
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMapping protein–polymer conformations in bioconjugates with atomic precision†
Kevin M.
Burridge‡
,
Ben A.
Shurina‡
,
Caleb T.
Kozuszek
,
Ryan F.
Parnell
,
Jonathan S.
Montgomery
,
Jamie L.
VanPelt
,
Nicholas M.
Daman
,
Robert M.
McCarrick
,
Theresa A.
Ramelot
,
Dominik
Konkolewicz
 * and
Richard C.
Page
*
* and
Richard C.
Page
*
Department of Chemistry and Biochemistry, Miami University, 651 E High St., Oxford, OH 45056, USA. E-mail: d.konkolewicz@MiamiOH.edu; pagerc@MiamiOH.edu
First published on 3rd June 2020
Abstract
Rational design of protein–polymer bioconjugates is hindered by limited experimental data and mechanistic understanding on interactions between the two. In this communication, nuclear magnetic resonance (NMR) paramagnetic relaxation enhancement (PRE) reports on distances between paramagnetic spin labels and NMR active nuclei, informing on the conformation of conjugated polymers. 1H/15N-heteronuclear single quantum coherence (HSQC) NMR spectra were collected for ubiquitin (Ub) modified with block copolymers incorporating spin labels at different positions along their backbone. The resultant PRE data show that the conjugated polymers have conformations biased towards the nonpolar β-sheet face of Ub, rather than behaving as if in solution. The bioconjugates are stabilized against denaturation by guanidine-hydrochloride, as measured by circular dichroism (CD), and this stabilization is attributed to the interaction between the protein and conjugated polymer.
Introduction
Protein–polymer bioconjugates have been known since 1952.1,2 In 1978, improved methods for attaching polyethylene glycol (PEG) to proteins were developed3 enabling a polymer to reduce immunogenicity and increase serum half-life of therapeutic proteins.4 Recently, advances in reversible deactivation radical polymerizations have led to an explosion in protein–polymer bioconjugates.5–10 However, mechanistic and structural understanding of these species remains elusive, largely due to the complexity of the polymers.Within the field of protein polymer conjugates there has been much seemingly conflicting data produced, without a satisfying unifying model. Intuitively, soluble proteins with a predominantly hydrophilic surface should prefer interactions with polar polymers featuring complementary functional groups. However, a recent publication showed that proteins such as glucose-oxidase can be modified with polystyrene to the point of becoming globular nanoparticles, with improved or retained activity.11 Recent work on surface-immobilization of enzymes has shown that lipase catalytic performance can be dramatically improved by attachment to hydrophilic surface-grafted polymers.12 In a subsequent publication, by the same group, it was found that there is a trade-off in this system – more protein–polymer attachments resulted in higher stability but lower activity due to decreased motion in the folded state of the protein.13 This experimental result conflicts with recent work on soluble chymotrypsin conjugates, where MD models showed more motion in the conjugate with the lowest enzymatic activity, a manifestation of a measured decrease in the substrate binding constant, Km.14
The degree, specificity, location, and timescale of protein–polymer associations are all open questions. It has been found that even small proteins such as insulin can have improved or decreased activity depending on the location of the modification. In all cases, insulin was stabilized against heat degradation.15 If the polymer is strongly interacting with the insulin surface, it would be expected to inhibit receptor binding no matter where it is attached, while the stabilization observed in these conjugates would intuitively be caused by some sort of physical association.
The understanding of interactions between proteins and polymers, and the proximities and conformations of polymers within bioconjugates have relied on molecular dynamics (MD) simulations, small-angle neutron scattering, and a single crystal structure. Although MD gives hypotheses on interactions and polymer conformations,14,16–20 the results are limited by the accuracy of the force fields used and an absence of data from physical samples. Neutron scattering data, while providing useful insights on polymer conformation and protein dynamics,21–23 are limited by: the quality of models and fits; specialized instrumentation; and resultant coarse-grained information. While the single existing crystal structure was determined from data collected on a protein–polymer bioconjugate, the polymer was not resolved, likely due to polymer flexibility.24 Other isolated coarse-grained experiments probing polymer behavior have been published, such as a 15A resolution Cryo-EM structure25 of a PEG-bottlebrush-modified virus-like particle (a privileged system with high symmetry), and motional dynamics probed by electron paramagnetic resonance (EPR) on the surface of polymer-modified self-assembling peptide amphiphiles.26
Finally, most structural information has been on PEG-conjugates, which constitute a subset of protein–polymer bioconjugates. Thus, experiments that report on conformations and proximities within protein–polymer bioconjugates are greatly needed.
There is no consensus on the impact of polymer–protein non-covalent associations and conformation on stability.16,17,27,28 One model proposes that protein–polymer associations are stabilizing through non-covalent bonds,17 others suggest protein–polymer associations are destabilizing by disrupting the hydration shell,28 or that the polymer acts as a chaperone.16 The dearth of experimental data and conflicting models limit the mechanistic understanding of protein–polymer hybrids and rational bioconjugate engineering. Often distinct metrics of a bioconjugate's ability to tolerate environmental challenges are used, such as stability measured by loss of activity or function in the bioconjugate upon exposure to a challenge, versus structural stability where the bioconjugate's fold or structure is lost upon exposure to a challenge. The various metrics of stability and lack of experimental data on conformations, interactions and proximities of the synthetic and biological components limit rational design of bioconjugates for enhanced performance. To address the limited information on the interplay of biological and synthetic components of a bioconjugate, this work applies paramagnetic relaxation enhancement (PRE) to bioconjugates, giving information on the conformation of these bioconjugates. PRE is a nuclear magnetic resonance (NMR) technique, providing up to 30 Å distance information.29,30 In PRE, paramagnetic species increase the relaxation rate of NMR-active nuclei,31 broadening resonances that either diminish (15–30 Å) or eliminate (0–15 Å) when the paramagnetic center approaches the NMR active nucleus (Scheme 1B). Through the PRE method, the proximity of a spin-labeled polymer to NMR active nuclei on a protein can be evaluated, creating a platform to correlate protein–polymer interactions and proximities with bioconjugate performance. This can open the door to rational bioconjugate design and engineering.
 | ||
| Scheme 1 (A) Synthesis of block polymers containing TEMPO spin label (magenta spheres) and conjugation to Ub. (B) Cartoon illustration of spin label distances in Ub conjugated to a short 5-unit polymer with TEMPO. Red residues will relax completely by PRE. | ||
Results and discussion
To conduct the PRE experiment, we prepared three 15N-human ubiquitin (Ub)–polymer conjugates, each with a different block copolymer containing TEMPO spin labels. The three block copolymers vary in placement of paramagnetic groups along the polymer backbone, enabling analysis of the localization of the polymer on or near Ub. NMR structural studies of bioconjugates give atomic resolution in dynamic systems, under biologically relevant conditions – conditions not achieved in neutron scattering or crystallography alone. Block copolymers containing TEMPO at approximately 20%, 50% or 80% from the polymer's α terminus were synthesized by reversible addition/fragmentation chain transfer (RAFT) polymerization, and oxidized using meta-chloroperbenzoic acid (mCPBA) (Scheme 1A).32Molecular weight and chain length data for each block are presented in Tables 1 and S1,† with number average molecular weight by NMR determined using eqn S1.† Four units of spin label precursor N-(2,2,6,6-tetramethyl-4-piperidinyl)-2-propenamide (TMPA) were targeted to account for (1) Poisson-like distribution of monomers incorporated during polymerization33 and (2) the non-quantitative yield of TEMPO in post-polymerization oxidation.
The dispersity (Mw/Mn) and length of the first blocks are critical as these parameters determine the distance between the spin label and Ub when conjugated. In general, the number average molecular weights (Mn) of each block agreed well with the theoretical values, where the Mn was determined using nuclear magnetic resonance (NMR) to calculate the number of DMAm and TMPA repeating units per polymer on average. Molar mass dispersities were determined by size exclusion chromatography (SEC) and we obtained Mw/Mn < 1.12 for the first block of all three block copolymers (Table 1), and Mw/Mn < 1.35 for all polymers overall.
| Block polymer | 0-Block | A-Block | B-Block | C-Block |
|---|---|---|---|---|
| 1st block (DMAm) | 34 | 5 | 15 | 25 |
| 2nd block (TMPA) | 0 | 4 | 4 | 4 |
| 3rd block (DMAm) | 0 | 25 | 15 | 5 |
| M n,theory-block1 | 3600 | 710 | 1700 | 2700 |
| M n,NMR-block1 | 3900 | 780 | 1900 | 3100 |
| M w/Mn,SEC-block1 | 1.10 | 1.12 | 1.12 | 1.12 |
| rTEMPO (Å) | — | 13 ± 3 | 21 ± 4 | 29 ± 6 |
| M n,theory-overall | 3600 | 4000 | 4000 | 4000 |
| M n,NMR-overall | 3900 | 4700 | 4400 | 4900 |
| M w/Mn,SEC-overall | 1.10 | 1.32 | 1.26 | 1.25 |
The chain lengths (Table 1) place the TEMPO spin label distances (rTEMPO) in key PRE-effect ranges of approximately rTEMPO = 13 ± 3 Å for A-block, rTEMPO = 21 ± 4 Å for B-block, and rTEMPO = 29 ± 6 Å for C-block, based on end-to-end distances (eqn S2–S8†). These values were estimated based on the end-to-end distance estimated from the weight average molecular weight (Mw) and the dispersity (Mw/Mn) of the poly(DMAm) block between the attachment site and the spin labeled block. This is because the spin label will be placed precisely one repeat unit after this first poly(DMAm) block. The dispersity of the first block would impact the placement of the spin labels most significantly, since the first spin label is found exactly 1 unit after the first block synthesized, therefore uncertainty in the placement of the spin label is relatively minimal, since the first poly(DMAm) blocks have dispersities in the range of 1.10–1.12.34
To account for the impact of molar mass dispersity on the placement of the spin label, the standard deviation (σM) in molecular weights of the first poly(DMAm) block was estimated for each of the A, B and C-block polymer.35 The uncertainty in the distance between the protein's surface and the spin label was found by estimating the end-to-end distance of the first poly(DMAm) block based: Mw, giving the mean value of rTEMPO; Mw + σM, giving the mean squared end-to-end distance of a polymer of molecular weight Mw + σM, which is denoted as 〈r(Mw + σM)2〉1/2; and finally Mw − σM, giving the mean squared end-to-end distance of a polymer of molecular weight Mw − σM: 〈r(Mw − σM)2〉1/2. The absolute difference between rTEMPO and 〈r(Mw + σM)2〉1/2 and the absolute distance between rTEMPO and 〈r(Mw − σM)2〉1/2 is calculated. The larger of |rTEMPO − 〈r(Mw + σM)2〉1/2| and |rTEMPO − 〈r(Mw − σM)2〉1/2| was used as the uncertainty in the placement of the spin label.
Oxidation by mCPBA gave TEMPO spin labeled polymers (Fig. S1†). A control DMAm homopolymer (0-block) of the same length as the block copolymers was also synthesized. This polymer was used to gauge the effect on the Ub spectrum from increased viscosity due to free polymer and structural perturbations from conjugation (Fig. 1b). Each polymer was conjugated to Ub using N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride/N-hydroxysuccinimide (EDC/NHS) targeting amine groups within Ub, such as the N-terminus and lysine side-chain ε-amines as indicated in Scheme 1.36 Conjugation was confirmed using polyacrylamide gel electrophoresis (PAGE) for the 0-block conjugate (Fig. 1a) and all spin labeled conjugates (Fig. S2†).
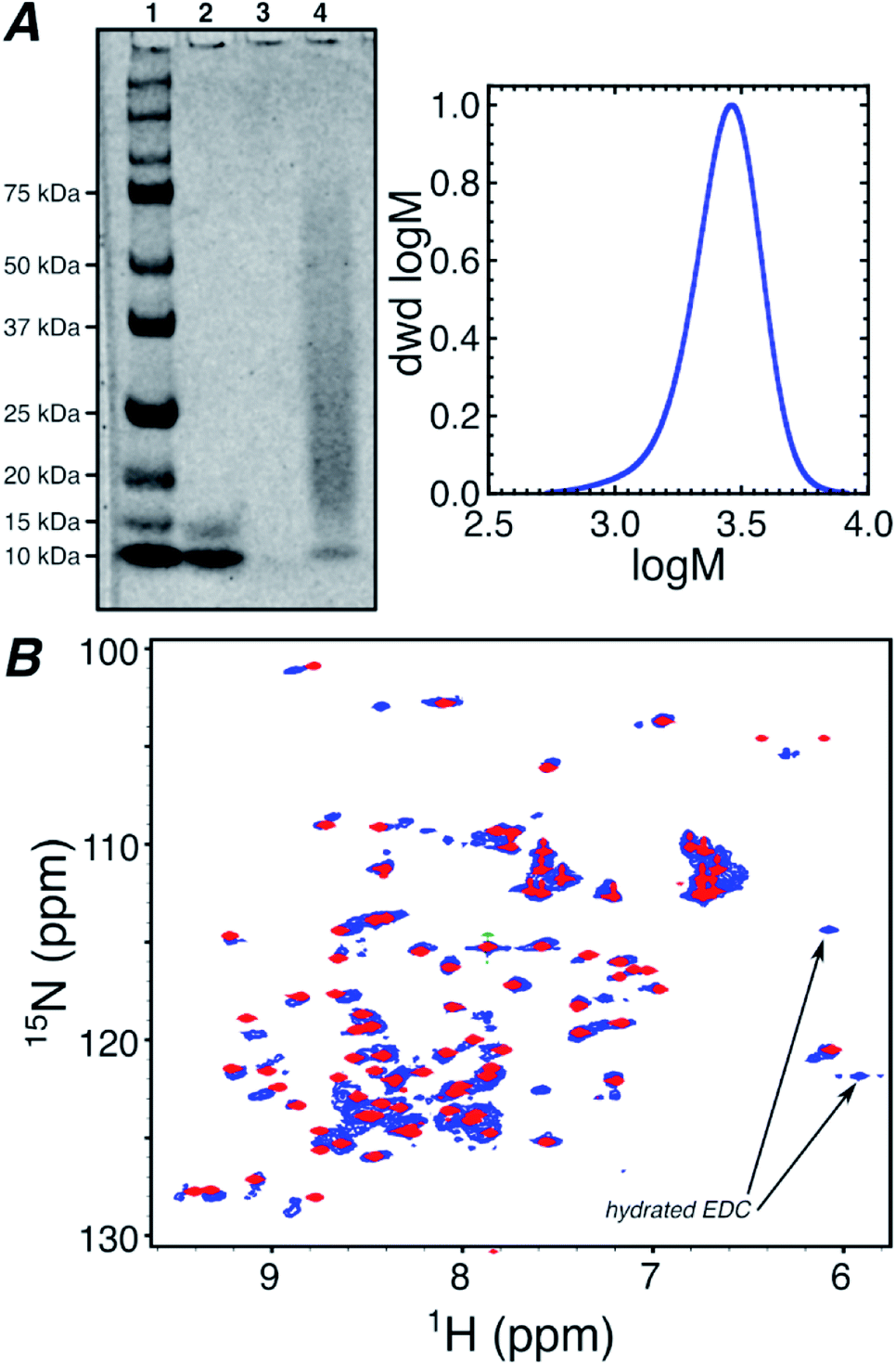 | ||
| Fig. 1 (A) SDS-PAGE of native Ub (lane 2) and Ub–0-block polymer conjugate (lane 4) compared to a standard ladder (lane 1, lane 3 is empty) and a gel permeation chromatography trace of 0-block polymer. (B) 1H/15N-HSQC spectra of native Ub mixed with free B-block polymer (red) and Ub conjugated with 0-block polymer (blue). | ||
To determine the location of polymers on Ub, conjugation with a 5 unit chain of DMAm, was used. This DMAm polymer gives peptide conjugates that ionize efficiently in MALDI-TOF-MS after trypsin digestion (Fig. S4†). Peaks resulting from polymer-modified species had masses differing by 99 m/z and isotopic signatures from natural-abundance versus15N-containing species (Fig. S4b†). Masses in Table S2† are the highest-intensity peak in each MALDI-TOF-MS series. Predicted mass matches are within 0.25 m/z of the observed mass. The MALDI-TOF-MS data indicate that conjugation can occur at K11, K48, and K63, however it is possible that not every protein will have precisely 3 conjugations. Instead there could be some variation where some proteins have a smaller number of attached polymers, still at one or two of the residues: K11, K48, and K63. Minor peaks observed in the MS data correspond to conjugation at K33, K6, or the N-terminus, indicating that a small number of proteins could also have additional polymers attached. The MALDI-TOF-MS data agree with a model developed by Russell et al.,37 which predicts K11, K48, and K63 to be fast-reacting, while all other amines are slower.
1H/15N-heteronuclear single quantum coherence (HSQC) spectroscopy was performed on all conjugates and native Ub. This NMR technique gives the proton and nitrogen chemical shift for each N–H bonded group in the protein, averaged over all molecules and conformations. Fig. S5† compares the 1H/15N-HSQC NMR spectrum of the native ubiquitin in aqueous media, to the protein in the presence of 40 equivalents of the B-block spin labeled polymer. The data in Fig. S5† indicate that although there are some small changes in the 1H/15N-HSQC spectrum of ubiquitin, the spectrum is overall preserved even in the presence of large excesses of spin labeled polymer. Therefore, unattached spin labelled polymers in solution only cause minimal perturbations in the protein's NMR spectrum, indicating that the polymer in solution does not have any specific affinity for the protein.
Fig. 1b overlays the NMR spectra of Ub conjugated with the 0-block polymer and native Ub in the presence of 40 equivalents of B-block polymer. Although the 1H/15N-HSQC of the 0-block Ub conjugate exhibits some differences compared to native Ub, due to heterogeneity in conjugation density, chain lengths, and conjugation sites, the similarity between the spectra suggests that perturbations in Ub structure caused by polymer conjugation are minor. However, due to the lack of spin label on the conjugated 0-block synthetic polymer, the data in Fig. 1b do not provide information on potential interactions, proximities, or conformations for the attached polymer and the protein.
To ascertain the possible conformation of the polymer with respect to the protein in the bioconjugates, PRE of protein residues by spin-labeled polymer was probed. If the spin label is within 15 Å of an amide proton, almost complete broadening and signal loss is observed for the corresponding resonance in the 1H/15N-HSQC. When the spin label is at a distance of 15–30 Å, partial broadening and reduced signal intensity occurs. If the spin label is greater than 30 Å from an amide proton, no change in signal intensity is observed (Scheme 1B).31 In this way, the proximities between the spin labeled polymer and the protein's individual amino acid residues can be evaluated, averaged over all bioconjugates in solution. However, it is important to note that substantial perturbations in the protein's NMR spectrum through PRE require the spin labeled polymer to have a dominant conformation in close proximity to the protein's surface. Fig. S5† shows that unconjugated polymers do not lead to notable PREs, suggesting that the poly(DMAm) in solution, without covalent bonding to the protein, is unlikely to have specific affinities or conformations on the protein's surface.
In A-block conjugate, the TEMPO is estimated to be within the total-signal-loss distance rTEMPO = 13 ± 3 Å (Fig. 2) for at least some residues (even if the polymer behaves in its solution form as an unperturbed random coil), and most residues are completely broadened, with only four protein peaks present. B-Block conjugate puts the TEMPO further from the protein with the expected distance in the weak PRE range with rTEMPO = 21 ± 4 Å, however, 47% of residues are completely broadened. Finally, 31% of C-block residues are completely broadened even though the TEMPO is expected to be rTEMPO = 29 ± 6 Å away from the nearest protein residue – almost double the distance that could enable complete PRE broadening. These data suggest the polymer, on average, adopts conformations that place TEMPO near the protein surface, much closer than solution behavior of DMAm would predict. NMR gives information of the ensemble averaged environment experienced by the NMR active nuclei. In this way, the 1H/15N HSQC NMR allows the environment of each of the protein's residue to be probed. Most of the residues experiencing PRE, which is attributed to close proximity of the NMR active residue to the polymer's spin label, located in the β-sheet region. This is most clear in B-block (Fig. 2), where there is a band of PRE-relaxed residues across the face of the β-sheet, but the helical region is mostly unaffected. The β-sheet region harbors surface-exposed hydrophobic and hydrogen-bond donating residues, providing complementary functional groups to those in DMAm. This information, combined with the gradual decrease in the number of PRE-relaxed residues moving from A-block to B-block to C-block leads to the following model: DMAm in the first block of the copolymer lays across or near the beta-sheet face of Ub, where its carbonyl groups may interact with hydrogen-bond donors and shield nonpolar residues from water. The end of the chain is still free to coil as normal. This puts TEMPO closer to the surface than expected, and explains the decrease in the number of PRE-relaxed resonances in C-block. If the polymer was not adopting conformations biased closer to the protein surface, only very weak or negligible PRE effects would be observed in C-block. In contrast, if the polymer was adopting a wider array of conformations that allowed for potential interactions across the entire Ub surface and were not biased to a particular face of Ub, nearly complete PRE would be expected for all residues of ubiquitin and there would likely be more substantial chemical-shift perturbations in the zero-block spectrum (Fig. 1).
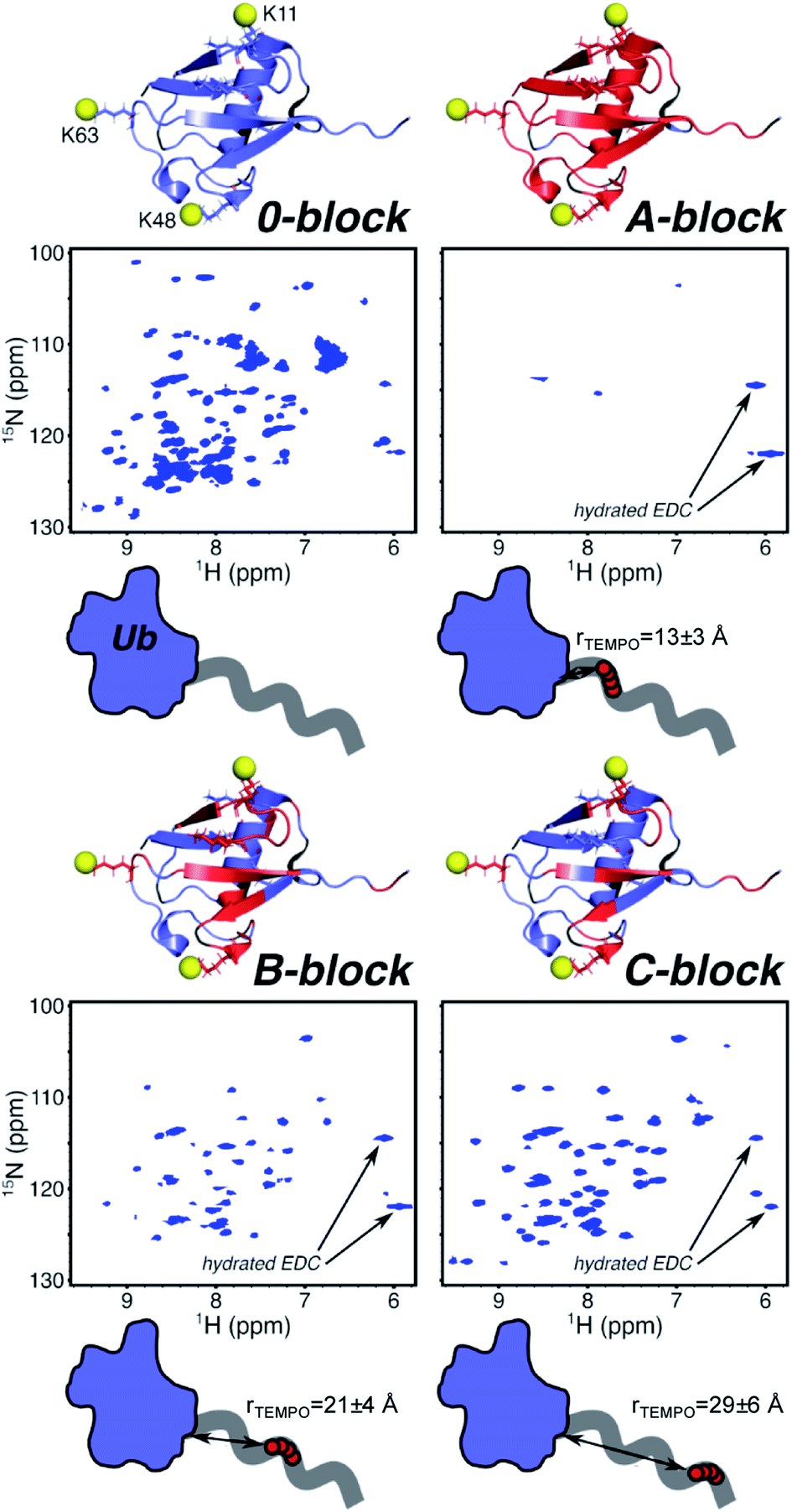 | ||
| Fig. 2 Structures of Ub (blue) with PRE-broadened residues within conjugates highlighted (red). Reactive amines (yellow spheres) are shown for lysine residues 11, 48, and 63. 1H/15N-HSQC spectra of each Ub conjugate are shown with annotated peaks resulting from reaction of EDC to form the urea. Schematic representations of conjugates with position of TEMPO (red circles) along the polymer chain (grey line) relative to Ub (blue). Average distances are estimated using Flory–Fox end-to-end distance based on the Mw of the first DMAm block and Mark–Houwink parameters for DMAm in water. | ||
To evaluate the impact of conjugation, stability against denaturation by guanidine hydrochloride (G-HCl) was performed (Fig. 3). Stability was monitored by circular dichroism (CD) on native Ub, Ub modified with 2–3 C-block polymers and Ub highly modified with 4–6 C-block polymers (SDS-PAGE in Fig. S3†). The number of polymers is estimated based on predicted reactivities37 and conjugation sites observed by MALDI-TOF (Table S2†). CD gives the mean density of secondary structural elements (α-helices, β-strands, and coils) in a given system. Monitoring the CD signal as a function of a chemical denaturant enables the averaged structural stability to be evaluated as the protein or conjugate faces an increasingly harsh environment. While native ubiquitin is completely denatured at 6 M G-HCl, both bioconjugates contain folded populations even at 7.2 M G-HCl. Based on a 4-parameter logistic symmetric sigmoidal fit, the inflection point for native ubiquitin is 4.0 M; C-block modified conjugate is 4.4 M; and C-block highly modified conjugate is >9.3 M. We attribute stabilization to protein–polymer interactions resulting from the conjugated polymer adopting conformations biasing its position to be near the Ub β-sheet face, conformations consistent with the PREs, occurring predominantly at residues located in the Ub β-sheet regions. Fig. S6† compares the 1H/15N-HSQC of C-block modified and highly modified species. The highly modified conjugate has many more PREs (Fig. S6†), suggesting that an increase in potential polymer–protein interactions are correlated with enhanced protein stability.
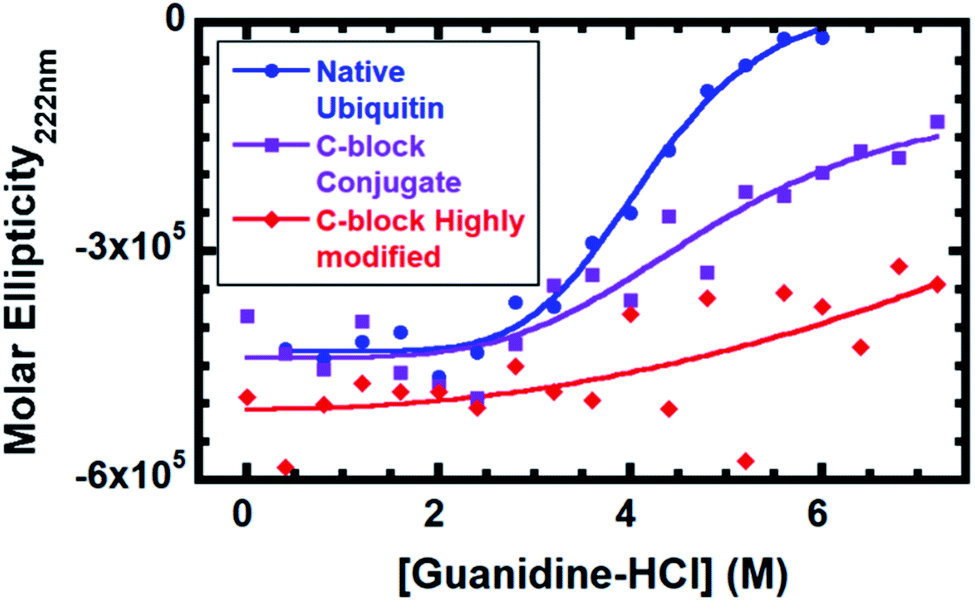 | ||
| Fig. 3 Guanidine denaturation monitored at 222 nm by circular dichroism for native Ub (blue), Ub conjugated with 80 eq. EDC and 40 eq. C-block (purple), and highly modified Ub conjugated with 240 eq. EDC and 40 eq. C-block (red). | ||
Conclusion
In summary, we report the first direct experimental observation of a conjugated polymer with conformations biased toward a single protein surface and correlated with an increase in protein stability. Solution behavior of C-block would not be expected to cause any complete PRE relaxation, yet 31% of residues in the C-block 1H/15N-HSQC are completely broadened. In B-block conjugate, 47% of residues are lost, and these are predominantly located on the β-sheet face of Ub, indicating that the polymer conformations are biased toward this predominant surface. Secondary structure stabilization supports our model, since the bioconjugate is stabilized against 10% higher concentration of guanidine than native Ub. Further polymer modification raises the stability past the saturation point of guanidine and correlates with increased protein–polymer proximity found by PRE. These data suggest that bioconjugate stability is enhanced when the polymer is in close proximity to the protein, suggesting that protein–polymer interactions result in enhanced stability of the bioconjugate. This would suggest that protein–polymer interactions lead to stabilizing non-covalent bonds that enhance the conjugates ability to withstand challenges. The experimental method developed here provides information of a bioconjugate's average conformations of synthetic and biological components with atomic-precision. This provides a link between polymer conjugation, conformation, proximity, and conjugate stability and performance. The technique can be extended to other synthetic or biological species in bioconjugates, paving the way for rational design of bioconjugates.Experimental section
Block copolymer synthesis (B-block)
To a 50 mL round-bottom flask was added DMAm (2.5 g, 25.2 mmol), PAETC (354 mg, 1.68 mmol), VA-044 (10.9 mg, 33.7 μmol), and 2.46 g of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water:methanol solution. The flask was sealed with a rubber septum, and deoxygenated, followed by reaction at 65 °C for 3 hours. Two post-reaction aliquots were taken to check conversion and chain length by NMR and SEC. With over 95% conversion, the next block was added as follows. TMPA-HCl (1.66 g, 6.72 mmol) was weighed out and added to the flask containing the polymer, followed by 10 mL of 1:1 water:methanol. Once the monomer had dissolved, VA-044 initiator (10.9 mg, 33.7 μmol) was added. Then, the flask was sealed and deoxygenated, followed by 18 h at 65 °C. Once again, 2 aliquots were taken for NMR and SEC. With over 95% conversion, the final block was added as follows. DMAm (2.5 g, 25.2 mmol), and VA-044 (10.9 mg, 33.7 μmol) were added and the flask was sealed and deoxygenate. Then the flask was heated to 65 °C for 3 h. 2 aliquots were again taken. With sufficient conversion, the polymer was then dried under reduced pressure with mild (ca. 50 °C) heat, reducing remaining methanol to trace (less than 0.1 equivalents versus PAETC).
:1 water:methanol solution. The flask was sealed with a rubber septum, and deoxygenated, followed by reaction at 65 °C for 3 hours. Two post-reaction aliquots were taken to check conversion and chain length by NMR and SEC. With over 95% conversion, the next block was added as follows. TMPA-HCl (1.66 g, 6.72 mmol) was weighed out and added to the flask containing the polymer, followed by 10 mL of 1:1 water:methanol. Once the monomer had dissolved, VA-044 initiator (10.9 mg, 33.7 μmol) was added. Then, the flask was sealed and deoxygenated, followed by 18 h at 65 °C. Once again, 2 aliquots were taken for NMR and SEC. With over 95% conversion, the final block was added as follows. DMAm (2.5 g, 25.2 mmol), and VA-044 (10.9 mg, 33.7 μmol) were added and the flask was sealed and deoxygenate. Then the flask was heated to 65 °C for 3 h. 2 aliquots were again taken. With sufficient conversion, the polymer was then dried under reduced pressure with mild (ca. 50 °C) heat, reducing remaining methanol to trace (less than 0.1 equivalents versus PAETC).
Oxidation of block copolymer with mCPBA (B-block)
To four, glass 20 mL vials was added dry polymer powder (125 mg, 30.0 μmol), which was dissolved in 1.12 mL tert-butanol. The vials were protected from ambient light by wrapping with duct tape or electrical tape. One vial at a time, mCPBA (58.8 mg, 263 μmol) was weighed out and added, followed immediately by 2 M NaOH (240 μL, 480 μmol). Then, the vials were stirred at 200 rpm for 1 h at room temperature. The oxidation results in viscous scarlet solutions. The yield of oxidation was measured by EPR on a Bruker NanoEMX benchtop EPR instrument, calibrated using TEMPO standards up to 300uM. The vial containing the polymer with highest oxidation yield (typically 55–60%) was then quantitatively transferred using t-BuOH to a 0.5–3 mL capacity, 2 kDa MWCO dialysis cassette. Then, the polymer was dialyzed at 4 °C into 3 L of 10 mM sodium dodecyl sulfate (SDS), followed by another 3 L of 10 mM SDS, followed by 3 L of water, followed by another 3 L of water, exchanging approximately every 12 hours. Upon completion, the polymer was transferred to a 3 kDa MWCO centrifugal filter. NMR confirmed the removal of contaminants. Then, the polymer solution was concentrated in the centrifuge at 4 °C to ca. 600 μL. The concentration of polymer in the upper solution was calculated by measuring its spin label concentration by EPR and then factoring in the oxidation yield and average number of TMPA per chain. The polymer solution was transferred to a dark brown Eppendorf tube and stored at −80 °C until use. The spin labels on polymers prepared and stored in this way show no decay for at least 40 days.Polymer conjugation to ubiquitin (B-block)
15N-ubiquitin, purchased from LifeSensors (catalog# NS101), was prepared by dissolving the lyophilized powder in 10 mM phosphate buffer (PB) at pH 7.5. Directly before use, the protein concentration was obtained by Pierce copper assay. The polymer was prepared by thawing and centrifuging at 17000 G for 20 minutes. To a 1.5 mL Eppendorf tube covered with tape were added 0.1 M PB at pH 7.5 (80.6 μL), N-hydroxysuccinimide (NHS) (4.57 μL of a 40 g L−1 PB solution – 1.6 μmol), and oxidized B-block solution (223 μL, 9.33 μmol). Then, EDC-HCl (7.16 mg, 37.3 μmol) was added and dissolved. Then, immediately, 15N-ubiquitin (192.3 μL, 233 μmol) was added and the tube was rotated at room temperature for 2 hours. Other conjugates differ only in the amount of EDC-HCl used; B-block is 160 eq. vs. ubiquitin, while A-block and C-block required 80 eq., 0-block required 120 eq., to reach sufficient conjugation, as determined by SDS-PAGE (Fig. S2†).
NMR sample preparation
Conjugate (360 μL) was transferred to a 1.5 mL Eppendorf tube, followed by D2O (40 μL), and mixed. All the sample was transferred to a D2O susceptibility-matched BMS-005B Shigemi NMR tube, and sealed with the corresponding matched plunger and parafilm.Protein and conjugate nuclear magnetic resonance spectroscopy
All protein NMR spectra were collected at 25 °C (298 K) on a Bruker 600 MHz Avance III spectrometer equipped with a 5 mm triple resonance (TXI) probe. 1H–15N HSQC spectra were acquired using the Bruker hsqcetf3gpsi pulse program using 2048 (proton) and 512 (nitrogen) complex points. All spectra of bioconjugates were acquired with 16 scans. Spectra were processed using NMRPipe38 and visualized using Sparky.39 Initial chemical shift assignments were pulled from the BMRB (entry 15410)40 and adjusted for pH and conjugation effects.
Chemical denaturation by circular dichroism
Using conditions adapted from Ibarra-Molero et al.,41 we compared the stability of native ubiquitin and ubiquitin conjugated with different densities of C-block. Briefly, native Ub or conjugate was added to solutions of G-HCl in 10 mM pH 4 acetate buffer, mixed, added to the rinsed CD cuvette (1 mm pathlength, 400 μL capacity), and mixed again. Then, the cuvette was inserted into the instrument (Aviv 435 CD Spectrometer) and measured. For conjugates, acceptable signal/noise was achieved using 10 scans with 5 s averaging times per point.Trypsin digestion of DP5 DMAm conjugate
To a 150 μL PCR tube was added 3.75 μL conjugate, 3.24 μL 10 mM phosphate buffer, and 2 μL water. The tube was heated to 95 °C for 2 minutes and then allowed to cool to room temperature. 1 μL of Pierce Trypsin protease (Thermo product # 90057) was added (such that a 1:15 trypsin:Ub weight ratio is achieved), the tube was mixed and incubated at 37 °C for 20 h. Upon completion, the sample was placed on ice and used for MALDI-TOF-MS within 2 hours.
MALDI-TOF-MS of DP5 DMAm conjugate
The DP5 DMAm conjugate digestion solution was mixed in a 1:2 ratio with α-cyano-4-hydroxycinnamic acid (CHCA) matrix and spotted on the target plate. The MALDI was calibrated with poly-alanine standards. The mass spectrum was acquired with 1000 laser shots on 50% laser power and random-walk sampling. The DP5 DMAm polymer was acquired in a similar manner. The obtained spectra are given in Fig. S3a and b.†
Conflicts of interest
The authors declare no conflicts.Acknowledgements
We thank Dr Macha for aid in collecting the MALDI-TOF data. This work was supported by US Army Research Office award (W911NF-19-1-0300 to DK and R.C.P), and Miami University.References
- M. A. Stahmann and R. R. Becker, J. Am. Chem. Soc., 1952, 74, 2695–2696 CrossRef CAS.
- R. R. Becker and M. A. Stahmann, J. Biol. Chem., 1953, 204, 745–752 CAS.
- F. F. Davis, A. Abuchowski, T. van Es, N. C. Palczuk, R. Chen, K. Savoca and K. Wieder, in Enzyme Engineering, Springer US, 1978, pp. 169–173 Search PubMed.
- J. Milton Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef PubMed.
- K. M. Burridge, T. A. Wright, R. C. Page and D. Konkolewicz, Macromol. Rapid Commun., 2018, 39, 1800093–1800114 CrossRef PubMed.
- Y. Qi and A. Chilkoti, Curr. Opin. Chem. Biol., 2015, 28, 181–193 CrossRef CAS PubMed.
- E. M. Pelegri-O’Day and H. D. Maynard, Acc. Chem. Res., 2016, 49, 1777–1785 CrossRef PubMed.
- J. Xu, K. Jung, N. A. Corrigan and C. Boyer, Chem. Sci., 2014, 5, 3568–3575 RSC.
- E. Daskalaki, B. Le Droumaguet, D. Gérard and K. Velonia, Chem. Commun., 2012, 48, 1586–1588 RSC.
- R. A. Olson, A. B. Korpusik and B. S. Sumerlin, Chem. Sci., 2020, 11, 5142–5156 RSC.
- A. Theodorou, E. Liarou, D. M. Haddleton, I. Georgia Stavrakaki, P. Skordalidis, R. Whitfield, A. Anastasaki and K. Velonia, Nat. Commun., 2020, 111(11), 1–11 Search PubMed.
- J. S. Weltz, D. F. Kienle, D. K. Schwartz and J. L. Kaar, ACS Catal., 2019, 9, 4992–5001 CrossRef CAS.
- J. S. Weltz, D. F. Kienle, D. K. Schwartz and J. L. Kaar, J. Am. Chem. Soc., 2020, 142, 3463–3471 CrossRef CAS PubMed.
- A. Munasinghe, S. L. Baker, P. Lin, A. J. Russell and C. M. Colina, Soft Matter, 2020, 16, 456–465 RSC.
- K. M. Mansfield and H. D. Maynard, ACS Macro Lett., 2018, 7, 324–329 CrossRef CAS PubMed.
- S. L. Baker, A. Munasinghe, H. Murata, P. Lin, K. Matyjaszewski, C. M. Colina and A. J. Russell, Biomacromolecules, 2018, 19, 3798–3813 CrossRef CAS PubMed.
- M. Lucius, R. Falatach, C. McGlone, K. Makaroff, A. Danielson, C. Williams, J. C. Nix, D. Konkolewicz, R. C. Page and J. A. Berberich, Biomacromolecules, 2016, 17, 1123–1134 CrossRef CAS PubMed.
- A. Munasinghe, A. Mathavan, A. Mathavan, P. Lin and C. M. Colina, Phys. Chem. Chem. Phys., 2019, 21, 25584–25596 RSC.
- P. Lin and C. M. Colina, Curr. Opin. Chem. Eng., 2019, 23, 44–50 CrossRef.
- B. Pérez, A. Coletta, J. N. Pedersen, S. V. Petersen, X. Periole, J. S. Pedersen, R. B. Sessions, Z. Guo, A. Perriman and B. Schiøtt, Sci. Rep., 2018, 8, 1–13 CrossRef PubMed.
- J. K. Pokorski and M. J. A. Hore, Curr. Opin. Colloid Interface Sci., 2019, 42, 157–168 CrossRef CAS.
- D. Russo, A. de Angelis, C. J. Garvey, F. R. Wurm, M.-S. Appavou and S. Prevost, Biomacromolecules, 2019, 20, 1944–1955 CrossRef CAS PubMed.
- D. Russo, A. De Angelis, A. Paciaroni, B. Frick, N. de Sousa, F. R. Wurm and J. Teixeira, Langmuir, 2019, 35, 2674–2679 CrossRef CAS PubMed.
- G. Cattani, L. Vogeley and P. B. Crowley, Nat. Chem., 2015, 7, 823–828 CrossRef CAS PubMed.
- P. W. Lee, S. A. Isarov, J. D. Wallat, S. K. Molugu, S. Shukla, J. E. P. Sun, J. Zhang, Y. Zheng, M. Lucius Dougherty, D. Konkolewicz, P. L. Stewart, N. F. Steinmetz, M. J. A. Hore and J. K. Pokorski, J. Am. Chem. Soc., 2017, 139, 3312–3315 CrossRef CAS PubMed.
- I. R. Smith, A. H. R. Charlier, A. M. Pritzlaff, A. Shishlov, B. Barnes, K. C. Bentz, C. P. Easterling, B. S. Sumerlin, G. E. Fanucci and D. A. Savin, ACS Macro Lett., 2018, 7, 1261–1266 CrossRef CAS.
- J. Lee, E.-W. Lin, U. Y. Lau, J. L. Hedrick, E. Bat and H. D. Maynard, Biomacromolecules, 2013, 14, 2561–2569 CrossRef CAS PubMed.
- C. S. Cummings, A. S. Campbell, S. L. Baker, S. Carmali, H. Murata and A. J. Russell, Biomacromolecules, 2017, 18, 576–586 CrossRef CAS PubMed.
- J. L. Battiste and G. Wagner, Biochemistry, 2000, 39, 5355–5365 CrossRef CAS PubMed.
- B. Liang, J. H. Bushweller and L. K. Tamm, J. Am. Chem. Soc., 2006, 128, 4389–4397 CrossRef CAS PubMed.
- P. A. Kosen, Methods Enzymol., 1989, 177, 86–121 CAS.
- N. Chikushi, H. Yamada, K. Oyaizu and H. Nishide, Sci. China Chem., 2012, 55, 822–829 CrossRef CAS.
- G. Gody, P. B. Zetterlund, S. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 10514 CrossRef CAS PubMed.
- R. Whitfield, N. P. Truong, D. Messmer, K. Parkatzidis, M. Rolland and A. Anastasaki, Chem. Sci., 2019, 10, 8724–8734 RSC.
- S. Harrisson, Polym. Chem., 2018, 9, 1366–1370 RSC.
- R. Falatach, C. McGlone, M. S. Al-Abdul-Wahid, S. Averick, R. C. Page, J. A. Berberich and D. Konkolewicz, Chem. Commun., 2015, 51, 5343–5346 RSC.
- S. Carmali, H. Murata, E. Amemiya, K. Matyjaszewski and A. J. Russell, ACS Biomater. Sci. Eng., 2017, 3, 2086–2097 CrossRef CAS.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS PubMed.
- W. Lee, M. Tonelli and J. L. Markley, Bioinformatics, 2015, 31, 1325–1327 CrossRef.
- V. A. Jaravine, A. V. Zhuravleva, P. Permi, I. Ibraghimov and V. Y. Orekhov, J. Am. Chem. Soc., 2008, 130, 3927–3936 CrossRef CAS PubMed.
- B. Ibarra-Molero, V. V. Loladze, G. I. Makhatadze and J. M. Sanchez-Ruiz, Biochemistry, 1999, 38, 8138–8149 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02200d |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2020 |