic system for engaging highly reactive radicals in highly enantioselective conjugate addition to α,β-unsaturated carbonyls is highly desirable. Direct photoexcitation of unbound substrates typically induces undesired background pathways for racemic products and remains a formidable challenge to be addressed in the area of enantioselective photocatalysis. Herein, we report a cooperative catalytic system comprising a chiral Cu(I) complex and an Ir(III) photocatalyst fueled by visible-light irradiation that allows for seamless integration of the catalytic formation of α-amino alkyl radicals and subsequent enantioselective addition to α,β-unsaturated amides. A 7-aza-6-MeO-indoline attachment on the amide substrates plays a pivotal role in suppressing the undesired pathways, resulting in excellent enantioselectivity and enabling expedited access to valuable γ-aminobutyramides. The indoline amide was readily diversified with full recovery of the azaindoline attachment, highlighting the synthetic utility of this cooperative catalytic system.
Introduction
Over the past decade, the field of visible-light photoredox catalysis has rapidly advanced as a sustainable methodology for exerting orthogonal activation modes to manipulate small organic molecules,1,2 in which photoinduced single-electron transfer triggers the in situ generation of active chemical species from organic substrates, thereby enabling a variety of unique chemical events to facilitate C–C and C–heteroatom bond formation.3–12 Recently, reaction promotion by combining photoexcitation and concomitant stereocontrol with the aid of catalytic chiral sources such as chiral Lewis acids or organocatalysts has become a topic of great interest and prompted the development of enantioselective photocatalysis.13–23 Meggers et al. developed chiral-at-metal Λ-configured or Δ-configured Ir(III) or Rh(III) photoactive complexes for a broad range of asymmetric photocatalytic transformations.24,25 Although several enantioselective photoredox reactions are now well established, a number of fundamental reactions such as Giese reactions26,27 could benefit from improved catalytic systems.28 In this particular case, the α,β-unsaturated carbonyl substrates may undergo direct [2 + 2]-photocycloaddition29–33 or E/Z-isomerization,34,35 which is often difficult to suppress.36 To overcome such shortcomings, elaborated catalytic systems were devised to streamline the desired catalytic scenario. In the late 20th century, Sibi and Porter described the Lewis acid-promoted enantioselective radical addition to the electron-deficient α,β-unsaturated compounds.37 Stoichiometric amounts of reagents were required, however, likely due to the undesired background reaction of the unbound substrate. Yet, this reaction set the standard for the future development of numerous enantioselective conjugate radical addition reactions.38–41 It was later demonstrated that visible light photocatalysts could be utilized to generate α-amino radicals, thereby obviating the mandatory use of external reagents.42–50 Catalytic enantioselective photocatalytic intramolecular conjugate addition of α-amino radicals to the quinolone skeleton was first disclosed by Bach's group using a chiral hydrogen-bonding photosensitizer.51 The work of Pandey et al.52–56 and others57–62 revealed that α-silylamines have a pivotal role in the photogeneration of α-amino radical species, which are integrated into a subsequent conjugate addition in an intermolecular fashion. Yoon et al. developed a dual catalytic system comprising a chiral Sc(III) complex and a Ru-based photocatalyst that promotes conjugate addition to form the chiral products with excellent enantiocontrol.63 Subsequently, Gong et al. and Meggers et al. independently provided alternative protocols for conjugate additions using ingeniously designed chiral photocatalysts such as Ni(II)-DBFOX64 and Δ-RhO,65 respectively. With a similar platform, few other groups attempted analogous reactions, and the dominant racemic background reaction could be overcome to the great extent.66,67Our recent efforts focusing on asymmetric catalysis led us to identify that 7-azaindoline amides are privileged substrates that drive a number of highly stereocontrolled C–C bond-forming reactions in the context of Cu(I) catalysis.68,69 However, the 7-azaindoline auxiliary had not been implemented in the field of photocatalysis and the use of inexpensive copper complexes is rarely explored in asymmetric photocatalysis.23,70–73 We reasoned that this auxiliary could also serve as a potential stereo-controlling unit in radical reactions and provide a powerful alternative to several challenging photocatalytic transformations. Herein, we established a photocatalytic additive-free protocol exerted by Cu(I)/Ir(III) dual catalysis to engage α,β-unsaturated 7-azaindoline amides and α-silylamines in a highly enantioselective radical conjugate addition. The intriguing substituent effect of the 7-azaindoline unit was first revealed by systematic studies and comparisons of the crystal structures of Cu(I)/amide complexes. The broad substrate generality is supported by divergent transformation of the 7-azaindoline moiety of the enantioenriched products and highlights the synthetic utility of the present catalytic protocol.
Results and discussion
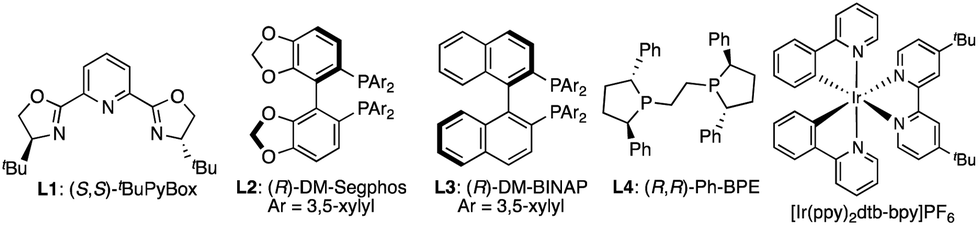
We initiated our investigation with the prototypical 7-azaindoline amide 1a and α-silylamine 2a as an electrophile and radical source, respectively, where 1 mol% of [Ir(ppy)2(dtb-bpy)]PF6 was used as a photocatalyst under blue-light irradiation (λmax 448/455 nm). The initial attempt using a Cu(I)/(S,S)-tBu-PyBox (L1) complex as a typical chiral Lewis acid in acetonitrile at room temperature resulted in poor conversion to give the desired product 3aa (Table 1, entry 1). This is likely due to the decomposition of the Cu(I) complex as indicated by the deeply colored reaction mixture, preventing photoexcitation of the Ir(III) photocatalyst. In contrast, Cu(I) phosphine complexes emerged as a compatible chiral Lewis acid under blue-light irradiation, affording 3aa in moderate yield (entries 2–6). Among the chiral phosphines tested, a commercially available biaryl-type (R)-DM-Segphos (L2) afforded the highest enantioselectivity (89% ee), which was sensitive to the reaction temperature (entries 2–4). A protic solvent like ethanol was beneficial for increasing the yield to 73% (vide infra, see mechanistic discussion), but a reduction in solubility hampered the smooth progress of the reaction at −20 °C (entries 7 and 8). Aiming to attain homogeneity, we next evaluated a mixed solvent system, leading to the identification of EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DME (1:3) as the optimal reaction media to give 3aa in 83% yield with 89% ee (entries 9–12). Lowering the reaction temperature to −30 °C did not further enhance the enantioselectivity, and the reaction became more sluggish (entry 13). No reaction occurred in the absence of a photocatalyst or light source, confirming that the photocatalytic activation of 2a is essential to the cooperative catalysis (entries 14 and 15). Conditions lacking a Cu(I) catalyst gave no product 3aa, indicating that the reaction system does not undergo a non-stereoselective background reaction (entry 16).
:DME (1:3) as the optimal reaction media to give 3aa in 83% yield with 89% ee (entries 9–12). Lowering the reaction temperature to −30 °C did not further enhance the enantioselectivity, and the reaction became more sluggish (entry 13). No reaction occurred in the absence of a photocatalyst or light source, confirming that the photocatalytic activation of 2a is essential to the cooperative catalysis (entries 14 and 15). Conditions lacking a Cu(I) catalyst gave no product 3aa, indicating that the reaction system does not undergo a non-stereoselective background reaction (entry 16).
| Entry | Ligand | Solvent | Temp (°C) | Yieldb (%) | eec (%) |
|---|---|---|---|---|---|
| a 1a: 0.1 mmol, 2a: 0.25 mmol, blue light (λmax 448/455 nm). b Determined by 1H NMR analysis of the crude reaction mixture with 1,1,2,2-tetrachloroethane as an internal standard. c Determined by chiral stationary phase HPLC analysis. d Isolated yield. e Without a photocatalyst. f Reaction in the dark. g Without a Cu(I) catalyst. | |||||
| 1 | L1 | MeCN | 23 | 10 | ND |
| 2 | L2 | MeCN | 23 | 72 | 30 |
| 3 | L2 | MeCN | 5 | 61 | 70 |
| 4 | L2 | MeCN | −20 | 62 | 89 |
| 5 | L3 | MeCN | 5 | 49 | 52 |
| 6 | L4 | MeCN | 5 | 43 | −44 |
| 7 | L2 | EtOH | 5 | 73 | 74 |
| 8 | L2 | EtOH | −20 | Trace | ND |
| 9 | L2 | EtOH:MeCN (1:1) |
−20 | 68 | 66 |
| 10 | L2 | EtOH:DME (1:1) |
−20 | 77 | 76 |
| 11 | L2 | EtOH:DME (3:1) |
−20 | 49 | 85 |
| 12 | L2 |
EtOH:DME (1:3) |
−20 | 83(80) | 89 |
| 13 | L2 | EtOH:DME (1:3) |
−30 | 67 | 89 |
| 14e | L2 | EtOH:DME (1:3) |
−20 | Trace | ND |
| 15f | L2 | EtOH:DME (1:3) |
−20 | Trace | ND |
| 16g | L2 | EtOH:DME (1:3) |
−20 | Trace | ND |
|
|||||
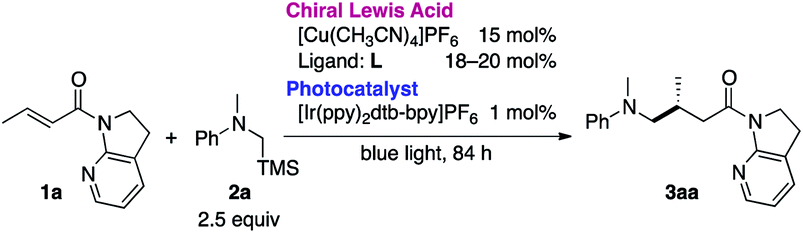
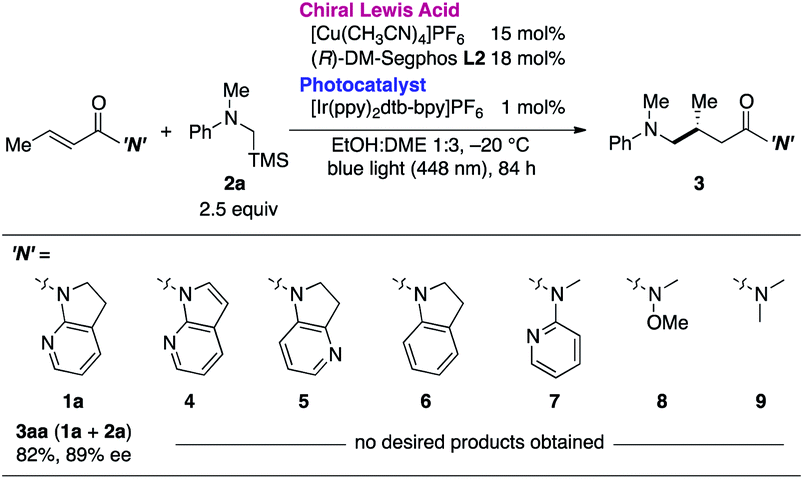
The necessity of the 7-azaindoline auxiliary was confirmed in a series of reactions using distinct α,β-unsaturated amides as acceptors of the in situ-generated radical (Table 2). Minute changing in the unsaturation of the heterocycle to 7-azaindole 4 resulted in no reaction. Isomeric 4-azaindoline 5 or indoline derivative 6 exhibited no reactivity, indicating that the coordination capability of the nitrogen atom at the 7-position is essential. Of note, a potentially chelating acyclic N-(2-pyridyl)amide 7 failed to promote the reaction, highlighting the exclusive nature of 7-azaindoline to engage the α,β-unsaturated carbonyl units in an asymmetric radical reaction. As expected, a Weinreb amide 8 and dimethyl amide 9 were incompatible substrates. Based on the apparent pivotal role of 7-azaindoline to control stereoselection via strong bidentate coordination to the Cu(I) complex, we conducted a systematic study of substituent effects (Table 3). Installation of non-coordinating methyl or phenyl groups at the 6-position, the most biasing position to interfere with the coordination to Cu(I), significantly decreased the enantioselectivity (3ba, 3ca). In sharp contrast, upon introducing a potentially coordinative substituent, e.g., methoxy or chloro substituents, at the same position enhanced the yield and improved enantioselectivity up to 98% ee (3da, 3ea).
| a Amide: 0.1 mmol, 2a: 0.25 mmol. Yields were determined by 1H NMR analysis of the crude reaction mixture with 1,1,2,2-tetrachloroethane as an internal standard. |
|---|
|
| a 1: 0.1 mmol, 2a: 0.25 mmol. Yields were determined by 1H NMR analysis of the crude reaction mixture with 1,1,2,2-tetrachloroethane as an internal standard. |
|---|

|
This finding was further supported by the negative effect of the 6-cyano substituent (3fa). This observation can be ascribed to the preferential end-on coordination mode of the cyano functionality, which hindered the coordination ability of the neighboring pyridyl nitrogen. Electronic effects on the pyridyl nitrogen were excluded on the basis of the minimal effect of methoxy or chloro substituents at the 4-position on the enantioselectivity (3ga, 3ha). Similarly, sterics at the 3-position had little effect on the reaction outcome (3ia), implying that delicate steric factors with coordination capabilities near the Cu(I) center are crucial toward achieving excellent enantioselectivity.
Reinvestigation of the reaction conditions with the proficient 7-aza-6-MeO-indoline attachment revealed that both the catalyst loading and the requisite amount of 2a could be decreased (Scheme 1). The stereoselectivity enhancement was maintained with α,β-unsaturated amides possessing different β-substituents, indicating that perturbation around the coordination sphere of the Cu(I) centre influenced the stereoselection at the β-position (Scheme 1a). Intriguingly, the beneficial effect of the 6-MeO substituent was also validated in the reaction using a different biaryl ligand, (R)-DM-BINAP L3, armed with identical aromatic biasing groups (3,5-xylyl) on the phosphorus atom (Scheme 1b). To gain more insight into this notable substituent effect, single crystals of L3/Cu(I)/7-azaindoline amide complexes were grown to determine the structural differences induced by the absence or presence of the 6-MeO substituent (Fig. 1). Consistent with the previous observations, the 7-azaindoline amide moiety adopted the Z-conformation for bidentate coordination to Cu(I), even in the presence of the 6-MeO substituent, as revealed by X-ray crystallographic analysis of the L3/Cu(I)/amide 1m complex (Fig. 1a).
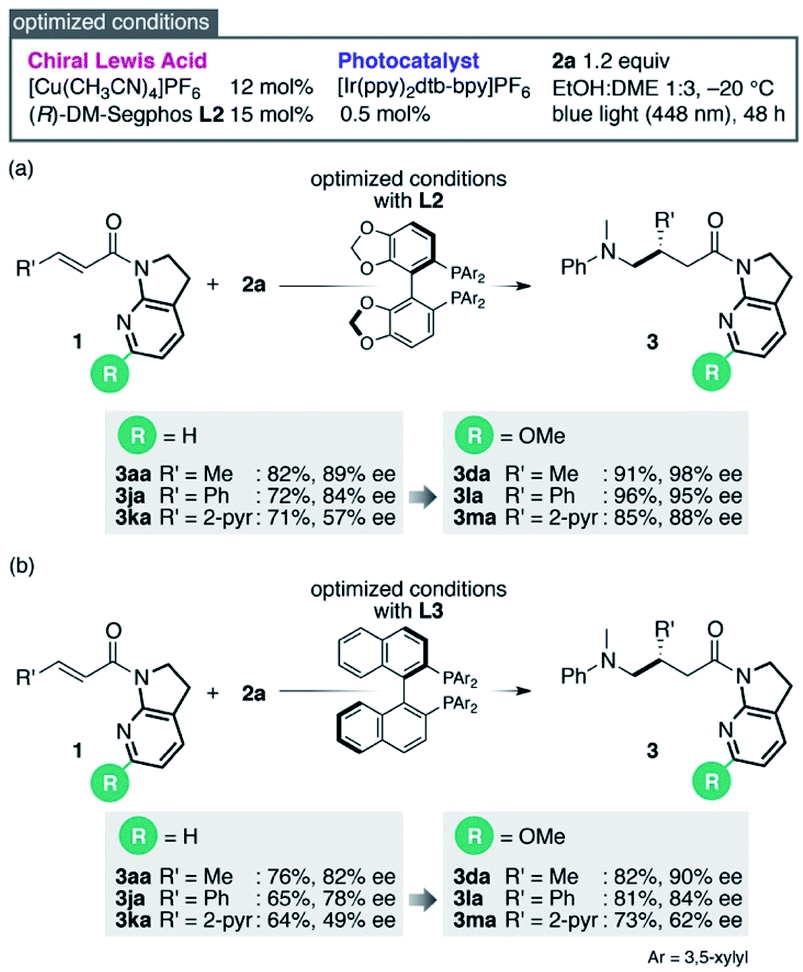 | ||
| Scheme 1 Optimized conditions and enhancement of stereoselection with a 6-MeO substituent. | ||
 | ||
| Fig. 1 Crystal structures of Cu(I)/amide complexes. (a) Crystal structure of a (R)-DM-BINAP (L3)/Cu(I)/amide 1m (with 6-MeO substituent) complex. (b) Front view of the L3/Cu(I)/amide 1m complex (with 6-MeO substituent). (c) Front view of the L3/Cu(I)/amide 1j complex (without 6-MeO substituent). The quadrant representation is applied to structures in (b and c), where the shaded area of the first and third quadrants represents the shielded area by 3,5-xylyl groups on the phosphorus. SD: standard deviation. Thermal ellipsoids are drawn at the 50% probability level. Hydrogens are omitted for clarity. Color code: gray, carbon; blue, nitrogen; red, oxygen; purple, phosphorus; green, copper. | ||
In-depth studies of the crystal structures of L3/Cu(I)/1m (with the 6-MeO substituent) and L3/Cu(I)/1j (without the 6-MeO substituent)74 elucidated the structural differences that may account for the positive effect of the 6-MeO substituent. In the absence of the 6-MeO substituent, the front view of the L3/Cu(I)/1j complex exhibited the ideal tetrahedral coordination around Cu(I), in which the amide 1i located perpendicular to the P–Cu–P fragment (Fig. 1c). Biasing 3,5-xylyl groups of L3 exist in the first and third quadrants, and the incoming radical predominantly approaches from the second quadrant to give the product with the observed absolute configuration (see the X-ray crystal structure of 3dd in Table 4). On the other hand, amide 1m with the 6-MeO substituent in the L3/Cu(I)/1m complex slightly rotated counterclockwise (Fig. 1b). Comparative analysis of the bond angles of the two complexes revealed a highly skewed tetrahedral coordination for the L3/Cu(I)/1m complex with deviated ∠P1–Cu–N (117.79°) and ∠P2–Cu–N (134.07°) angles. While the distance between Cu(I) and the 6-MeO substituent (2.88 Å) exceeded the sum of the van der Waals radii, the narrower ∠Cu–N–C(6) angle (117.83°) compared with that of the L3/Cu(I)/1j complex (119.74°) suggested that the weak attractive interaction between Cu(I) and the 6-MeO group results from the skewed coordination mode. Indeed, a competitive binding study using an equimolar mixture of the L3/Cu(I) complex, 1j, and 1m resulted in a nearly 1:1 mixture of L3/Cu(I)/1j and L3/Cu(I)/1m complexes, suggesting that the 6-MeO group is not merely imparting a steric bias to disfavor complexation (see Section 9 in the ESI for 1H-NMR analyses of these complexes†).

The deviation of the position of amide 1m from the horizontal line rendered the β-position of the amide more likely to be shielded by the 3,5-xylyl group at the third quadrant, leading to higher enantioselectivity. We believe this mechanism is also operative with 6-Cl-substituted 7-azaindoline amide 1e, in which the enantioselectivity was similarly increased despite the smaller magnitude of enhancement (Table 3, 3ea).
Having determined the optimal cooperative catalytic system and incorporating the newly generated the 7-aza-6-MeO-indoline attachment, we next investigated the substrate scope of the catalytic asymmetric aminomethylation (Table 4). The reaction of archetypal β-Me-substituted unsaturated amide could be performed on a gram scale with lower catalyst loading (3da, Cu: 10 mol%, Ir: 0.3 mol%), albeit with a marginal loss in enantioselectivity. Linear, branched, and cyclic β-alkyl substituents were largely accommodated to deliver the corresponding products in high yield and with high enantioselectivity (3na–3ra). β-Aromatic amides were successfully accommodated as tractable substrates to deliver the corresponding γ-aminomethylated products with high enantioselectivity, irrespective of the presence of electron-donating or -withdrawing substituents (3la, 3sa–3za). Potentially coordinative heteroaromatic units did not interfere with the catalysis (3ma, 3aaa), although the reaction was complicated by the presence of an α-Me substituent on the amide substrate, giving a mixture of diastereomers (81:19) in moderate yield with moderate enantioselectivity (3aba). Sterically more (N-Bn, iPr) or less (N-H) demanding α-silylamines 2b–d as well as 2e bearing an o-F substituent were compatible, affording the desired γ-amino amides (3db–3de) in high yield with high enantioselectivity.
The proposed catalytic cycle is delineated in Fig. 2. 7-Aza-6-MeO-indoline amide 1d readily interacts with the L2/Cu(I) complex to form complex I, mitigating the direct excitation of 1d for [2 + 2]-photocycloaddition. Independently, α-silylamine 2a is oxidized by the photoexcited Ir(III)* complex generated by blue-light irradiation, catalytically producing α-amino radical II and TMS cations. At low temperature, the coordinated amide in complex I is sufficiently more electrophilic than non-ligated 1d, and undergoes coupling with radical II with excellent stereocontrol, as described in Fig. 1.
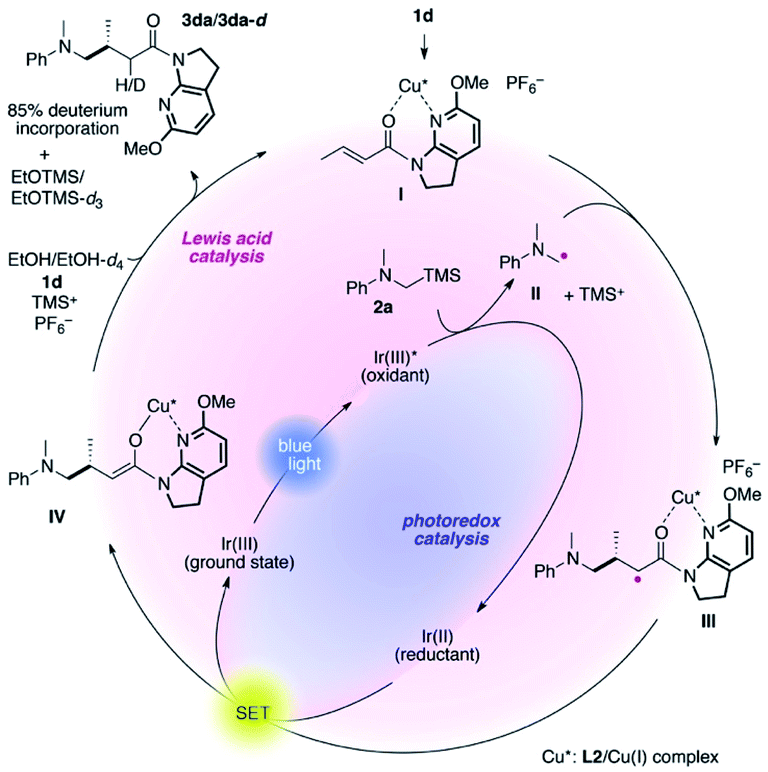 | ||
| Fig. 2 Plausible catalytic cycle. | ||
The thus-formed α-amide radical III was quenched with the Ir(II) complex via single-electron transfer to give amide enolate IV while concomitantly regenerating the ground state Ir(III) complex to close the photocatalytic cycle. The co-solvent EtOH protonated the amide enolate to liberate γ-amino amide product 3da, as evidenced by the 85% deuterium incorporation (see Fig. S4 and S5 in the ESI for HRMS and 1H NMR analyses, respectively†) when using EtOH-d4 in place of EtOH. The 6-MeO-7-azaindoline attachment was readily removed after taming the reactivity of the cooperative asymmetric catalysis, allowing for diverse functional group transformations (Scheme 2). Simple acidic hydrolysis of product 3da gave Me ester 11 or acid 12 depending on the acid concentration and reaction media. This class of compounds provides potentially useful intermediates for γ-aminobutyric acid derivatives, a key structural motif shared in several marketed therapeutics for central nervous system disorders.75,76 The 7-azaindoline unit served to stabilize the tetrahedral intermediate formed upon reaction with an organolithium reagent, delivering methyl ketone 13 without over-alkylated product. For secondary amine product 3dd, simple treatment with KOtBu at 0 °C afforded lactam 14. In all cases, the 6-MeO-7-azaindoline attachment was recovered in over 95% yield.
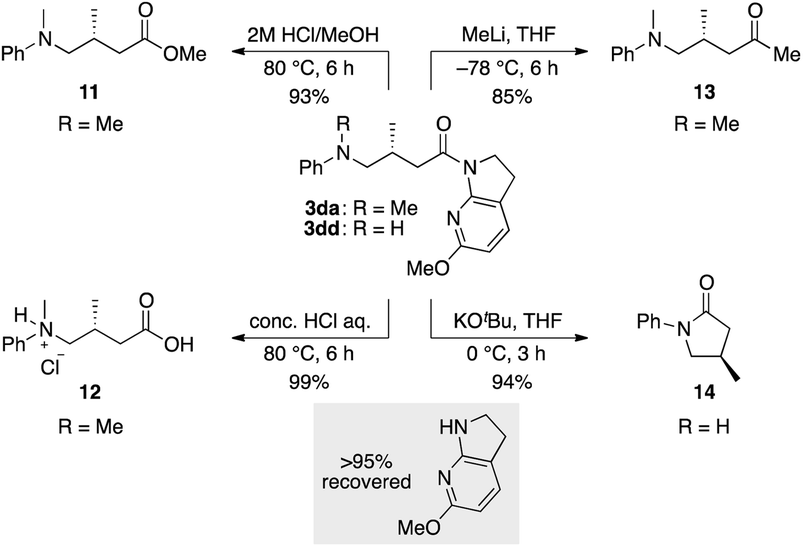 | ||
| Scheme 2 Transformation of the products. | ||
Conclusions
In conclusion, we developed a new protocol for a highly enantioselective catalytic conjugate addition of α-amino radicals fueled by visible light irradiation. A chiral Cu(I) catalyst and an Ir-based photocatalyst exert their influence orthogonally to promote the smooth reaction, where the 7-azaindoline unit of the α,β-unsaturated amides plays a key role in both the reaction progress and stereoselection. Substituents at the 6-position of the 7-azaindoline attachment had a significant effect on the reaction, shedding light on the stereocontrol of the privileged Cu(I)/7-azaindoline combination in asymmetric catalysis. The substrate generality and functional group transformation of the products will be highly applicable to practical organic synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by KAKENHI (17H03025 and 18H04276 in Precisely Designed Catalysts with Customized Scaffolding) from JSPS and MEXT. S. K. P. thanks the German Research Foundation (DFG: PA 3350/1-1; Project Number: 407495001) for the postdoctoral research fellowship. Dr Roman Pluta is gratefully acknowledged for his fruitful discussions during the project. We are grateful to Dr Kiyoko Iijima, Dr Ryuichi Sawa, Yumiko Kubota, and Yuko Takahashi at the Institute of Microbial Chemistry for technical assistance in NMR and MS analyses. We especially thank Dr Tomoyuki Kimura for the single crystal X-ray analyses for 1w (CCDC 1985034), 3dd (CCDC 1985035), and a (R)-DM-BINAP (L3)/Cu(I)/amide 1m (with 6-MeO substituent) complex (CCDC 1985036).References
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed
.
- K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef CAS PubMed
- M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed
- L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC
- J. Xuan and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed
- D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2013, 4, 355–360 CrossRef
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed
- D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed
- M. H. Shaw, J. Twilton and D. W. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed
- L. Marzo, S. K. Pagire, O. Reiser and B. Konig, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed
- S. K. Pagire, T. Föll and O. Reiser, Acc. Chem. Res., 2020, 53, 782–791 CrossRef CAS PubMed
- M. N. Hopkinson, B. Sahoo, J. L. Li and F. Glorius, Chem.–Eur. J., 2014, 20, 3874–3886 CrossRef CAS PubMed
- E. Meggers, Chem. Commun., 2015, 51, 3290–3301 RSC
- M. Pena-Lopez, A. Rosas-Hernandez and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 5006–5008 CrossRef CAS PubMed
- C. Wang and Z. Lu, Org. Chem. Front., 2015, 2, 179–190 RSC
- X. Lang, J. Zhao and X. Chen, Chem. Soc. Rev., 2016, 45, 3026–3038 RSC
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed
- C. Jiang, W. Chen, W. H. Zheng and H. Lu, Org. Biomol. Chem., 2019, 17, 8673–8689 RSC
- J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed
- G. C. Fu, ACS Cent. Sci., 2017, 3, 692–700 CrossRef CAS PubMed
- Q. Lu and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 49–51 CrossRef CAS PubMed
- K. Wang and W. Kong, Chin. J. Chem., 2018, 36, 247–256 CrossRef CAS
- A. G. Amador and T. P. Yoon, Angew. Chem., Int. Ed., 2016, 55, 2304–2306 CrossRef CAS PubMed
- L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320–330 CrossRef CAS PubMed
- D. A. Evans, T. Rovis and J. S. Johnson, Pure Appl. Chem., 1999, 71, 1407–1415 CAS
- J. Christoffers, G. Koripelly, A. Rosiak and M. Rössle, Synthesis, 2007, 9, 1279–1300 CrossRef
- J. H. Wu, R. Radinov and N. A. Porter, J. Am. Chem. Soc., 1995, 117, 11029–11030 CrossRef CAS
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392–396 CrossRef CAS PubMed
- T. R. Blum, a. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391–1395 CrossRef CAS PubMed
- M. E. Daub, H. Jung, B. J. Lee, J. Won, M. H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 9543–9547 CrossRef CAS PubMed
- S. K. Pagire, A. Hossain, L. Traub, S. Kerres and O. Reiser, Chem. Commun., 2017, 53, 12072–12075 RSC
- S. Kerres, E. Plut, S. Malcherek, J. Rehbein and O. Reiser, Adv. Synth. Catal., 2019, 361, 1400–1407 CrossRef CAS
- K. Zhan and Y. Li, Catalysts, 2017, 7, 337 CrossRef
- J. B. Metternich, D. G. Artiukhin, M. C. Holland, M. von Bremen-Kuhne, J. Neugebauer and R. Gilmour, J. Org. Chem., 2017, 82, 9955–9977 CrossRef CAS PubMed
- R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872–3890 CrossRef CAS PubMed
- M. P. Sibi, J. G. Ji, J. H. Wu, S. Gurtler and N. A. Porter, J. Am. Chem. Soc., 1996, 118, 9200–9201 CrossRef CAS
- M. P. Sibi and S. Manyem, Tetrahedron, 2000, 56, 8033–8061 CrossRef CAS
- J. H. Wu, G. Zhang and N. A. Porter, Tetrahedron Lett., 1997, 38, 2067–2070 CrossRef CAS
- M. P. Sibi, J. Ji, J. B. Sausker and C. P. Jasperse, J. Am. Chem. Soc., 1999, 121, 7517–7526 CrossRef CAS
- M. P. Sibi and J. Zimmerman, J. Am. Chem. Soc., 2006, 128, 13346–13347 CrossRef CAS PubMed
- A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed
- P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672–675 CrossRef CAS PubMed
- Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338–3341 CrossRef CAS PubMed
- J. W. Beatty and C. R. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS PubMed
- M. El Khatib, R. A. Serafim and G. A. Molander, Angew. Chem., Int. Ed., 2016, 55, 254–258 CrossRef CAS PubMed
- J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563–2575 CrossRef CAS PubMed
- Y. Yin, Y. Dai, H. Jia, J. Li, L. Bu, B. Qiao, X. Zhao and Z. Jiang, J. Am. Chem. Soc., 2018, 140, 6083–6087 CrossRef CAS PubMed
- K. Ohmatsu, R. Suzuki, Y. Furukawa, M. Sato and T. Ooi, ACS Catal., 2020, 10, 2627–2632 CrossRef CAS
- J. L. Schwarz, R. Kleinmans, T. O. Paulisch and F. Glorius, J. Am. Chem. Soc., 2020, 142, 2168–2174 CrossRef CAS PubMed
- A. Bauer, F. Westkämper, S. Grimme and T. Bach, Nature, 2005, 436, 1139–1140 CrossRef CAS PubMed
- G. Pandey, G. Kumaraswamy and U. T. Bhalerao, Tetrahedron Lett., 1989, 30, 6059–6062 CrossRef CAS
- G. Pandey, G. Devi Reddy and G. Kumaraswamy, Tetrahedron, 1994, 50, 8185–8194 CrossRef CAS
- G. Pandey and D. Chakrabarti, Tetrahedron Lett., 1996, 37, 2285–2288 CrossRef CAS
- G. Pandey, G. D. Reddy and D. Chakrabarti, J. Chem. Soc., Perkin Trans. 1, 1996, 219–224 RSC
- G. Pandey and M. Kapur, Synthesis, 2001, 8, 1263–1267 Search PubMed
- M. A. Brumfield, S. L. Quillen, U. C. Yoon and P. S. Mariano, J. Am. Chem. Soc., 1984, 106, 6855–6856 CrossRef CAS
- E. Hasegawa, W. Xu, P. S. Mariano, U. C. Yoon and J. U. Kim, J. Am. Chem. Soc., 1988, 110, 8099–8111 CrossRef CAS
- W. Xu, Y. T. Jeon, E. Hasegawa, U. C. Yoon and P. S. Mariano, J. Am. Chem. Soc., 1989, 111, 406–408 CrossRef CAS
- Y. S. Jung and P. S. Mariano, Tetrahedron Lett., 1993, 34, 4611–4614 CrossRef CAS
- X. Zhang, S.-R. Yeh, S. Hong, M. Freccero, A. Albini, D. E. Falvey and P. S. Mariano, J. Am. Chem. Soc., 1994, 116, 4211–4220 CrossRef CAS
- Z. Su, P. S. Mariano, D. E. Falvey, U. C. Yoon and S. W. Oh, J. Am. Chem. Soc., 1998, 120, 10676–10686 CrossRef CAS
- L. R. Espelt, I. S. McPherson, E. M. Wiensch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452–2455 CrossRef PubMed
- X. Shen, Y. J. Li, Z. R. Wen, S. Cao, X. Y. Hou and L. Gong, Chem. Sci., 2018, 9, 4562–4568 RSC
- J. Ma, J. Lin, L. Zhao, K. Harms, M. Marsch, X. Xie and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 11193–11197 CrossRef CAS PubMed
- J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS PubMed
- M. Silvi, C. Verrier, Y. P. Rey, L. Buzzetti and P. Melchiorre, Nat. Chem., 2017, 9, 868–873 CrossRef CAS PubMed
- N. Kumagai and M. Shibasaki, Synthesis, 2019, 51, 185–193 CrossRef CAS
- N. Kumagai, M. Kanai and H. Sasai, ACS Catal., 2016, 6, 4699–4709 CrossRef CAS
- Y. Li, K. Zhou, Z. Wen, S. Cao, X. Shen, M. Lei and L. Gong, J. Am. Chem. Soc., 2018, 140, 15850–15858 CrossRef CAS PubMed
- B. Han, Y. Li, Y. Yu and L. Gong, Nat. Commun., 2019, 10, 3804 CrossRef PubMed
- F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS PubMed
- Q. S. Gu, Z. L. Li and X. Y. Liu, Acc. Chem. Res., 2020, 53, 170–181 CrossRef CAS PubMed
- M. Zhang, N. Kumagai and M. Shibasaki, Chem.–Eur. J., 2016, 22, 5525–5529 CrossRef CAS PubMed
- E. Boonstra, R. de Kleijn, L. S. Colzato, A. Alkemade, B. U. Forstmann and S. Nieuwenhuis, Front. Psychol., 2015, 6, 1520 Search PubMed
- R. B. Silverman, Angew. Chem., Int. Ed., 2008, 47, 3500–3504 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1985034–1985036. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01890b |
| This journal is © The Royal Society of Chemistry 2020 |