
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“CLipP”ing on lipids to generate antibacterial lipopeptides†
Victor
Yim
 ac,
Iman
Kavianinia
abc,
Melanie K.
Knottenbelt
d,
Scott A.
Ferguson
d,
Gregory M.
Cook
d,
Simon
Swift
e,
Aparajita
Chakraborty
ab,
Jane R.
Allison
ab,
Alan J.
Cameron
ab,
Paul W. R.
Harris
*abc and
Margaret A.
Brimble
*abc
ac,
Iman
Kavianinia
abc,
Melanie K.
Knottenbelt
d,
Scott A.
Ferguson
d,
Gregory M.
Cook
d,
Simon
Swift
e,
Aparajita
Chakraborty
ab,
Jane R.
Allison
ab,
Alan J.
Cameron
ab,
Paul W. R.
Harris
*abc and
Margaret A.
Brimble
*abc
aSchool of Biological Sciences, University of Auckland, 3A Symonds Street, Auckland 1010, New Zealand
bMaurice Wilkins Centre for Molecular Biodiscovery, University of Auckland, 3A Symonds Street, Auckland 1010, New Zealand
cSchool of Chemical Sciences, University of Auckland, 23 Symonds Street, Auckland 1010, New Zealand
dDepartment of Microbiology and Immunology, School of Medical Sciences, University of Otago, 720 Cumberland Street, Dunedin 9054, New Zealand
eDepartment of Molecular Medicine and Pathology, School of Medical Sciences, University of Auckland, 85 Park Road, Grafton, Auckland 1023, New Zealand
First published on 20th May 2020
Abstract
We herein report the synthesis and biological and computational evaluation of 12 linear analogues of the cyclic lipopeptide battacin, enabled by Cysteine Lipidation on a Peptide or Amino Acid (CLipPA) technology. Several of the novel “CLipP”ed lipopeptides exhibited low micromolar MICs and MBCs against both Gram-negative and Gram-positive bacteria. The mechanism of action was then simulated with the MIC data using computational methods.
Introduction
Naturally occurring antimicrobial peptides (AMPs) form part of the innate immune system and are recognised as potentially useful therapeutic agents. Due to the unique and non-specific bactericidal mechanism of action (MOA) of AMPs, it is believed that AMPs have a lower tendency to elicit antibiotic resistance than conventional antibiotics.1–3 Lipopeptides, a subclass of AMPs, have emerged as a new class of antibiotics, exemplified by the clinically-approved daptomycin (Cubicin) and polymyxin B which are used to treat Gram-positive and Gram-negative bacterial infections, respectively.2 The polymyxins are last-line defence agents against Gram-negative “superbugs”.4 However, their clinical use is limited by significant nephrotoxicity and the rise of polymyxin-resistant bacterial strains.5Battacin 1 (Fig. 1), a member of the octapeptin family of antibiotics, isolated by Qian et al.,6 was found to be less toxic than polymyxin B while exhibiting potent activity against multidrug-resistant and extremely drug-resistant Gram-negative clinical isolates, including hospital strains of E. coli with minimum inhibitory concentrations (MICs) of 2 to 4 μM. Structurally, battacin is a cationic lipopeptide antibiotic composed of a heptapeptide ring and a single amino acid exocyclic tail capped by a fatty acid. Battacin 1 is cationic, arising from the five α,γ-diaminobutyric acid (Dab) residues. The lactam ring is formed between a Dab side chain amine and a Leu carboxyl. Due to its similarity, battacin 1 is postulated to act similarly to polymxyin antibiotics, whereby electrostatic interactions occur with the negatively charged lipid A head groups of lipopolysaccharides (LPS) on the surface of the bacterial outer membrane (OM), leading to accumulation at the bacterial surface. Insertion of the lipid tail into the OM then leads to membrane destabilisation, self-promoted uptake and transit to the inner membrane, whereby insertion and membrane disruption are again thought to elicit the antimicrobial effects.6
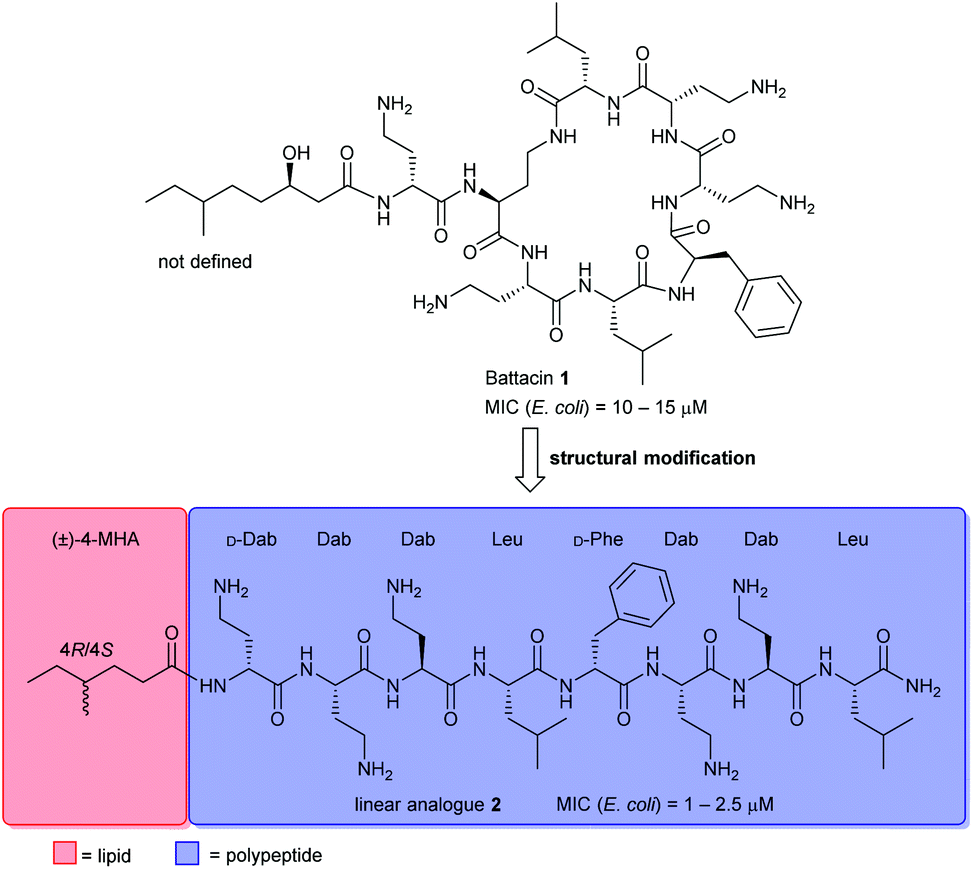 | ||
| Fig. 1 Chemical structure of battacin 1 and linear analogue 2 with improved activity. | ||
This class of lipopeptide antibiotics represents a rich source of new therapeutic agents in an otherwise scarce pipeline.7,8 To date, some structure–activity relationship (SAR) studies have been reported such as D-amino acid substitutions and C-terminal modifications, to address the potential therapeutic importance of battacin.9–13 De Zoysa et al. synthesised battacin and a series of lipidated analogues by solid-phase peptide synthesis (SPPS) and conducted a SAR study of the most active lipidated battacin analogues by alanine scanning.9,12 Among their findings was the linear analogue of battacin 2 with a shorter fatty acid, 4R/4S-methylhexanoic acid (MHA), which was more active than the native cyclic structure.9 Interestingly, battacin analogue 2 was also found to be highly potent against the Gram-positive pathogen S. aureus, while the native cyclic structure 1 was not, akin to its structural analogues in the polymyxin family. Furthermore, like polymyxin antibiotics, battacin analogue 2 was also shown to induce damage to both the outer and inner bacterial membranes.
To further develop battacin analogues for combatting multidrug-resistant Gram-negative bacteria, we were particularly interested in generating a series of linear analogues that differ in the chemical composition of the lipid chain. We recently reported a novel lipidation technology, based on the thiol–ene reaction, termed “CLipPA”—Cysteine Lipidation on a Peptide or Amino acid that enables a lipid to be installed selectively on a peptide under UV irradiation.14–18 This strategy has been applied to the synthesis of biologically active self-adjuvanting antigenic peptides14 and calcitonin gene-related peptide (CGRP) receptor antagonists.19 More recently, we reported a novel strategy for the preparation of cyclic lipopeptides employing CLipPA S-lipidation and intramolecular native chemical ligation (NCL) mediated cyclisation, but were disappointed by the inactivity of these iturin A analogues.20 We herein report the preparation of a focused library of novel linear battacin analogues using our thiol–ene CLipPA technology and their activity against Gram-negative and Gram-positive bacteria. Several of these analogues possessed broad-spectrum antibacterial properties.
The CLipPA reaction is carried out by irradiating (365 nm) a NMP solution of a peptide with a thiol handle 3, a vinyl ester containing a lipid of the desired chain length and photoinitiator, 2,2-dimethoxy-2-phenylacetophenone (DMPA). The generated sulfur radical species quickly reacts with the vinyl group of the ester to give a stabilised radical intermediate 4. Hydride transfer by tert-butylthiol (tBuSH) or tert-nonyl thiol (tNonylSH) and triisopropylsilane (TIPS) then affords the desired, lipidated peptide 5 (Scheme 1). In the absence of the hydride reagent, an undesired bis-adduct 6 forms alongside 5. The pH of the reaction is lowered using trifluoroacetic acid (TFA) to protonate electron rich side-chain residues, thus preventing the propagation of radical species by single-electron transfer from 4.15,21 The reaction conditions are mild, generally high-yielding, atom economical, and chemoselective. Thus, fully deprotected peptides are amenable to CLipPA.
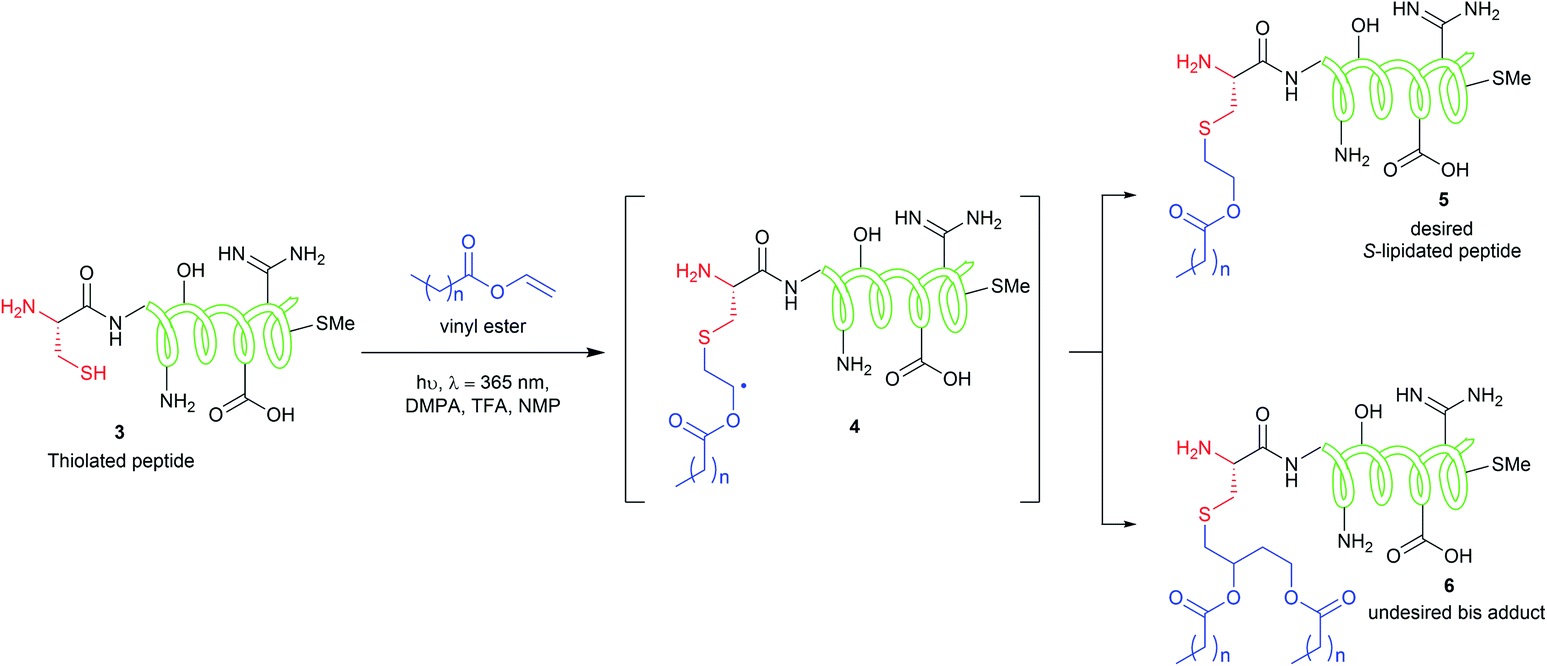 | ||
| Scheme 1 CLipPA thiol–ene reaction on a polypeptide with a vinyl ester yielding site selective S-linked lipopeptides. | ||
We envisaged the functionalisation of the N-terminus of linear battacin analogue 2 with a suitable thiol handle, which would serve as the site for the chemoselective conjugation of vinyl esters using CLipPA technology to generate a library of lipidated linear battacin analogues.
Results and discussion
The synthesis of the CLipPA analogues (Scheme 2) of linear battacin 2 entailed the use of (9H-fluoren-9-yl)methoxycarbonyl (Fmoc) based SPPS to construct two N-terminal thiol-containing sequences, with either a Cys or 3-mercaptopropionate (MPA). Subsequent CLipPA-enabled conjugation of the lipid afforded two sets of lipidated analogues – C and M series, respectively.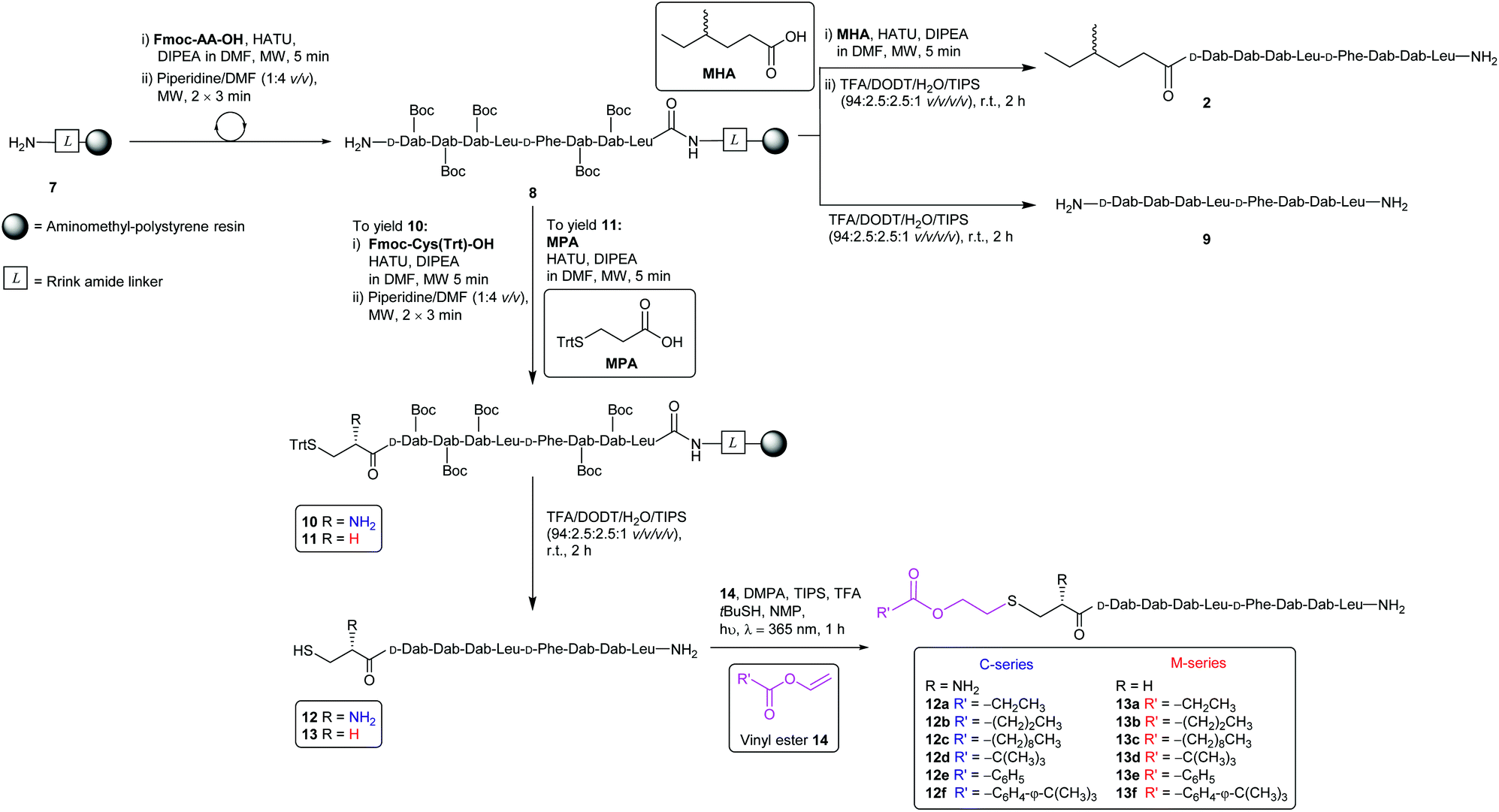 | ||
| Scheme 2 Synthesis of battacin analogues 2, 9, 12a–12f and 13a–13f. MW = microwave irradiation. | ||
Synthesis of the linear battacin analogues began with the formation of linker-resin 7 by loading Fmoc-protected Rink amide linker onto aminomethyl-polystyrene resin using 6-chloro-1-hydroxybenzotriazole (6-Cl-HOBt) and N,N′-diisopropylcarbodiimide (DIC) followed by Fmoc removal using piperidine/DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 v/v). The peptide sequence was then elongated using Fmoc-SPPS under microwave irradiation. Briefly, condensation of each subsequent amino acid was carried out using 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) as the coupling reagent and N,N-diisopropylethylamine (DIPEA) as a base, in DMF (5 min, 25 W, 50 °C). Iterative removals of the N-terminal Fmoc protecting group were performed by treating the resin with piperidine/DMF (1:4 v/v) (2 × 3 min, 50 W, 75 °C). The completion of each coupling step was confirmed by a negative result from the ninhydrin test.22 Following successful assembly of the resin-bound peptide 8, a portion was treated with a TFA cocktail (TFA/DODT/H2O/TIPS, 94:2.5:2.5:1 v/v/v/v) to yield linear sequence 9 for use as a non-lipidated reference compound in the antimicrobial assays. To prepare an additional reference compound, MHA was coupled onto another portion of peptidyl resin 8. Subsequent treatment with a TFA cocktail afforded the linear battacin analogue 2.
:4 v/v). The peptide sequence was then elongated using Fmoc-SPPS under microwave irradiation. Briefly, condensation of each subsequent amino acid was carried out using 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) as the coupling reagent and N,N-diisopropylethylamine (DIPEA) as a base, in DMF (5 min, 25 W, 50 °C). Iterative removals of the N-terminal Fmoc protecting group were performed by treating the resin with piperidine/DMF (1:4 v/v) (2 × 3 min, 50 W, 75 °C). The completion of each coupling step was confirmed by a negative result from the ninhydrin test.22 Following successful assembly of the resin-bound peptide 8, a portion was treated with a TFA cocktail (TFA/DODT/H2O/TIPS, 94:2.5:2.5:1 v/v/v/v) to yield linear sequence 9 for use as a non-lipidated reference compound in the antimicrobial assays. To prepare an additional reference compound, MHA was coupled onto another portion of peptidyl resin 8. Subsequent treatment with a TFA cocktail afforded the linear battacin analogue 2.
The remaining peptidyl resin 8 was functionalised with a thiol-containing residue. Coupling of trityl-protected Cys and subsequent Fmoc removal yielded 10. Similarly, coupling trityl-protected MPA yielded 11. Subsequent treatment with the TFA cocktail described above afforded CLipPA precursor sequences with a Cys thiol handle 12 or with an MPA thiol handle 13, the progenitors of the analogues classified as the C- or M-series respectively. The two series enabled the elucidation of the impact of the presence/absence of an additional amino group at the N-terminus upon antimicrobial activity.
Purified linear peptides 12 or 13, photoinitiator DMPA and a series of commercially available vinyl esters 14 bearing diverse lipid moieties were dissolved in a solution of NMP with CLipPA additives. The reaction mixture was then irradiated under UV at 365 nm to effect S-lipidation. Conjugating vinyl esters 14 onto peptide 12 afforded C-series battacin analogues 12a–12f, while conjugation onto peptide 13 afforded M-series battacin analogues 13a–13f (Fig. 2). The masses as determined by LC-MS for the analogues and their yields are tabulated below (Table 1).
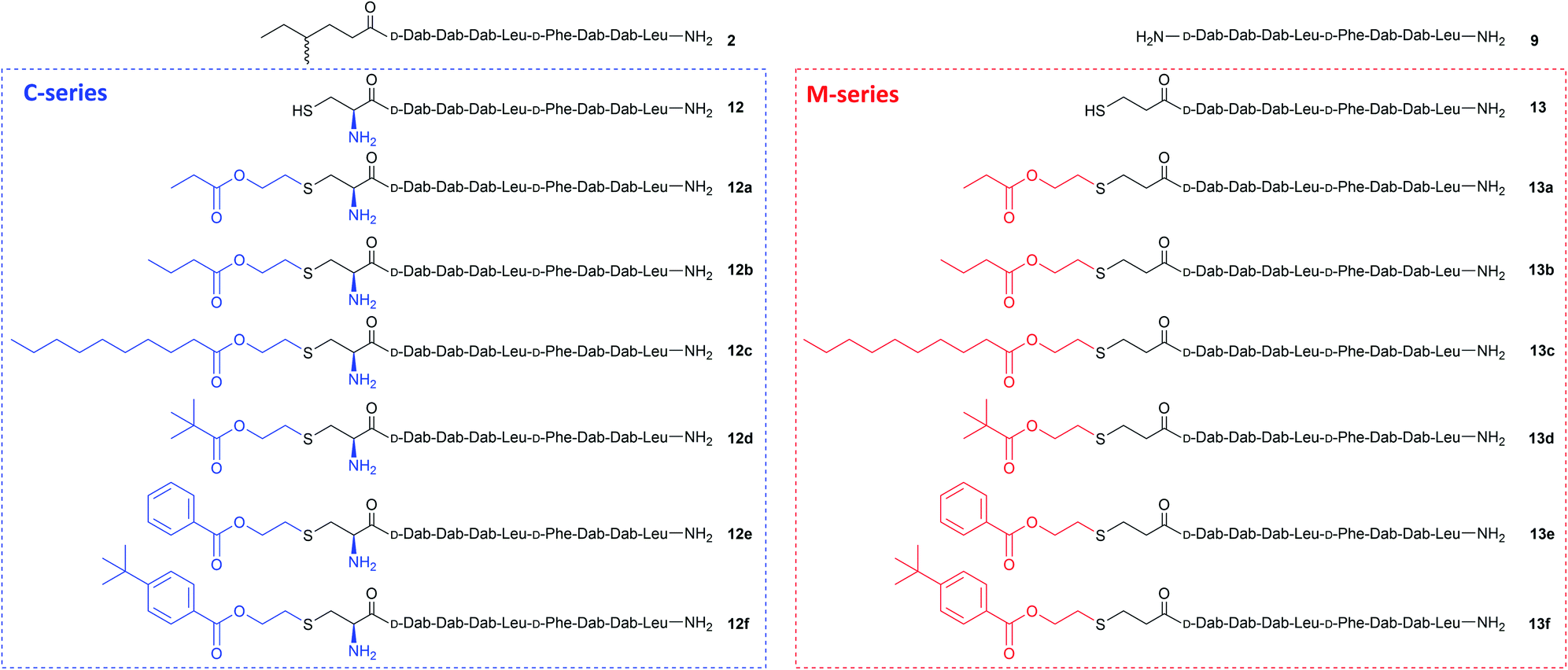 | ||
| Fig. 2 Structures of battacin analogues used for elucidating MICs against bacteria. 9 was the negative control and 2 was the lead compound. The remaining analogues were accessed through CLipPA using free thiols from cysteine 12 in C-series (12a–12f, blue) and from MPA 13 in M-series (13a–13f, red). | ||
| Peptide | Mass found | Yield (% purity) |
|---|---|---|
| 9 | 891.5 | 77 (96) |
| 2 | 1004.6 | 58 (99) |
| 12 | 994.5 | 30 (98) |
| 12a | 1094.6 | 10 (97) |
| 12b | 1108.6 | 5 (99) |
| 12c | 1192.6 | 1 (97) |
| 12d | 1122.6 | 3 (99) |
| 12e | 1142.6 | 1 (99) |
| 12f | 1198.6 | 6 (99) |
| 13 | 979.5 | 32 (95) |
| 13a | 1079.6 | 16 (98) |
| 13b | 1093.6 | 17 (99) |
| 13c | 1177.7 | 6 (94) |
| 13d | 1107.6 | 15 (96) |
| 13e | 1127.7 | 8 (96) |
| 13f | 1183.6 | 5 (96) |
The antimicrobial profiles of the synthetic analogues were evaluated by examining their minimum inhibitory concentration (MIC) values against the Gram-negative bacteria E. coli, P. aeruginosa and A. calcoaceticus and against the Gram-positive bacteria S. aureus (Table 2), and insight into the mechanism of action of selected analogues was provided through molecular dynamics (MD) simulations. As expected, non-lipidated linear battacin 9, showed very poor activity (128 μM against E. coli) across all four strains of bacteria. In our MD simulations, it did not insert into our model E. coli inner membrane (Fig. 3 and 4) and had only weak interactions with the membrane lipids (Fig. S17†). Despite lacking a lipid tail, 12 and 13 exhibited moderate inhibitory activity, potentially deriving from reactive thiols.23
| Peptide | E. coli | P. aeruginosa | A. calcoaceticus | S. aureus |
|---|---|---|---|---|
| MIC (μM) | ||||
| 9 | 128 | >128 | >128 | >128 |
| 2 | 8 | >128 | 128 | 64 |
| 12 | 32 | 64 | 64 | 128 |
| 12a | 32 | >128 | >128 | >128 |
| 12b | 16 | >128 | >128 | 128 |
| 12c | 16 | 32 | 32 | 8 |
| 12d | 16 | 128 | >128 | >128 |
| 12e | 8 | 64 | 128 | 64 |
| 12f | 8 | 8 | 32 | 8 |
| 13 | 32 | 32 | 64 | 64 |
| 13a | 16 | >128 | >128 | >128 |
| 13b | 16 | >128 | >128 | 128 |
| 13c | 8 | 8 | 32 | 8 |
| 13d | 16 | 64 | 128 | 64 |
| 13e | 8 | 64 | 64 | 32 |
| 13f | 8 | 16 | 32 | 8 |
| Tetracycline (μg ml−1) | — | 8 | 2 | — |
| Ampicillin (μg ml−1) | 2 | — | — | 0.125 |
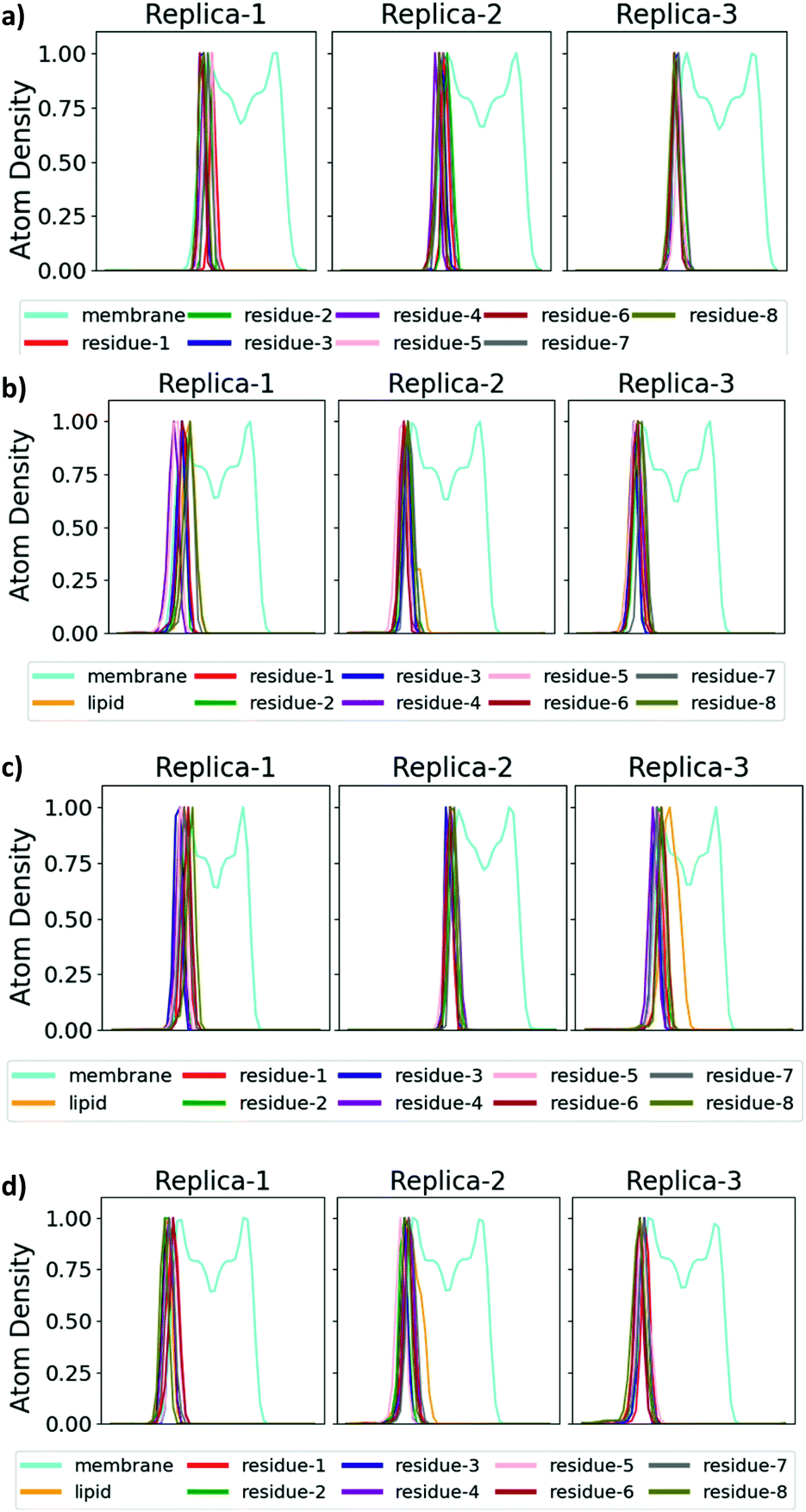 | ||
| Fig. 3 Location of the lipid tail and amino acid residues of battacin with respect to the membrane normal. The penetration of the lipid tails into a model E. coli inner membrane is illustrated by plotting the projection of the atom density onto the membrane normal (x-axis) for the membrane lipids (cyan) and for each residue of battacin, as labelled, for analogues (a) 9, (b) 2, (c) 13c, and (d) 13f. The centre of the membrane is indicated by a dashed line. The atom density was averaged across the entire 500 ns simulation for each of the three replicate simulations. ‘Lipid’ refers to the entire lipid tail, including the linker region. Where there is a lipid tail present, it penetrates deeper into the membrane than the remainder of the battacin residues, which co-locate with the head groups of the membrane lipids. | ||
 | ||
| Fig. 4 Binding mode of battacin analogues to an E. coli inner membrane. Snapshots showing the penetration of the lipid tails into a model E. coli inner membrane for one of the three replicate simulations of each of (a) 9, (b) 2, (c) 13c, and (d) 13f. The membrane lipids are coloured according to atom type (carbon: cyan; oxygen: red; nitrogen: blue; hydrogen: white) and the residues of battacin are coloured as in Fig. 3. Where there is a lipid tail present, it penetrates deeper into the membrane than the remainder of the battacin residues, which co-locate with the head groups of the membrane lipids. | ||
To our surprise, linear battacin analogue 2 exhibited only moderate or no inhibition of growth and was strikingly less potent than the previous report.9 In one of our three replicate simulations, the lipid portion and residues 6 (Dab) and 7 (Dab) reach a short way into the membrane and interact favourably with the lipid core, and in another, only the lipid portion penetrates the membrane (Fig. 3 and S17†). The differences in activity may be due to differences in susceptibility between different strains of the bacterial species tested. Because a direct comparison of MICs cannot be made, tetracycline and ampicillin were also tested alongside as reference compounds.
Comparing analogues that are S-lipidated with the same vinyl ester (e.g.12a against 13a) indicated that the M-series of analogues inhibited growth at either the same or lower concentrations. Hence, this suggests that the N-terminal amino group in the C-series introduced a positive charge that was generally unfavourable for activity. Given the proximity of the charge to the lipid tail, we suggest it may hinder insertion of the lipid tail upon membrane interaction. Moreover, it could impede the aggregation of monomers. Unfortunately, however, we could not detect any consistent differences in the insertion profiles or interaction energies of the M- and C-series of compounds in our MD simulations.
For the CLipPA analogues, the lowest MIC values (8 μM) were obtained from analogues containing long alkyl chains (12c and 13c) and substituted aromatic moieties (12f and 13f). In the MD simulations (Fig. 3 and 4), these lipid tails insert into and exhibit favourable interactions with (Fig. S17†) the membrane core, suggesting that these alkyl chains encourage insertion into the membrane and thus anchoring of the battacin to the membrane, increasing its activity.
The MIC values of the linear alkyl chain analogues (12a–12c and 13a–13c) showed that increasing the chain length enhanced potency. While the shortest chain analogues 13a and 13b (MIC > 128 μM) showed no inhibition of P. aeruginosa growth, long chain analogue 13c was the most potent with an MIC of 8 μM. Peptide 13c was also the most potent compound against all four species evaluated.
For the analogues conjugated with the bulky pivalate ester (12d and 13d), the MIC values were moderate to high (16 to >128 μM). Potentially, the steric bulk of the tert-butyl group could hinder insertion into the membrane, as well as aggregation between monomers. The benzoate analogues (12e and 13e), where aggregation could be facilitated through π-stacking, also showed moderate or no inhibitory activity against most test species. However, the tert-butyl substituted benzoate analogues (12f and 13f) showed significantly more potent activity, with 12f being equipotent to 13c across all test species and 13f being similarly active. The enhanced activity of the substituted benzoates suggests that the tert-butyl or aromatic groups alone do not confer sufficient hydrophobicity to the peptide and the tert-butyl bulk is actually well-tolerated.
The minimum bactericidal concentrations (MBCs) of these compounds were all equivalent to or within 2-fold of the respective MICs (Table S1†).24 This suggests that these peptides are also bactericidal.
The hydrophobicity of a peptide has been suggested to strongly affect its antimicrobial activity and toxicity. The most direct way for quantifying hydrophobicity is by comparing the compound retention times in reverse-phase high-performance liquid-chromatography (RP-HPLC).25 However, RP-HPLC was performed under acidic conditions (pH = 2), which is far from physiological pH of 7.4 where biological evaluations are relevant. Hence, to complement the HPLC data we calculated logD values at pH 7.4 for all compounds. LogD refers to the distribution coefficient of an ionisable compound in a buffered aqueous/organic system, where the higher value refers to more hydrophobicity.26 LogD calculations has been shown to be a better predictor for hydrophobicity than retention time. The logD values and retention times are compiled in Table 3 with the most active analogues of each series displaying increased hydrophobicity. In particular, 13c of the M-series being the most hydrophobic and most potent overall.27 This suggests that overall hydrophobicity is an important property for battacin analogues for forming pores in bacterial membranes. In terms of potential mammalian toxicity of the battacin molecules prepared here, it was reported that the linear battacins containing an N-terminal Fmoc or myristyl group caused a significant amount of haemolysis in mouse red blood cells.9 As the most potent compounds prepared here (12f and 13c) have an altered N-terminus with an aromatic lipid (12f) and long fatty acyl chain (13c) but a retained linear pharmacophore, we suggest that these compounds would also exhibit similar toxicities.
D values for synthetic battacin analogues presented in this work
| Peptide | t R (min) | logD |
Chemical formula |
|---|---|---|---|
| 9 | 11.3 | −13.36 | C48H86N14O9 |
| 2 | 14.7 | −11.05 | C44H79N15O9S |
| 12 | 12.5 | −13.88 | C49H87N15O11S |
| 12a | 13.4 | −13.62 | C50H89N15O11S |
| 12b | 13.9 | −13.17 | C56H101N15O11S |
| 12c | 17.7 | −10.50 | C49H87N15O11S |
| 12d | 14.4 | −12.52 | C49H87N15O11S |
| 12e | 17.8 | −12.26 | C49H87N15O11S |
| 12f | 16.6 | −10.72 | C 44 H 78 N 14 O 9 S |
| 13 | 16.8 | −12.44 | C49H86N14O11S |
| 13a | 14.3 | −12.12 | C50H88N14O11S |
| 13b | 14.8 | −11.68 | C56H100N14O11S |
| 13c | 18.5 | −9.01 | C 51 H 90 N 14 O 11 S |
| 13d | 14.7 | −11.02 | C53H86N14O11S |
| 13e | 15.6 | −10.77 | C49H86N14O11S |
| 13f | 17.5 | −9.22 | C48H86N14O9 |
Conclusion
In summary, we prepared a series of novel lipopeptides based on the antimicrobial linear battacin analogue 2 by introducing lipid-bearing vinyl esters onto this framework. Conjugating long alkyl chains or tert-butyl substituted benzoates provided the most potent antimicrobial compounds and resulted in the strongest interaction with the cell membrane. The demonstrated antibacterial properties exhibited by the synthetic analogues described in this work show that active antimicrobial lipopeptides can be synthesised by employing CLipPA technology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Biocide Toolbox (contract UOA1410), funded by New Zealand Ministry of Business, Innovation and Employment, the Health Research Council of New Zealand (Grant HRC 16/010), the Maurice Wilkins Centre for Molecular Biodiscovery, Lottery Health Research Fellowship (AJC) and a Rutherford Discovery Fellowship (15-MAU-001 to JRA) for financial support.Notes and references
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2016, 6, 1–12 CrossRef PubMed.
- I. W. Hamley, Chem. Commun., 2015, 51, 8574–8583 RSC.
- J. L. Anaya-López, J. E. López-Meza and A. Ochoa-Zarzosa, Crit. Rev. Microbiol., 2013, 39, 180–195 CrossRef PubMed.
- T. Velkov, C. Dai, G. D. Ciccotosto, R. Cappai, D. Hoyer and J. Li, Pharmacol. Ther., 2018, 181, 85–90 CrossRef CAS PubMed.
- T. Velkov, P. E. Thompson, R. L. Nation and J. Li, J. Med. Chem., 2010, 53, 1898–1916 CrossRef CAS PubMed.
- C.-D. Qian, X.-C. Wu, Y. Teng, W.-P. Zhao, O. Li, S.-G. Fang, Z.-H. Huang and H.-C. Gao, Antimicrob. Agents Chemother., 2012, 56, 1458–1465 CrossRef CAS PubMed.
- B. Becker, M. S. Butler, K. A. Hansford, A. Gallardo-Godoy, A. G. Elliott, J. X. Huang, D. J. Edwards, M. A. T. Blaskovich and M. A. Cooper, Bioorg. Med. Chem. Lett., 2017, 27, 2407–2409 CrossRef CAS PubMed.
- Swarbrick, M. A. T. Blaskovich, A. G. Elliott, M. Han, P. E. Thompson, K. D. Roberts, J. X. Huan, T. Velkov, A. Gallardo-Godoy, J. D. g, B. Becker, M. S. Butler, L. H. Lash, S. T. Henriques, R. L. Nation, S. Sivanesan, M.-A. Sani, F. Separovic, H. Mertens, D. Bulach, T. Seemann, J. Owen, J. Li and M. A. Cooper, Cell Chem. Biol., 2018, 25, 380–391 CrossRef PubMed.
- G. H. De Zoysa, A. J. Cameron, V. V. Hegde, S. Raghothama and V. Sarojini, J. Med. Chem., 2015, 58, 625–639 CrossRef CAS PubMed.
- G. H. De Zoysa and V. Sarojini, ACS Appl. Mater. Interfaces, 2017, 9, 1373–1383 CrossRef CAS PubMed.
- G. H. De Zoysa, H. D. Glossop and V. Sarojini, Eur. J. Med. Chem., 2018, 146, 344–353 CrossRef CAS PubMed.
- H. D. Glossop, E. Pearl, G. H. De Zoysa and V. Sarojini, in Advances in Protein Chemistry and Structural Biology, ed. R. Donev, Academic Press, Cambridge, MA, 2018, vol. 112, pp. 385–394 Search PubMed.
- H. D. Glossop, G. H. De Zoysa, Y. Hemar, P. Cardoso, K. Wang, J. Lu, C. Valéry and V. Sarojini, Biomacromolecules, 2019, 20, 2515–2529 CrossRef CAS PubMed.
- T. H. Wright, A. E. S. Brooks, A. J. Didsbury, G. M. Williams, P. W. R. Harris, P. R. Dunbar and M. A. Brimble, Angew. Chem., Int. Ed., 2013, 52, 10616–10619 CrossRef CAS PubMed.
- S. Yang, P. W. R. Harris, G. M. Williams and M. A. Brimble, Eur. J. Org. Chem., 2016, 2016, 2608–2616 CrossRef CAS.
- M. A. Brimble, T. H. Wright, P. R. Dunbar and G. M. Williams, Amino acid and peptide conjugates and conjugation process, Google Patents, 2017 Search PubMed.
- S.-H. Yang, Y. O. J. Hermant, P. W. R. Harris and M. A. Brimble, Eur. J. Org. Chem., 2020 Search PubMed , advance article.
- Y. O. Hermant, A. J. Cameron, P. W. R. Harris and M. A. Brimble, in Peptide Synthesis: Methods and Protocols, ed. W. M. Hussein, M. Skwarczynski and I. Toth, Springer US, New York, NY, 2020, pp. 263–274 Search PubMed.
- E. T. Williams, P. W. R. Harris, M. A. Jamaluddin, K. M. Loomes, D. L. Hay and M. A. Brimble, Angew. Chem., Int. Ed., 2018, 57, 11640–11643 CrossRef CAS PubMed.
- V. V. Yim, I. Kavianinia, A. J. Cameron, P. W. R. Harris and M. A. Brimble, Org. Biomol. Chem., 2020 Search PubMed , advance article.
- F. Li, A. Allahverdi, R. Yang, G. B. J. Lua, X. Zhang, Y. Cao, N. Korolev, L. Nordenskiöld and C.-F. Liu, Angew. Chem., Int. Ed., 2011, 50, 9611–9614 CrossRef CAS PubMed.
- V. K. Sarin, S. B. H. Kent, J. P. Tam and R. B. Merrifield, Anal. Biochem., 1981, 117, 147–157 CrossRef CAS PubMed.
- T. Oguri, B. Schneider and L. Reitzer, J. Bacteriol., 2012, 194, 4366–4376 CrossRef CAS PubMed.
- G. L. French, J. Antimicrob. Chemother., 2006, 58, 1107–1117 CrossRef CAS PubMed.
- C. T. Mant, N. E. Zhou and R. S. Hodges, J. Chromatogr. A, 1989, 476, 363–375 CrossRef CAS PubMed.
- M. Kah and C. D. Brown, Chemosphere, 2008, 72, 1401–1408 CrossRef CAS PubMed.
-
LogD calculator, https://disco.chemaxon.com/calculators/demo/plugins/chemicalterms/?expression=logd(%277.4%27), accessed 14 May 2020 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, data for new compounds and biological data. See DOI: 10.1039/d0sc01814g |
| This journal is © The Royal Society of Chemistry 2020 |