
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLate-stage and strain-accelerated oxidation enabled synthesis of haouamine A†
Kun Ho (Kenny)
Park
,
Antonio
Rizzo
and
David Y.-K.
Chen
 *
*
Department of Chemistry, Seoul National University, Gwanak-1 Gwanak-ro, Gwanak-gu, Seoul 08826, South Korea. E-mail: davidchen@snu.ac.kr
First published on 24th June 2020
Abstract
Herein we report a new synthetic entry to the strained cyclophane alkaloid natural product, haouamine A. The successful strategy featured a rhodium-catalyzed diazo-insertion reaction to install the all-carbon quaternary center and a rhodium-catalyzed intramolecular aziridination reaction to establish the nitrogen-bearing stereocenter, of the target molecule. Most notably, a late-stage, site-selective and strain-accelerated oxidation of a “deoxygenated” macrocyclic intermediate was successfully implemented, and in doing so provided a novel solution to the infamous biphenol cyclophane system of haouamine A.
Introduction
Strained cyclophanes are intriguing structural motifs with unusual physical properties, chemical reactivities, and methods for their preparation.1 Due to the strain imposed by the macrocyclic framework, the constituent aryl ring(s) within the cyclophane often experiences stress that leads to a distortion from planarity. Consequently, one of the most common tactics for the construction of strained cyclophanes exploits a conformational flexible “masked” aryl precursor bearing sp3 hybridized carbon center(s) for the macrocyclization event, followed by an aromatization-driven generation of the aryl ring(s) to render the targeted strained cyclophane system. In two instructive examples, Baran and co-workers disclosed contrasting approaches in their first syntheses of the indeno-tetrahydropyridine natural product, haouamine A (1, Scheme 1).2 In their first-generation synthesis,3a an intramolecular [4 + 2] cycloaddition followed by an enthalpic and entropic driven retro-[4 + 2]/aromatization event successfully delivered the strained cyclophane system of haouamine A for the first time. A second-generation synthesis followed shortly after where an intramolecular N-alkylation of a cyclohexenone precursor bearing sp3 hybridized carbon centers, followed by oxidative aromatization, rendered a more practical solution.3b In view of this state of affairs,4 we hypothesized a “late-stage” oxidation5 of a “deoxygenated” macrocyclic precursor (2) may provide an alternative solution to the biphenol cyclophane system of haouamine A (Scheme 2a). While this strategic maneuver benefits from greatly simplified synthetic precursors and broadens the selection of synthetic transformations for their preparation, site-selectivity of this unprecedented late-stage oxidation/oxygenation is expected to pose a serious challenge. Furthermore, we also envisaged the application of two rhodium-catalyzed processes ((a) diazo-insertion6 and (b) intramolecular aziridination;7Scheme 2b) starting from two readily accessible building blocks 4 and 5 to provide a novel and practical synthetic entry to the indeno-tetrahydropyridine core of haouamine A via amino-alcohol 7.4d,k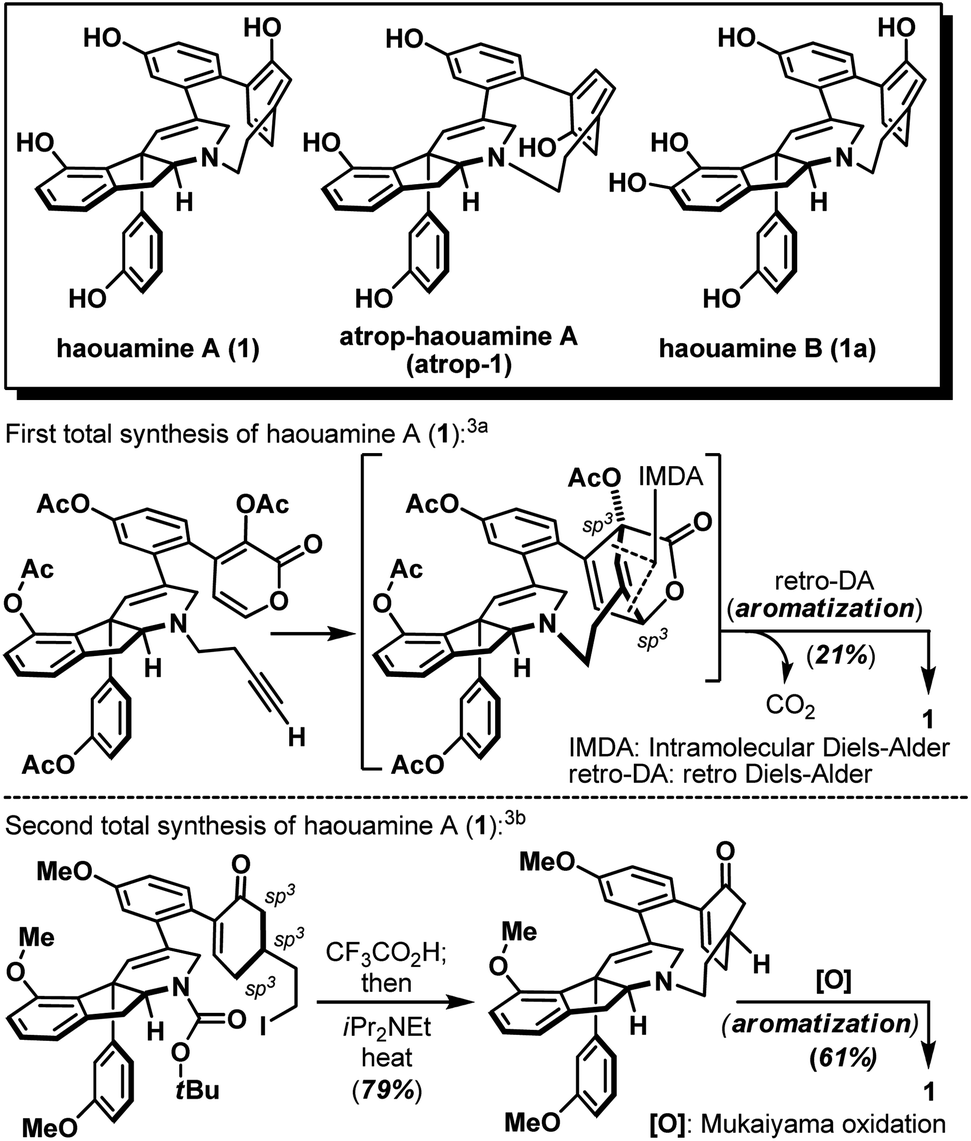 | ||
| Scheme 1 Structures of haouamine A (1), atrop-haouamine A (atrop-1), haouamine B (1a) and reported syntheses of the strained cyclophane. | ||
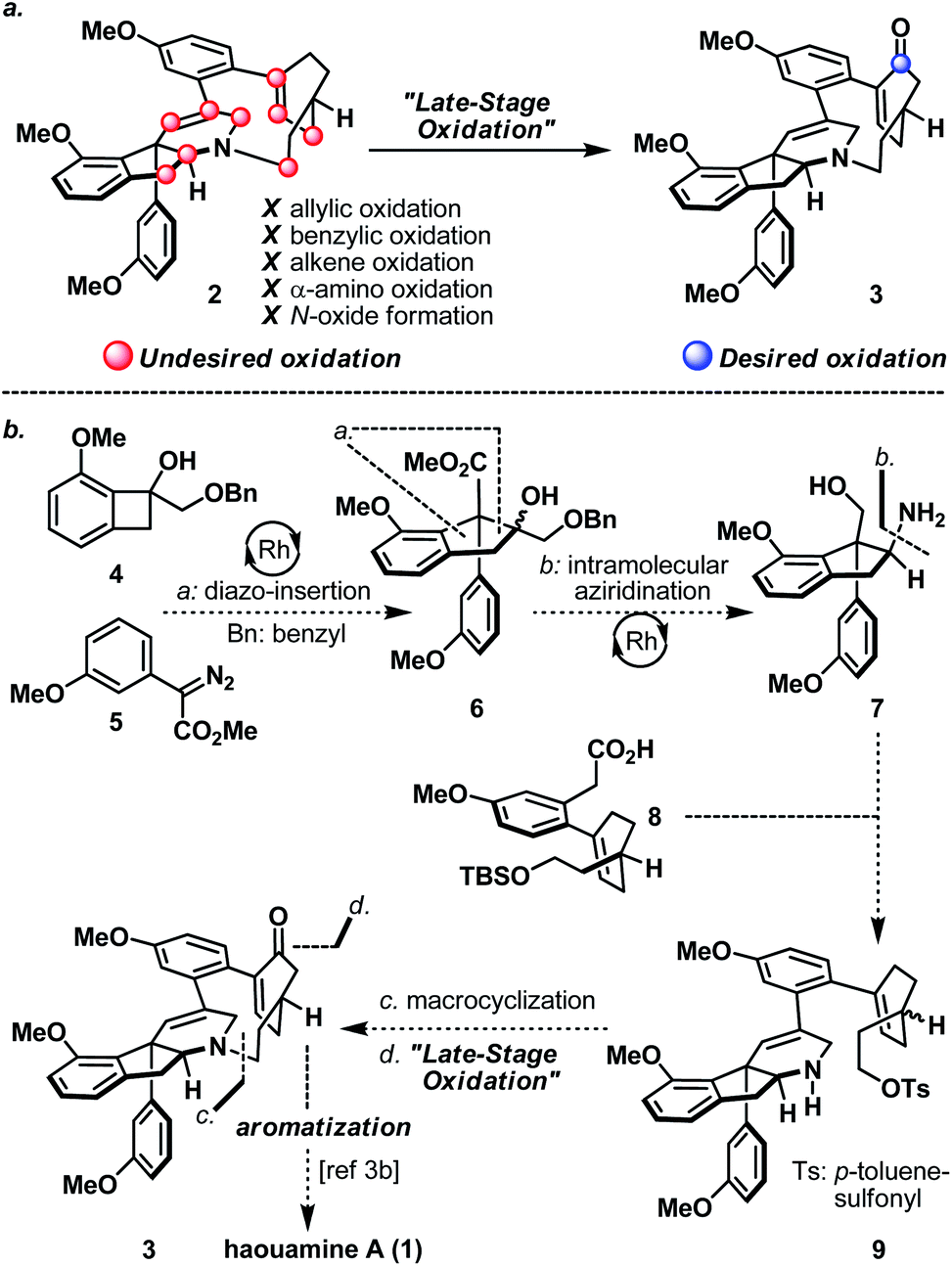 | ||
| Scheme 2 (a) Desired and undesired oxidation of “Deoxygenated” macrocycle 2; (b) proposed synthesis of haouamine A (1) in this work from building blocks 4, 5, and 8. | ||
Results and discussion
As shown in Scheme 3a, the synthesis of haouamine A (1) commenced with the preparation of amino-alcohol 7. Inspired by the protocol originally developed by Wang and co-workers,6 rhodium-catalyzed diazo-insertion reaction engaging benzocyclobutanol 4 (ref. 8) and diazoester 5 (ref. 9) proceeded smoothly to afford tertiary alcohol 6 in good yield. Notably, this reaction took place with significantly improved yield at room temperature instead of the elevated temperature (100 °C) initially reported by Wang, and was routinely performed on multi-gram scale with further reduced catalyst loading (2 mol% to 0.8 mol%). In preparation for the ensuing intramolecular aziridination (16 to 17), hydroxy methyl ester 6 was elaborated to alkenyl alcohol 15via oxidative cleavage of diol 12, a two-step deoxygenation of keto ester 13 and reduction of alkenyl methyl ester 14. On treatment with chlorosulfonyl isocyanate, primary alcohol 15 was converted to sulfamate 16 in readiness for the intramolecular aziridination. Analogous to the reaction conditions originally developed by the Du Bois laboratory,7 rhodium-catalyzed intramolecular aziridination of 16 took place at a slightly elevated temperature (40 °C) to furnish aziridine 17 in high yield as the sole product (e.g. nitrene CH-insertion product(s) was not observed). Sequential reductive transformations on 17 that involved rupture of its aziridine (Pd/C, H2) and cleavage of the sulfamate (AlH3·EtNMe2) yielded amino-alcohol 7 with spectroscopic data in full accordance with literature reports,4d thereby validated our developed reaction sequence. Furthermore, practicality of the developed sequence has been demonstrated in generating multi-gram quantities (>7 grams. For details, see ESI†) of amino-alcohol 7 for the ensuing synthetic investigations.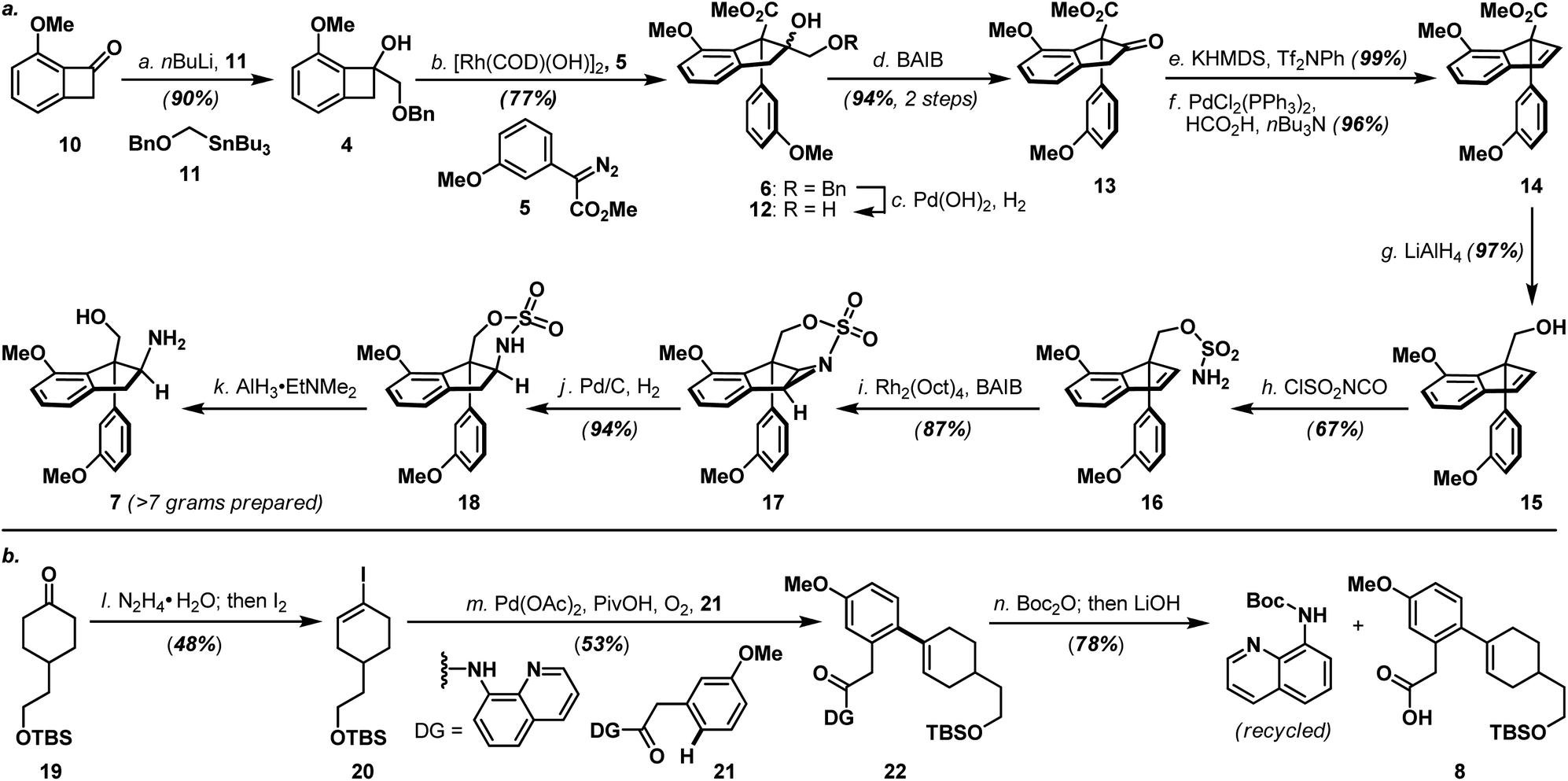 | ||
Scheme 3 (a) Preparation of amino-alcohol 7; (b) preparation of bicyclic carboxylic acid 8. Reagents and conditions: (a) 11 (1.05 equiv.), nBuLi (2.5 M in hexanes, 1.05 equiv.), THF, −78 °C, 50 min, 90%; (b) 5 (1.05 equiv.), [Rh(COD)(OH)]2 (0.008 equiv.), toluene, 0 to 25 °C, 3 h, 77%; (c) Pd(OH)2 (5 wt% on carbon, 18% wt/wt), H2 (1 atm, balloon), MeOH, 25 °C, 15 h; (d) BAIB (1.0 equiv.), CH2Cl2, 25 °C, 3.5 h, 94% over 2 steps; (e) KHMDS (0.7 M in toluene, 1.1 equiv.), Tf2NPh (1.3 equiv.), THF, −78 °C, 2 h, 99%; (f) PdCl2(PPh3)2 (0.05 equiv.), nBu3N (3.0 equiv.), HCO2H (2.0 equiv.), DMF, 60 °C, 1.5 h, 96%; (g) LiAlH4 (1.1 equiv.), Et2O, 0 to 25 °C, 1.5 h, 97%; (h) ClSO2NCO (2.5 equiv.), HCO2H (2.5 equiv.), pyridine (2.5 equiv.), CH3CN/DMA (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9), 25 °C, 7.5 h, 67%; (i) Rh2(Oct)4 (0.02 equiv.), MgO (2.3 equiv.), BAIB (1.2 equiv.), CH2Cl2, 25 to 40 °C, 4 h, 87%; (j) Pd/C (10 wt% on carbon, 45% wt/wt), H2 (1 atm, balloon), EtOAc, 25 °C, 3 h, 94%; (k) AlH3·EtNMe2 (0.5 M in toluene, 6.0 equiv.), toluene, 25 to 110 °C, 6 h; (l) N2H4·H2O (4.0 equiv.), MeOH, 25 °C, 14 h; then I2 (2.0 equiv.), Et3N (10.0 equiv.), Et2O, 0 to 25 °C, 25 min, 48% over 2 steps; (m) 20 (1.5 equiv.), 21 (1.0 equiv.), Pd(OAc)2 (0.1 equiv.), PivOH (0.2 equiv.), KHCO3 (2.0 equiv.), 1,2-dichloroethane, O2, 100 °C, 14 h, 53% (2 cycles); (n) Boc2O (3.0 equiv.), DMAP (0.5 equiv.), CH3CN, 60 °C, 16 h; then LiOH·H2O (8.5 equiv.), THF/H2O (3:1), 25 to 60 °C, 2 h, 78% over 2 steps. BAIB = (diacetoxyiodo)benzene; COD = cyclooctadienyl; DMA = N,N′-dimethylacetamide; DMAP = N,N′-dimethylaminopyridine; DMF = N,N′-dimethylformamide; EtOAc = ethyl acetate; KHMDS = potassium bis(trimethylsilyl)amide; Rh2(Oct)4 = rhodium(II) octanoate, dimer; Tf2NPh = N-phenyl-bis(trifluoromethanesulfonimide). :9), 25 °C, 7.5 h, 67%; (i) Rh2(Oct)4 (0.02 equiv.), MgO (2.3 equiv.), BAIB (1.2 equiv.), CH2Cl2, 25 to 40 °C, 4 h, 87%; (j) Pd/C (10 wt% on carbon, 45% wt/wt), H2 (1 atm, balloon), EtOAc, 25 °C, 3 h, 94%; (k) AlH3·EtNMe2 (0.5 M in toluene, 6.0 equiv.), toluene, 25 to 110 °C, 6 h; (l) N2H4·H2O (4.0 equiv.), MeOH, 25 °C, 14 h; then I2 (2.0 equiv.), Et3N (10.0 equiv.), Et2O, 0 to 25 °C, 25 min, 48% over 2 steps; (m) 20 (1.5 equiv.), 21 (1.0 equiv.), Pd(OAc)2 (0.1 equiv.), PivOH (0.2 equiv.), KHCO3 (2.0 equiv.), 1,2-dichloroethane, O2, 100 °C, 14 h, 53% (2 cycles); (n) Boc2O (3.0 equiv.), DMAP (0.5 equiv.), CH3CN, 60 °C, 16 h; then LiOH·H2O (8.5 equiv.), THF/H2O (3:1), 25 to 60 °C, 2 h, 78% over 2 steps. BAIB = (diacetoxyiodo)benzene; COD = cyclooctadienyl; DMA = N,N′-dimethylacetamide; DMAP = N,N′-dimethylaminopyridine; DMF = N,N′-dimethylformamide; EtOAc = ethyl acetate; KHMDS = potassium bis(trimethylsilyl)amide; Rh2(Oct)4 = rhodium(II) octanoate, dimer; Tf2NPh = N-phenyl-bis(trifluoromethanesulfonimide). | ||
The synthesis of bicyclic carboxylic acid 8 is outlined in Scheme 3b. Inspired by the recent advances in CH-functionalization of phenylacetic acid derivatives, Pd(OAc)2-catalyzed cross-coupling between quinolinamide 21 (ref. 10) and cyclohexanone 19 (ref. 11) derived vinyl iodide 20 under the aerobic ortho-alkenylation conditions described by Chen and co-workers12 smoothly delivered bicycle 22 as the only detectable product. Hydrolytic amide-bond cleavage through the Boc derivative of quinolinamide 22 completed the synthesis of carboxylic acid 8 together with recovered 8-aminoquinoline directing group.13
Annulation of the tetrahydropyridine domain of haouamine A onto amino-alcohol 7 was realized through an adaptation of the reaction sequence described by Weinreb4d and Wipf groups,4k through the intermediacy primary alcohol 23 and intramolecular aldol-condensation of aldehyde 24, to deliver lactam 25 uneventfully (Scheme 4).14 In preparation for the macrocyclization event and the completion of macrocycle 2/2a, TBS ether 25 was converted to its corresponding tosylate 27 followed by a Ru-catalyzed amide reduction15 to afford amine 9. Intramolecular N-alkylation of amino-tosylate 9 under high-dilution conditions3,4a–c (where the inclusion of NaI proved crucial) proceeded smoothly to deliver macrocycle 2/2a as a mixture of diastereoisomers (Scheme 4). Notably, diastereoisomeric amino-tosylates (9, d.r. 1:1) exhibited different rate of macrocyclization that resulted the formation of diastereoisomerically enriched macrocycle (2:2a ∼ 2.8:1) together with unreacted and diastereoisomerically enriched amino-iodide intermediate (which could be re-subjected to the macrocyclization condition to afford additional supply of macrocycle 2/2a) after 16 hours at 90 °C. On the other hand, inspired by the recently reported palladium-catalyzed intramolecular cross-coupling16 featured in the herquline syntheses,17 macrocyclic Suzuki reaction of boronic ester-aryl bromide 29 was also attempted but failed to deliver macrocycle 2/2a (Scheme 5a). Notwithstanding the conformational and mechanistic differences between intramolecular N-alkylation and Suzuki cross-coupling, these results appear to substantiate the importance of site selection for a successful macrocyclization event. This finding is particularly noteworthy and path-pointing for future synthetic investigations in this field since it demonstrated for the first time that by simply replacing a constituent aromatic ring of the haouamine biphenol cyclophane system with a sp3 hybridized “masked” aryl precursor may not guarantee the desired ring closure to take place.
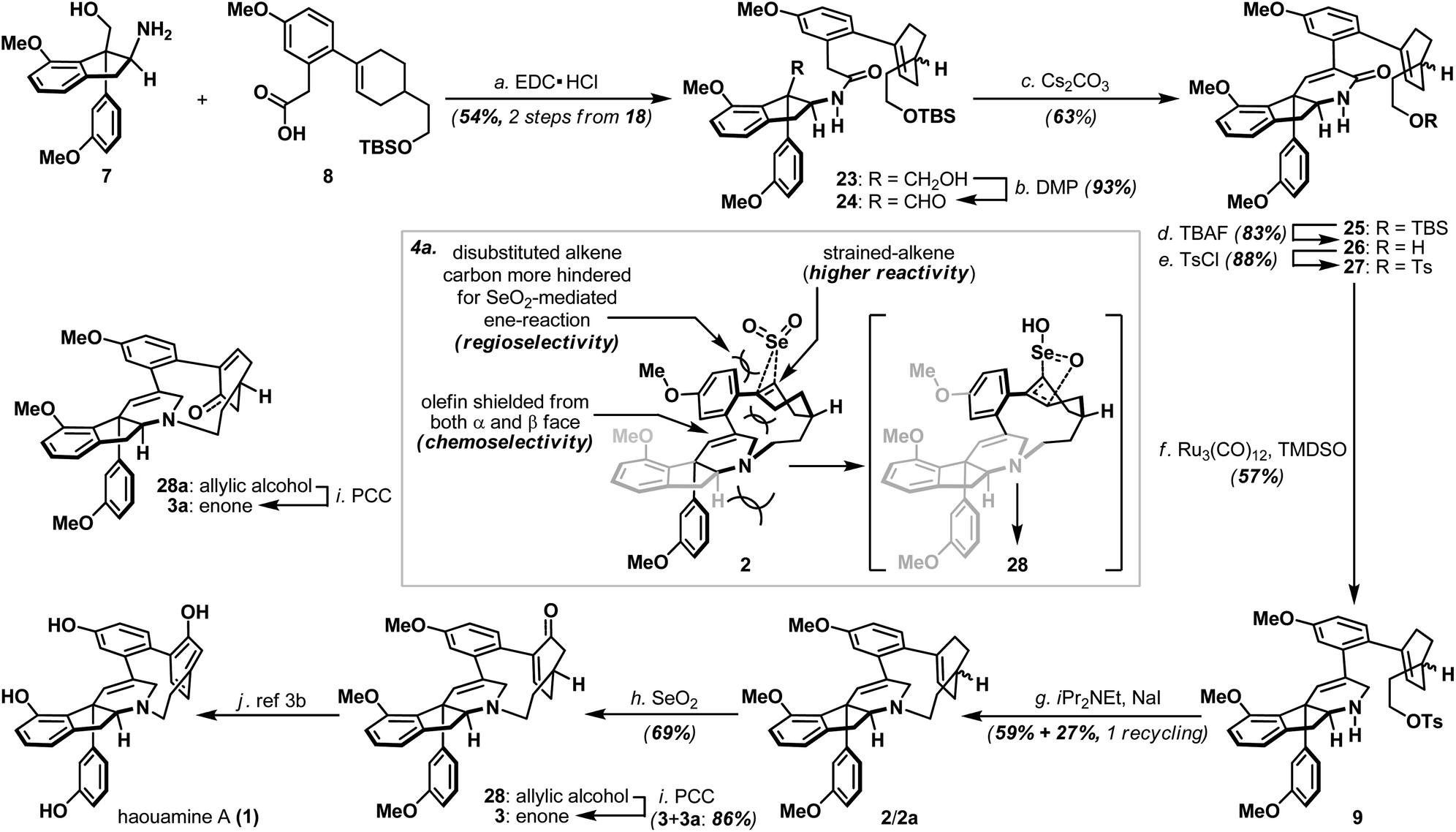 | ||
| Scheme 4 Late-stage oxidation enabled synthesis of haouamine A (1); (a) 3-dimensional rendering and hypothetical transition-state structure to rationalize the highly effective late-stage oxidation/oxygenation of macrocycle 2. Reagents and conditions: (a) EDC·HCl (1.1 equiv.), iPr2NEt (2.2 equiv.), 8 (1.05 equiv.), CH2Cl2, 25 °C, 14 h, 54% for 2 steps from 18; (b) DMP (1.4 equiv.), CH2Cl2, 0 to 25 °C, 4 h, 93%; (c) Cs2CO3 (4.0 equiv.), CH3CN/MeOH (10:1), 60 °C, 8 h, 63%; (d) TBAF (1.0 M in THF, 2.0 equiv.), THF, 0 to 25 °C, 14 h, 83%; (e) TsCl (2.0 equiv.), DMAP (0.2 equiv.), Et3N (5.0 equiv.), CH2Cl2, 25 °C, 4 h, 88%; (f) Ru3(CO)12 (0.1 equiv.), TMDSO (10.0 equiv.), toluene, 60 °C, 14 h, 57% (+27 10%); (g) NaI (10 equiv.), iPr2NEt (10 equiv.), CH3CN, 90 °C, 16 h + 16 h, 59% + 27% with one recycling; (h) SeO2 (1.8 equiv.), NaHCO3 (5.0 equiv.), 1,4-dioxane, 45 °C, 5 h, 69%; (i) PCC (2.0 equiv.), Celite®, CH2Cl2, 25 °C, 4 h, 3: 58%, 3a: 28%. DMP = Dess–Martin Periodinane; EDC = N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide; EtOAc = ethyl acetate; PCC = pyridinium chlorochromate; TBAF = tetra-n-butylammonium fluoride; TMDSO = 1,1,3,3-tetramethyldisiloxane. | ||
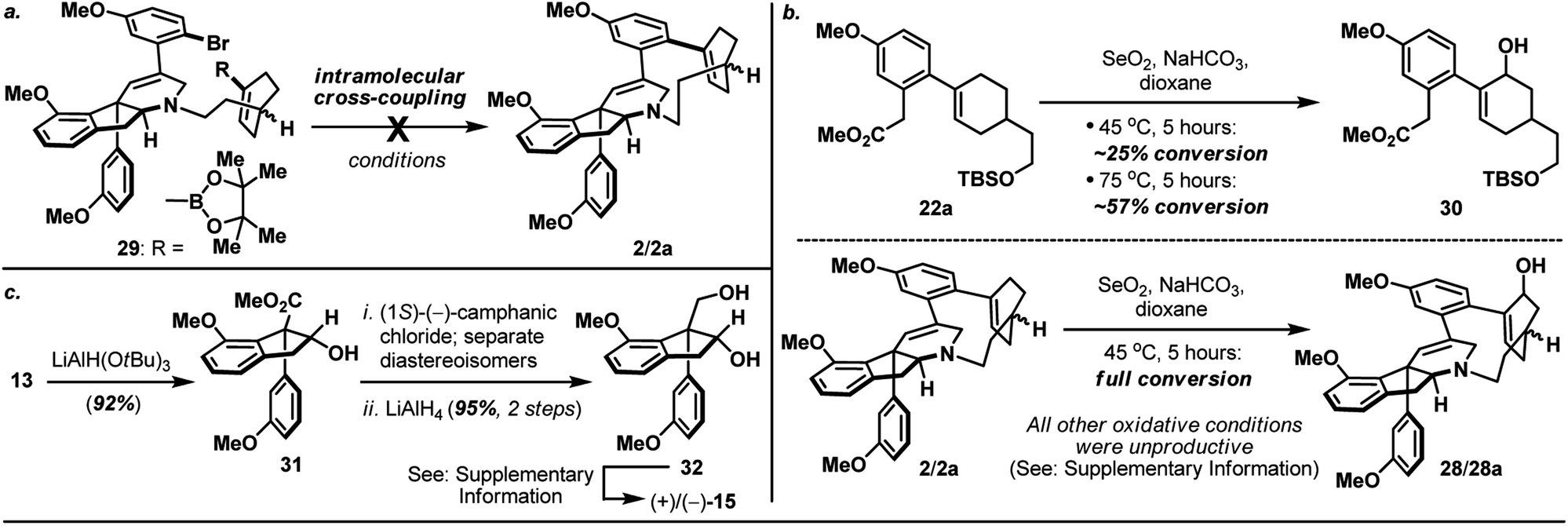 | ||
| Scheme 5 (a) Attempted formation of macrocycle 2/2avia intramolecular Suzuki cross-coupling of boronic ester-aryl bromide 29; (b) SeO2-mediated allylic oxidation of bicyclic substrate 22aversus macrocycle 2/2a; (c) Synthesis of optically active alkenyl alcohol 15. For details, see ESI.† | ||
With macrocycle 2/2a in hand, the highly anticipated site-selective oxidation was pursued in earnest (Scheme 4). Having conducted an exhaustive study of conventional oxidation protocols (osmium-catalyzed dihydroxylation, peracid-mediated epoxidation, hydroboration–oxidation, metal-catalyzed and SeO2-mediated allylic oxidation. For details, see ESI†), and recognizing the possibility to directly access the previously reported enone intermediate 3/3a,3b we opted the SeO2-mediated allylic oxidation as the focal point of our investigations on macrocycle 2/2a. After extensive experimentations, we discovered that while prolonged treatment with SeO2 at elevated temperature (100 °C) indeed generated analytically detectable amounts of enones 3 and 3a, this condition proved highly capricious and difficult to obtain chromatographically pure material. Alternatively, performing the reaction at 45 °C for 5 hours cleanly afforded the allylic alcohol intermediate (28 and 28a) that could be easily isolated, and subsequent oxidation with PCC smoothly delivered a readily separable mixture of enones 3 and 3a. It is worth-noting that allylic oxidation of model substrate 22a under the identical reaction condition only proceeded in ∼25% conversion (Scheme 5b), suggesting the enhanced reactivity of olefin 2/2a may be a consequence of its strained macrocyclic system.18 This mechanism-based selection of oxidation/oxygenation protocol proved crucial to achieve the overall selectivity for this challenging late-stage transformation (Scheme 4a).19 Furthermore, conversion of allylic alcohols 28/28a to enones 3/3a was ineffective under Dess–Martin periodinane, Swern, and MnO2 oxidation conditions. Enones 3 and 3a exhibited spectroscopic data in complete accordance to those reported in the literature, and their conversion to haouamine A and atrop-haouamine A, respectively, have been reported.3b Finally, optically active alkenyl alcohol 15 could be conveniently obtained through a resolution process to provide an asymmetric entry to haouamine A (Scheme 5c).
Conclusions
In conclusion, a new synthetic entry to the cyclophane alkaloid natural product haouamine A (1) has been realized. Most notably, a late-stage, site-selective, and strain-accelerated oxidation/oxygenation of macrocycle 2 was successfully implemented to render a novel solution to the biphenol cyclophane domain of haouamine A. In doing so, a simplified precursor has been identified for the first time to facilitate future chemical investigations of the haouamines by the synthetic community. The construction of the indeno-tetrahydropyridine core of haouamine A (1) developed herein also showcased two highly efficient and practical rhodium-catalyzed carbon–carbon and carbon–nitrogen bond forming processes, namely a diazo-ester (5) insertion to benzocyclobutanol 4 and an intramolecular aziridination of sulfamate 16, respectively. Collectively, the modular synthetic approach developed herein and ample supply of amino-alcohol 7 should enable a ready access to other members of the haouamine family and designed analogues, which is currently under investigation in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (No. 2013R1A1A2057837; and 2014R1A5A1011165, Center for New Directions in Organic Synthesis), and Novartis. Antonio Rizzo was supported by the BK21Plus Program, Ministry of Education. We thank Geun Seok Lee and Suyong Goh for preliminary synthetic studies toward the synthesis of amino-alcohol 7.Notes and references
- For reviews, see: (a) T. Gulder and P. S. Baran, Strained Cyclophane Natural Products: Macrocyclization at Its Limits, Nat. Prod. Rep., 2012, 29, 899–934 RSC; (b) T. Tsuji, Highly Strained Cyclophanes, in Mod. Cyclophane Chem., ed. R. Gleiter and H. Hopf, Wiley-VCH, Weinheim, 2004, pp. 81–104 Search PubMed; (c) T. Tsuji, Extremely Strained Paracyclophanes: Preparation, Structures, and Properties, Adv. Strained Interesting Org. Mol., 1999, 7, 103–152 CrossRef CAS; (d) F. Bickelhaupt and W. H. de Wolf, Unusual Reactivity of Highly Strained Cyclophanes, J. Phys. Org. Chem., 1998, 11, 362–376 CrossRef CAS; (e) F. Bickelhaupt and W. H. de Wolf, Small and Strained Cyclophanes, Adv. Strain Org. Chem., 1993, 3, 185–227 CAS; (f) D. J. Cram and J. M. Cram, Cyclophane Chemistry: Bent and Battered Benzene Rings, Acc. Chem. Res., 1971, 4, 204–213 CrossRef CAS.
- L. Garrido, E. Zubia, M. J. Ortega and J. Salva, Haouamines A and B: A New Class of Alkaloids from the Ascidian Aplidium haouarianum, J. Org. Chem., 2003, 68, 293–299 CrossRef CAS PubMed.
- (a) P. S. Baran and N. Z. Burns, Total Synthesis of (±)-Haouamine A, J. Am. Chem. Soc., 2006, 128, 3908–3909 CrossRef CAS PubMed; (b) N. Z. Burns, I. N. Krylova, R. N. Hannoush and P. S. Baran, Scalable Total Synthesis and Biological Evaluation of Haouamine A and Its Atropisomer, J. Am. Chem. Soc., 2009, 131, 9172–9173 CrossRef CAS PubMed.
- For total syntheses, see: (a) M. Matveenko, G. Liang, E. M. W. Lauterwasser, E. Zubia and D. Trauner, A Total Synthesis Prompts the Structure Revision of Haouamine B, J. Am. Chem. Soc., 2012, 134, 9291–9295 CrossRef CAS PubMed; (b) Y. Momoi, K.-i. Okuyama, H. Toya, K. Sugimoto, K. Okano and H. Tokuyama, Total Synthesis of (−)-Haouamine B Pentaacetate and Structural Revision of Haouamine B, Angew. Chem., Int. Ed., 2014, 53, 13215–13219 CrossRef CAS PubMed; (c) H. Tsukamoto, S. Nakamura, A. Tomida and T. Doi, Scalable Total Syntheses and Structure-Activity Relationships of Haouamine A, B, and their Derivatives as Stable Formate Salts, Chem.–Eur. J. DOI:10.1002/chem.202001756.For formal synthesis and synthetic studies, see: (d) J. H. Jeong and S. M. Weinreb, Formal Total Synthesis of the Cytotoxic Marine Ascidian Alkaloid Haouamine A, Org. Lett., 2006, 8, 2309–2312 CrossRef CAS PubMed; (e) A. Furstner and J. Ackerstaff, Formal Total Synthesis of (−)-Haouamine A, Chem. Commun., 2008, 2870–2872 RSC; (f) T. Taniguchi, H. Zaimoku and H. Ishibashi, Formal Total Synthesis of Haouamine A, J. Org. Chem., 2009, 74, 2624–2626 CrossRef CAS PubMed; (g) N. D. Smith, J. Hayashida and V. H. Rawal, Facile Synthesis of the Indeno-Tetrahydropyridine Core of Haouamine A, Org. Lett., 2005, 7, 4309–4312 CrossRef CAS PubMed; (h) P. Wipf and M. Furegati, Synthesis of the 3-Aza-[7]-paracyclophane Core of Haouamine A and B, Org. Lett., 2006, 8, 1901–1904 CrossRef CAS PubMed; (i) T. Tanaka, H. Inui, H. Kida, T. Kodama, T. Okamoto, A. Takeshima, Y. Tachi and Y. Morimoto, Diastereoselective Synthesis of the Indeno-tetrahydropyridine Core Bearing a Diaryl-substituted Stereogenic Quaternary Carbon Center of Haouamine B, Chem. Commun., 2011, 47, 2949–2951 RSC; (j) E. Fenster, C. Fehl and J. Aubé, Use of a Tandem Prins/Friedel–Crafts Reaction in the Construction of the Indeno-Tetrahydropyridine Core of the Haouamine Alkaloids: Formal Synthesis of (−)-Haouamine A, Org. Lett., 2011, 13, 2614–2617 CrossRef CAS PubMed; (k) L. Cao, C. Wang and P. Wipf, Grob-Type Fragmentation Releases Paracyclophane Ring Strain in a Late-Stage Precursor of Haouamine A, Org. Lett., 2019, 21, 1538–1541 CrossRef CAS PubMed; (l) T. J. Idzik, A. Borzyszkowska-Ledwig, L. Struk and J. G. Sosnicki, Magnesiate-Utilized/Benzyne-Mediated Approach to Indenopyridones from 2-Pyridones: An Attempt to Synthesize the Indenopyridine Core of Haouamine, Org. Lett., 2019, 21, 9667–9671 CrossRef CAS PubMed.
- M. C. White and J. Zhao, Aliphatic C–H Oxidations for Late-Stage Functionalization, J. Am. Chem. Soc., 2018, 140, 13988–14009 CrossRef CAS PubMed.
- Y. Xia, Z. Liu, Z. Liu, R. Ge, F. Ye, M. Hossain, Y. Zhang and J. Wang, Formal Carbene Insertion into C–C Bond: Rh(I)-Catalyzed Reaction of Benzocyclobutenols with Diazoesters, J. Am. Chem. Soc., 2014, 136, 3013–3015 CrossRef CAS PubMed.
- For intramolecular aziridination leading to an analogous ring system, see: P. M. Wehn and J. Du Bois, A Stereoselective Synthesis of the Bromopyrrole Natural Product (−)-Agelastatin A, Angew. Chem., Int. Ed., 2009, 48, 3802–3805 CrossRef CAS PubMed.
- For preparation of ketone 10, see: P. H. Chen, N. A. Savage and G. Dong, Concise Synthesis of Functionalized Benzocyclobutenones, Tetrahedron, 2014, 70, 4135–4146 CrossRef CAS PubMed . For preparation of (benzyloxymethyl)tri-n-butylstannane (11), see: V. Di Bussolo, A. Fiasella, M. R. Romano, L. Favero, M. Pineschi and P. Crotti, Stereoselective Synthesis of 2,3-Unsaturated-aza-O-glycosides via New Diastereoisomeric N-Cbz-imino Glycal-Derived Allyl Epoxides, Org. Lett., 2007, 9, 4479–4482 CrossRef PubMed.
- For preparation of diazo ester 5, see: W. Chan, S. Yeung, Z. Zhou, A. S. C. Chan and W. Yu, Ruthenium Catalyzed Directing Group-Free C2-Selective Carbenoid Functionalization of Indoles by α-Aryldiazoesters, Org. Lett., 2010, 12, 604–607 CrossRef CAS PubMed.
- For preparation of quinolinamide 21, see: Q. Zhao, T. Poisson, X. Pannecoucke, J.-P. Bouillon and T. Besset, Pd-Catalyzed Diastereoselective Trifluoromethylthiolation of Functionalized Acrylamides, Org. Lett., 2017, 19, 5106–5109 CrossRef CAS PubMed.
- For preparation of cyclohexanone 19, see: (a) S. Desrat, C. Remeur and F. Roussi, Development of an Efficient Route Toward Meiogynin A-Inspired Dual Inhibitors of Bcl-xL and Mcl-1 Anti-Apoptotic Proteins, Org. Biomol. Chem., 2015, 13, 5520–5531 RSC; (b) M. Ihara, T. Taniguchi, K. Makita, M. Takano, M. Ohnishi, N. Taniguchi, K. Fukumoto and C. Kabuto, Synthesis of Polycyclic Cyclobutane Derivatives by Tandem Intramolecular Michael-Aldol Reaction Under Two Complementary Conditions: TBDMSOTf-Et3N and TMSI-(TMS)2NH, J. Am. Chem. Soc., 1993, 115, 8107–8115 CrossRef CAS.
- Y. Zhao, G. He, W. A. Nack and G. Chen, Palladium-Catalyzed Alkenylation and Alkynylation of ortho-C(sp2)–H Bonds of Benzylamine Picolinamides, Org. Lett., 2012, 14, 2948–2951 CrossRef CAS PubMed.
- In comparison, Suzuki cross-coupling based synthesis of bicyclic carboxylic acid 8 involved more costly reagents and stringent experimental conditions, particularly in the triflation of ketone 19 and subsequent boronic ester formation. The simplicity of building block 20 and the implementation of aerobic CH-activation based cross-coupling both represented significant improvements compared to the reported total syntheses of haouamines (ref. 3 and 4a–c).
- Notably, intramolecular aldol condensation of aldehyde 24 under the conditions described by Weinreb and Wipf groups (K2CO3, MeOH, 60 °C, ref. 4d and k) afforded lactam 25 in significantly lower yield.
- H. Nagashima, Efficient Transition Metal-Catalyzed Reactions of Carboxylic Acid Derivatives with Hydrosilanes and Hydrosiloxanes, Afforded by Catalyst Design and the Proximity Effect of Two Si–H Groups, Synlett, 2015, 26, 866–890 CrossRef CAS.
- T. O. Ronson, R. J. K. Taylor and I. J. S. Fairlamb, Palladium Catalysed Macrocyclisations in the Total Synthesis of Natural Products, Tetrahedron, 2015, 71, 989–1009 CrossRef CAS.
- (a) J. B. Cox, A. Kimishima and J. L. Wood, Total Synthesis of Herquline B and C, J. Am. Chem. Soc., 2019, 141, 25–28 CrossRef CAS PubMed; (b) C. He, T. P. Stratton and P. S. Baran, Concise Total Synthesis of Herqulines B and C, J. Am. Chem. Soc., 2019, 141, 29–32 CrossRef CAS PubMed; (c) X. Zhu, C. C. McAtee and C. S. Schindler, Total Syntheses of Herqulines B and C, J. Am. Chem. Soc., 2019, 141, 3409–3413 CrossRef CAS PubMed.
- M. R. Wilson and R. E. Taylor, Strained Alkenes in Natural Product Synthesis, Angew. Chem., Int. Ed., 2013, 52, 4078–4087 CrossRef CAS PubMed.
- In addition to experimental evidences (Scheme 5a and b), preliminary computation studies suggested the alkene (which participated in SeO2-mediated allylic oxidation) in substrate 2 exhibited a notable “twist” in contrast with the alkene in substrate 22a. For details, see ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02299c |
| This journal is © The Royal Society of Chemistry 2020 |