
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A fluorescent ESIPT-based benzimidazole platform for the ratiometric two-photon imaging of ONOO−in vitro and ex vivo†
Maria L.
Odyniec
 a,
Sang-Jun
Park
b,
Jordan E.
Gardiner
a,
Emily C.
Webb
a,
Adam C.
Sedgwick
c,
Juyoung
Yoon
d,
Steven D.
Bull
*a,
Hwan Myung
Kim
*b and
Tony D.
James
*a
a,
Sang-Jun
Park
b,
Jordan E.
Gardiner
a,
Emily C.
Webb
a,
Adam C.
Sedgwick
c,
Juyoung
Yoon
d,
Steven D.
Bull
*a,
Hwan Myung
Kim
*b and
Tony D.
James
*a
aDepartment of Chemistry, University of Bath, BA2 7AY, UK. E-mail: T.D.James@bath.ac.uk; S.D.Bull@bath.ac.uk
bDepartment of Chemistry, Ajou University, 16499, Suwon, Korea. E-mail: kimhm@ajou.ac.kr
cDepartment of Chemistry, University of Texas at Austin, 105 E, 24th Street, A5300, Austin, USA
dDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 120-750, Korea
First published on 16th June 2020
Abstract
In this work, we have developed an ESIPT-based benzimidazole platform (MO-E1 and MO-E2) for the two-photon cell imaging of ONOO− and a potential ONOO−-activated theranostic scaffold (MO-E3). Each benzimidazole platform, MO-E1–3, were shown to rapidly detect ONOO− at micromolar concentrations (LoD = 0.28 μM, 6.53 μM and 0.81 μM respectively). The potential theranostic MO-E3 was shown to release the parent fluorophore and drug indomethacin in the presence of ONOO− but unfortunately did not perform well in vitro due to low solubility. Despite this, the parent scaffold MO-E2 demonstrated its effectiveness as a two-photon imaging tool for the ratiometric detection of endogenous ONOO− in RAW264.7 macrophages and rat hippocampus tissue. These results demonstrate the utility of this ESIPT benzimidazole-based platform for theranostic development and bioimaging applications.
Introduction
Fluorescent probes using bespoke chemical architectures are rationally designed to elicit a fluorescent response after reacting with a target molecule. Peroxynitrite (ONOO−) is a reactive nitrogen species (RNS) of interest. Low levels of ONOO− are constituently produced under basal conditions, which are tolerated through thiol-dependent antioxidant detoxification pathways.1 However, high levels of ONOO− are implicated in the pathological and physiological processes of multiple oxidative stress-related diseases, including neurodegenerative and inflammatory diseases.2 ONOO− is a strong oxidant with a short half-life which can react with various biomolecules such as DNA and proteins, causing major oxidative injury.3 Due to this high reactivity and short lifetime, accurate detection of ONOO− remains a challenging task.One successfully implemented approach for the detection of ONOO− uses boronate-based fluorescent probes.4,5 Previously, such probes have been used to detect hydrogen peroxide (H2O2). However, given the reaction rates of boronate esters with ONOO− are 106 times faster than H2O2, probes for the former have recently been developed.6 A variety of probes to detect ONOO−via different mechanisms are reported and are discussed elegantly in reviews by Shen et al. and Chan et al.7,8 Perhaps one of the most useful non-boronate probes for monitoring ONOO− is the reversible near-IR cyanine dye modulated by a PET processes through selenium oxidation; designed by the Han group. A reversible signal is particularly useful for continuous monitoring real time changes in the levels of a biological analyte.9
Excited state intramolecular proton transfer (ESIPT) probes are attractive systems for developing reaction-based fluorescence systems since they have excellent photophysical properties, including good photostability and large Stokes shifts.10 In addition ratiometric responses can also be observed. A dual-emission ratiometric response is desirable as it can provide direct information about the concentration of a target analyte using internal calibration, which is not possible with a single emission system.10 ESIPT probes often incorporate; 2-(2′-hydroxyphenyl)benzimidazole (HBI), 2-(2′-hydroxyphenyl)benzoxazole (HBO) or 2-(2′-hydroxyphenyl)benzothiazole (HBT) in the framework.
Four fluorescent probes (1–4) for ONOO− detection are illustrated in Fig. 1. Each of these probes use an HBT core. The first reported example, probe 1 designed by Kim et al., used a benzothiazolyl iminocoumarin scaffold and a boronate ester activating group to enable endogenous imaging of ONOO− in J774A.1 macrophages.11 Probes 2 and 3 designed by the James group incorporated the boronate ester onto a more simple benzothiazole scaffold for detection of ONOO−in vitro. Moreover, probe 3 was targeted to the endoplasmic reticulum (ER).5,12 Following a different mechanism of activation, probe 4 designed by Li et al. was used for visualisation of ONOO− in neurovascular ischemia progression in the brain of a live mouse.13 As such, benzothiazoles have been shown to be good fluorescent probes for ONOO− detection, with high selectivity, good cell permeability and potential to transport across the blood brain barrier.13
 | ||
| Fig. 1 Structures of previously reported ONOO− activatable probes. | ||
Due to the favourable properties of ESIPT based probes, we were interested in developing a potential ESIPT-based theranostic. Theranostics are the portmanteau of diagnostics and therapy. Typically, a theranostic will include an activating group, therapeutic group and a targeting group connected via a single linker.12,14
A common feature of theranostic linker building blocks are multiple primary alcohol functional groups, which need to be derivatised independently. This can lead to long tedious synthetic routes with multiple protection and deprotection steps. However, with our design strategy, we use the fluorophore as the ‘linker’ (Fig. 2).15,16 In this work, we have used HBI as the fluorophore ‘core’ and as the theranostic linker. Indomethacin was chosen as the therapeutic agent. Prominent HBI-based examples in the literature are probes which detect changes in pH. The Kim group developed a two-photon benzimidazole system to estimate acidic pH values in lysosomal compartments in vitro and in vivo.17 Liang and colleagues developed 3-benzimidazole-7-hydroxycoumarin which was able to detect two distinct pH ranges and monitor pH changes in the mitochondria of HeLa cells.18 From this work, we anticipated that our proposed benzimidazole system would provide a suitable platform for two-photon imaging of ONOO− and provide the potential for theranostic applications (Fig. 2).
 | ||
| Fig. 2 Design strategy towards ESIPT theranostics. | ||
Results and discussion
In brief, for the synthesis of MO-E1–3 (Fig. 3), commercially available HBI was subjected to duff reaction conditions (hexamethylenetetramine (HTMA) and trifluoroacetic acid (TFA)) to form intermediate 4 in a modest yield (35%); next, a mixture of 4,4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl bromide, and K2CO3 in dry DMF afforded MO-E1 in 35% yield. The aldehyde functionality of MO-E1 was reduced using NaBH4 to afford the corresponding alcohol MO-E2 in 62% yield. Finally, MO-E2 was conjugated to our chosen drug indomethacin using EDCI, in DMF to give MO-E3 in 40% yield (Scheme S1, ESI†).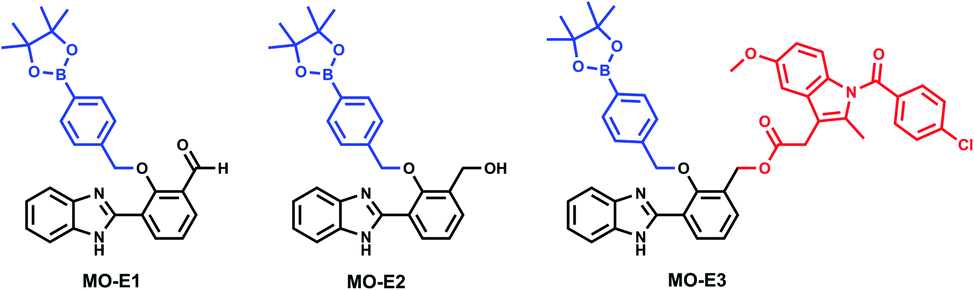 | ||
| Fig. 3 Structures of MO-E1–3. | ||
Fluorescence analysis
The synthetic route to MO-E3 provided a novel theranostic compound and two new fluorescent probes MO-E1 and MO-E2. With these molecules in hand, spectroscopic analysis was performed. Data was collected in PBS buffer pH = 8.2 (52% w/w H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH) at ambient temperature. Measurements were taken immediately after ONOO− addition. The UV-Vis spectra of MO-E1–3 (5 μM) were shown to have an absorption peak at 325 nm. The addition of ONOO− led to an increase in absorption at 325 nm (Fig S2–S4, ESI†).
:MeOH) at ambient temperature. Measurements were taken immediately after ONOO− addition. The UV-Vis spectra of MO-E1–3 (5 μM) were shown to have an absorption peak at 325 nm. The addition of ONOO− led to an increase in absorption at 325 nm (Fig S2–S4, ESI†).
The mechanism of activation requires ONOO− induced oxidation of an aryl boronate and subsequent elimination of the therapeutic. The boron atom of the boronate ester is sp2 hybridised and Lewis acidic. ONOO− attacks the boron atom resulting in the formation of a peroxyborate intermediate. Subsequent aryl migration to the oxygen atom gives the boronate intermediate, which in the presence of water, undergoes quantitative hydrolysis to the corresponding phenol. Finally, a spontaneous 1,4-elimination results in release of the therapeutic leaving a reactive quinone methide, which on reaction with water forms the desired fluorescent adduct. The full mechanism can be found in the ESI (Fig S5, ESI†). Mass spectrometric analysis confirmed the formation of the expected fluorescent molecule (Fig S6–S9, ESI†).
Following confirmation of the expected products, fluorescence studies were performed. Each probe (5 μM) was incubated with varying concentrations of ONOO− and the fluorescence response was monitored (Fig. 4a–c). Initially, MO-E3 was shown to be non-fluorescent. As the concentration of ONOO− increased, the fluorescence response increased proportionally up to a plateau of 20 μM. The maximum fluorescence response was observed at λmax = 450 nm (Fig. 4a). The limit of detection for ONOO− is 0.28 μM (Fig. S13 and S14, ESI†). To ensure indomethacin was released from the system, LC-MS studies were performed using H2O2 as the reactive species. The data revealed the release of indomethacin, thus confirming MO-E3 as a potential theranostic platform (Fig. S10–S12, ESI†). The cleavage of the ester was as expected from previous research in the James group.19
 | ||
| Fig. 4 (a–c) Fluorescence spectra of MO-E1–3 (5 μM) in the presence of ONOO− (a) MO-E3: 0–30 μM, (b) MO-E2: 1–7 μM and (c) MO-E1: 1–5 μM. The dashed line represents sensor only and measurements were taken instantly after ONOO− addition. All fluorescence measurements were measured in PBS buffer pH = 8.2 (52% w/w H2O:MeOH) at ambient temperature where λex = 325 (bandwidth: 16) nm on a BMG Labtech CLARIOstar® plate reader. (d) ESIPT excited state diagram of HBI. | ||
Unlike MO-E3, MO-E1–2 elicited ratiometric responses in the presence of ONOO− (Fig. 3b and c). Initially, both probes displayed a fluorescence emission intensity at 355 nm. This emission can be attributed to an ESIPT ‘enol’ form as the ESIPT process is blocked by the benzyl boronate ester. As the concentration of ONOO− increased, (0–7 μM), a decrease in fluorescence emission at 355 nm with a concomitant increase at 450 nm was observed, characteristic of the ESIPT-based emission of HBI (Fig. 3e). The limit of detection for MO-E1–2 were calculated to be 6.53 μM and 0.81 μM respectively (Fig S15–S18†). To date, the estimated steady state concentration of ONOO− is in the nanomolar range.20–22 There are limited fluorescence methods for direct quantitation of ONOO−. Recently Wang and colleagues used a ratiometric BODIPY probe which measured average basal ONOO− levels as 550–700 nM.23 Hence, MO-E3 and MO-E2 are well placed to monitor endogenous basal changes in ONOO− as well as oxidative stress events where the concentration of ONOO− is likely to increase significantly.
To test the probes ability to be activated by ONOO− over other reactive oxygen species (ROS), selectivity studies were performed. MO-E1–3 were tested against ROS (100 μM), ˙OH, O2˙−, 1O2, ClO−, ROO˙ and H2O2. As expected, the most significant fluorescence response was shown towards ONOO−. A 98-fold, 20-fold and 10-fold fluorescent enhancement was observed for MO-E3, MO-E2 and MO-E1 respectively at 450 nm (Fig. S19–S21†).
Unfortunately, MO-E1–3 were unsuitable for traditional cell imaging and confocal imaging experiments due to low excitation wavelengths (<360 nm). Therefore, to overcome this limitation each probe was evaluated towards two-photon microscopy (TPM). Unlike traditional fluorescence microscopy in which the excitation wavelength is shorter than the emission wavelength, in TPM the two photons excite at a longer wavelength than the resulting emitted light. This TPM strategy reduces short-wavelength induced phototoxicity and increases light penetration through living tissues with minimal light scattering.24 Before conducting TPM cell imaging experiments, two-photon action cross sections (TPA) of each ESIPT probe was measured (Fig. 5). TPA spectra for each probe before and after the addition of ONOO− (50 μM) in PBS buffer (pH = 8.2, 52% w/w H2O:MeOH) were measured over a 700 nm–880 nm wavelength range. MO-E3 and MO-E2 reacted with ONOO− and an increase in TPA was observed. All probes exhibited the highest TPA at 740 nm. However, MO-E1 was unsuccessful for TPM imaging as only a minimal change was observed before and after the addition of ONOO−. The TPA values of each probe are shown in Table S1, ESI.†
 | ||
| Fig. 5 Two-photon action spectra of MO-E probes (5 μM) without and with ONOO− (50 μM) in PBS buffer pH = 8.2 (52% w/w H2O:MeOH) at 25 °C. | ||
TPM imaging of MO-E3 was performed using RAW264.7 macrophages. RAW264.7 macrophages were incubated with MO-E3 (5 μM) for 30 min and with a 740 nm excitation wavelength, MO-E3 was successfully imaged (Fig S22†). However, intense spots on the outside of the cells were observed suggesting poor cell permeability/solubility for MO-E3. In addition, fluorescence intensities of intracellular MO-E3 were almost unchanged, despite pre-treatment with 3-morpholinosydnonimine (SIN-1, 50 μM, 30 min), a ONOO− donor, and exogenous ONOO− (50 μM, 30 min). SIN-1 is the most widely used ONOO− generator under physiological conditions. The sydnonimine ring of SIN-1 hydrolyses under aerobic aqueous conditions and releases nitric oxide and superoxide radicals, these two molecules combine to generate the ONOO−.25 Overall, we believe the limited solubility of MO-E3 has resulted in its unsuccessful in vitro ONOO− imaging. Nevertheless, MO-E3 represents a proof of principle system which can allow for development and exploration into novel ratiometric ESIPT-based theranostics.
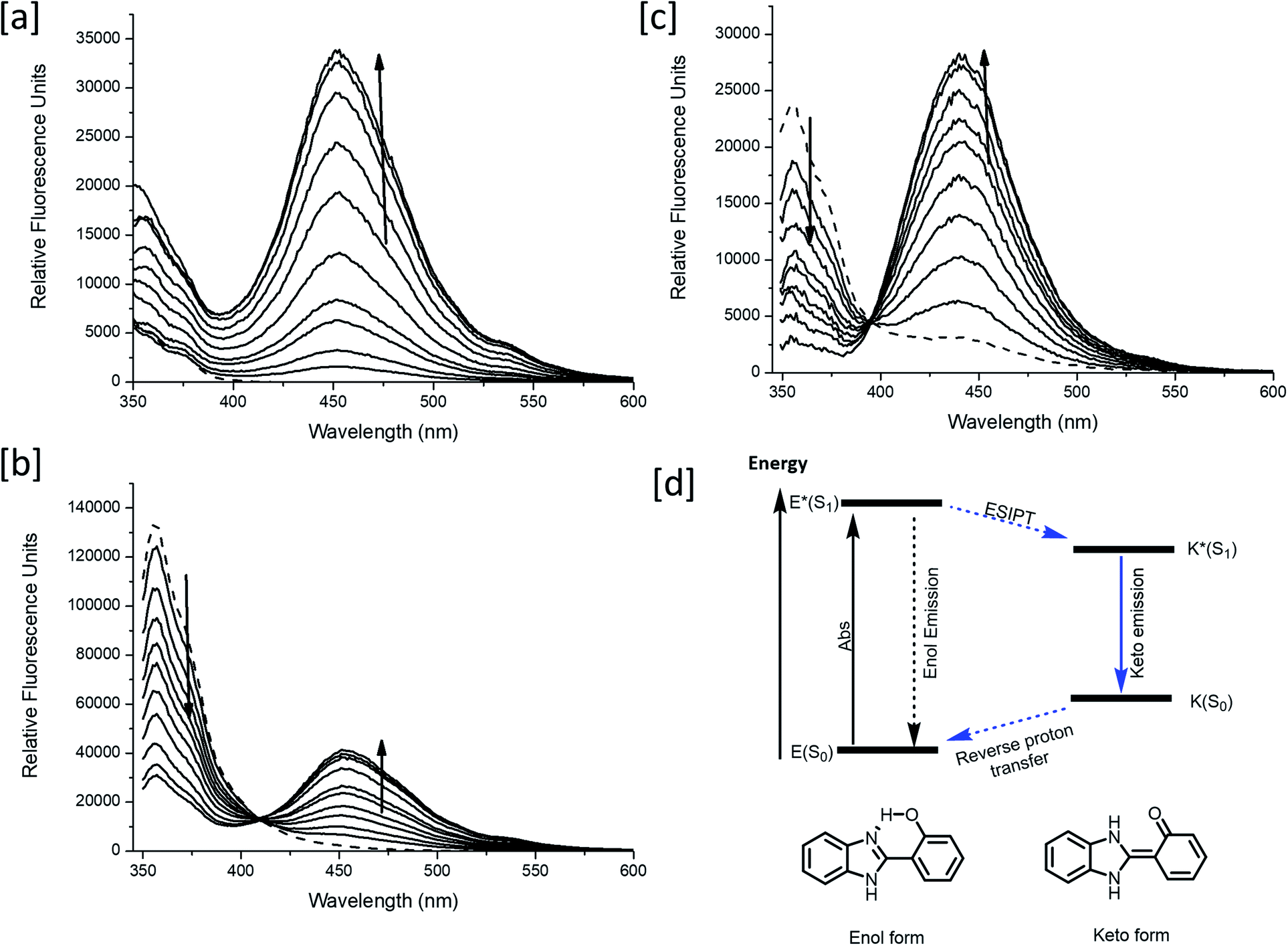
Meanwhile, blue (380–430 nm, Fblue) and green (470–520 nm, Fgreen) regions were chosen as the detection windows for the ratiometric images (Fgreen/Fblue) of MO-E2. Unlike MO-E3, MO-E2 (5 μM, 30 min) reacted with ONOO− and displayed a large fluorescence change (Fig. 6). When pre-treated with exogenous ONOO− (50 μM, 30 min) and SIN-1 (50 μM, 30 min), the average ratio values were significantly enhanced from 2.0 to 5.0 and 3.5, respectively (Fig. 6a–c). Additionally, cells were pre-treated with lipopolysaccharide (LPS, 1 μg mL−1, 4 h) and interferon gamma (INF-γ, 50 ng mL−1, 1 h), which are known to produce nitric oxide and superoxide to generate ONOO− through induced iNOS expression and activated NADPH oxidase,26 and the average ratio value increased to 2.6 (Fig. 6e). Ebselen, an organoselenium compound, is a known ONOO− scavenger that rapidly catalyses the reduction of ONOO−.27 When the stimulated macrophages were treated with ebselen (50 μM, 30 min), the average ratio values were unchanged and were at a similar level to those of the control (Fig. 6d and f). These results successfully demonstrate that MO-E2 can be used to visualise ONOO− in living cells. Furthermore, a CCK-8 assay demonstrated that MO-E2 exhibited almost no cytotoxicity. When RAW264.7 cells were treated with MO-E2 (50 μM) 80% of the cells survived (Fig. 7). The IC50 of MO-E2 in RAW 264.7 cells for 24, 48, and 72 h were 164, 125 and 75 μM (Fig. S23†) respectively, confirming that MO-E2 has negligible cytotoxicity under the imaging conditions. In addition, MO-E2 has high photostability (Fig. 8). This was shown by incubating RAW264.7 cells with MO-E2 followed by irradiation. The fluorescence intensities were obtained from 1800 signals with 2 s intervals for 1 h. Remarkably, the fluorescence emission intensity remained constant at 740 nm excitation wavelength over 1 h.
 | ||
| Fig. 6 TPM ratiometric images of RAW264.7 macrophages labelled with MO-E2 (5 μM) for 30 min. (a) Control image. (b–f) Cells were pre-treated with (b) exogenous ONOO− (50 μM, 30 min), (c) SIN-1 (50 μM, 30 min), (d) SIN-1 and ebselen (50 μM, 30 min), (e) LPS (1 μg mL−1, 4 h) and IFN-γ (50 ng mL−1, 1 h), (f) LPS, IFN-γ, and ebselen (g) average ratio values in the corresponding TPM ratiometric images. Excitation wavelength was 740 nm and emission windows of each regions were 380–430 nm (Fblue) and 470–520 nm (Fgreen). Scale bars = 20 μm. Asterisks stand for the statistical significance (p < 0.001) and n is number of counted cells. | ||
 | ||
| Fig. 7 Cytotoxicity assays of MO-E2 labelled RAW264.7 macrophages using CCK-8. Cells were incubated with 0–50 μM of MO-E2 for 2 h. | ||
 | ||
| Fig. 8 (a) TPM fluoresce images of RAW264.7 macrophages labelled with MO-E2. (b) The fluorescence intensities of areas A–C as a function of time from (a). The fluorescence intensity was obtained for 60 min at 2 s intervals. Excitation wavelength and detection windows were 740 nm and 380–600 nm, respectively. | ||
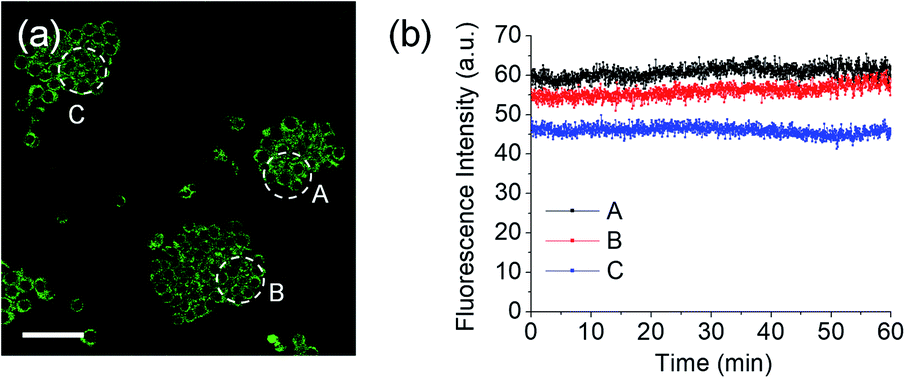
For the detection of ONOO− in tissue, TPM is a powerful tool because of its lower excitation energy (>700 nm), deep tissue penetration depth, low photo damage and longer observation times. Due to the excellent photophysical properties of TPM and compatibility with MO-E2, we turned our attention towards the ex vivo imaging of ONOO− using rat hippocampal tissue slices (Fig. 9). The average ratio values of tissues that had been stained with MO-E2 (50 μM) for 1.5 h was 2.0, which was comparable to the macrophage control (Fig. 9a). When the hippocampal slice was pre-treated with SIN-1 (200 μM) for 30 min, the average ratio value was dramatically increased to 4.8 (Fig. 9b). When endogenous ROS was generated by treatment with PMA (20 μM, 30 min), the average ratio value was significantly increased to 3.6. While the addition ebselen suppressed the value similar-to that observed for the control (Fig. 9c and d). These results demonstrate the potential of MO-E2 to be used as a two-photon ratiometric tool for the imaging of endogenous ONOO− in living systems.
 | ||
| Fig. 9 TPM ratiometric images of rat hippocampal slices stained with MO-E2 (50 μM) for 1.5 h. (a) Control image. (b–d) Tissues were pre-treated with (b) SIN-1 (200 μM, 30 min), (c) PMA (20 μM, 30 min) (d) PMA and ebselen (200 μM, 30 min), (e) average ratio values in the corresponding TPM ratiometric images. Excitation wavelength was 740 nm and emission windows of each regions were 380–430 nm (Fblue) and 470–520 nm (Fgreen). Scale bars = 35 μm. Asterisks stand for the statistical significance (p < 0.001) and n is number of ROI from three samples for each imaging condition. | ||
Conclusions
With this work we demonstrate the utility of benzimidazoles for two-photon fluorescence imaging and as a reactive linker scaffold for the design of theranostic systems. Our approach used a simple synthetic route to prepare two-photon activatable probes, MO-E1–3. The probes were evaluated using two-photon microscopy. MO-E1 did not produce large fluorescence changes on the addition of ONOO− and as such was not evaluated further. Unfortunately, despite MO-E3 illustrating the successful release of the fluorophore and therapeutic agent (mass spectrometry and LC-MS analysis), the system did not perform well in vitro. We believe this was due to the highly hydrophobic nature of indomethacin facilitating aggregation in the cell medium before the probe could enter cells. However, MO-E2 excelled in visualising ONOO− endogenously in RAW 264.7 macrophages and rat hippocampus tissue. The success of MO-E2 in imaging ONOO− using two-photon excitation indicates that a benzimidazole ‘linker’ is suitable for the construction of novel theranostic molecules incorporating more suitable drug candidates. Towards that end, we are currently exploring the development of such improved theranostic systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MLO, JEG, SDB and TDJ would like to thank the University of Bath for support. MLO and JEG thank the EPSRC for studentships. TDJ wishes to thank the Royal Society for a Wolfson Research Merit Award. H. M. K. acknowledges a grant from the National Leading Research Lab Program of the National Research Foundation of Korea (NRF), funded by the Korean government (2019R1A2B5B03100278). JY acknowledges a grant from the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (No. 2012R1A3A2048814). NMR Characterisation facilities were provided through the Chemical Characterisation and Analysis Facility (CCAF) at the University of Bath (https://www.bath.ac.uk/ccaf). All data supporting this study are provided as ESI† accompanying this paper.Notes and references
- R. E. Huie and S. Padmaja, Free Radical Res. Commun., 1993, 18, 195–199 CrossRef CAS PubMed.
- P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS PubMed.
- R. P. Patel, J. McAndrew, H. Sellak, C. R. White, H. Jo, B. A. Freeman and V. M. Darley-Usmara, Biochim. Biophys. Acta, 1999, 1411, 385–400 CrossRef CAS.
- A. C. Sedgwick, J. E. Gardiner, G. Kim, M. Yevglevskis, M. D. Lloyd, A. T. A. Jenkins, S. D. Bull, J. Yoon and T. D. James, Chem. Commun., 2018, 54, 4786–4789 RSC.
- A. C. Sedgwick, X. L. Sun, G. Kim, J. Yoon, S. D. Bull and T. D. James, Chem. Commun., 2016, 52, 12350–12352 RSC.
- A. Z. Sikora, J. Zielonka, M. Lopez, J. Joseph and B. Kalyanaraman, Free Radical Biol. Med., 2009, 47, 1401–1407 CrossRef CAS PubMed.
- X. Chen, H. Chen, R. Deng and J. Shen, Biomed. J., 2014, 37, 120–126 CrossRef PubMed.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed.
- F. Yu, P. Li, G. Li, G. Zhao, T. Chu and K. Ha, J. Am. Chem. Soc., 2011, 133(29), 11030–1103310 CrossRef CAS PubMed.
- A. C. Sedgwick, L. L. Wu, H. H. Han, S. D. Bull, X. P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC.
- J. Kim, J. Park, H. Lee, Y. Choi and Y. Kim, Chem. Commun., 2014, 56, 9353–9356 RSC.
- L. L. Wu, Y. Wang, M. Weber, L. Y. Liu, A. C. Sedgwick, S. D. Bull, C. S. Huang and T. D. James, Chem. Commun., 2018, 54, 9953–9956 RSC.
- X. Li, R. R. Tao, L. J. Hong, J. Cheng, Q. Jiang, Y. M. Lu, M. H. Liao, W. F. Ye, N. N. Lu, F. Han, Y. Z. Hu and Y. H. Hu, J. Am. Chem. Soc., 2015, 137, 12296–12303 CrossRef CAS PubMed.
- R. Weinstain, E. Segal, R. Satchi-Fainaro and D. Shabat, Chem. Commun., 2010, 46, 553–555 RSC.
- (a) M. H. Lee, J. L. Sessler and J. S. Kim, Acc. Chem. Res., 2015, 48, 2935–2946 CrossRef CAS PubMed; (b) R. Weinstain, E. Segal, R. Satchi-Fainaro and D. Shabat, Chem. Commun., 2010, 46, 553–555 RSC.
- M. L. Odyniec, H. H. Han, J. E. Gardiner, A. C. Sedgwick, X.-P. He, S. D. Bull and T. D. James, Front. Chem., 2019, 7, 775–778 CrossRef CAS PubMed.
- H. J. Kim, C. H. Heo and H. M. Kim, J. Am. Chem. Soc., 2013, 135, 17969–17977 CrossRef CAS PubMed.
- X. Q. Jiang, Z. J. Liu, Y. Z. Yang, H. Li, X. Y. Qi, W. X. Ren, M. M. Deng, M. H. Lu, J. M. Wu and S. C. Liang, Spectrochim. Acta, Part A, 2020, 22 Search PubMed.
- M. L. Odyniec, A. C. Sedgwick, A. H. Swan, M. Weber, T. M. S. Tang, J. E. Gardiner, M. Zhang, Y. B. Jiang, G. Kociok-Kohn, R. B. P Elmes, S. D. Bull, X. P. He and T. D. James, Chem. Commun., 2018, 54(61), 8466–8469 RSC.
- G. Ferrer-Sueta and R. Radi, ACS Chem. Biol., 2009, 4(3), 161–177 CrossRef CAS PubMed.
- N. Nalwaya and W. M. Dean, Chem. Res. Toxicol., 2005, 18(3), 486–493 Search PubMed.
- L. VirLág, E. Szabób, P. Gergelya and C. Szabóc, Toxicol. Lett., 2003, 11(140–141), 113–124 CrossRef.
- Z. Li, S.-H. Yan, C. Chen, Z.-R. Geng, J.-Y. Chang, C.-X. Chen, B.-H. Huang and Z.-L. Wang, Biosens. Bioelectron., 2017, 90, 75–82 CrossRef CAS PubMed.
- R. K. P. Benninger and D. W. Piston, Curr. Protoc. Cell Biol., 2013, 59, 4.11.1–4.11.24 CrossRef PubMed.
- S. F. Peteu, R. Boukherroub and S. Szunerits, Biosens. Bioelectron., 2014, 58, 359–373 CrossRef CAS PubMed.
- G. K. Azad and R. S. Tomar, Mol. Biol. Rep., 2014, 41, 4865–4879 CrossRef CAS PubMed.
- B. K. Yoo, J. W. Choi, C. Y. Shin, S. J. Jeon, S. J. Park, J. H. Cheong, S. Y. Han, J. R. Ryu, M. R. Song and K. H. Ko, Neurochem. Int., 2008, 52, 1188–1197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02347g |
| This journal is © The Royal Society of Chemistry 2020 |