
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insight into hydroxamate transfer reaction mimicking the inhibition of zinc-containing enzymes†
Nam
Kwon‡
a,
Jong-Min
Suh‡
b,
Mi Hee
Lim
 *b,
Hajime
Hirao
*c and
Jaeheung
Cho
*a
*b,
Hajime
Hirao
*c and
Jaeheung
Cho
*a
aDepartment of Emerging Materials Science, DGIST, Daegu 42988, Korea. E-mail: jaeheung@dgist.ac.kr
bDepartment of Chemistry, KAIST, Daejeon 34141, Korea
cDepartment of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong
First published on 13th August 2020
Abstract
A hydroxamate transfer reaction between metal complexes has been investigated by a combination of experimental and theoretical studies. A hydroxamate-bound cobalt(II) complex bearing a tetradentate macrocyclic ligand, [CoII(TBDAP)(CH3C(–NHO)O)]+ (1), is prepared by the reduction of a hydroximatocobalt(III) complex with a biological reductant. Alternatively, 1 is accessible via a synthetic route for the reaction between the cobalt(II) complex and acetohydroxamic acid in the presence of a base. 1 was isolated and characterized by various physicochemical methods, including UV-vis, IR, ESI-MS, and X-ray crystallography. The hydroxamate transfer reactivity of 1 was examined with a zinc complex, which was followed by UV-vis and ESI-MS. Kinetic and activation parameter data suggest that the hydroxamate transfer reaction occurs via a bimolecular mechanism, which is also supported by DFT calculations. Moreover, 1 is able to inhibit the activity against a zinc enzyme, i.e., matrix metalloproteinase-9. Our overall investigations of the hydroxamate transfer using the synthetic model system provide considerable insight into the final step involved in the inhibition of zinc-containing enzymes.
Introduction
Hydroxamic acids are important pharmacophores in diverse biological functions such as antimicrobial activity and metal detoxification.1–5 The derivatives of hydroxamic acids are well known as zinc-binding groups; thus, they can inhibit the activity of zinc-containing enzymes, including carbonic anhydrase, histone deacetylase and carboxypeptidase.6–11 So far, hydroxamic acids have also been considered as highly promising anticancer reagents: they can serve as inhibitors of matrix metalloproteinases (MMPs) that are overexpressed in cancer cells and cause tumor invasion and metastasis.12–17 The use of hydroxamate functional groups as traditional anticancer reagents, however, is rather restricted within tumor cells because the environment disrupts drug supply.18–21Recently, numerous hydroxamate-bound metal complexes have been explored as prodrugs targeting cancer cells. For example, cobalt(III) complexes of hydroxamic acids are reported to be potential prodrugs for hypoxia-selective anticancer agents.22–25 As shown in Scheme 1, Hambley and co-workers have provided experimental support for two possible pathways of releasing the hydroxamate group (i.e., bio-reduction and endogenous ligand exchange pathways).22,26 Recent studies on the characterization and reactivity of cobalt(III)-hydroximato and -hydroxamato complexes bearing TPA ligands revealed their redox behaviors and ligand exchange reactions.27,28 Little is known about the molecular-level mechanism of the hydroxamate transfer, which is the final step of inhibiting the activity of zinc-containing enzymes by hydroxamate, however.
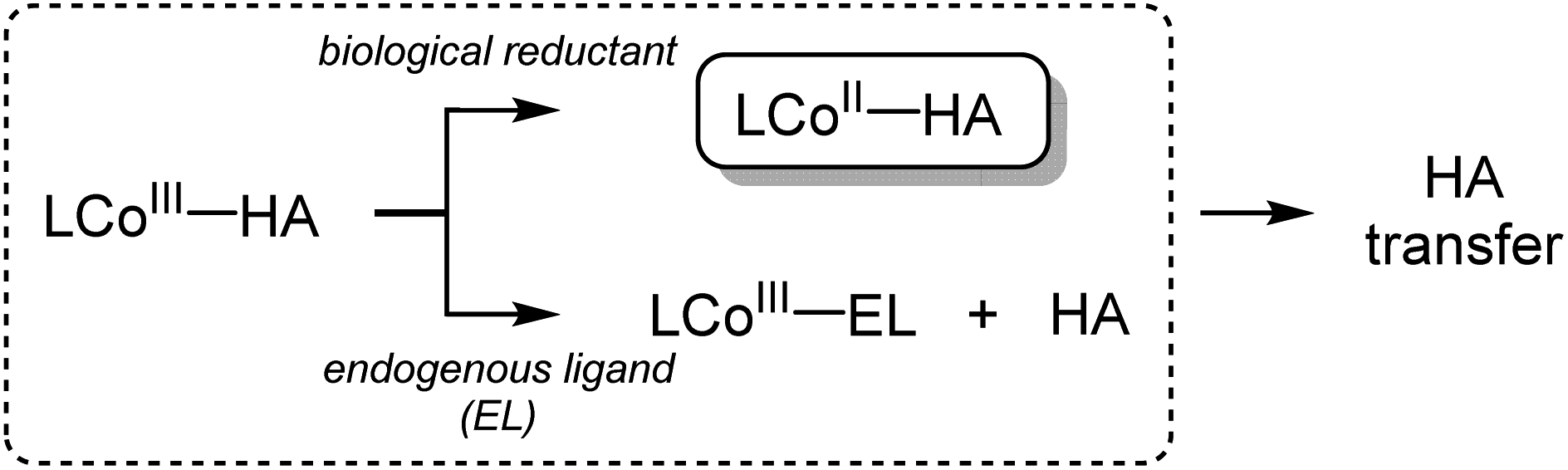 | ||
| Scheme 1 Proposed pathways for the release of hydroxamate (HA). | ||
Herein, we report a novel approach to investigate the hydroxamate transfer activity of a hydroxamatocobalt(II) complex, [CoII(TBDAP)(CH3C(–NHO)O)]+ (1; TBDAP = N,N-di-tert-butyl-2,11-diaza[3.3](2,6)-pyridinophane), which is derived from the reduction of [CoIII(TBDAP)(CH3C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NO)O)]+ (2).291 was characterized by X-ray crystallography and multiple spectroscopic methods. To the best of our knowledge, 1 represents a rare example of a structurally characterized cobalt(II) complex bearing an acetohydroxamate ligand that has been prepared by the reaction of a hydroximatocobalt(III) complex with a biological reductant. In this work, we have examined the mechanism of the hydroxamate transfer from 1 towards a zinc complex which is a model of zinc-containing active sites in enzymes.30 Kinetic studies and density functional theory (DFT) calculations support the notion that the hydroxamate transfer occurs through a bimolecular mechanism. Moreover, 1 performs the better inhibitory activity against matrix metalloproteinase-9 (MMP-9), compared to 2.
NO)O)]+ (2).291 was characterized by X-ray crystallography and multiple spectroscopic methods. To the best of our knowledge, 1 represents a rare example of a structurally characterized cobalt(II) complex bearing an acetohydroxamate ligand that has been prepared by the reaction of a hydroximatocobalt(III) complex with a biological reductant. In this work, we have examined the mechanism of the hydroxamate transfer from 1 towards a zinc complex which is a model of zinc-containing active sites in enzymes.30 Kinetic studies and density functional theory (DFT) calculations support the notion that the hydroxamate transfer occurs through a bimolecular mechanism. Moreover, 1 performs the better inhibitory activity against matrix metalloproteinase-9 (MMP-9), compared to 2.
Results and discussion
The hydroxamatocobalt(II) complex, 1, was synthesized by reacting 1 equiv. of acetohydroxamic acid in CH3CN with a starting CoII complex, [CoII(TBDAP)(NO3)(H2O)]+, in the presence of 2 equiv. of triethylamine (TEA) under ambient conditions, where the solution color changed from pink to orange. The UV-vis spectrum of 1 in CH3CN at 25 °C revealed two characteristic absorption bands at λmax = 361 (ε = 1900 M−1 cm−1) and 468 nm (ε = 100 M−1 cm−1). The electrospray ionization mass spectrometry (ESI-MS) spectrum of 1 showed a single signal at mass-to-charge (m/z) ratio of 485.3 (calcd m/z 485.2); the mass and isotope distribution pattern correspond to [Co(TBDAP)(CH3C(–NHO)O)]+ (Fig. S1†). The FT-IR spectrum of 1 revealed the existence of N–H vibrational frequency at 3206 cm−1, which also corroborates the hypothesis that acetohydroxamic acid is bound in the form of singly deprotonated monoanionic hydroxamate rather than doubly deprotonated dianionic hydroximate (Fig. S2†).31 The effective magnetic moment of 1 (μeff = 4.41 B.M.) was determined using the 1H NMR spectroscopy method of Evans in CD3CN at 25 °C,32 suggesting the high spin state (S = 3/2) of the CoII ion (see ESI†). 1 has a slightly higher effective magnetic moment due to spin–orbit coupling.33 Thus, 1 is characterized as the cobalt(II) complex with the singly deprotonated hydroxamate ligand.The X-ray crystal structure of 1 reveals a mononuclear acetohydroxamate cobalt complex in a distorted octahedral geometry, in which the hydroxamate group coordinated in a bidentate mode (Fig. 1). The average Co–O bond length (2.034 Å) in 1 is similar to that of the hydroxamatocobalt(II) complex with 6-(Me2Ph)2TPA ligand (2.038 Å)34 but is longer than that in 2 (1.856 Å) and other hydroximatocobalt(III) complexes.24,25,27–29,351 is the rare example of a structurally characterized hydroxamatocobalt(II) complex, which would be a reactive species towards inhibition of metalloenzymes.
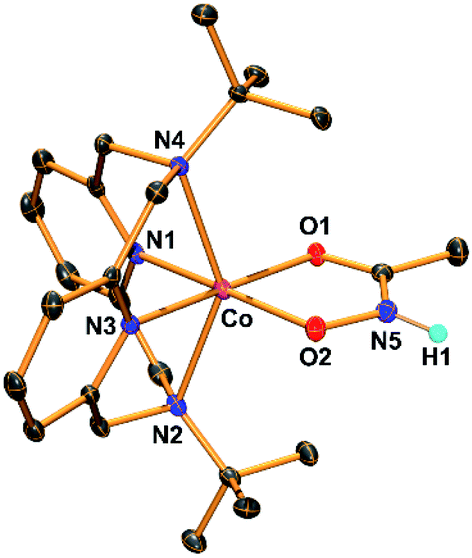 | ||
| Fig. 1 ORTEP diagram of the hydroxamatocobalt(II) complex, [CoII(TBDAP)(CH3C(–NHO)O)]+ (1), with thermal ellipsoids drawn at the 30% probability level. All hydrogen atoms except H1 are omitted for clarity. H1 was found in the Fourier difference map. | ||
It has been proposed that the hydroxamatocobalt(II) species is a key intermediate in the inhibition against MMP.22,23 We investigated the intermolecular transfer of the hydroxamate group from 1 to a zinc complex, [ZnII(Me3-TACN)(NO3)]+ (3), which is a model of the active site of MMP (Scheme S1†).30 Upon addition of 3 to 1, the characteristic absorption band of 1 disappeared (Fig. 2a). The hydroxamate transfer from 1 to 3 was confirmed by ESI-MS analysis in the course of the reaction, where the mass peak at m/z 485.3 corresponding to 1 vanished with a concomitant appearance of the mass peak at m/z 309.2 corresponding to [ZnII(Me3-TACN)(CH3C(–NHO)O)]+ (4) (Fig. 2b). Many attempts to isolate the product as single crystals have been unsuccessful. The structural information was obtained from an alternative synthetic route: the complex 4 was crystallized from the solution of the reaction mixture of 3 and excess acetohydroxamic acid in the presence of TEA (see ESI and Fig. S3†). Although the equilibrium constant (Keq = 5.9 × 10−2) of the transfer reaction determined by optical titrations is small (Fig. S4†), the reaction readily occurs upon the addition of an excess amount of 3.
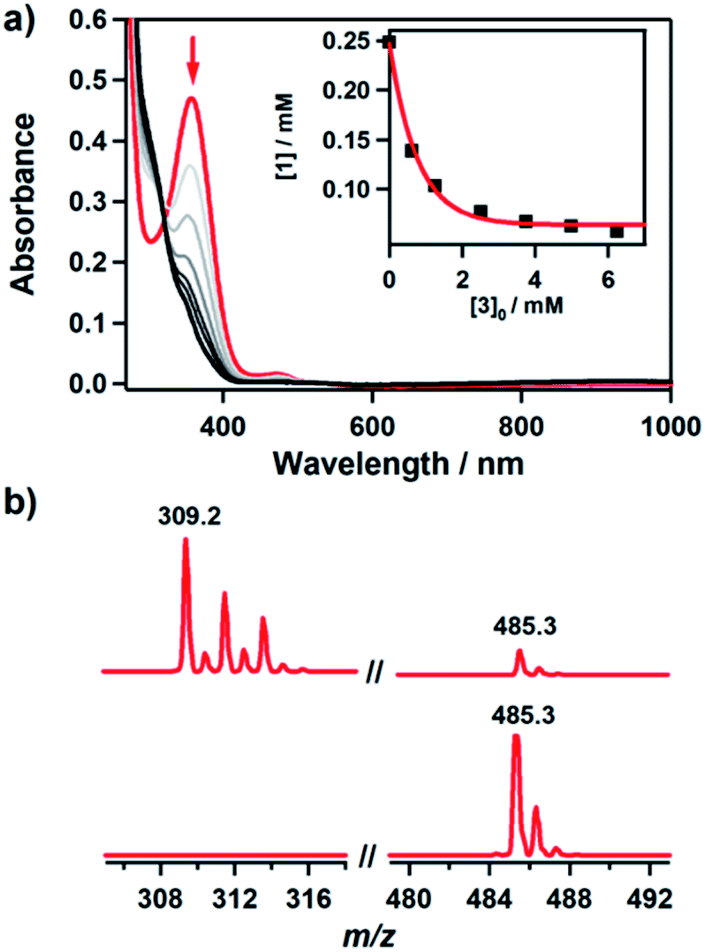 | ||
Fig. 2 Reaction of 1 with 3. (a) UV-vis spectral change during the reaction of 1 (0.25 mM) with 3 (10 mM) in CH3CN/H2O (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at −10 °C. Inset shows the decrease of concentration of 1 with increasing concentration of 3. (b) ESI-MS spectra obtained for the reaction of 1 (0.25 mM) with 3 (25 mM) before (lower) and after (upper) the reaction. The peaks at m/z 309.2 and 485.3 are assigned to [ZnII(Me3-TACN)(CH3C(–NHO)O)]+ (calcd m/z 309.1) and [CoII(TBDAP)(CH3C(–NHO)O)]+ (calcd m/z 485.2), respectively. :1) at −10 °C. Inset shows the decrease of concentration of 1 with increasing concentration of 3. (b) ESI-MS spectra obtained for the reaction of 1 (0.25 mM) with 3 (25 mM) before (lower) and after (upper) the reaction. The peaks at m/z 309.2 and 485.3 are assigned to [ZnII(Me3-TACN)(CH3C(–NHO)O)]+ (calcd m/z 309.1) and [CoII(TBDAP)(CH3C(–NHO)O)]+ (calcd m/z 485.2), respectively. | ||
Kinetic studies of the hydroxamate group transfer reaction from 1 to 3 were carried out in a mixture of CH3CN and H2O (99:1). Upon addition of 3 (2.5 mM) to the solution of 1 (0.25 mM) at −10 °C, the characteristic absorption bands of 1 decayed (Fig. 2). The rate constant increased with the concentration of 3, providing a second-order rate constant (k2) of 4.0(2) M−1 s−1 (Fig. 3a). The temperature dependence of the k2 value was investigated in the range of 253–283 K, from which a linear Eyring plot was obtained with activation parameters of ΔH‡ = 59(3) kJ mol−1 and ΔS‡ = −5(1) J mol−1 K−1 (Fig. 3b). The observed data suggest that the hydroxamate transfer reaction occurs through a bimolecular mechanism, in which the formation of an undetected [(TBDAP)Co–(CH3C(–NHO)O)–Zn(Me3-TACN)]3+ species is the rate-determining step.
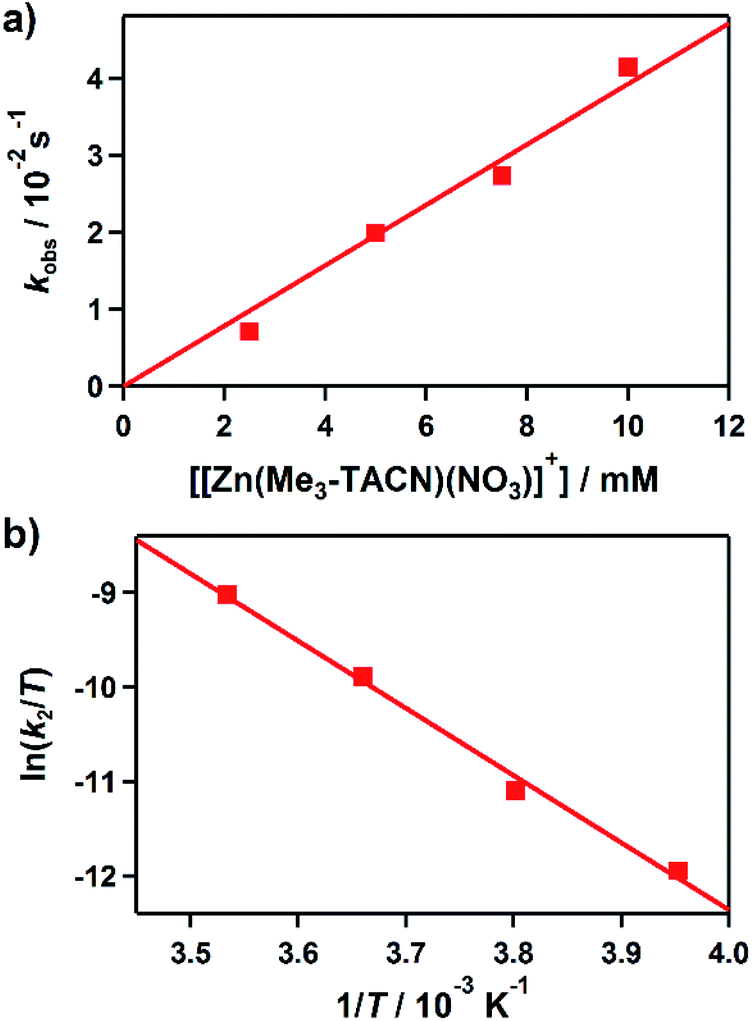 | ||
| Fig. 3 Rate and activation parameters for the reaction of 1 with 3 in CH3CN/H2O (99:1). (a) Plot of kobs against the concentration of 3 at −10 °C to determine a second-order rate constant (k2). (b) Eyring plot of ln(k2/T) against 1/T to obtain the activation parameters. | ||
DFT calculations were performed for the hydroxamate transfer reaction (see ESI†). According to the DFT results (Fig. 4), the oxygen atom in the NHO− moiety of hydroxamate in the Co(II) complex is first coordinated by the Zn(II) complex at Int1. Subsequently, the carbonyl oxygen of hydroxamate is transferred from the Co(II) center to the Zn(II) center via a transition state (TS1) to form a product complex (Int3). The free energy profile also suggests that the reaction is not a very favorable process thermodynamically, which is reasonably consistent with the small equilibrium constant (vide supra).
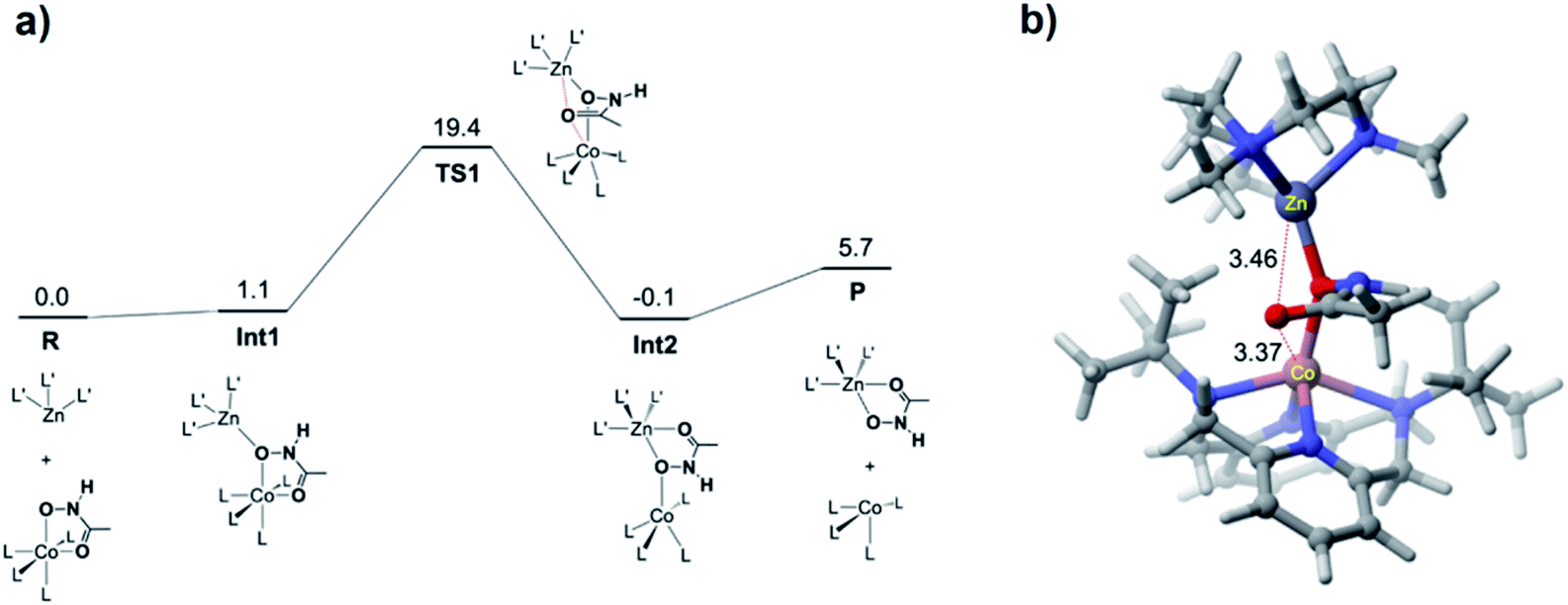 | ||
| Fig. 4 (a) DFT-calculated free energy profile (in kcal mol−1) for the hydroxamate transfer reaction. L and L′ denote the ligands for the Co and Zn complexes, respectively. (b) Optimized geometry of TS1, with key distances shown in Å. | ||
On the other hand, upon addition of 3 to a solution of 2 (0.5 mM), 2 remained intact without showing any absorption spectral change (Fig. S5a†), indicating that 2 is not able to conduct the hydroxamate group transfer reaction. The ESI-MS spectrum of the reaction solutions confirmed that no transferred product was formed (Fig. S5b†). Notably, the transfer reaction occurs by adding a biological reductant such as ascorbic acid, which is a 2H+/2e− donor. Addition of 0.5 equiv. of ascorbic acid to a reaction mixture of 2 and 3 resulted in the conversion of 2 to 1, and then the hydroxamate in 1 was transferred to 3 (Fig. S6a†). After the reaction had been completed, 4 was produced, which was confirmed by ESI-MS (Fig. S6b†).
The cyclic voltammograms of 1 and 2 in CH3CN exhibit a reversible couple between the Co(II) and Co(III) complexes (Fig. S7†). From the E1/2 values, the one-electron redox potentials of 1 and 2 were determined to be 0.28 and −0.47 V (versus SCE), respectively. The redox potential of 2 is more negative than that of ascorbic acid.36 In earlier studies, however, the protonation of hydroximatocobalt(III) complexes resulted in more positive potential affording facile reduction.27 The proton-assisted reduction process was confirmed by the cyclic voltammetry (CV) experiments, where the redox signal of 2 disappeared with the concomitant generation of the redox signal of 1 upon addition of proton (Fig. S8†). Alternatively, the hydroxamatocobalt(III) complex, [CoIII(TBDAP)(CH3C(–NHO)O)]2+ (5), which is not only a protonated form of 2 but also an one electron oxidized species of 1, was prepared by the reaction of 2 with 1 equiv. of HClO4 (Fig. S11†). The formation of 5 was confirmed by UV-vis and ESI-MS (Fig. S12†). 2 and 5 are interconvertible through the acid–base chemistry.
In order to verify the influence of 1, relative to acetohydroxamic acid or 2, on a zinc-containing enzyme, their inhibitory activity against MMP-9 was evaluated. After incubation of activated MMP-9 with a peptide as a substrate in the presence of 1, 2, or acetohydroxamic acid, the amount of the substrate that was not cleaved by the enzyme was analyzed. Inhibition against MMP-9 by 1 and 2, relative to that by acetohydroxamic acid, was 77(2)% and 24(3)%, respectively. The more noticeable inhibitory activity of 1 than 2 against MMP-9 was expected from our reactivity and mechanistic studies (vide supra).
To visualize possible interactions between 1 and MMP-9 at the molecular level, docking studies were carried out employing the catalytic domain of MMP-9 (PDB 4H3X).37 As illustrated in Fig. 5, 1 could access to the catalytic cleft of MMP-9 where three histidine residues (i.e., H226, H230, and H236) are coordinated to the active Zn(II) center. According to the docked structures, 1 may have multiple contacts with the catalytic domain of MMP-9: (i) hydrogen bonding {[C–H from 1 and oxygen (O) donor atoms from A189, A191, E227, and D235; a nitrogen (N) donor atom from H236] and [C–H from H236 and an O donor atom from 1]} and (ii) C–H⋯π interaction (C–H from the side chain of L187 and H236 and pyridine groups from 1). In addition, the representative conformations exhibited the possibility of the hydroxamate moiety onto 1 to be located close to the Zn(II) center in the catalytic domain of MMP-9. Collectively, 1 may interact with the catalytic site of MMP-9.
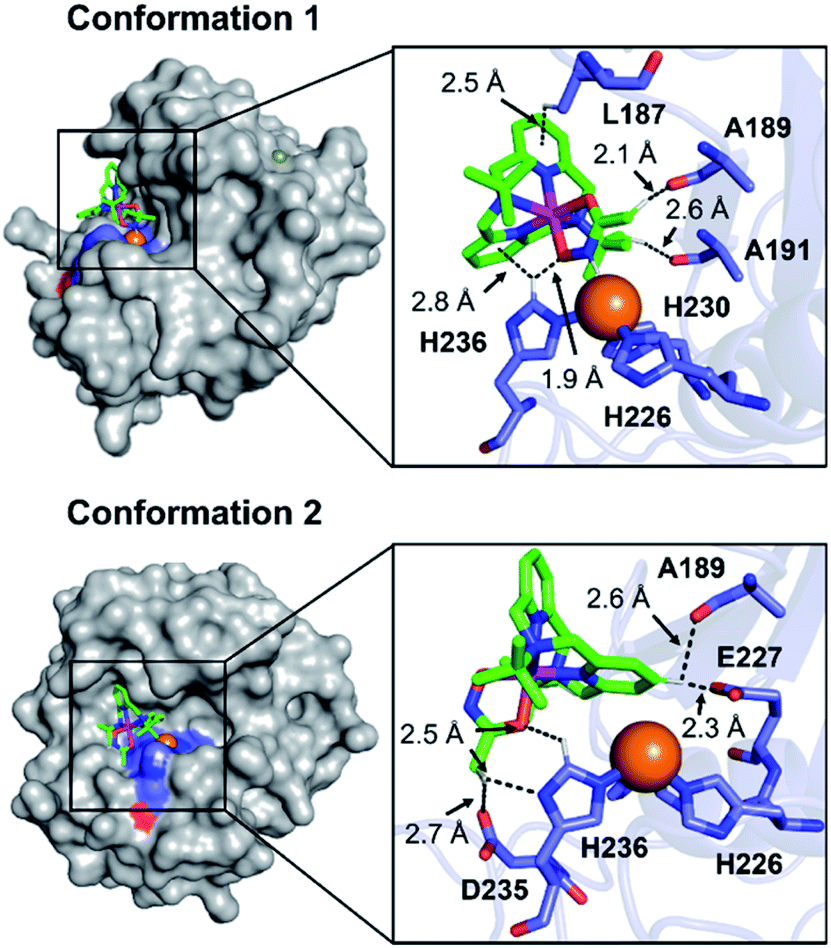 | ||
| Fig. 5 Two possible representative conformations of 1 docked with the catalytic domain of MMP-9 (PDB 4H3X) by AutoDock Vina [surface (left) and cartoon (right) versions]. These conformations exhibited the calculated binding energies in a range from −5.5 to −3.5 kcal mol−1. Hydrogen bonding and C–H⋯π interactions within 3.0 Å are indicated with dashed lines. The Zn(II) center in the catalytic domain of MMP-9 is highlighted as an orange sphere. | ||
To explore how the substituent onto the ligand affects the hydroxamate transfer reactivity towards 3 and the inhibitory activity of Co(II) complexes against MMP-9, a benzohydroxamatocobalt(II) complex, [CoII(TBDAP)(C6H5C(–NHO)O)]+ (6), was prepared (see ESI†). Kinetic studies for the reaction of 6 with 3 were carried out, affording k2 of 4.9(5) × 10−1 M−1 s−1 at −10 °C (Fig. S16 and S17†). The equilibrium constant for the transfer reaction from 6 was also determined as 4.4 × 10−2 (Fig. S18†). These results are similar to those of the hydroxamate transfer reaction of 1. Under our experimental conditions, the inhibitory activity of 6 towards MMP-9 was same as that of 1 (data not shown). Thus, replacing a structural moiety of the ligand from acetohydroxamate to benzohydroxamate may not significantly change the hydroxamate transfer reactivity towards 3 and the inhibitory activity of its corresponding Co(II) complex against MMP-9.
Based on the experimental and computational studies, it is suggested that the hydroxamate transfer can be accelerated in the presence of a weak acid, which reduces the coordination ability of the hydroxamate group in the hydroxamatocobalt(II) complex. Thus, we further investigated the equilibrium between 1 and 4 under acidic conditions. By adding acetic acid to the solution of 1 and 3, the absorption band of 1 exponentially decreased with increasing concentration of the acetic acid (Fig. S19†). This result indicates that, in the presence of acetic acid, the equilibrium is shifted in favor of 4, which was also confirmed by ESI-MS (Fig. S20†). Taken together, the hydroxamate transfer reaction effectively occurs in relatively acidic conditions.
Conclusions
We have succeeded in the isolation and structural characterization of a hydroxamatocobalt(II) complex bearing macrocyclic tetradentate N4 ligand, [CoII(TBDAP)(CH3C(–NHO)O)]+ (1), in which the acetohydroxamate ligand is bound in a bidentate mode. The intermediate is further characterized by various physicochemical methods such as FT-IR, UV-vis, and ESI-MS. 1 exhibited hydroxamate group transfer reactivity towards a zinc(II) complex. The observation of the hydroxamate transfer between metal complexes is unprecedented. In addition, the proton-assisted reduction mechanism was examined by the CV measurements. Kinetic studies suggest that the transfer reaction proceeds by a bimolecular mechanism, which is supported by DFT calculations. Moreover, the inhibitory activity of 1 towards a zinc-containing enzyme, MMP-9, was confirmed. The interaction and accessibility of 1 to MMP-9 as well as the substitution effect onto the hydroxamate ligand were also examined.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. C. at DGIST acknowledges the financial support from the NRF (2019R1A2C2086249 and 2018R1A5A1025511) and the Ministry of Science, ICT and Future Planning (MSIP) (CGRC 2016M3D3A01913243) of Korea. M. H. L was supported by the NRF funded by the MSIP (2017R1A2B3002585). H. H. acknowledges the support of City University of Hong Kong (9610369).Notes and references
- C. Apfel, D. W. Banner, D. Bur, M. Dietz, T. Hirata, C. Hubschwerlen, H. Locher, M. G. P. Page, W. Pirson, G. Rosse and J.-L. Specklin, J. Med. Chem., 2000, 43, 2324–2331 CrossRef CAS PubMed.
- M. J. Miller, Chem. Rev., 1989, 89, 1563–1579 CrossRef CAS.
- E. M. Muri, M. J. Nieto, R. D. Sindelar and J. S. Williamson, Curr. Med. Chem., 2002, 9, 1631–1653 CrossRef CAS PubMed.
- C. J. Marmion, D. Griffith and K. B. Nolan, Eur. J. Inorg. Chem., 2004, 2004, 3003–3016 CrossRef.
- S. Parvathy, I. Hussain, E. H. Karran, A. J. Turner and N. M. Hooper, Biochemistry, 1998, 37, 1680–1685 CrossRef CAS PubMed.
- V. Alterio, A. Di Fiore, K. D'Ambrosio, C. T. Supuran and G. De Simone, Chem. Rev., 2012, 112, 4421–4468 CrossRef CAS PubMed.
- C. T. Supuran, A. Scozzafava and A. Casini, Med. Res. Rev., 2003, 23, 146–189 CrossRef CAS PubMed.
- A. Scozzafava and C. T. Supuran, J. Med. Chem., 2000, 43, 3677–3687 CrossRef CAS PubMed.
- T. A. Miller, D. J. Witter and S. Belvedere, J. Med. Chem., 2003, 46, 5097–5116 CrossRef CAS PubMed.
- M. S. Finnin, J. R. Donigian, A. Cohen, V. M. Richon, R. A. Rifkind, P. A. Marks, R. Breslow and N. P. Pavletich, Nature, 1999, 401, 188–193 CrossRef CAS PubMed.
- W. L. Mock and H. Cheng, Biochemistry, 2000, 39, 13945–13952 CrossRef CAS PubMed.
- M. Whittaker, C. D. Floyd, P. Brown and A. J. H. Gearing, Chem. Rev., 1999, 99, 2735–2776 CrossRef CAS PubMed.
- R. P. Verma, Matrix Metalloproteinase Inhibitors: Specificity of Binding and Structure-Activity Relationships, ed. S. P. Gupta, Springer, Basel, 2012, pp. 137–176 Search PubMed.
- S. D. Shapiro, Curr. Opin. Cell Biol., 1998, 10, 602–608 CrossRef CAS PubMed.
- N. Johansson, M. Ahonen and V.-M. Kähäri, Cell. Mol. Life Sci., 2000, 57, 5–15 CrossRef CAS PubMed.
- E. Kerkelä and U. Saarialho-Kere, Exp. Dermatol., 2003, 12, 109–125 CrossRef PubMed.
- M. A. Rudek, J. Venitz and W. D. Figg, Pharmacotherapy, 2002, 22, 705–720 CrossRef CAS PubMed.
- V. P. Chauhan, T. Stylianopoulos, Y. Boucher and R. K. Jain, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 281–298 CrossRef CAS PubMed.
- Y. Boucher, L. T. Baxter and R. K. Jain, Cancer Res., 1990, 50, 4478–4484 CAS.
- A. I. Minchinton and I. F. Tannock, Nat. Rev. Cancer, 2006, 6, 583–592 CrossRef CAS PubMed.
- R. E. Vandenbroucke and C. Libert, Nat. Rev. Drug Discovery, 2014, 13, 904–927 CrossRef CAS PubMed.
- T. W. Hambley, Science, 2007, 318, 1392–1393 CrossRef CAS PubMed.
- C. R. Munteanu and K. Suntharalingam, Dalton Trans., 2015, 44, 13796–13808 RSC.
- T. W. Failes, C. Cullinane, C. I. Diakos, N. Yamamoto, J. G. Lyons and T. W. Hambley, Chem.–Eur. J., 2007, 13, 2974–2982 CrossRef CAS PubMed.
- T. W. Failes and T. W. Hambley, Dalton Trans., 2006, 6, 1895–1901 RSC.
- N. Yamamoto, S. Danos, P. D. Bonnitcha, T. W. Failes, E. J. New and T. W. Hambley, J. Biol. Inorg Chem., 2008, 13, 861–871 CrossRef CAS PubMed.
- P. D. Bonnitcha, B. J. Kim, R. K. Hocking, J. K. Clegg, P. Turner, S. M. Neville and T. W. Hambley, Dalton Trans., 2012, 41, 11293–11304 RSC.
- M. Alimi, A. Allam, M. Selkti, A. Tomas, P. Roussel, E. Galardon and I. Artaud, Inorg. Chem., 2012, 51, 9350–9356 CrossRef CAS PubMed.
- H. Noh, D. Jeong, T. Ohta, T. Ogura, J. S. Valentine and J. Cho, J. Am. Chem. Soc., 2017, 139, 10960–10963 CrossRef CAS PubMed.
- F. E. Jacobsen, J. A. Lewis and S. M. Cohen, J. Am. Chem. Soc., 2006, 128, 3156–3157 CrossRef CAS PubMed.
- B. J. Brennan, J. Chen, B. Rudshteyn, S. Chaudhuri, B. Q. Mercado, V. S. Batista, R. H. Crabtree and G. W. Brudvig, Chem. Commun., 2016, 52, 2972–2975 RSC.
- D. F. Evans and D. A. Jakubovic, J. Chem. Soc., Dalton Trans., 1988, 2927–2933 RSC.
- F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, Wiley, New York, 1988 Search PubMed.
- M. M. Makowska-Grzyska, E. Szajna, C. Shipley, A. M. Arif, M. H. Mitchell, J. A. Halfen and L. M. Berreau, Inorg. Chem., 2003, 42, 7472–7488 CrossRef CAS PubMed.
- R. Codd, Coord. Chem. Rev., 2008, 252, 1387–1408 CrossRef CAS.
- T. Matsui, Y. Kitagawa, M. Okumura and Y. Shigeta, J. Phys. Chem. A, 2015, 119, 369–376 CrossRef CAS PubMed.
- C. Antoni, L. Vera, L. Devel, M. P. Catalani, B. Czarny, E. Cassar-Lajeunesse, E. Nuti, A. Rossello, V. Dive and E. A. Stura, J. Struct. Biol., 2013, 182, 246–254 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1847955 and 1864483. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02676j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |