
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRu-catalyzed isomerization of ω-alkenylboronates towards stereoselective synthesis of vinylboronates with subsequent in situ functionalization†
Guo-Ming
Ho
 ,
Lucas
Segura
and
Ilan
Marek
*
,
Lucas
Segura
and
Ilan
Marek
*
Schulich Faculty of Chemistry, Technion – Israel Institute of Technology, Technion City 3200009, Haifa, Israel. E-mail: chilanm@technion.ac.il
First published on 26th May 2020
Abstract
The stereoselective preparation of synthetically versatile vinylboronates from ω-alkenylboronates is achieved through a ruthenium-catalyzed isomerization reaction. A variety of di- and trisubstituted vinylboronates were conveniently produced and could be used as a new starting point for subsequent in situ remote functionalization through either a sequential Ru/Pd or Ru/Cu double catalytic system.
Introduction
The distant and selective functionalization of unreactive C–H and C–C bonds stands as one important challenge in organic chemistry.1 In recent years, the remote functionalization of alkenes by metal-walk along a hydrocarbon chain has attracted increased attention as it allows the functionalization of a distant and generally unreactive position.2 This concept paved the way to new approaches to efficiently build molecular complexity from easily accessible starting materials. From the pioneering metal-walk using stoichiometric zirconium complexes3 to catalytic systems employing palladium,4 nickel,5 ruthenium,6 rhodium,7 iridium8 or cobalt complexes,9 many attractive protocols have been reported in the last decade. These chain-walking processes are usually leading either to sp3 post-functionalization reactions2 or to the generation of a carbonyl group10 (Scheme 1a, path a). However, the preparation of a distant functionalized alkenyl organometallic species from an original sp3 carbon center remains less explored (Scheme 1a, path b).2g In this regard, we11 and Mazet12 have independently reported an efficient Pd–Ni and Ru–Ni double catalytic system transforming readily available ω-alkenyl ethers into a large variety of styrenyl products (Scheme 1b). Despite these significant advances, several challenges remain to be addressed such as the moderate level of stereocontrol over the geometry of the formed enol ether as well as the low E/Z-selectivity of the final cross-coupled products.13 Apart from the control of stereochemistry, the process is incompatible with sensitive functionalities and the final coupling products generally lack the diversity of functional groups that one might expect due to the nature of the Grignard reagent required for the final cross-coupling reactions.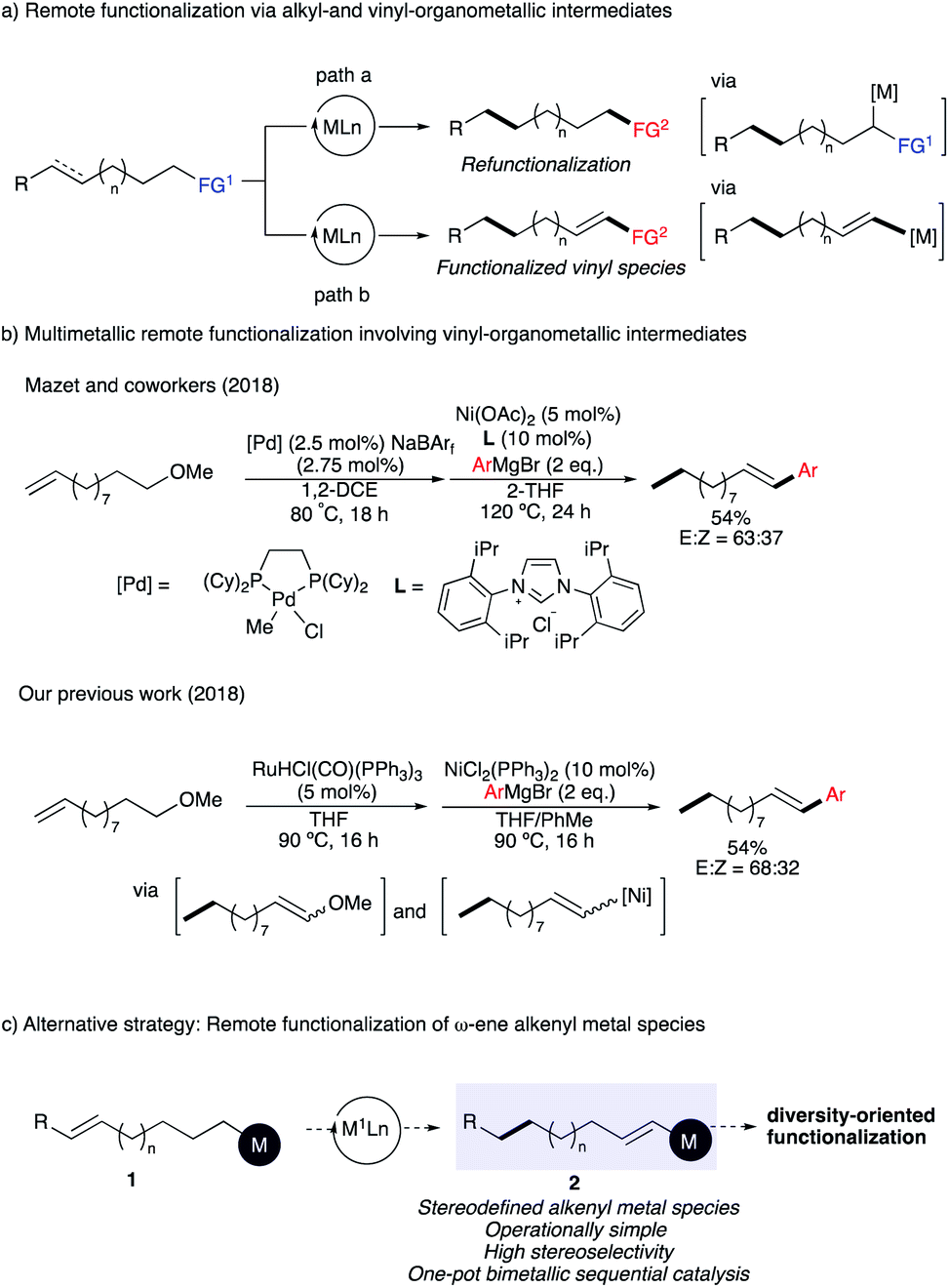 | ||
| Scheme 1 Strategies for remote functionalization of ω-alkenes. | ||
To address these shortcomings, we hypothesized that a straightforward alternative should involve a metal-catalyzed remote functionalization of ω-alkenyl organometallic derivative 1 into a synthetically versatile alkenyl organometallic species 2 with excellent stereocontrol of the olefin geometry (Scheme 1c). Alkenyl organometallic reagents are important and versatile intermediates in organic chemistry that were abundantly used for cross-coupling reactions. Among all possible organometallic candidates for this transformation, we concentrate our initial study on the formation of alkenyl organoboronates 2 [M = B(OR)2]14 that have been recognized as the most widely used representatives due to their diverse reactivity profile, excellent functional-group tolerance, stability to air, and non-toxic nature (Scheme 1c), unique characteristics among all other organometallic reagents.15
However, it was also clear that this transformation would be challenging to perform as the Ru,16 Ir,17 Pd,18 or Ni-catalyzed19 isomerization of 1-alkenylboronate provide the corresponding 2-alkenylboronate(allylboronate) intermediate (the opposite of what we are planning to achieve), which react with a carbonyl group in a stereospecific way, delivering homoallylic alcohol products with excellent stereoselectivity.20 In addition, upon proper choice of the coupling partners, the synthesis of branched-selective hydrofunctionalization could be achieved to produce α-aryl alkylboronates and 1,1-diboron compounds, respectively.9c,21 In other words, isomerization towards the preparation of synthetically versatile vinylboronate species is highly challenging, but of high synthetic utility given the prevalence and importance of vinylboronates in cross-coupling reactions. To date, only two examples were reported and restricted to a 1-carbon migration of allyl pinacol boronates.22 With the aim of developing an efficient alternative towards functionalized vinyl species, we decided to embark on a study and identify a metal that would be able to catalyze the migration of a remote double bond of ω-alkenylboronates 1 to stereodefined vinylboronates 2en route to diversely functionalized alkenes and α-functionalized alkylboronates (Scheme 1c).
Results and discussion
We started our study by evaluating the isomerization of our model substrate, 2-(but-3-en-1-yl)-5,5-dimethyl-1,3,2-dioxaborinane 1a, with a selection of various commercially available well-established isomerization catalysts Cat1–4 (Table 1). Gratifyingly, the formation of the desired vinylboronate product 2a was observed in all cases, albeit along with partial allylboronate isomers. For instance, the ruthenium-based “alkene zipper” Grotjahn's catalyst Cat1,23 successfully used for successive isomerizations of a remote double bond combined with a retro-ene reaction of ω-alkenyl cyclopropyl carbinols,24 afforded the vinylboronate 2a in satisfactory yield with a perfect E-selectivity (Table 1, entry 1).|
|
|||||
|---|---|---|---|---|---|
| Entry | [Cat] | Solvent | T (°C) | Yieldb (%) |
E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zc :Zc |
| a All reactions were carried out using 1a (0.3 mmol) and catalyst (5 mol%) in 1.5 mL of solvent under Ar. b Yields were determined after filtration of the crude reaction mixture through a short pad of silica gel (pentane/ether, 20/1). c The E/Z ratio was determined by 1H NMR of the crude reaction mixture. d The catalytic loading could be decreased to 2.5 mol% without altering the reaction. | |||||
| 1 | Grotjahn's catalyst Cat1 | DCE | 40 | 62 | >95:5 |
| 2 | RhH(CO)(PPh3)3Cat2 | Toluene | 100 | 29 | 93:7 |
| 3 | RuH2(CO)(PPh3)3Cat3 | Toluene | 100 | 58 | 91:9 |
| 4 | RuHCl(CO)(PPh3)3Cat4 | Toluene | 100 | 64 | 93:7 |
| 5 | RuHCl(CO)(PPh3)3Cat4 | DCE | 60 | 38 | 93:7 |
| 6 | RuHCl(CO)(PPh3)3Cat4 | Pinacolone | 60 | 11 | 95:5 |
| 7 | RuHCl(CO)(PPh3)3Cat4 | THF | 60 | 76 | 94:6d |

|
|||||

The neutral rhodium hydride complex Cat2 required higher temperature but produced 2a in low yield, whereas ruthenium dihydride catalyst Cat3 afforded 2a in acceptable yields with good levels of stereocontrol (Table 1, entries 2 and 3). On the other hand, the ruthenium monohydride [RuH(Cl)(CO)(PPh3)3, Cat4] was shown to be as effective catalyst as Cat1. Having catalyst Cat4 in hand, the nature of the solvent was briefly investigated and when toluene was replaced by dichloroethane or pinacolone, lower yields were observed (Table 1, entries 5 and 6). Ultimately, the presence of THF at 60 °C allowed the formation of 2a in 76% yield with an excellent E/Z ratio (Table 1, entry 7). Further reduction of the catalyst loading to 2.5 mol% didn't alter the transformation (72%, E:Z 94:6). It should be noted that subjecting pure E-2a (prepared by an independent route) to the optimized catalytic conditions (Table 1, entry 7), leads to a minor quantity of E- and Z-allylboronates indicating that the isomerization reaction of 1a is reversible with vinylboronate product 2a still being the major isomer.
With the optimal conditions in hand (Table 1, entry 7), we set out to explore the scope of this transformation as summarized in Scheme 2. Terminal alkenyl boronates 1a–d with various masked boron groups were all compatible under this experimental condition, providing the corresponding isomerized products 2a–d in good yields with good E/Z selectivities (Scheme 2). As expected, both cis- and trans-internal alkenyl boronates (Z)- and (E)-1e appeared to be equally tolerated, delivering the same product 2d in similar yields and selectivity (Scheme 2). Substrates with longer tether (n = 1–4) between the unsaturation and the sp3-centered boron atom still afforded the desired products 2e, 2f and 2g in excellent to moderate yields with high E/Z selectivities. However, the isomerization of 1h, possessing 9 methylene units between the unsaturation and the boron atom, provides 2g in lower yield despite the good stereoselectivity. A significant amount of internal double-bond isomers was observed in the crude reaction mixture.
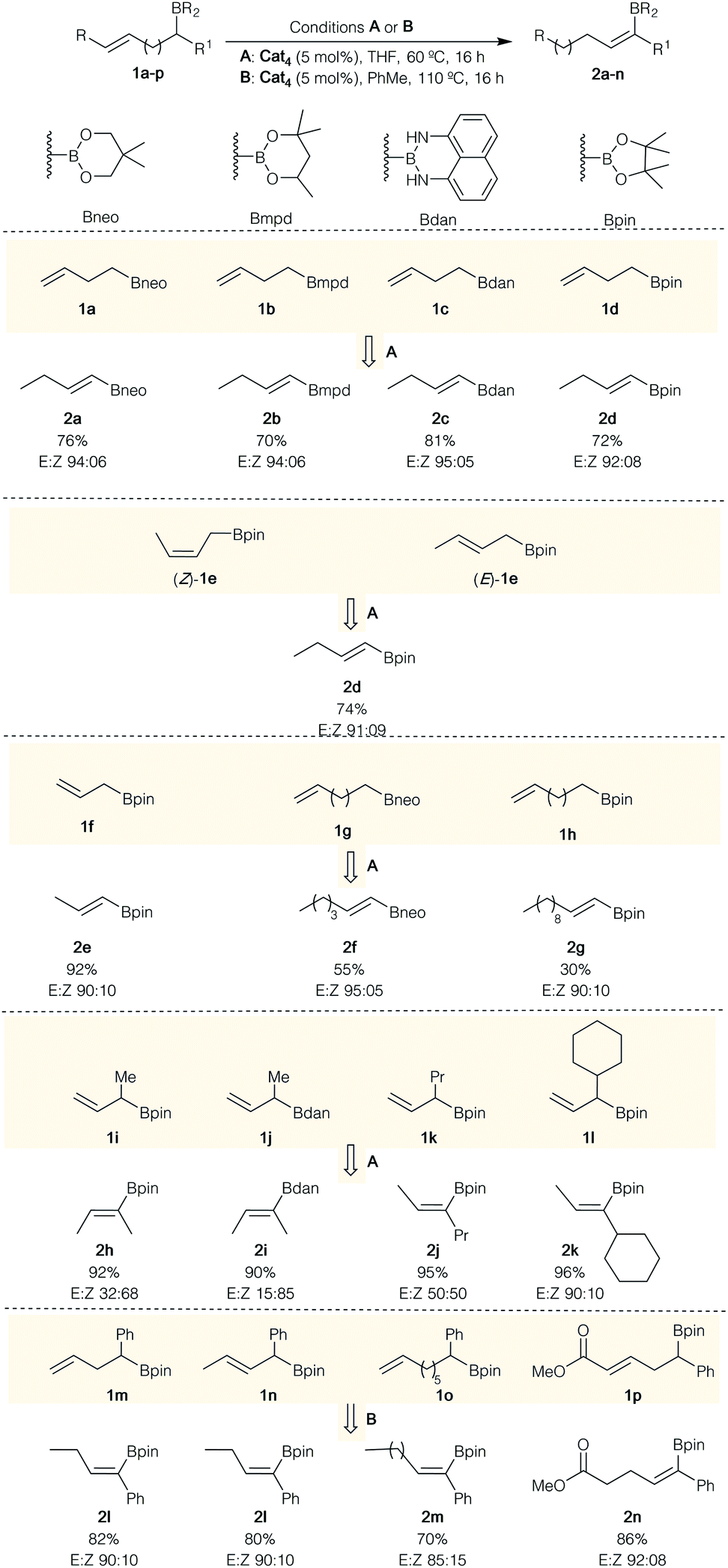 | ||
| Scheme 2 Substrate scope for the Ru-catalyzed isomerization of ω-alkenylboronates. | ||
α-Branched allylboronates 1i–o with different alkyl groups were also successfully engaged in this transformation for two different types of boronates species, (i.e., Bpin, Bdan). Although Bdan presents a slightly higher selectivity (Scheme 2, 2hversus2i), Bpin has been preferentially investigated as easier to manipulate for subsequent transformation. It is interesting to note that the selectivity of the formed double bond is very dependent of the steric hindrance of either the substituent on the boron atom (Scheme 2, compare 2h and 2i) or of the nature of the alkyl group (Scheme 2, compare 2h with 2j and 2k).
When α-phenyl ω-alkenylboronates 1m–o having different olefin geometry and chain lengths were subjected to the condition B in toluene at 110 °C, the synthetically challenging (E)-trisubstituted vinylboronates 2l–m were obtained in good yields with high stereoselectivities (Scheme 2). When α-phenyl alkenylboronates 1m–p were subjected to the condition A (THF, 60 °C), the desired vinylboronates were obtained in acceptable yields with high levels of stereocontrol but with a significant amount of allyl isomers. These results suggested that the α-phenyl substituent has a pronunced influence on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond reactivity towards Ru–H insertion during the transformation of allylboronate intermediate into vinylboronate. Switching the solvent to toluene at 110 °C (condition B) was required to convert allylboronate intermediate into conjugated vinylboronate. The stereochemistry of (E)-2l was determined by comparison with reported data25 and could be rationalized by the relative bulkiness of substituents in the Ru–H β-syn elimination (Scheme 3, D). It should be noted that metal-catalyzed hydroboration of internal alkynes and allenes usually produces the (Z)-vinylboronates26 whereas the (E)-isomer could only be obtained through the elegant non-classical trans-hydroboration of internal alkynes recently reported by Fürstner (excluding metal-catalyzed diboration reactions of allenes and propargyl alcohols).27 Hence, these results demonstrated an alternative regio- and stereoselective synthesis of E-trisubstituted vinylboronates by ruthenium-catalyzed isomerization.
C bond reactivity towards Ru–H insertion during the transformation of allylboronate intermediate into vinylboronate. Switching the solvent to toluene at 110 °C (condition B) was required to convert allylboronate intermediate into conjugated vinylboronate. The stereochemistry of (E)-2l was determined by comparison with reported data25 and could be rationalized by the relative bulkiness of substituents in the Ru–H β-syn elimination (Scheme 3, D). It should be noted that metal-catalyzed hydroboration of internal alkynes and allenes usually produces the (Z)-vinylboronates26 whereas the (E)-isomer could only be obtained through the elegant non-classical trans-hydroboration of internal alkynes recently reported by Fürstner (excluding metal-catalyzed diboration reactions of allenes and propargyl alcohols).27 Hence, these results demonstrated an alternative regio- and stereoselective synthesis of E-trisubstituted vinylboronates by ruthenium-catalyzed isomerization.
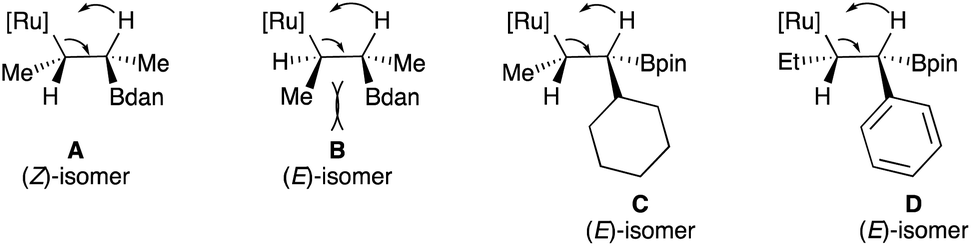 | ||
| Scheme 3 Mechanistic hypothesis to rationalize the stereochemistry of vinylboronates. | ||
Finally, the deconjugative/reconjugative isomerization of 1p was successfully achieved to afford the reconjugative product E-2n in good yield with excellent control over the double-bond geometry (Scheme 2). It is proposed that the steric preference of Bdan versus Bpin to produce the (Z)-isomer when R1 = Me (2hversus2i) as well as the constant formation of the E-vinyl boronates 2k–n as major isomer for bulkier R1 substituents relates to the steric interactions in the syn Ru–H β-elimination reaction (Scheme 3). Comparing intermediates A and B, less steric interactions exist in A than in B, explaining why the isomer Z-is preferentially formed. When the size of the substituent increases, the trend is reversed and now intermediate C should be the favored intermediate before the syn-[Ru] β-elimination reaction (Scheme 3). Although the same explanation should hold for the intermediate D, one could not exclude additional potential stabilizing interactions between the aromatic ring and the [Ru] complex.28 It should be noted that the opposite isomer (Z)-2l, prepared by an independent route (see the ESI† for details), treated in our standard reaction conditions, does not provide any other isomeric products including (E)-2l, indicating that the formation of (E)-2l doesn't result from subsequent isomerization processes.
Motivated by merging transition-metal-catalyzed chain-walking processes with subsequent remote functionalization, we further explored the possibility of combining the ruthenium-catalyzed isomerization with a palladium-catalyzed Suzuki–Miyaura cross-coupling reaction in a single-pot operation as described in Scheme 4a. To our delight, this [Ru/Pd] catalytic combination enabled a highly E-selective cross-coupling reaction of the in situ generated (E)-vinylboronates that was not possible to get with our previous procedure on ω-alkenyl ethers.11,12 Various aryl halides and alkenyl triflate were compatible furnishing the coupling products 3a–f in satisfying yields (based on two chemical steps) with excellent E:Z ratios. It should be highlighted that the present methodology allows the cross-coupling with partners bearing electron-withdrawing substituents, heteroaromatic or enone, which was also not allowed by our previously reported protocol (Scheme 4b).11 An additional example to illustrate the power of this transformation is to combine the Ru-walk with a copper-catalyzed hydroallylation reaction.29 For instance, when ω-alkenylboronate 1c was first isomerized into 2c, and then in situ treated with an allylphosphonate in the presence of a catalytic amount of copper salt, the sp3gem-dialkylated boron species 4 was obtained in 68% yield, illustrating the compatibility of this new double catalytic system (Scheme 4c). It is remarkable that the presence of Ru catalyst doesn't interfere in the second catalytic cycle.
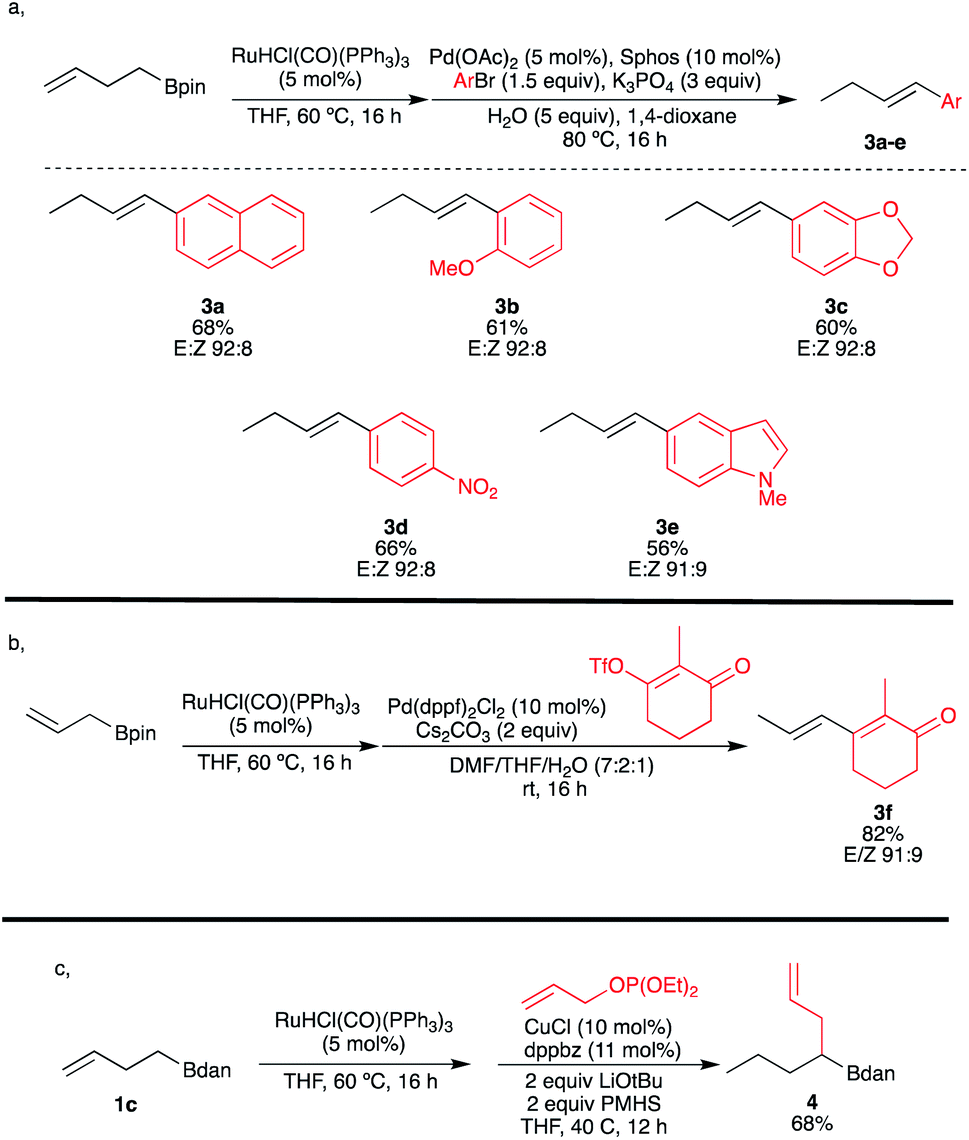 | ||
| Scheme 4 Combined [Ru]-catalyzed isomerization with subsequent functionalization. | ||
Conclusions
In conclusion, we have developed a regio- and stereoselective ruthenium-catalyzed isomerization of ω-alkenyl boronates into stereodefined di- and trisubstituted alkenylboronate derivatives. This method provides not only a new access to a variety of synthetically valuable alkenyl organoboronates, not always easily accessible, but serve also as an entry point for subsequent remote functionalization. These sequential catalytic processes, in a one-pot operation, could be achieved by either a subsequent palladium-catalyzed Suzuki–Miyaura cross coupling or by a copper-catalyzed hydroallylation reactions. These transformations delivered the products in practical yields with a high degree of olefin stereocontrol in the former case.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under Grant Agreement No. 786976.Notes and references
- (a) R. Breslow, Acc. Chem. Res., 1980, 13, 170 CrossRef CAS; (b) H. Schwarz, Acc. Chem. Res., 1989, 22, 282 CrossRef CAS; (c) P. B. Reese, Steroids, 2001, 66, 481 CrossRef CAS PubMed.
- For reviews on remote functionalization and isomerization of alkenes, see: (a) E. Larionov, H. Li and C. Mazet, Chem. Commun., 2014, 50, 9816 RSC; (b) I. Franzoni and C. Mazet, Org. Biomol. Chem., 2014, 12, 233 RSC; (c) A. Vasseur, J. Bruffaerts and I. Marek, Nat. Chem., 2016, 8, 209 CrossRef CAS PubMed; (d) H. Sommer, F. Juliá-Hernández, R. Martin and I. Marek, ACS Cent. Sci., 2018, 4, 153 CrossRef CAS PubMed; (e) D. Janssen-Müller, B. Sahoo, S.-Z. Sun and R. Martin, Isr. J. Chem., 2019, 60, 195 CrossRef; (f) T. Kochi, S. Kanno and F. Kakiuchi, Tetrahedron Lett., 2019, 60, 150938 CrossRef; (g) J. J. Molloy, T. Morack and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 13654 CrossRef CAS PubMed.
- (a) N. Chinkov, A. Levin and I. Marek, Angew. Chem., Int. Ed., 2006, 45, 465 CrossRef CAS PubMed; (b) N. Chinkov, S. Majumdar and I. Marek, J. Am. Chem. Soc., 2002, 124, 10282 CrossRef CAS PubMed; (c) N. Chinkov, S. Majumdar and I. Marek, J. Am. Chem. Soc., 2003, 125, 13258 CrossRef CAS PubMed; (d) A. Masarwa, D. Didier, T. Zabrodski, M. Schinkel, L. Ackermann and I. Marek, Nature, 2014, 505, 199 CrossRef CAS PubMed; (e) A. Vasseur, L. Perrin, O. Eisenstein and I. Marek, Chem. Sci., 2015, 6, 2770 RSC; (f) L. Mola, M. Sidera and S. P. Fletcher, Aust. J. Chem., 2015, 68, 401 CrossRef CAS.
- For selected Pd-catalyzed remote functionalization, see: (a) E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455 CrossRef CAS PubMed; (b) S. Aspin, A.-S. Goutierre, P. Larini, R. Jazzar and O. Baudoin, Angew. Chem., Int. Ed., 2012, 51, 10808 CrossRef CAS PubMed; (c) T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340 CrossRef CAS PubMed; (d) E. Larionov, L. Lin, L. Guénée and C. Mazet, J. Am. Chem. Soc., 2014, 136, 16882 CrossRef CAS PubMed; (e) T. Kochi, T. Hamasaki, Y. Aoyama, J. Kawasaki and F. Kakiuchi, J. Am. Chem. Soc., 2012, 134, 16544 CrossRef CAS PubMed; (f) S. Dupuy, K.-F. Zhang, A.-S. Goutierre and O. Baudoin, Angew. Chem., Int. Ed., 2016, 55, 14793 CrossRef CAS PubMed; (g) L. Lin, C. Romano and C. Mazet, J. Am. Chem. Soc., 2016, 138, 10344 CrossRef CAS PubMed; (h) S. Singh, J. Bruffaerts, A. Vasseur and I. Marek, Nat. Commun., 2017, 8, 14200 CrossRef PubMed; (i) D. G. Kohler, S. N. Gockel, J. L. Kennemur, P. J. Waller and K. L. Hull, Nat. Chem., 2018, 10, 333 CrossRef CAS PubMed; (j) J. Bruffaerts, D. Pierrot and I. Marek, Nat. Chem., 2018, 10, 1164 CrossRef CAS PubMed; (k) C. Han, Z. Fu, S. Guo, X. Fang, A. Lin and H. Yao, ACS Catal., 2019, 9, 4196 CrossRef CAS; (l) C. Romano, D. Fiorito and C. Mazet, J. Am. Chem. Soc., 2019, 141, 16983 CrossRef CAS PubMed; (m) T. Kochi, K. Ichinose, M. Shigekane, T. Hamasaki and F. Kakiuchi, Angew. Chem., Int. Ed., 2019, 58, 5261 CrossRef CAS PubMed.
- For selected Ni-catalyzed remote functionalization, see: (a) W.-C. Lee, C.-H. Wang, Y.-H. Lin, W.-C. Shih and T.-G. Ong, Org. Lett., 2013, 15, 5358 CrossRef CAS PubMed; (b) J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 13098 CrossRef CAS PubMed; (c) I. Busolv, J. Becouse, S. Mazza, M. Montandon-Clerc and X. Hu, Angew. Chem., Int. Ed., 2015, 54, 14523 CrossRef PubMed; (d) I. Buslov, F. Song and X. Hu, Angew. Chem., Int. Ed., 2016, 55, 12295 CrossRef CAS PubMed; (e) Y. He, Y. Cai and S. Zhu, J. Am. Chem. Soc., 2017, 139, 1061 CrossRef CAS PubMed; (f) F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84 CrossRef PubMed; (g) M. Gaydou, T. Moragas, F. Juliá-Hernández and R. Martin, J. Am. Chem. Soc., 2017, 139, 12161 CrossRef CAS PubMed; (h) F. Chen, K. Chen, Y. Zhang, Y. He, Y.-M. Wang and S. Zhu, J. Am. Chem. Soc., 2017, 139, 13929 CrossRef CAS PubMed; (i) J. Xiao, Y. He, F. Ye and S. Zhu, Chem, 2018, 4, 1645 CrossRef CAS; (j) L. Peng, Y. Li, Y. Li, W. Wang, H. Pang and G. Yin, ACS Catal., 2018, 8, 310 CrossRef CAS; (k) S.-Z. Sun, M. Börjesson, R. Martin-Montero and R. Martin, J. Am. Chem. Soc., 2018, 140, 12765 CrossRef CAS PubMed; (l) Z. Wang, H. Yin and G. C. Fu, Nature, 2018, 563, 379 CrossRef CAS PubMed; (m) F. Zhou, Y. Zhang, X. Xu and S. Zhu, Angew. Chem., Int. Ed., 2019, 58, 1754 CrossRef CAS PubMed; (n) J. He, P. Song, X. Xu, S. Zhu and Y. Wang, ACS Catal., 2019, 9, 3253 CrossRef CAS; (o) Y. Zhang, X. Xu and S. Zhu, Nat. Commun., 2019, 10, 1752 CrossRef PubMed; (p) B. Liu, P. Hu, F. Xu, L. Cheng, M. Tan and W. Han, Commun. Chem., 2019, 2, 5 CrossRef; (q) L. Zhou, C. Zhu, P. Bi and C. Feng, Chem. Sci., 2019, 10, 1144 RSC.
- For selected Ru-catalyzed remote functionalization, see: (a) H. Wakamatsu, M. Nishida, N. Adachi and M. Mori, J. Org. Chem., 2000, 65, 3966 CrossRef CAS PubMed; (b) T. Doi, T. Fukuyama, J. Horiguchi, T. Okamura and I. Ryu, Synlett, 2006, 3013 CAS; (c) T. Doi, T. Fukuyama, S. Minamino, G. Husson and I. Ryu, Chem. Commun., 2006, 1875 RSC; (d) D. B. Grotjahn, C. R. Larsen, J. L. Gustafson, R. Nair and A. Sharma, J. Am. Chem. Soc., 2007, 129, 9592 CrossRef CAS PubMed; (e) T. Fukuyama, T. Doi, S. Minamino, S. Omura and I. Ryu, Angew. Chem., Int. Ed., 2007, 46, 5559 CrossRef CAS PubMed; (f) S. Omura, T. Fukuyama, J. Horiguchi, Y. Murakami and I. Ryu, J. Am. Chem. Soc., 2008, 130, 14094 CrossRef CAS PubMed; (g) K. Sorimachi and M. Terada, J. Am. Chem. Soc., 2008, 130, 14452 CrossRef CAS PubMed; (h) C. L. Hansen, J. W. Clausen, R. G. Ohm, E. Ascic, S. T. Le Quement, D. Tanner and T. E. Nielsen, J. Org. Chem., 2013, 78, 12545 CrossRef CAS PubMed; (i) J. R. Clark, J. R. Griffiths and S. T. Diver, J. Am. Chem. Soc., 2013, 135, 3327 CrossRef CAS PubMed; (j) D. Lazzari, M. C. Cassani, M. A. Brucka, G. Solinas and M. Prettoa, New J. Chem., 2014, 38, 641 RSC; (k) Y. Toda and M. Terada, Synlett, 2013, 24, 752 CrossRef CAS; (l) E. Ascic, R. G. Ohm, R. Petersen, M. R. Hansen, C. L. Hansen, D. Madsen, D. Tanner and T. E. Nielsen, Chem.–Eur. J., 2014, 20, 3297 CrossRef CAS PubMed.
- For selected Rh-catalyzed remote functionalization, see: (a) I. Matsuda, T. Kato, S. Sato and Y. Izumi, Tetrahedron Lett., 1986, 27, 5747 CrossRef CAS; (b) L.-G. Zhuo, Z.-K. Yao and Z.-X. Yu, Org. Lett., 2013, 15, 4634 CrossRef CAS PubMed; (c) R.-Z. Huang, K.-K. Lau, Z.-F. Li, T.-L. Liu and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 14647 CrossRef CAS PubMed.
- For selected Ir-catalyzed remote functionalization, see: (a) T. Ohmura, Y. Yamamoto and N. Miyaura, Chem. Commun., 1998, 1337 RSC; (b) T. Ohmura, Y. Yamamoto and N. Miyaura, Organometallics, 1999, 18, 413 CrossRef CAS; (c) S. G. Nelson, C. J. Bungard and K. Wang, J. Am. Chem. Soc., 2003, 125, 13000 CrossRef CAS PubMed; (d) H. J. Lim, C. R. Smith and T. V. RajanBabu, J. Org. Chem., 2009, 74, 4565 CrossRef CAS PubMed; (e) H. Li and C. Mazet, J. Am. Chem. Soc., 2015, 137, 10720 CrossRef CAS PubMed; (f) H. Li and C. Mazet, Acc. Chem. Res., 2016, 49, 1232 CrossRef CAS PubMed; (g) H. Sommer, T. Weissbrod and I. Marek, ACS Catal., 2019, 9, 2400 CrossRef CAS; (h) I. Massad and I. Marek, ACS Catal., 2020, 10, 5793–5804 CrossRef CAS.
- For selected Co-catalyzed remote functionalization, see: (a) J. V. Obligacion and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 19107 CrossRef CAS PubMed; (b) T. Yamakawa and N. Yoshikai, Chem.–Asian J., 2014, 9, 1242 CrossRef CAS PubMed; (c) M. L. Scheuermann, E. J. Johnson and P. J. Chirik, Org. Lett., 2015, 17, 2716 CrossRef CAS PubMed; (d) X. Chen, Z. Cheng, J. Guo and Z. Lu, Nat. Commun., 2018, 9, 3939 CrossRef PubMed.
- (a) J. Liu, Q. Yuan, F. D. Toste and M. S. Sigman, Nat. Chem., 2019, 11, 710 CrossRef CAS PubMed; (b) A. Bahamonde, B. Al Rifaie, V. Martín-Heras, J. R. Allen and M. S. Sigman, J. Am. Chem. Soc., 2019, 141, 8708 CrossRef CAS PubMed; (c) N. J. Race, C. S. Schwalm, T. Nakamuro and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 15881 CrossRef CAS PubMed; (d) H. H. Patel and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 14226 CrossRef CAS PubMed; (e) Z. Chen, M. J. Hilton and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 11461 CrossRef CAS PubMed; (f) M. J. Hilton, B. Cheng, B. R. Buckley, L. Xu, O. Wiest and M. S. Sigman, Tetrahedron, 2015, 71, 6513 CrossRef CAS PubMed; (g) H. H. Patel and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 3462 CrossRef CAS PubMed; (h) M. J. Hilton, L. Xu, P. Norrby, Y. Wu, O. Wiest and M. S. Sigman, J. Org. Chem., 2014, 79, 11841 CrossRef CAS PubMed; (i) T. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830 CrossRef CAS PubMed.
- G.-M. Ho, L. Judkele, J. Bruffaerts and I. Marek, Angew. Chem., Int. Ed., 2018, 57, 8012 CrossRef CAS PubMed.
- C. Romano and C. Mazet, J. Am. Chem. Soc., 2018, 140, 4743 CrossRef CAS PubMed.
- G.-M. Ho, H. Sommer and I. Marek, Org. Lett., 2019, 21, 2913 CrossRef CAS PubMed.
- J. Carreras, A. Caballero and P. J. Perez, Chem.–Asian J., 2019, 14, 329 CrossRef CAS PubMed.
- D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis Medicine and Materials, Wiley-VCH, Weinheim, 2nd edn, 2011, vol. 1 and 2 Search PubMed.
- (a) T. Moriya, A. Suzuki and N. Miyaura, Tetrahedron Lett., 1995, 36, 1887 CrossRef CAS; (b) T. Miura, J. Nakahashi, W. Zhou, Y. Shiratori, S. G. Stewart and M. Murakami, J. Am. Chem. Soc., 2017, 139, 10903 CrossRef CAS PubMed.
- (a) Y. Yamamoto, T. Miyairi, T. Ohmura and N. Miyaura, J. Org. Chem., 1999, 64, 296 CrossRef CAS PubMed; (b) T. Miura, Y. Nishida, M. Morimoto and M. Murakami, J. Am. Chem. Soc., 2013, 135, 11497 CrossRef CAS PubMed.
- T. Miura, J. Nakahashi and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 6989 CrossRef CAS PubMed.
- F. Weber, M. Ballmann, C. Kohlmeyer and G. Hilt, Org. Lett., 2016, 18, 548 CrossRef CAS PubMed.
- S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS PubMed.
- Y. Zhang, B. Han and S. Zhu, Angew. Chem., Int. Ed., 2019, 58, 13860 CrossRef CAS PubMed.
- (a) F. Weber, A. Schmidt, P. Röse, M. Fischer, O. Burghaus and G. Hilt, Org. Lett., 2015, 17, 2952 CrossRef CAS PubMed; (b) A. Kapat, T. Sperger, S. Guven and F. Schoenebeck, Science, 2019, 363, 391 CrossRef CAS PubMed.
- (a) G. Erdogan and D. B. Grotjahn, J. Am. Chem. Soc., 2009, 131, 10354 CrossRef CAS PubMed; (b) C. R. Larsen and D. B. Grotjahn, J. Am. Chem. Soc., 2012, 134, 10357 CrossRef CAS PubMed; (c) G. E. Dobereiner, G. Erdogan, C. R. Larsen, D. B. Grotjahn and R. R. Schrock, ACS Catal., 2014, 4, 3069 CrossRef CAS; (d) D. B. Grotjahn, C. R. Larsen and G. Erdogan, Top. Catal., 2014, 57, 1483 CrossRef CAS.
- J. Bruffaerts, A. Vasseur and I. Marek, Adv. Synth. Catal., 2018, 360, 1389 CrossRef CAS.
- Y. Hu, W. Sun, T. Zhang, N. Xu, J. Xu, Y. Lan and C. Liu, Angew. Chem., Int. Ed., 2019, 58, 15813 CrossRef CAS PubMed.
- For selected examples, see: (a) H. R. Kim and J. Yun, Chem. Commun., 2011, 47, 2943 RSC; (b) W. Yuan, X. Zhang, Y. Yu and S. Ma, Chem.–Eur. J., 2013, 19, 7193 CrossRef CAS PubMed; (c) K. Semba, M. Shinomiya, T. Fujihara, J. Terao and Y. Tsuji, Chem.–Eur. J., 2013, 19, 7125 CrossRef CAS PubMed; (d) K. Semba, N. Bessho, T. Fujihara, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2014, 53, 9007 CrossRef CAS PubMed; (e) J. Zhao, Z. Niu, H. Fu and Y. Li, Chem. Commun., 2014, 50, 2058 RSC; (f) Y. D. Bidal, F. Lazreg and C. S. J. Cazin, ACS Catal., 2014, 4, 1564 CrossRef CAS; (g) M. Espinal-Viguri, C. R. Woof and R. L. Webster, Chem.–Eur. J., 2016, 22, 11605 CrossRef CAS PubMed.
- (a) T. Biberger, C. P. Gordon, M. Leutzsch, S. Peil, A. Guthertz, C. Coperet and A. Fürstner, Angew. Chem., Int. Ed., 2019, 58, 8845 CrossRef CAS PubMed; (b) H. Jin and A. Fürstner, Org. Lett., 2019, 21, 3446 CrossRef CAS PubMed; (c) A. Fürstner, J. Am. Chem. Soc., 2019, 141, 11 CrossRef PubMed; (d) X. B. Mo, A. Letort, D. A. Rosca, K. Higashida and A. Fürstner, Chem.–Eur. J., 2018, 24, 9667 CrossRef CAS PubMed; (e) A. Guthertz, M. Leutzsch, L. M. Wolf, P. Gupta, S. M. Rummelt, R. Goddard, C. fares, W. Thiel and A. Fürstner, J. Am. Chem. Soc., 2018, 140, 3156 CrossRef CAS PubMed; (f) B. Sundararaju and A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 14050 CrossRef CAS PubMed.
- S. Krompiec, M. Pigulla, T. Bieg, W. Szczepankiewicz, N. Kuźnik, M. Krompiec and M. Kubicki, J. Mol. Catal. A: Chem., 2002, 189, 169 CrossRef CAS.
- J. T. Han, W. J. Jang, N. Kim and J. Yun, J. Am. Chem. Soc., 2016, 138, 15146 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02542a |
| This journal is © The Royal Society of Chemistry 2020 |