
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile benzene reduction promoted by a synergistically coupled Cu–Co–Ce ternary mixed oxide†
Hao
Chen‡
ab,
Wenwen
Lin‡
a,
Zihao
Zhang‡
 a,
Zhenzhen
Yang
b,
Kecheng
Jie
b,
Jie
Fu
*a,
Shi-ze
Yang
*d and
Sheng
Dai
*bc
a,
Zhenzhen
Yang
b,
Kecheng
Jie
b,
Jie
Fu
*a,
Shi-ze
Yang
*d and
Sheng
Dai
*bc
aKey Laboratory of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: jiefu@zju.edu.cn
bDepartment of Chemistry, University of Tennessee, Knoxville, TN 37996, USA
cChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: dais@ornl.gov
dEyring Materials Center, Arizona State University, Tempe, 85257, USA. E-mail: shize.yang@asu.edu
First published on 22nd May 2020
Abstract
Hydrogenation of aromatic rings promoted by earth-abundant metal composites under mild conditions is an attractive and challenging subject in the long term. In this work, a simple active site creation and stabilization strategy was employed to obtain a Cu+-containing ternary mixed oxide catalyst. Simply by pre-treatment of the ternary metal oxide precursor under a H2 atmosphere, a Cu+-derived heterogeneous catalyst was obtained and denoted as Cu1Co5Ce5Ox. The catalyst showed (1) high Cu+ species content, (2) a uniform distribution of Cu+ doped into the lattices of CoOx and CeO2, (3) formation of CoOx/CuOx and CeO2/CuOx interfaces, and (4) a mesoporous structure. These unique properties of Cu1Co5Ce5Ox endow it with pretty high hydrogenation activity for aromatic rings under mild conditions (100 °C with 5 bar H2), which is much higher than that of the corresponding binary counterparts and even exceeds the performance of commercial noble metal catalysts (e.g. Pd/C). The synergetic effect plays a crucial role in the catalytic procedure with CeO2 functioning as a hydrogen dissociation and transfer medium, Cu+ hydrogenating the benzene ring and CoOx stabilizing the unstable Cu+ species. This will unlock a new opportunity to design highly efficient earth-abundant metal-derived heterogeneous catalysts via interface interactions.
Hydrogenation is one of the central themes of petrochemical, coal chemical, fine chemical and environmental industries and is one of the most intensively investigated topics in catalysis.1–4 In addition, in the synthesis of fine chemicals, reduction of various functional groups, such as –C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, –C
C, –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, –CO, –NO2, –CN, –COOH, and –CONH2, is required to afford the corresponding alkanes, alkenes, alcohols, and amine products that are key intermediates for the fine chemical, polymer, agrochemical, and pharmaceutical industries, especially using H2 as a clean and cheap hydrogen source.5,6 Among all these transformations, hydrogenation of benzene is a direct and important approach to afford cycloalkane intermediates for petrochemical and agrochemical production, and has received enormous attention over the past few decades.7–9 However, the π-conjugation in aromatic rings makes it one of the most robust chemical bonds due to high aromaticity and non-polarity.10–12 Over the past few decades, technologies mainly depending on expensive and precious metal-based catalysts, such as Pd, Ru, Pt, and Ir, have been extensively investigated to facilitate this transformation. Concerns over the scarcity and high cost of noble metals have driven the search for nonprecious earth-abundant alternatives with comparable activity, selectivity and stability, which are greatly desired for scalable and cost-effective chemical transformations.11–18 To date, there have been a few reports on Ni/Al2O3, Ni/SiO2, Co/SiO2 and Ni–Al alloys that can partially or fully hydrogenate benzene with transition metal-based catalysts, but harsh reaction conditions (e.g. high reaction temperature up to 200 °C and high H2 pressure up to 8 MPa) and low weight hourly space velocity (WHSV) limit their further application.19–21 Therefore, despite intensive studies on the subject of benzene hydrogenation, catalytic systems based on nonprecious metals capable of promoting the reaction under mild conditions are still rarely reported. There are still significant challenges in developing cheap, easily synthesized and highly efficient heterogeneous catalysts derived from earth-abundant alternatives by rational design.
C, –CO, –NO2, –CN, –COOH, and –CONH2, is required to afford the corresponding alkanes, alkenes, alcohols, and amine products that are key intermediates for the fine chemical, polymer, agrochemical, and pharmaceutical industries, especially using H2 as a clean and cheap hydrogen source.5,6 Among all these transformations, hydrogenation of benzene is a direct and important approach to afford cycloalkane intermediates for petrochemical and agrochemical production, and has received enormous attention over the past few decades.7–9 However, the π-conjugation in aromatic rings makes it one of the most robust chemical bonds due to high aromaticity and non-polarity.10–12 Over the past few decades, technologies mainly depending on expensive and precious metal-based catalysts, such as Pd, Ru, Pt, and Ir, have been extensively investigated to facilitate this transformation. Concerns over the scarcity and high cost of noble metals have driven the search for nonprecious earth-abundant alternatives with comparable activity, selectivity and stability, which are greatly desired for scalable and cost-effective chemical transformations.11–18 To date, there have been a few reports on Ni/Al2O3, Ni/SiO2, Co/SiO2 and Ni–Al alloys that can partially or fully hydrogenate benzene with transition metal-based catalysts, but harsh reaction conditions (e.g. high reaction temperature up to 200 °C and high H2 pressure up to 8 MPa) and low weight hourly space velocity (WHSV) limit their further application.19–21 Therefore, despite intensive studies on the subject of benzene hydrogenation, catalytic systems based on nonprecious metals capable of promoting the reaction under mild conditions are still rarely reported. There are still significant challenges in developing cheap, easily synthesized and highly efficient heterogeneous catalysts derived from earth-abundant alternatives by rational design.
From this aspect, we focus our attention on one of the most challenging kinds of metal species based on copper, which is pretty cheap and abundant. On the other hand, copper-based catalysts have been widely investigated for the hydrogenation of biomass22,23 and CO2,24–27 with the activity being mainly attributed to the Cu0 species in vapor-phase reactions.23,28 For instance, Ma et al. revealed that the formation rate of alcohol is strongly correlated with the density of surface Cu0 sites.25,29 Notably, compared with Cu0 and Cu2+, Cu+ has a higher hydrogenation activity considering its intrinsic ability to facilitate electron transfer through gaining or losing an electron.25,30,31 Many studies have reported that Cu0/Cu+ leads to an enhanced catalytic activity for hydrogenation, which is attributed to the activation of the ester groups by Cu+ species in the production of alcohols.30,32–34 However, these conjectures are inconclusive as obtaining Cu+ is synthetically challenging due to its tendency to easily oxidize to Cu2+ or reduce to Cu0 during catalyst preparation and processing. The key to success lies in the design and fabrication of copper-containing composites capable of stabilizing the highly active Cu+ species through interface interactions. This will also enable a deeper understanding of the catalytic contributions from the Cu+ species, which is significant for the rational design of active hydrogenation catalysts.
As previously reported, the CoOy in Cu catalyst not only enhances the metallic Cu dispersion and H2 activation ability, but also modifies the chemical states of Cu to create suitable surface Cu0/Cu+ distributions due to strong electronic interactions at the Cu/CoOx interface.35 This inspires us to fabricate multi-component heterogeneous catalysts containing Cu+ species. It is known that the adsorption and activation of H2 constitutes another critical step in the hydrogenation reactions. Various kinds of materials have been reported to activate H2 by homolytic or heterolytic dissociation.36,37 Recently, Sai et al. created solid frustrated Lewis pairs (FLPs) on the surface of CeO2 by regulating their surface defects. The resultant catalysts exhibited H2 dissociation ability with a low activation barrier and delivered a high catalytic activity for hydrogenation of alkenes and alkynes, as well as transformation of CO2.38,39 However, the catalytic activity of these CeO2-based materials is still insufficient to achieve hydrogenation of aromatic rings. Therefore, we expect that the combination of Cu, Co and Ce species will create enhanced H2 activation capability, realize the hydrogenation of aromatic rings under mild conditions through a synergistic effect, and lead to further understanding of the interface interaction during the catalytic procedure.
In previous studies, our group developed a simple fabrication procedure to obtain a ternary CuO–Co3O4–CeO2 catalyst, which showed excellent catalytic activity for CO oxidation.40 Herein, a simple active site creation and stabilization strategy was employed to obtain a Cu+-containing ternary mixed oxide catalyst. Simply by pre-treatment of the ternary metal oxide precursor under a H2 atmosphere, a Cu+-derived heterogeneous catalyst was obtained and denoted as Cu1Co5Ce5Ox. The catalyst showed (1) high Cu+ species content, (2) a uniform distribution of Cu+ doped into the lattices of CoOx and CeO2, (3) formation of CoOy/CuOx and CeO2/CuOx interfaces, and (4) a mesoporous structure. These unique properties of Cu1Co5Ce5Ox endow it with pretty high hydrogenation activity for aromatic rings under mild conditions (100 °C with 5 bar H2), which is much higher than that of the corresponding binary counterparts and even exceeds the performance of commercial noble metal catalysts (e.g. Pd/C). The synergetic effect plays a crucial role in the catalytic procedure with CeO2 functioning as a hydrogen dissociation and transfer medium, Cu+ hydrogenating the benzene ring and CoOx stabilizing the unstable Cu+ species. This will unlock a new opportunity to design highly efficient earth-abundant metal-derived heterogeneous catalysts via interface interactions.
The ternary Cu1Co5Ce5Ox catalyst was prepared via a two-step approach involving co-precipitation and heat-treatment. Cu1Co5Ce5Oy with a Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co:Ce atomic ratio of 1:5:5 was first synthesized using a co-precipitation method,40,41 and further pre-treatment at 100 °C with 5 bar H2 for 24 h leads to the formation of Cu1Co5Ce5Ox. The ICP result shown in Table S1† confirmed that the Cu:Co:Ce atomic ratio was almost the same as that calculated from the raw ratio. The XRD pattern of Cu1Co5Ce5Oy in Fig. 1a suggests that the as-synthesized ternary oxides are composed of crystalline CeO2 and Co3O4. After H2 heat-treatment, no change in the diffraction peaks of CeO2 and Co3O4 was found in Cu1Co5Ce5Ox. Then, we compared the XRD pattern of CeO2 (PDF#81-0792),42 Co3O4 (PDF-74-1657)43 and Cu1Co5Ce5Ox as shown in Fig. 1(a) to further prove that Cu is doped into the lattices of CoOy and CeO2 in Cu1Co5Ce5Ox. It was found that the XRD peaks for Ce and Co in Cu1Co5Ce5Ox shifted to lower 2 theta angle, which means a larger lattice parameter of the CeO2 and Co3O4 in Cu1Co5Ce5Ox than the pure CeO2 and Co3O4 after the introduction of copper species, as well as providing evidence to show that the Cu ions are incorporated into the CeO2 and Co3O4 crystallites in Cu1Co5Ce5Ox.42,44 Then we investigated the structure of CuCoOx (Cu:Co = 1:10 mole ratio), CuCeOx (Cu:Ce = 1:10 mole ratio) and CoCeOx (Ce:Co = 1:1 mole ratio) by XRD and the results are shown in Fig. S1.† The results showed that the 10 mol% Cu dispersed well in the CoOx and CeOx with almost no Cu XRD peak observed. Thus, in this work, the Cu uniformly distributed among CoOy and CeO2, respectively. XPS was performed to probe the oxidation states of Co and Cu on the surface of the ternary oxides. As shown in Fig. 1b–d, both Cu 2p and Co 2p peaks exhibit peak shifts towards lower energies, indicating that the H2 pre-treatment significantly reduces the surface of Cu1Co5Ce5Oy and partially lowers the oxidation states of the metal species (Cu2+ 934.1 eV, Co3+ 781.2 eV, Cu+/Cu0 932.0 eV and Co2+ 779.2 eV, Table S2†).45 Considering the overlap of the XPS peaks corresponding to Cu+ and Cu0, we turned to Cu LMM to determine the Cu oxidation states in the ternary oxide catalysts (Fig. 1d and S2†), where only Cu2+ (569.3 eV) was observed in the Cu1Co5Ce5Oy catalyst while only Cu+ (573.2 eV) was observed in the Cu1Co5Ce5Ox catalyst.46–48 Therefore, the spectroscopic results indicate that Cu+ and Co2+ are formed on the surface of Cu1Co5Ce5Ox after the H2 pre-treatment. CeO2 exhibits hydrogenation activity for unsaturated compounds as the oxidation states of Ce can change reversibly between Ce4+ under oxidizing conditions and Ce3+ under reducing conditions.49,50 As shown in Fig. 1e, XPS has been performed for Cu1Co5Ce5Oy and Cu1Co5Ce5Ox to distinguish between the Ce4+ and Ce3+ species, where the shifting of peaks to lower binding energy may be due to a higher proportion of Ce3+.50 Chen et al.51 reported that active Cu clusters consist of Cu0 at the top layer and Cu+ species at the Cu/CeO2 interface due to electron depletion caused by the oxygen vacancies (Ov) in CeO2. In our case, the H2 pre-treatment may create more oxygen vacancies which stabilize Cu+ through forming Cu+–Ov–Ce3+ interfacial bonds. It was found that the Cu species in CuCoCeOy after pre-treatment for 6 h and 18 h were almost maintained at the Cu2+ state (Fig. S3†). However, it seemed that the Co was easily reduced and more Co2+ was formed compared with the CuCoCeOy after pre-treatment for 6 h and 18 h. And for Ce, the Ce in CuCoCeOy after pre-treatment for 6 h have no significant change compared with the starting material, with all the Ce species in the state of Ce4+. After pre-treatment for 18 h, a small amount of Ce3+ formed.
:Co:Ce atomic ratio of 1:5:5 was first synthesized using a co-precipitation method,40,41 and further pre-treatment at 100 °C with 5 bar H2 for 24 h leads to the formation of Cu1Co5Ce5Ox. The ICP result shown in Table S1† confirmed that the Cu:Co:Ce atomic ratio was almost the same as that calculated from the raw ratio. The XRD pattern of Cu1Co5Ce5Oy in Fig. 1a suggests that the as-synthesized ternary oxides are composed of crystalline CeO2 and Co3O4. After H2 heat-treatment, no change in the diffraction peaks of CeO2 and Co3O4 was found in Cu1Co5Ce5Ox. Then, we compared the XRD pattern of CeO2 (PDF#81-0792),42 Co3O4 (PDF-74-1657)43 and Cu1Co5Ce5Ox as shown in Fig. 1(a) to further prove that Cu is doped into the lattices of CoOy and CeO2 in Cu1Co5Ce5Ox. It was found that the XRD peaks for Ce and Co in Cu1Co5Ce5Ox shifted to lower 2 theta angle, which means a larger lattice parameter of the CeO2 and Co3O4 in Cu1Co5Ce5Ox than the pure CeO2 and Co3O4 after the introduction of copper species, as well as providing evidence to show that the Cu ions are incorporated into the CeO2 and Co3O4 crystallites in Cu1Co5Ce5Ox.42,44 Then we investigated the structure of CuCoOx (Cu:Co = 1:10 mole ratio), CuCeOx (Cu:Ce = 1:10 mole ratio) and CoCeOx (Ce:Co = 1:1 mole ratio) by XRD and the results are shown in Fig. S1.† The results showed that the 10 mol% Cu dispersed well in the CoOx and CeOx with almost no Cu XRD peak observed. Thus, in this work, the Cu uniformly distributed among CoOy and CeO2, respectively. XPS was performed to probe the oxidation states of Co and Cu on the surface of the ternary oxides. As shown in Fig. 1b–d, both Cu 2p and Co 2p peaks exhibit peak shifts towards lower energies, indicating that the H2 pre-treatment significantly reduces the surface of Cu1Co5Ce5Oy and partially lowers the oxidation states of the metal species (Cu2+ 934.1 eV, Co3+ 781.2 eV, Cu+/Cu0 932.0 eV and Co2+ 779.2 eV, Table S2†).45 Considering the overlap of the XPS peaks corresponding to Cu+ and Cu0, we turned to Cu LMM to determine the Cu oxidation states in the ternary oxide catalysts (Fig. 1d and S2†), where only Cu2+ (569.3 eV) was observed in the Cu1Co5Ce5Oy catalyst while only Cu+ (573.2 eV) was observed in the Cu1Co5Ce5Ox catalyst.46–48 Therefore, the spectroscopic results indicate that Cu+ and Co2+ are formed on the surface of Cu1Co5Ce5Ox after the H2 pre-treatment. CeO2 exhibits hydrogenation activity for unsaturated compounds as the oxidation states of Ce can change reversibly between Ce4+ under oxidizing conditions and Ce3+ under reducing conditions.49,50 As shown in Fig. 1e, XPS has been performed for Cu1Co5Ce5Oy and Cu1Co5Ce5Ox to distinguish between the Ce4+ and Ce3+ species, where the shifting of peaks to lower binding energy may be due to a higher proportion of Ce3+.50 Chen et al.51 reported that active Cu clusters consist of Cu0 at the top layer and Cu+ species at the Cu/CeO2 interface due to electron depletion caused by the oxygen vacancies (Ov) in CeO2. In our case, the H2 pre-treatment may create more oxygen vacancies which stabilize Cu+ through forming Cu+–Ov–Ce3+ interfacial bonds. It was found that the Cu species in CuCoCeOy after pre-treatment for 6 h and 18 h were almost maintained at the Cu2+ state (Fig. S3†). However, it seemed that the Co was easily reduced and more Co2+ was formed compared with the CuCoCeOy after pre-treatment for 6 h and 18 h. And for Ce, the Ce in CuCoCeOy after pre-treatment for 6 h have no significant change compared with the starting material, with all the Ce species in the state of Ce4+. After pre-treatment for 18 h, a small amount of Ce3+ formed.
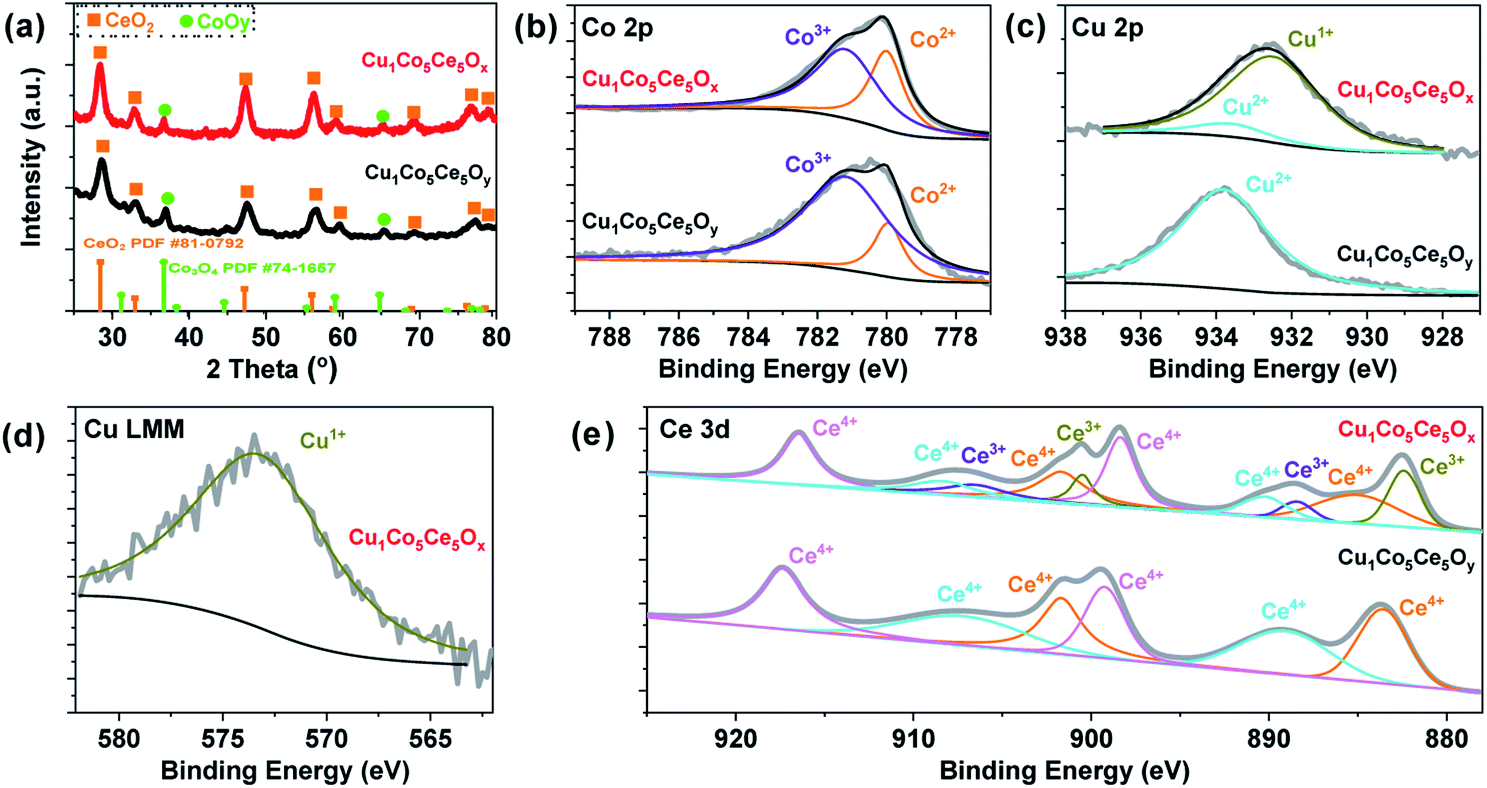 | ||
| Fig. 1 (a) XRD patterns, and (b) XPS results of Co 2p, (c) Cu 2p, (d) Cu-LMM, and (e) Ce 3d in Cu1Co5Ce5Oy and Cu1Co5Ce5Ox obtained before and after pre-treatment under 5 bar H2 and 100 °C temperature for 24 h. | ||
As reported in our previous work,40,41 the as-synthesized Cu1Co5Ce5Oy without H2 treatment showed a structure with copper–ceria and cobalt–ceria interfaces (Fig. S4†). To further probe the structural details of the ternary Cu1Co5Ce5Ox as well as the interface of CoOy(Cu2O)–CeO2(Cu2O), STEM-HAADF with EDS elemental mapping was conducted as shown in Fig. 2. The absence of the diffraction peaks corresponding to Cu, Cu2O, or CuO in the XRD pattern (Fig. 1a), together with a uniform distribution of Cu among CoOy and CeO2 in the STEM-EDS elemental maps (Fig. 2), suggests that Cu is doped into the lattices of CoOy and CeO2 for Cu1Co5Ce5Ox and forms CoOy/CuOx and CeO2/CuOx interfaces.
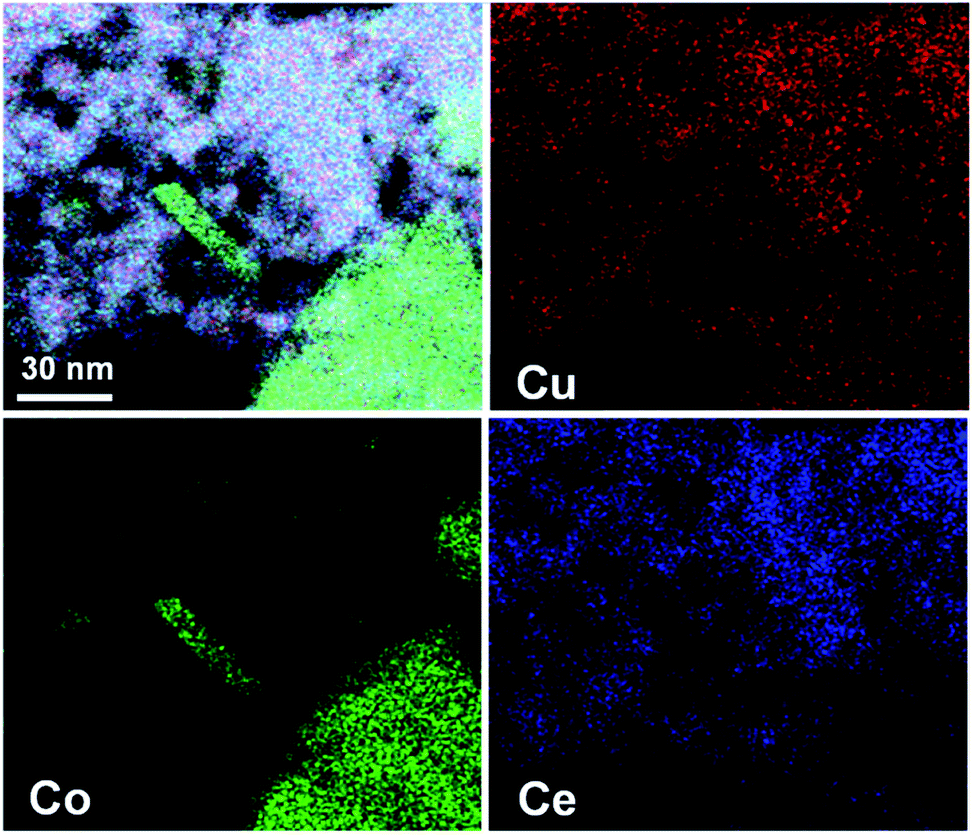 | ||
| Fig. 2 STEM-HAADF images and EDS elemental mapping images of Cu, Co and Ce in the Cu1Co5Ce5Ox catalyst. | ||
The N2 adsorption–desorption isotherm at 77 K is shown in Fig. 3a and the sample exhibits a typical type IV shape isotherm, suggesting the existence of mesopores with 2–10 nm pore diameters in Cu1Co5Ce5Ox after H2 treatment. The Brunauer–Emmett–Teller (BET) surface area of the Cu1Co5Ce5Ox is estimated to be 82 m2 g−1 with a total pore volume of 0.13 m3 g−1 (Fig. 3), which is a little higher compared with that of the Cu1Co5Ce5Oy before H2 treatment (78 m2 g−1). Notably, mesopores played a dominant role in the pore structure, contributing ∼99% of the total pore volume (Vmicro = 0 m3 g−1, calculated using the t-plot method). For catalytic or adsorptive materials, a high surface area together with a mesoporous structure can dramatically enhance their reactivity due to an improved mass transfer effect.52,53
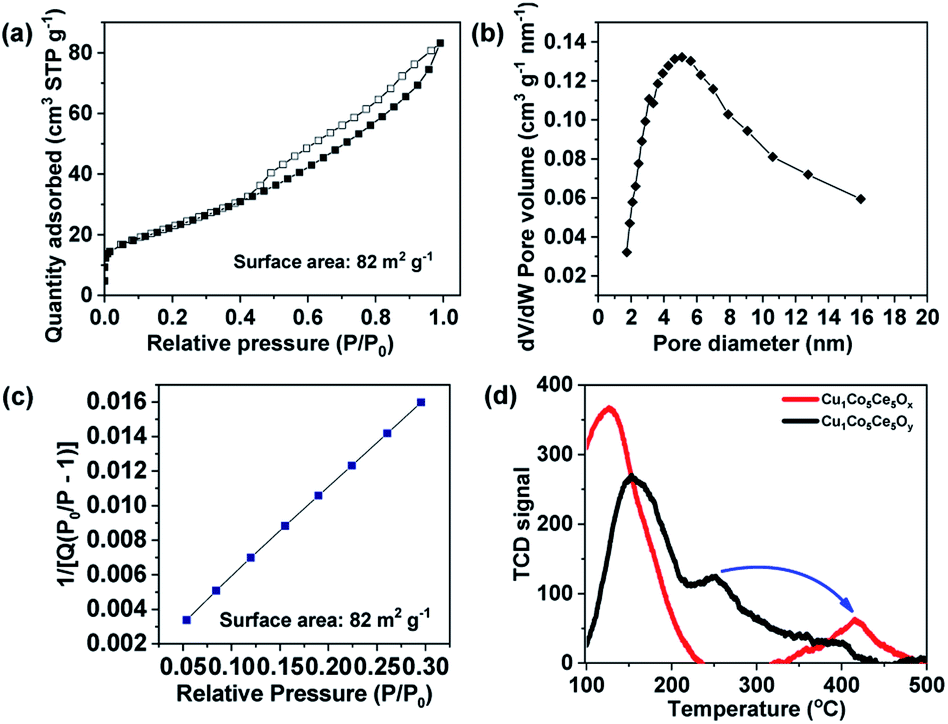 | ||
| Fig. 3 (a) N2 adsorption/desorption isotherm at 77 K, (b) pore size distribution curve, (c) BET plot, and (d) benzene temperature programmed desorption of Cu1Co5Ce5Ox and Cu1Co5Ce5Oy catalysts. | ||
Acetyl benzene is selected as a model substrate to evaluate the catalytic properties of Cu1Co5Ce5Ox (Fig. 4a). In a typical catalytic experiment, 100 mg of Cu1Co5Ce5Oy was pre-treated at 100 °C with 5 bar H2 for 24 h. Then 150 mg acetyl benzene (A) and 5 mL hexane were added to the reaction solution for hydrogenation under the same conditions. The H2-pretreated ternary oxide catalyst Cu1Co5Ce5Ox exhibits excellent activity towards complete hydrogenation of both acetyl and benzene groups, producing ethylcyclohexane (D) with 100% conversion and 97% yield, which even exceeds the performance of commercial 5 wt% Pd/C catalysts (74% conversion and 72% yield) under the same reaction conditions (Fig. 4b). Further control experiments show that the untreated ternary oxide catalyst Cu1Co5Ce5Oy exhibited much inferior hydrogenation capability, with ethylbenzene (C) being obtained as the sole product (Fig. S5†). To further investigate the synergetic effect of the ternary oxides, H2-pretreated binary oxide catalysts including CuCeOx, CuCoOx, and CoCeOx were prepared and the catalytic results indicated that ethylbenzene (C) was obtained in the presence of CuCeOx and CuCoOx, and when using CoCeOx as the catalyst, only reduction of the carbonyl group can be achieved, affording 1-phenylethanol as the product. That is, none of them showed the ability to hydrogenate the benzene ring. In addition, selective hydrogenation was realized using catalysts obtained by pre-treating Cu1Co5Ce5Oy under hydrogen with different times. Then, the XPS spectra of the CuCoOx and CuCeOx were measured (Fig. S6†). It can be found that without the synergistic effect of CoOx and CeOx, the Cu species in CuCoOx and CuCeOx after H2 pre-treatment all existed as Cu2+ and Cu0, and controlled reduction to Cu+ cannot be achieved. This was also proved by CODRIFTS as shown in Fig. S7.† Therefore, Cu+ was the key factor in this work to achieve successful hydrogenation of the benzene ring. The generated Cu1Co5Ce5Ox catalysts exhibited distinct hydrogenation capabilities. As summarized in Fig. 4c, H2-pretreatments for 6 h, 18 h, and 24 h lead to the formation of 96% ethyl benzene (C), 98% ethyl benzene (C), and 97% ethyl cyclohexane (D), respectively. Therefore, the H2-pretreated ternary Cu–Co–Ce oxides show an excellent capability towards the hydrogenation of aromatic rings.
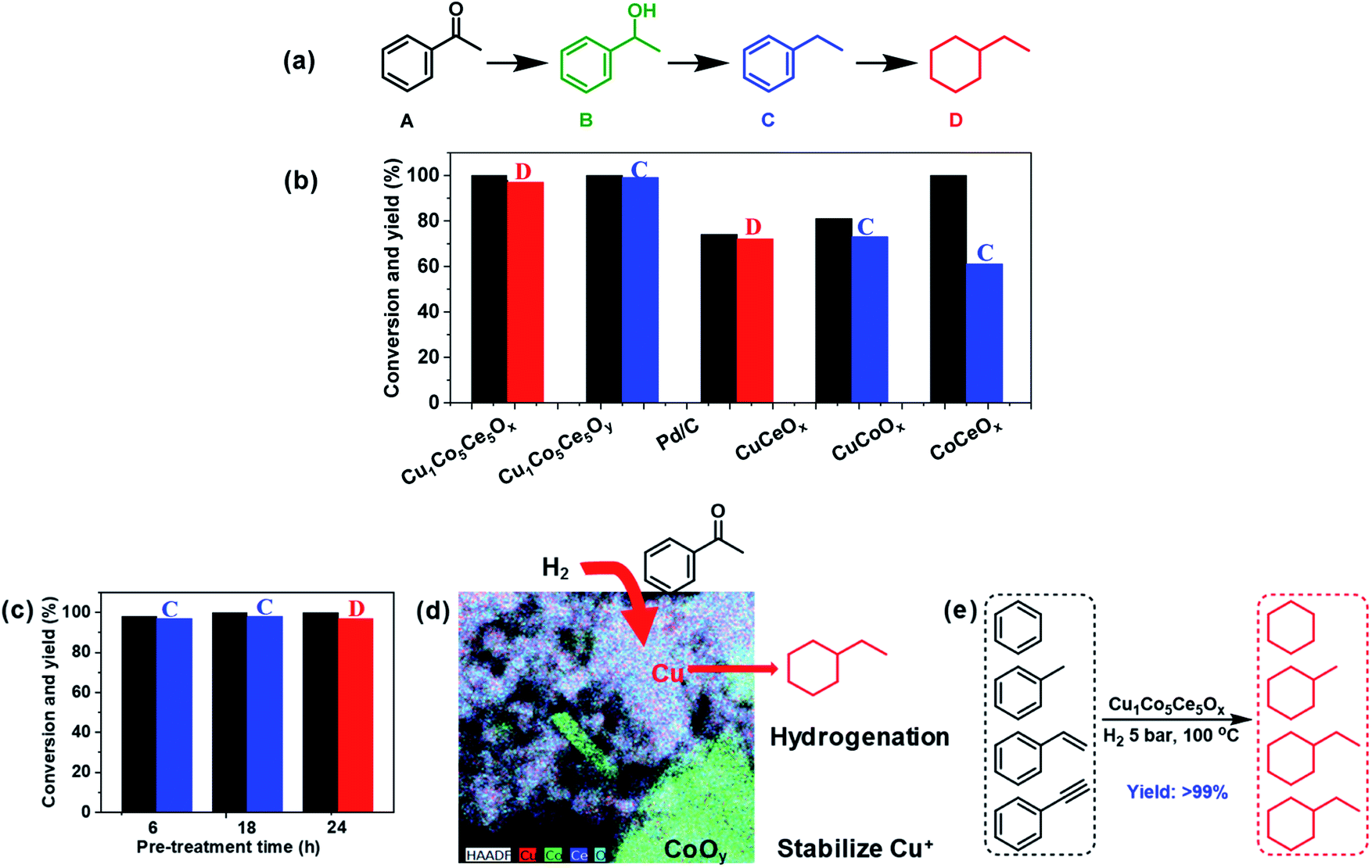 | ||
| Fig. 4 (a) Catalytic hydrodeoxygenation of acetyl benzene. (b) Comparison of the catalytic activity using Cu1Co5Ce5Ox, Cu1Co5Ce5Oy, Pd/C, CuOCeO2, CoOyCeO2, and CuOCoOy after H2 pretreatment. (c) Catalytic hydrodeoxygenation of acetyl benzene over the Cu1Co5Ce5Oy catalyst with different pre-treatment times. (d) Synergistic effect of Cu2O, CoOy and CeO2 in the CCC catalyst. (e) Hydrogenation of other aromatic compounds containing the benzene ring. All the reactions were performed under the following conditions: catalyst (100 mg), hexane (5 mL), substrate (1.25 mmol), reaction time (24 h), temperature (100 °C), H2 (5 bar). Acetyl benzene was used as the substrate for the results in (a) and (b). | ||
As previously reported, compared with Cu0 and Cu2+, Cu+ has a higher hydrogenation activity considering its intrinsic ability to facilitate electron transfer through gaining or losing an electron.25,30,31 Many studies have reported that Cu0/Cu+ leads to an enhanced catalytic activity for hydrogenation, which is attributed to the activation of the ester groups by Cu+ species.30,33,34 However, it is difficult to isolate Cu+ species for direct comparison, as it can rapidly convert to Cu0 or Cu2+ during catalyst preparation and processing. Here we report a ternary oxide system Cu1Co5Ce5Ox where Cu+ can be formed and stabilized through a simple pretreatment under H2. As previously reported, the CoOy in Cu catalyst not only enhances the metallic Cu dispersion and H2 activation ability, but also modifies the chemical states of Cu to create suitable surface Cu0/Cu+ distributions due to strong electronic interactions at the Cu/CoOx interface.35 In our case, Cu is doped into the lattices of CoOy and CeO2 for Cu1Co5Ce5Ox and CoOy exists as a promoter to stabilize Cu+ under 5 bar H2 and 100 °C through interfacial effects with CeO2. This is further supported by the emergence of Cu0 after the pretreatment of CuCeOx (Fig. S6 and S7†), indicating that Cu2+ will be reduced to Cu0 without cobalt. The stabilized Cu+ in Cu1Co5Ce5Ox exhibits an excellent catalytic performance for the hydrogenation of both benzene and CO, while CuCeOx, CuCoOx, and CoCeOx exhibit limited conversion and selectivity of converting acetyl benzene to ethylcyclohexane. It was also reported that the defect-enriched CeO2 constructed interfacial frustrated Lewis pairs (Ce3+⋯O2−) that effectively activate the H2 and CO2 (ref. 38 and 39) and XPS results show that the H2 pretreatment led to the formation of Ce3+ with oxygen vacancies on the surface. Chen et al.51 also reported that the Cu+ species directly bonded to the oxygen vacancy in CeO2 exhibits a high activity for the water-gas shift reaction, where the Cu+ site chemically adsorbs CO while the neighbouring Ov–Ce3+ site activates H2O. Thus, CeO2 functions as a hydrogen dissociation and transfer medium via the Ce3+⋯O2− frustrated Lewis pairs54 and then the neighbouring Cu+ hydrogenates the benzene as shown in Fig. 4d. In addition, benzene temperature programmed desorption (Ben–TPD) was performed to study the adsorption capacity of the benzene ring on Cu1Co5Ce5Oy and Cu1Co5Ce5Ox catalysts as shown in Fig. 3d. The two observed peaks at 100–200 and 250–450 °C are attributed to physical and chemical adsorption of benzene on the two oxide catalysts, respectively. Clearly, chemisorption of benzene on Cu1Co5Ce5Oy is enhanced after H2 pretreatment as evidenced by the increase in desorption temperature from 250 °C to 420 °C, probably due to the strong interaction between benzene and a withdrawing Cu+ from the oxygen ring.55 Besides acetyl benzene, a series of benzene and benzene derivatives including benzene, phenylacetylene and methylbenzene are also fully hydrogenated to the corresponding alkanes using the Cu1Co5Ce5Ox catalyst under mild conditions (Fig. 4e), demonstrating the wide applicability of the ternary oxides for efficient benzene hydrogenation. In summary, a new type of Cu–Co–Ce ternary mixed oxide catalyst with remarkable hydrogenation activity of benzene is reported. Formation of Cu+ during a simple pretreatment process is considered to be key to the activity promotion, while CoOx functions as the Cu+ stabilizer and CeO2 facilitates the dissociation and transfer of hydrogen. Demonstration of Cu+ in Cu1Co5Ce5Ox as the key component leading to extraordinary hydrogenation activity of substituted benzenes provides new insights into the design and modification of noble-metal-free catalysts for a wide scope of heterogeneous transformations. The resultant turnover number (TON) using Cu1Co5Ce5Oy, Cu1Co5Ce5Ox and commercial 5 wt% Pd/C in this work was compared. The TON obtained in different catalytic systems was estimated based on the following equation: TON = mmol (ethylbenzene)/mmol (active site).56,57 As a result, the TON of the Cu1Co5Ce5Oy, Cu1Co5Ce5Ox and commercial 5 wt% Pd/C was calculated to be 0, 38.8 and 18.6. It was found that the Cu1Co5Ce5Ox obtained after H2 pre-treatment exhibited a TON value double that obtained using a Pd/C catalyst. The results of stability during five cycles of reuse as well as the XPS and XRD measurements are shown in Fig. S8–S10.† It was found that the Cu+ remained in the 1+ valence state after the hydrogenation (Fig. S8†). This is because the catalyst was pre-treated under the same conditions as the reaction conditions. Thus, during the reaction process within 24 h, the catalyst was stable and there should be no change of valence state during the catalytic recycling (Fig. S10†). The catalytic results revealed that the catalyst showed very good reusability for at least five cycles without any decrease in the catalytic activity, with >99% conversion of acetyl benzene and >95% yield of ethylcyclohexane being obtained in the fifth run. And the XRD results showed that the structure of the recycled catalyst was maintained well after catalyzing the hydrogenation reaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
HC, ZZY, KCJ, and SD were supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, U.S. Department of Energy. JF was supported by the National Natural Science Foundation of China (No. 21978259 and 21706228), the Zhejiang Provincial Natural Science Foundation of China (No. LR17B060002) and the Fundamental Research Funds for the Central Universities. The STEM characterization used resources of the Center for Functional Nanomaterials, which is a U.S. DOE Office of Science Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704.Notes and references
- R. A. Johnstone, A. H. Wilby and I. D. Entwistle, Chem. Rev., 1985, 85, 129–170 CrossRef CAS.
- F. Meemken and A. Baiker, Chem. Rev., 2017, 117, 11522–11569 CrossRef CAS PubMed.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2018, 119, 2681–2751 CrossRef PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- S. Miao, Z. Liu, B. Han, J. Huang, Z. Sun, J. Zhang and T. Jiang, Angew. Chem. Int. Ed., 2006, 45, 266–269 CrossRef CAS.
- C. P. Rader and H. A. Smith, J. Am. Chem. Soc., 1962, 84, 1443–1449 CrossRef.
- L. Foppa and J. Dupont, Chem. Soc. Rev., 2015, 44, 1886–1897 RSC.
- X. Kang, G. Luo, L. Luo, S. Hu, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2016, 138, 11550–11559 CrossRef CAS PubMed.
- H. Liu, R. Fang, Z. Li and Y. Li, Chem. Eng. Sci., 2015, 122, 350–359 CrossRef CAS.
- C. Hubert, E. G. Bilé, A. Denicourt-Nowicki and A. Roucoux, Green Chem., 2011, 13, 1766–1771 RSC.
- H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250–1252 CrossRef CAS PubMed.
- P. Zhang, T. Wu, T. Jiang, W. Wang, H. Liu, H. Fan, Z. Zhang and B. Han, Green Chem., 2013, 15, 152–159 RSC.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097–3101 CrossRef CAS PubMed.
- C. Vangelis, A. Bouriazos, S. Sotiriou, M. Samorski, B. Gutsche and G. Papadogianakis, J. Catal., 2010, 274, 21–28 CrossRef CAS.
- P. Tomkins, E. Gebauer-Henke, W. Leitner and T. E. Müller, ACS Catal., 2014, 5, 203–209 CrossRef.
- J.-F. Yuan, C.-Q. Luo, Q. Yu, A.-P. Jia, G.-S. Hu, J.-Q. Lu and M.-F. Luo, Catal. Sci. Technol., 2016, 6, 4294–4305 RSC.
- X. Kang, H. Liu, M. Hou, X. Sun, H. Han, T. Jiang, Z. Zhang and B. Han, Angew. Chem., Int. Ed., 2016, 55, 1080–1084 CrossRef CAS PubMed.
- R. Molina and G. Poncelet, J. Catal., 2001, 199, 162–170 CrossRef CAS.
- L. Lu, Z. Rong, W. Du, S. Ma and S. Hu, ChemCatChem, 2009, 1, 369–371 CrossRef CAS.
- Z. Zhang, Q. Yang, H. Chen, K. Chen, X. Lu, O. Pingkai, J. Fu and J. G. Chen, Green Chem., 2017, 20, 197–205 RSC.
- Z. Zhang, S. Yao, C. Wang, M. Liu, F. Zhang, X. Hu, H. Chen, X. Gou, K. Chen, Y. Zhu, X. Lu, P. Ouyang and J. Fu, J. Catal., 2019, 373, 314–321 CrossRef CAS.
- B. An, J. Zhang, K. Cheng, P. Ji, C. Wang and W. Lin, J. Am. Chem. Soc., 2017, 139, 3834–3840 CrossRef CAS PubMed.
- J. Gong, H. Yue, Y. Zhao, S. Zhao, L. Zhao, J. Lv, S. Wang and X. Ma, J. Am. Chem. Soc., 2012, 134, 13922–13925 CrossRef CAS PubMed.
- Z.-Q. Wang, Z.-N. Xu, S.-Y. Peng, M.-J. Zhang, G. Lu, Q.-S. Chen, Y. Chen and G.-C. Guo, ACS Catal., 2015, 5, 4255–4259 CrossRef CAS.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. R. Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- S. Zhao, H. Yue, Y. Zhao, B. Wang, Y. Geng, J. Lv, S. Wang, J. Gong and X. Ma, J. Catal., 2013, 297, 142–150 CrossRef CAS.
- X. Chang, T. Wang, Z. J. Zhao, P. Yang, J. Greeley, R. Mu, G. Zhang, Z. Gong, Z. Luo, J. Chen, Y. Cui, G. A. Ozin and J. Gong, Angew. Chem. Int. Ed., 2018, 57, 15415–15419 CrossRef CAS.
- W. Chen, T. Song, J. Tian, P. Wu and X. Li, Catal. Sci. Technol., 2019, 9, 6749–6759 RSC.
- C. Wen, A. Yin, Y. Cui, X. Yang, W.-L. Dai and K. Fan, Appl. Catal., A, 2013, 458, 82–89 CrossRef CAS.
- Y. Wang, Y. Shen, Y. Zhao, J. Lv, S. Wang and X. Ma, ACS Catal., 2015, 5, 6200–6208 CrossRef CAS.
- S. Zhang, Y. Ma, H. Zhang, X. Zhou, X. Chen and Y. Qu, Angew. Chem. Int. Ed., 2017, 56, 8245–8249 CrossRef CAS PubMed.
- J. Wu, G. Gao, P. Sun, X. Long and F. Li, ACS Catal., 2017, 7, 7890–7901 CrossRef CAS.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Hävecker, A. Knop-Gericke, S. D. Jackson and R. Schlögl, Science, 2008, 320, 86–89 CrossRef CAS PubMed.
- T. Mitsudome, Y. Mikami, M. Matoba, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem. Int. Ed., 2012, 51, 136–139 CrossRef CAS PubMed.
- S. Zhang, Z. Xia, Y. Zou, F. Cao, Y. Liu, Y. Ma and Y. Qu, J. Am. Chem. Soc., 2019, 141, 11353–11357 CrossRef CAS PubMed.
- S. Zhang, Z. Q. Huang, Y. Ma, W. Gao, J. Li, F. Cao, L. Li, C. R. Chang and Y. Qu, Nat. Commun., 2017, 8, 15266 CrossRef CAS PubMed.
- A. J. Binder, T. J. Toops, R. R. Unocic, J. E. Parks and S. Dai, Angew. Chem. Int. Ed., 2015, 54, 13263–13267 CrossRef CAS.
- W. Xiao, S. Yang, P. Zhang, P. Li, P. Wu, M. Li, N. Chen, K. Jie, C. Huang, N. Zhang and S. Dai, Chem. Mater., 2018, 30, 2924–2929 CrossRef CAS.
- C. He, Y. Yu, L. Yue, N. Qiao, J. Li, Q. Shen, W. Yu, J. Chen and Z. Hao, Appl. Catal., B, 2014, 147, 156–166 CrossRef CAS.
- Y. Tak and K. Yong, J. Phys. Chem. C, 2008, 112, 74–79 CrossRef CAS.
- N. R. Radwan, M. Mokhtar and G. A. El-Shobaky, Appl. Catal., A, 2003, 241, 77–90 CrossRef CAS.
- H. Yan, H. Qin, W. Liang, X. Jin, Y. Zhang, X. Feng, Y. Liu, X. Chen and C. Yang, Catal. Sci. Technol., 2019, 9, 4909–4919 RSC.
- Y. Tanaka, R. Kikuchi, T. Takeguchi and K. Eguchi, Appl. Catal., B, 2005, 57, 211–222 CrossRef CAS.
- S. Zhu, X. Gao, Y. Zhu, W. Fan, J. Wang and Y. Li, Catal. Sci. Technol., 2015, 5, 1169–1180 RSC.
- C. Yang, Z. Miao, F. Zhang, L. Li, Y. Liu, A. Wang and T. Zhang, Green Chem., 2018, 20, 2142–2150 RSC.
- L. Vivier and D. Duprez, ChemSusChem, 2010, 3, 654–678 CrossRef CAS PubMed.
- D. Mullins, S. Overbury and D. Huntley, Surf. Sci., 1998, 409, 307–319 CrossRef CAS.
- A. Chen, X. Yu, Y. Zhou, S. Miao, Y. Li, S. Kuld, J. Sehested, J. Liu, T. Aoki and S. Hong, Nat. Catal., 2019, 2, 334 CrossRef CAS.
- H. Chen, Q. Wang, X. Zhang and L. Wang, Appl. Catal., B, 2015, 166–167, 327–334 CrossRef CAS.
- H. Chen, Q. Wang, X. Zhang and L. Wang, Ind. Eng. Chem. Res., 2014, 53, 19916–19924 CrossRef CAS.
- M. García-Melchor and N. López, J. Phys. Chem. C, 2014, 118, 10921–10926 CrossRef.
- E. Kukulska-Zając, P. Kozyra and J. Datka, Appl. Catal., A, 2006, 307, 46–50 CrossRef.
- A. Dubey, L. Nencini, R. R. Fayzullin, C. Nervi and J. R. Khusnutdinova, ACS Catal., 2017, 7, 3864–3868 CrossRef CAS.
- X. Shao, X. Yang, J. Xu, S. Liu, S. Miao, X. Liu, X. Su, H. Duan, Y. Huang and T. Zhang, Chem, 2019, 5, 693–705 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02238a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |