
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective aromatic C–H λ3-iodanation with a cyclic iodine(III) electrophile in solution and solid phases†
Wei
Ding
a,
Chen
Wang
 ab,
Jie Ren
Tan
a,
Chang Chin
Ho
a,
Felix
León
a,
Felipe
García
*a and
Naohiko
Yoshikai
*a
ab,
Jie Ren
Tan
a,
Chang Chin
Ho
a,
Felix
León
a,
Felipe
García
*a and
Naohiko
Yoshikai
*a
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: fgarcia@ntu.edu.sg; nyoshikai@ntu.edu.sg
bZhejiang Key Laboratory of Alternative Technologies for Fine Chemicals Process, Shaoxing University, Shaoxing 312000, China
First published on 23rd June 2020
Abstract
An efficient and site-selective aromatic C–H λ3-iodanation reaction is achieved using benziodoxole triflate (BXT) as an electrophile under room temperature conditions. The reaction tolerates a variety of electron-rich arenes and heteroarenes to afford the corresponding arylbenziodoxoles in moderate to good yields. The reaction can also be performed mechanochemically by grinding a mixture of solid arenes and BXT under solvent-free conditions. The arylbenziodoxoles can be used for various C–C and C–heteroatom bond formations, and are also amenable to further modification by electrophilic halogenation. DFT calculations suggested that the present reaction proceeds via a concerted λ3-iodanation–deprotonation transition state, where the triflate anion acts as an internal base.
Introduction
Direct and site-selective C–H functionalization of arenes and heteroarenes with a leaving group that can allow aryl–carbon and aryl–heteroatom bond formations offers a highly attractive strategy to access diverse (hetero)aromatic compounds. Transition metal-catalyzed arene C–H borylations, which install a boryl group as an electrofugal leaving group on arenes, are among the most powerful C–H functionalization methods of this type.1 The installation of a nucleofugal leaving group on arenes can be equally attractive and complementary to C–H borylation. However, traditional electrophilic halogenation reactions often suffer from drawbacks such as incomplete site selectivity and harsh reaction conditions.2,3 As such, methods for site-selective installation of synthetically versatile nucleofuges other than halogens are highly desirable.4Trivalent iodine (λ3-iodane) moieties represent a class of useful nucleofuges in arene transformations. Thus, vast arrays of aryl–carbon and aryl–heteroatom bond forming reactions have been developed using diaryliodonium salts as electrophilic aryl-transfer agents.5 Particularly attractive among diaryliodonium salts are the unsymmetrical ones that allow selective transfer of one of the aryl groups over the other, “dummy” aryl group (e.g., mesityl).6,7 A direct C–H λ3-iodanation approach has proved feasible for the synthesis of such compounds from electron-rich (hetero)arenes and bulky aryl-λ3-iodane reagents such as MesI(OH)OTs with an acidic activator, or from (hetero)arenes, bulky aryl iodide, oxidants and acids.8 However, diaryliodonium salts are potentially unstable for isolation and prolonged storage and are often used without isolation.8a–c In this context, aryl-λ3-iodane compounds bearing a cyclic benziodoxol(on)e (BX) moiety could be a valuable alternative or complement to diaryliodonium salts, as their non-ionic nature and rigid structure would endow them with enhanced stability for facile isolation and long-term storage as well as the ability to tolerate a wider range of reaction conditions. The BX group has already played an important role in the development of reagents for transferring groups such as trifluoromethyl, alkynyl, azido, and cyano groups.9 On the other hand, access to analogous (hetero)aryl–BX reagents remains relatively limited and their reactivity less explored. The direct preparation of aryl–BXs from arenes and 2-iodobenzoic acid was reported by Olofsson and Zhdankin (Scheme 1a).10 However, these compounds have not been utilized as aryl transfer agents. Instead, they were reported to produce 2-functionalized benzoic acids in the reaction with nucleophiles.10b More recently, Waser developed indole- and pyrrole-BX reagents via zinc-catalyzed BX transfer to indoles and pyrroles with acetoxy benziodoxolone and demonstrated their utility in C–H functionalization reactions (Scheme 1b),11,12 while the reaction required relatively high dilution and was not extended to arenes.
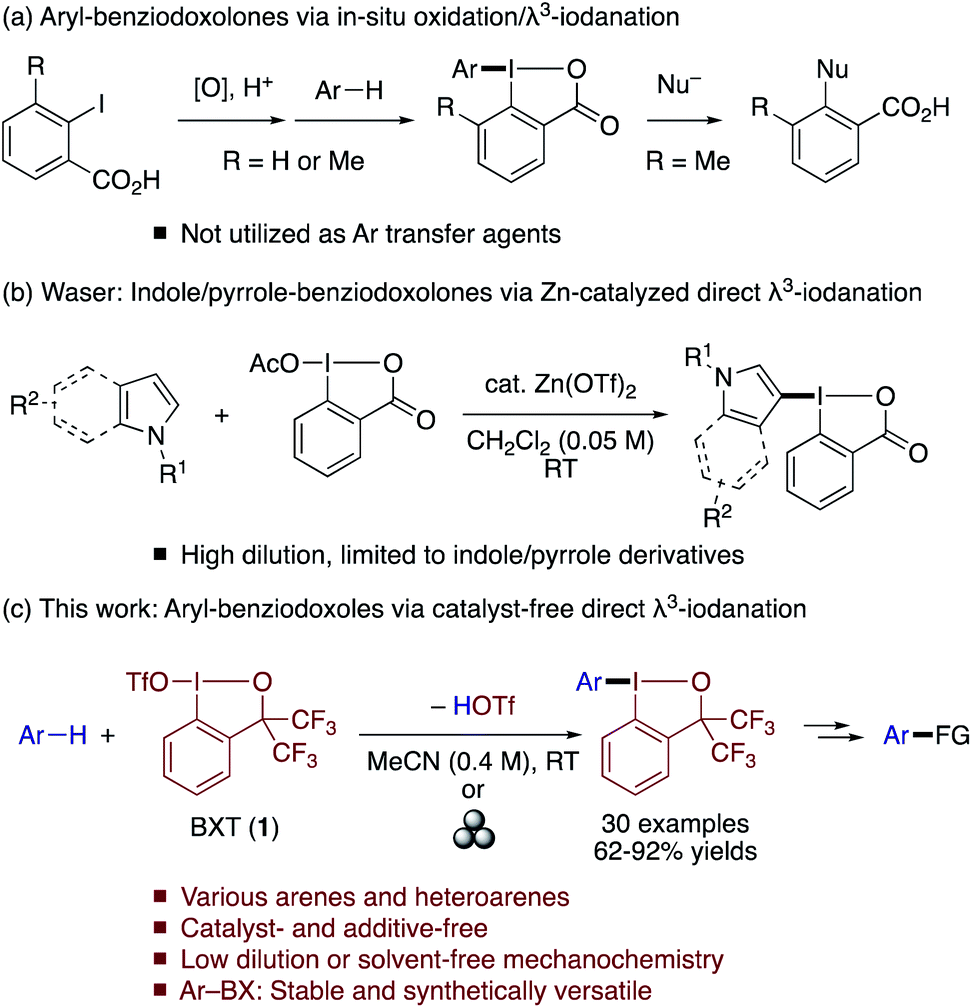 | ||
| Scheme 1 Synthetic approaches to (hetero)arylbenziodoxol(on)es via C–H λ3-iodanation. | ||
Herein, we report that Zhdankin's benziodoxole triflate (BXT; 1) derived from α,α-bis(trifluoromethyl)-2-iodobenzyl alcohol13 facilitates C–H λ3-iodanation of (hetero)arenes with high site selectivity (Scheme 1c). This BX transfer reaction tolerates various electron-rich arenes and heteroarenes, allowing facile preparation of the corresponding aryl–BX derivatives in moderate to excellent yields under simple and mild conditions without any catalyst or promoter. Moreover, the reaction can also be performed mechanochemically using solid arenes and BXT under solvent-free conditions. The aryl–BX compounds serve as aryl donors for a variety of C–C and C–heteroatom bond formations, and also tolerate further modification by electrophilic halogenation. Owing to the superior leaving group ability of the BX group, iodinated aryl–BX compounds can be used for chemoselective sequential cross-couplings.
Results and discussion
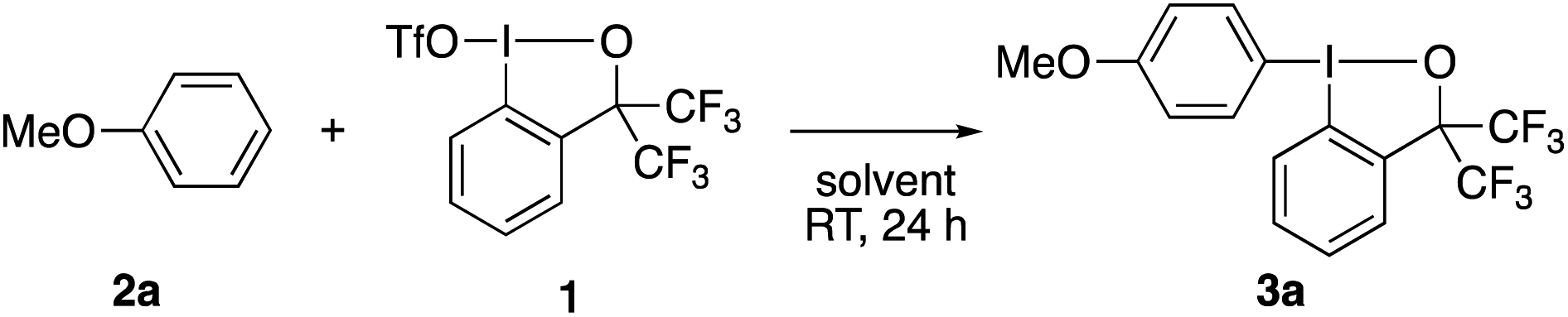
Pursuing our continuing interest in hypervalent iodine chemistry14 and encouraged by our recent success in using 1 as a λ3-iodane electrophile,15 the present study commenced with the exploration of the reaction between anisole (2a, 1.5 equiv.) and 1 (Table 1). The reaction proceeded smoothly in MeCN at room temperature, affording the aryl–BX product 3a with exclusive para-selectivity in 94% yield, which could be purified by routine chromatography and is stable to air and moisture (entry 1). No decomposition was observed upon storage at room temperature for at least one month. The reaction proved highly solvent-dependent. Aromatic and chlorinated solvents could be used albeit with a sizable decrease in the yield (entries 2–4). On the other hand, no product was observed in polar coordinating (DMF), ethereal (Et2O), and protic (MeOH) solvents (entries 5–7). The yield slightly dropped upon using an equimolar mixture of 2a and 1 or using 1 as the excess reagent (entries 8 and 9). Note that an analogous BXT reagent derived from 2-iodobenzoic acid13 failed to promote the desired λ3-iodanation of 2a. While the above and the following experiments to explore the substrate scope were all set up in an Ar-filled glove box, the reaction actually proved to be unaffected by air and moisture. Thus, the model reaction between 1 and 2a could be run in air to afford 3a in an equally high yield (95%).|
|
||
|---|---|---|
| Entry | Solvent | Yieldb (%) |
| a Unless otherwise noted, the reaction of 1 (0.10 mmol) and 2a (0.15 mmol) was carried out in the solvent (0.25 mL) at room temperature. b Determined by 19F NMR using 1,4-bis(trifluoromethyl)benzene as an internal standard. c 1 (0.10 mmol) and 2a (0.10 mmol) were used. d 1 (0.20 mmol) and 2a (0.10 mmol) were used. | ||
| 1 | MeCN | 94 |
| 2 | CH2Cl2 | 83 |
| 3 | Toluene | 65 |
| 4 | Chlorobenzene | 69 |
| 5 | DMF | 0 |
| 6 | Et2O | 0 |
| 7 | MeOH | 0 |
| 8c | MeCN | 84 |
| 9d | MeCN | 86 |
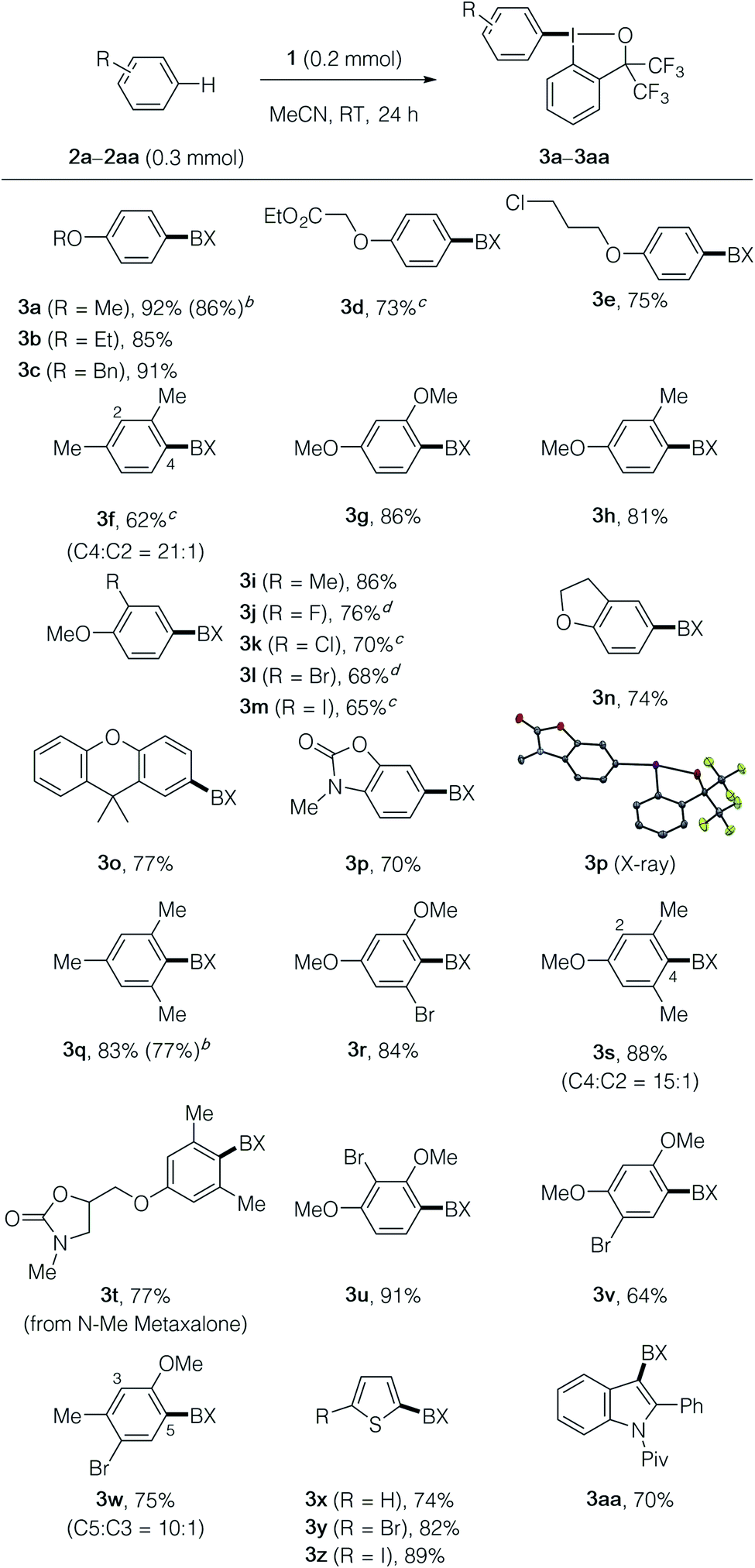
With the optimized reaction conditions in hand, we explored the scope of the λ3-iodanation reaction (Table 2). A variety of monoalkoxyarenes took part in the reaction to afford the desired aryl–BX products 3a–3e in high yields with exclusive para-selectivity. 1,3-Disubstituted arenes such as m-xylene, resorcinol dimethyl ether, and 3-methoxytoluene regioselectively afforded aryl–BX products 3f–3h with a 1,2,4-substitution pattern. The latter reacted exclusively at the para-position of the methoxy group. 1-Alkoxy-2-substituted benzene derivatives, including dihydrobenzofuran and 9,9-dimethylxanthene, were functionalized at the para-position of the alkoxy group, thus affording the products 3i–3o in moderate to good yields, while elevated temperatures (60–80 °C) were necessary for 1-methoxy-2-halobenzenes. 3-Methylbenzoxazol-2(3H)-one reacted exclusively at the para-position of the nitrogen atom, as unambiguously supported by X-ray crystallographic analysis of product 3p.16 Electron-rich 1,3,5-trisubstituted arenes such as mesitylene, 1-bromo-3,5-dimethoxybenzene, 3,5-dimethylanisole, and N-methyl metaxalone17 took part in the reaction to afford the products 3q–3t in good yields, where, for the latter three, the para-position of the alkoxy group was selectively functionalized. Other trisubstituted methoxyarenes with different substitution patterns were also amenable to the present λ3-iodanation (see the products 3u–3w). Electron-rich heteroarenes such as thiophene and indole derivatives could be employed as substrates for the present reaction, producing the products 3x–3aa in good yields. The scalability of the present λ3-iodanation reaction was demonstrated by gram-scale (3 mmol) synthesis of 3a and 3q, which could be achieved without a significant decrease in the yield (86% and 77% yields, respectively). It should be noted that the present method allows access to unique aryl–BX compounds such as 3m and 3z that contain iodonium and iodide moieties, which would offer opportunities for selective and sequential functionalizations (vide infra).
| a The reaction was performed on a 0.2 mmol scale according to the standard conditions described in Table 1, entry 1. BX in the product formula refers to the benziodoxole moiety. b The yield for a 3 mmol-scale reaction is given in the parentheses. c The reaction was performed at 60 °C. d The reaction was performed at 80 °C. |
|---|
|
Besides arenes and heteroarenes, 1,1-diphenylethene (2ab) and its para-fluoro-substituted derivative (2ac) reacted with BXT under the standard conditions to afford the corresponding alkenyl–BXs 3ab and 3ac in good yields (Scheme 2a).14c–e,18 The reaction of 1,1-di(p-tolyl)ethene (2ac) did not take place under the standard conditions but resulted in the decomposition of 1, while the addition of Na2CO3 and the use of chlorobenzene as the solvent allowed us to obtain the desired product 3ad in a moderate yield (Scheme 2b). Other alkenes such as styrene, α-methylstyrene, and vinylcyclohexane failed to participate in the present C–H λ3-iodanation.
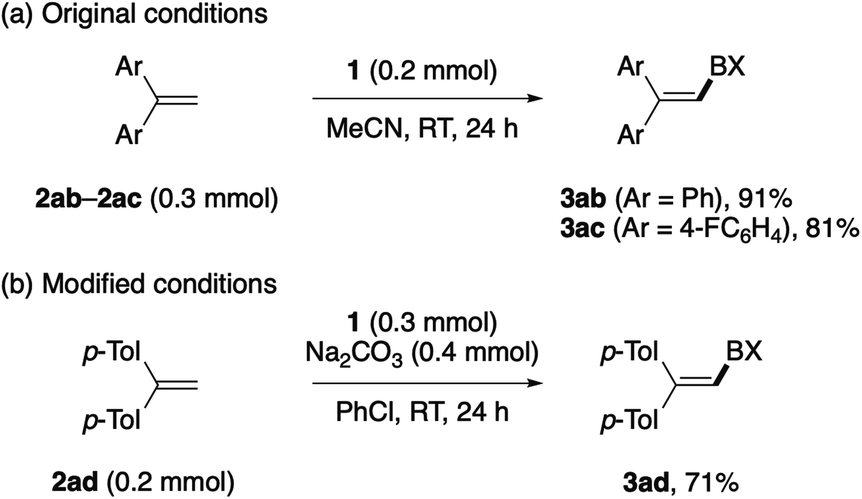 | ||
| Scheme 2 C–H λ3-iodanation of 1,1-diarylethylenes. | ||
Recently, mechanochemistry has emerged as a powerful tool in synthetic chemistry due to its attractive merits such as a solvent-free process, reduced reaction times, ability to engage poorly soluble solid compounds, and unique reactivity and selectivity.19 Notably, mechanochemistry has proved to offer an ideal means to accelerate the present λ3-iodanation under solvent-free conditions. Thus, by simply subjecting a mixture of an arene (0.3 mmol) and 1 (0.2 mmol) to ball milling (30 Hz for 2 h), aryl–BX derivatives 3o, 3q, 3r, and 3u were obtained in good yields (Scheme 3a). Note that, except for 3q, the starting arenes of these aryl–BXs are solid compounds. Additionally, the mechanochemical synthesis of 3r could be performed on a gram scale using a slight excess of the arene 2r, yielding an analytically pure product without chromatographic purification (Scheme 3b).
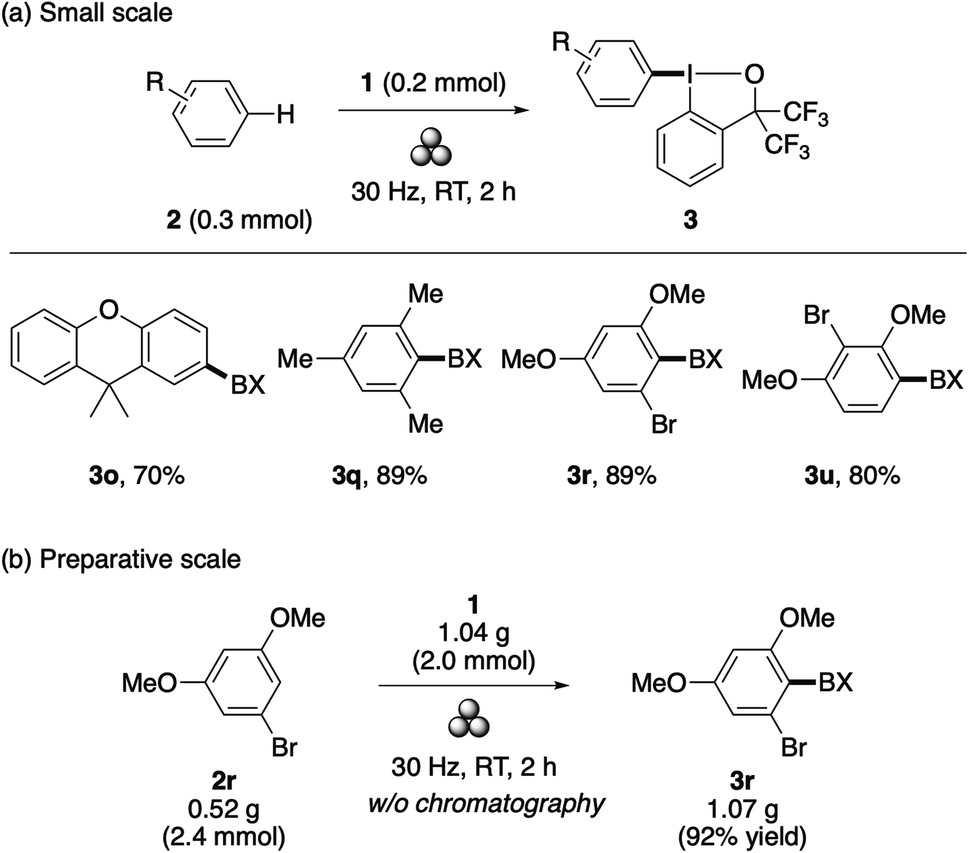 | ||
| Scheme 3 Mechanochemical λ3-iodanation of arenes. | ||
Some important limitations of the present λ3-iodanation should be noted. Less electron-rich, electron-neutral or electron-poor arenes did not participate well in the reaction with 1 even at elevated temperatures (up to 80 °C), and were largely recovered. However, the reaction of moderately electron-rich o-xylene, which was sluggish under the original conditions (28% yield at 60 °C), could be promoted by the addition of a Sc(OTf)3 catalyst (20 mol%) and the use of CH2Cl2 as the solvent to afford the corresponding aryl–BX product 3ae in 55% yield (Scheme 4a). Meanwhile, aryl–BXs bearing an electron-neutral phenyl (3af) or electron-deficient 4-bromophenyl (3ag) or 4-cyanophenyl (3ah) group could be synthesized in good yield via silicon-to-iodine(III) or boron-to-iodine(III) aryl transfer20,21 using the corresponding aryltrimethylsilane or aryltrifluoroborate (Scheme 4b and c). Highly electron-rich arenes, such as 1,2- and 1,4-dimethoxybenzenes and N,N-dimethylaniline, failed to give the desired aryl–BX products with 1 and decomposed to α,α-bis(trifluoromethyl)-2-iodobenzyl alcohol. This reduction of the I(III) reagent is likely caused by the ability of these electron-rich arenes to undergo single electron transfer.
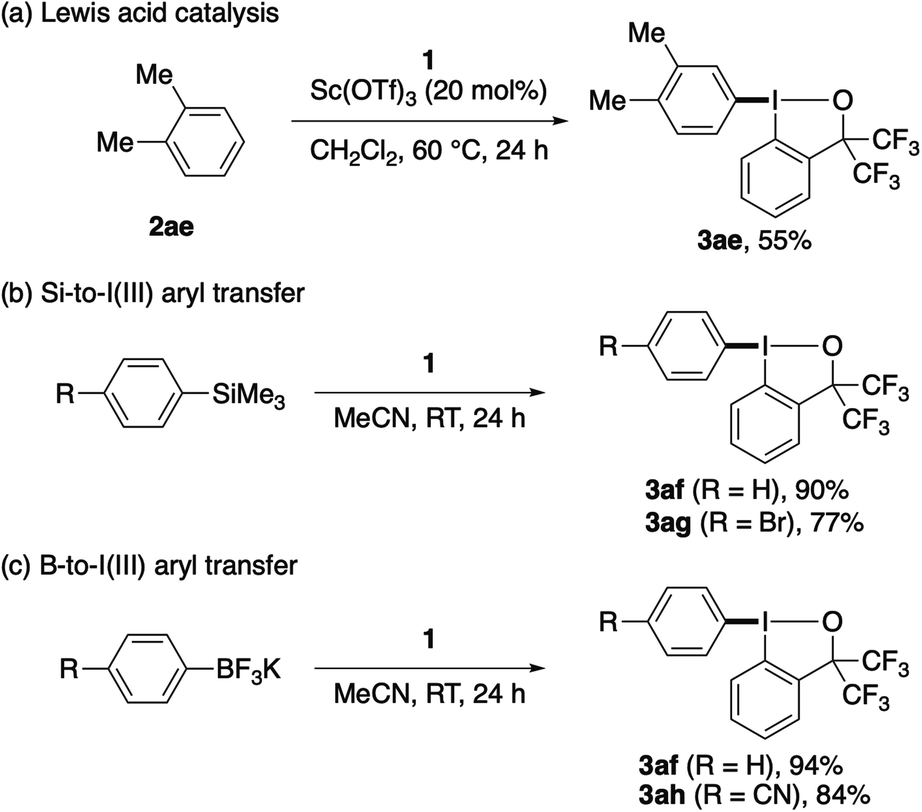 | ||
| Scheme 4 Preparation of aryl–BXs via Lewis acid-catalyzed C–H λ3-iodanation or aryl transfer from aryl silicon or boron reagents. | ||
The functionalized aryl–BX products obtained by the present reaction could be used as versatile building blocks for further transformations via selective transfer of the arene-derived aryl groups (Scheme 5). As illustrated in Scheme 5a, the benziodoxole moiety in 3a and 3q served as an excellent leaving group in Pd-catalyzed Suzuki–Miyaura, Sonogashira, and Stille couplings, and Miyaura borylation,22 which afforded biaryl 4, aryl alkyne 5, styrene 6, and aryl boronate 7, respectively, in good yields. In addition, aryl–BX 3a was amenable to Cu-catalyzed sulfonylation23 and cyanation,24 affording the corresponding aryl sulfone 8 and aryl nitrile 9, respectively. 3-Iodo-4-methoxyphenyl–BX 3m and 5-iodothien-2-yl–BX 3z underwent site-selective Sonogashira coupling with trimethylsilylacetylene on the aryl–BX moiety over the aryl–iodide moiety to afford the monoalkynylated products 10 and 12, respectively, which illustrated the superior leaving group ability of the BX group in oxidative addition to Pd(0) (Scheme 5b). Subsequent Sonogashira coupling of 10 and 12 with phenylacetylene provided unsymmetrically dialkynylated arenes 11 and 13, respectively. Finally, the compounds 3g and 3o could be regioselectively brominated or chlorinated without affecting the BX group to afford the products 3v, 14, and 15 in good yields (Scheme 5c).3e The sequential installation of BX and halogens may offer a means for chemoselective difunctionalization of aromatic compounds.
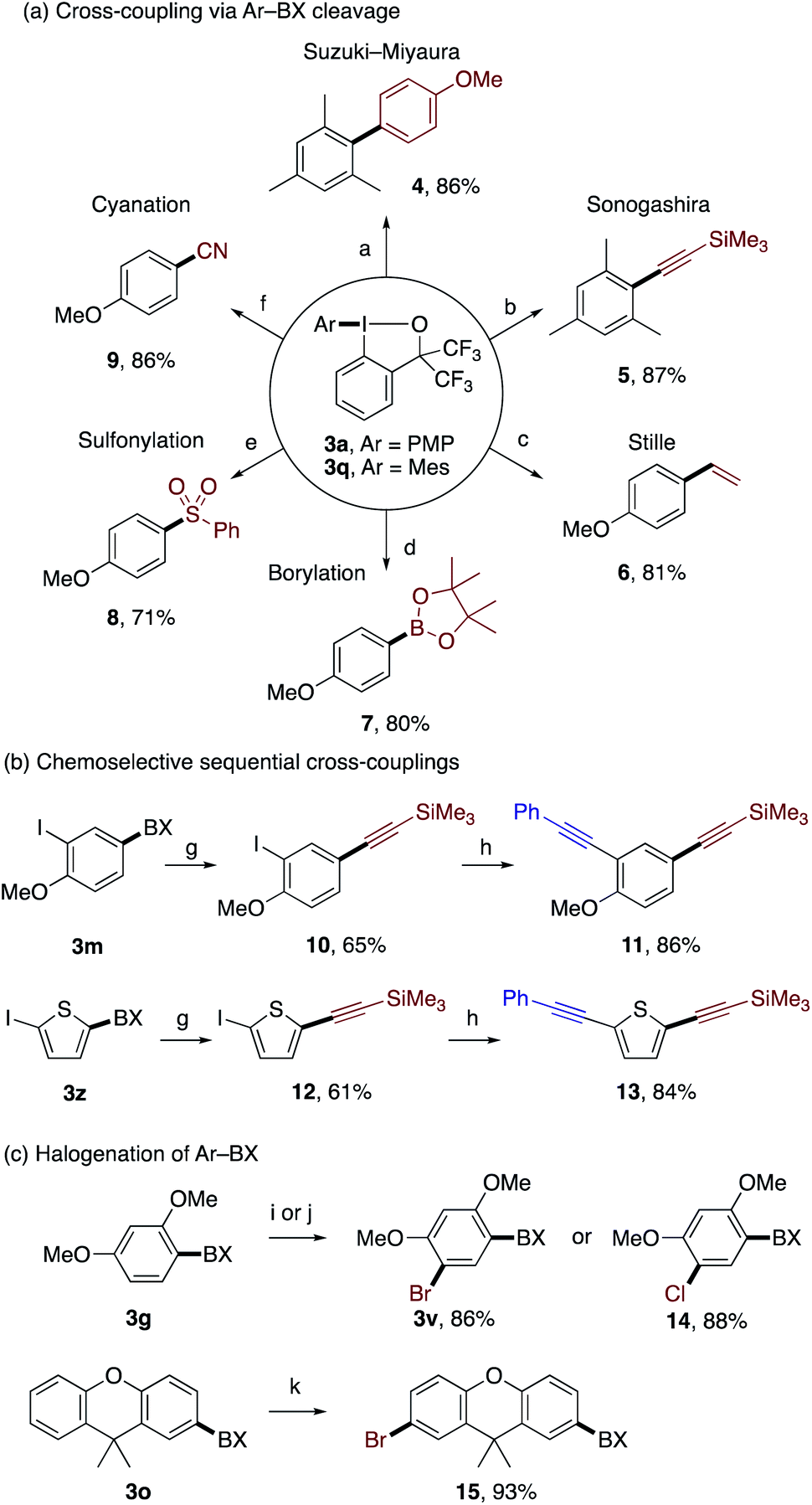 | ||
Scheme 5 Product transformations. Reaction conditions: (a) Pd(PPh3)4, 4-MeOC6H4B(OH)2, K2CO3, DMF/H2O, 100 °C, 8 h; (b) PdCl2(PPh3)2, CuI, trimethylsilylacetylene, Et3N, DMF, rt, 12 h; (c) PdCl2(PPh3)2, (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)SnBu3, DMF, 60 °C, 12 h; (d) Pd(OAc)2, PPh3, CuI, B2Pin2, Cs2CO3, MeCN, rt, 24 h; (e) CuI, L-proline sodium salt, PhSO2Na, DMSO, 90 °C, 24 h; (f) CuCN, L-proline, DMF, 90 °C, 24 h; (g) PdCl2(PPh3)2, CuI, trimethylsilylacetylene, Et3N, DMF, 0 °C, 12 h; (h) PdCl2(PPh3)2, CuI, phenylacetylene, THF/Et3N, rt, 12 h; (i) NBS, HFIP, 40 °C, 24 h; (j) NCS, HFIP, 80 °C, 24 h; and (k) NBS, HFIP, rt, 4 h. CH)SnBu3, DMF, 60 °C, 12 h; (d) Pd(OAc)2, PPh3, CuI, B2Pin2, Cs2CO3, MeCN, rt, 24 h; (e) CuI, L-proline sodium salt, PhSO2Na, DMSO, 90 °C, 24 h; (f) CuCN, L-proline, DMF, 90 °C, 24 h; (g) PdCl2(PPh3)2, CuI, trimethylsilylacetylene, Et3N, DMF, 0 °C, 12 h; (h) PdCl2(PPh3)2, CuI, phenylacetylene, THF/Et3N, rt, 12 h; (i) NBS, HFIP, 40 °C, 24 h; (j) NCS, HFIP, 80 °C, 24 h; and (k) NBS, HFIP, rt, 4 h. | ||
To gain insight into the mechanism of the C–H λ3-iodanation, DFT calculations on the reaction between 1 and anisole (2a) were performed (Fig. 1; see the ESI† for the computational details). Prior to the iodanation event, the triflate in 1 slipped to the trans position of the aryl group (1 to 1′viaTS1), which allows 2a to bind to the cis position of the aryl ligand (CP1).25 This is followed by a six-centered TS for concerted λ3-iodanation/deprotonation of the para-position (TS2p, ΔG‡ = 19.3 kcal mol−1) to give the product 3a and TfOH, which is reminiscent of the concerted metalation–deprotonation mechanism in aromatic C–H activation by transition metal carboxylates.25,26 Note that λ3-iodanation without prior isomerization of 1 requires an unreasonably high activation energy (37.3 kcal mol−1; Fig. S4†). The calculated endergonicity (2.1 kcal mol−1) might be a reflection of a computational artifact, which does not take the interaction between TfOH and MeCN (in the solution-phase reaction) or the intermolecular hydrogen bonding between TfOH molecules (in the solid-phase reaction) into account. Consistent with the experiment, analogous transition states for the λ3-iodanation of the ortho- and meta-positions were found to require higher activation energies (20.5 kcal mol−1 and 27.5 kcal mol−1, respectively; Fig. S5†).
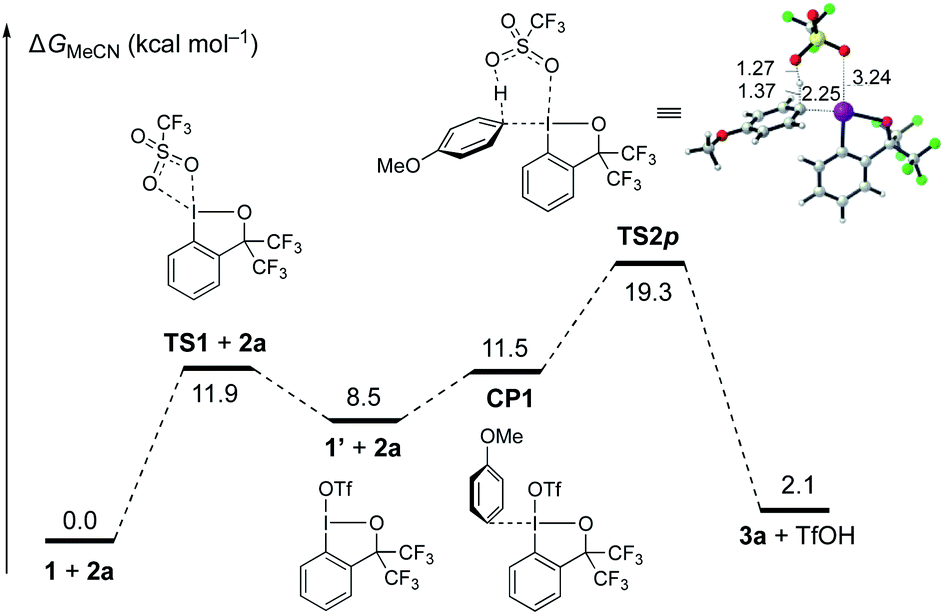 | ||
| Fig. 1 Gibbs free energy diagram for the para C–H λ3-iodanation of anisole (2a) with BXT (1). The bond distances are in Å. | ||
Conclusions
In summary, we have reported site-selective λ3-iodanation of aromatic compounds with benziodoxole triflate under simple and mild conditions. The reaction tolerates a variety of electron-rich arenes and heteroarenes, affording arylbenziodoxole derivatives in moderate to excellent yields. Mechanochemistry has proved to offer further improvement, enabling an expedient and solvent-free reaction between solid reactants. The scope of the arylbenziodoxole synthesis based on this metal-free C–H λ3-iodanation may be complemented by the Lewis acid-assisted λ3-iodanation or the aryl group transfer from the corresponding silicon and boron compounds. The thus-synthesized arylbenziodoxoles serve as versatile synthetic intermediates for C–C and C–heteroatom bond formations and for electrophilic aromatic substitution. Further studies on the use of benziodoxole triflate and related compounds as iodane transfer agents and the transformation of organo–BX compounds are currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Singapore Ministry of Education (Academic Research Fund Tier 2 (MOE2016-T2-2-043) and Tier 1 (RG114/18) to N. Y.) and Nanyang Technological University. F. G. and F. L. acknowledge A*STAR (AME IRG A1783c0003) and the NTU Start-Up Grant (M4080552) for financial support. We thank Dr Yongxin Li (Nanyang Technological University) for his assistance with the X-ray crystallographic analysis.Notes and references
- (a) I. A. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (b) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef CAS; (c) A. Ros, R. Fernandez and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC; (d) Y. Kuroda and Y. Nakao, Chem. Lett., 2019, 48, 1092–1100 CrossRef CAS.
- (a) G. A. Olah, Acc. Chem. Res., 1971, 4, 240–248 CrossRef CAS; (b) R. Taylor, Electrophilic Aromatic Substitution, John Wiley & Sons, New York, 1990 Search PubMed.
- (a) F. Mo, J. M. Yan, D. Qiu, F. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 2028–2032 CrossRef CAS PubMed; (b) S. M. Maddox, C. J. Nalbandian, D. E. Smith and J. L. Gustafson, Org. Lett., 2015, 17, 1042–1045 CrossRef CAS PubMed; (c) R. C. Samanta and H. Yamamoto, Chem.–Eur. J., 2015, 21, 11976–11979 CrossRef CAS PubMed; (d) X. Xiong, F. Tan and Y. Y. Yeung, Org. Lett., 2017, 19, 4243–4246 CrossRef CAS PubMed; (e) R. J. Tang, T. Milcent and B. Crousse, J. Org. Chem., 2018, 83, 930–938 CrossRef CAS PubMed.
- F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223–228 CrossRef CAS PubMed.
- (a) N. R. Deprez and M. S. Sanford, Inorg. Chem., 2007, 46, 1924–1935 CrossRef CAS PubMed; (b) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed; (c) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- For selected examples, see: (a) Y. Kita, K. Morimoto, M. Ito, C. Ogawa, A. Goto and T. Dohi, J. Am. Chem. Soc., 2009, 131, 1668–1669 CrossRef CAS PubMed; (b) N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 11234–11241 CrossRef CAS PubMed; (c) T. Dohi, M. Ito, N. Yamaoka, K. Morimoto, H. Fujioka and Y. Kita, Angew. Chem., Int. Ed., 2010, 49, 3334–3337 CrossRef CAS PubMed; (d) E. Cahard, H. P. Male, M. Tissot and M. J. Gaunt, J. Am. Chem. Soc., 2015, 137, 7986–7989 CrossRef CAS PubMed; (e) M. S. McCammant, S. Thompson, A. F. Brooks, S. W. Krska, P. J. H. Scott and M. S. Sanford, Org. Lett., 2017, 19, 3939–3942 CrossRef CAS PubMed; (f) D. H. Lukamto and M. J. Gaunt, J. Am. Chem. Soc., 2017, 139, 9160–9163 CrossRef CAS PubMed; (g) J. Rae, J. Frey, S. Jerhaoui, S. Choppin, J. Wencel-Delord and F. Colobert, ACS Catal., 2018, 8, 2805–2809 CrossRef CAS; (h) W. Shang, Z. D. Mou, H. Tang, X. Zhang, J. Liu, Z. Fu and D. Niu, Angew. Chem., Int. Ed., 2018, 57, 314–318 CrossRef CAS PubMed.
- D. R. Stuart, Chem.–Eur. J., 2017, 23, 15852–15863 CrossRef CAS PubMed.
- (a) I. Sokolovs, D. Lubriks and E. Suna, J. Am. Chem. Soc., 2014, 136, 6920–6928 CrossRef CAS PubMed; (b) B. Berzina, I. Sokolovs and E. Suna, ACS Catal., 2015, 5, 7008–7014 CrossRef CAS; (c) I. Sokolovs and E. Suna, J. Org. Chem., 2016, 81, 371–379 CrossRef CAS PubMed; (d) G. Laudadio, H. P. L. Gemoets, V. Hessel and T. Noel, J. Org. Chem., 2017, 82, 11735–11741 CrossRef CAS PubMed; (e) T. Dohi, T. Hayashi, S. Ueda, T. Shoji, K. Komiyama, H. Takeuchi and Y. Kita, Tetrahedron, 2019, 75, 3617–3627 CrossRef CAS; (f) E. Lindstedt, M. Reitti and B. Olofsson, J. Org. Chem., 2017, 82, 11909–11914 CrossRef CAS PubMed.
- (a) V. V. Zhdankin, Curr. Org. Synth., 2005, 2, 121–145 CrossRef CAS; (b) J. P. Brand, D. F. Gonzalez, S. Nicolai and J. Waser, Chem. Commun., 2011, 47, 102–115 RSC; (c) J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165–4179 RSC; (d) J. Charpentier, N. Fruh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed; (e) Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436–4454 CrossRef CAS PubMed; (f) J. Waser, Synlett, 2016, 27, 2761–2773 CrossRef CAS; (g) D. P. Hari, P. Caramenti and J. Waser, Acc. Chem. Res., 2018, 51, 3212–3225 CrossRef CAS PubMed.
- (a) E. A. Merritt and B. Olofsson, Eur. J. Org. Chem., 2011, 3690–3694 CrossRef CAS; (b) M. S. Yusubov, R. Y. Yusubova, V. N. Nemykin and V. V. Zhdankin, J. Org. Chem., 2013, 78, 3767–3773 CrossRef CAS PubMed.
- (a) P. Caramenti, S. Nicolai and J. Waser, Chem.–Eur. J., 2017, 23, 14702–14706 CrossRef CAS PubMed; (b) P. Caramenti and J. Waser, Helv. Chim. Acta, 2017, 100, e1700221 CrossRef; (c) P. Caramenti, R. K. Nandi and J. Waser, Chem.–Eur. J., 2018, 24, 10049–10053 CrossRef CAS PubMed; (d) E. Grenet, A. Das, P. Caramenti and J. Waser, Beilstein J. Org. Chem., 2018, 14, 1208–1214 CrossRef CAS PubMed; (e) E. Grenet and J. Waser, Org. Lett., 2018, 20, 1473–1476 CrossRef CAS PubMed.
- (a) K. Ishida, H. Togo and K. Moriyama, Chem.–Asian J., 2016, 11, 3583–3588 CrossRef CAS PubMed; (b) K. Morimoto, Y. Ohnishi, D. Koseki, A. Nakamura, T. Dohi and Y. Kita, Org. Biomol. Chem., 2016, 14, 8947–8951 RSC.
- V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, J. T. Bolz and A. J. Simonsen, J. Org. Chem., 1996, 61, 6547–6551 CrossRef CAS PubMed.
- (a) B. Lu, J. Wu and N. Yoshikai, J. Am. Chem. Soc., 2014, 136, 11598–11601 CrossRef CAS PubMed; (b) J. Wu and N. Yoshikai, Angew. Chem., Int. Ed., 2015, 54, 11107–11111 CrossRef CAS PubMed; (c) J. Wu, X. Deng, H. Hirao and N. Yoshikai, J. Am. Chem. Soc., 2016, 138, 9105–9108 CrossRef CAS PubMed; (d) J. Wu, K. Xu, H. Hirao and N. Yoshikai, Chem.–Eur. J., 2017, 23, 1521–1525 CrossRef CAS PubMed; (e) J. Wu, X. Deng and N. Yoshikai, Chem.–Eur. J., 2019, 25, 7839–7842 CrossRef CAS PubMed.
- (a) B. Wu, J. Wu and N. Yoshikai, Chem.–Asian J., 2017, 12, 3123–3127 CrossRef CAS PubMed; (b) W. Ding, J. Chai, C. Wang, J. Wu and N. Yoshikai, J. Am. Chem. Soc., 2020, 142, 8619–8624 CrossRef CAS PubMed.
- CCDC 1981452 (3p) contains crystallographic data for this paper.
- S. See and R. Ginzburg, Pharmacotherapy, 2012, 28, 207–213 CrossRef PubMed.
- (a) T. Kitamura, T. Fukuoka and Y. Fujiwara, Synlett, 1996, 659–660 CrossRef CAS; (b) E. Stridfeldt, A. Seemann, M. J. Bouma, C. Dey, A. Ertan and B. Olofsson, Chem.–Eur. J., 2016, 22, 16066–16070 CrossRef CAS PubMed; (c) P. Caramenti, N. Declas, R. Tessier, M. D. Wodrich and J. Waser, Chem. Sci., 2019, 10, 3223–3230 RSC.
- (a) J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed; (b) J. G. Hernandez and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef CAS PubMed; (c) J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC; (d) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC; (e) D. Tan and F. García, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC; (f) T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed.
- (a) G. F. Koser, R. H. Wettach and C. S. Smith, J. Org. Chem., 1980, 45, 1543–1544 CrossRef CAS; (b) M. Ochiai, K. Sumi, Y. Takaoka, M. Kunishima, Y. Nagao, M. Shiro and E. Fujita, Tetrahedron, 1988, 44, 4095–4112 CrossRef CAS; (c) P. J. Stang, V. V. Zhdankin, R. Tykwinski and N. S. Zefirov, Tetrahedron Lett., 1991, 32, 7497–7498 CrossRef CAS; (d) P. J. Stang and V. V. Zhdankin, J. Am. Chem. Soc., 1993, 115, 9808–9809 CrossRef CAS.
- (a) M. Ochiai, M. Toyonari, T. Nagaoka, D.-W. Chen and M. Kida, Tetrahedron Lett., 1997, 38, 6709–6712 CrossRef CAS; (b) M. A. Carroll, V. W. Pike and D. A. Widdowson, Tetrahedron Lett., 2000, 41, 5393–5396 CrossRef CAS; (c) M. Yoshida, K. Osafune and S. Hara, Synthesis, 2007, 1542–1546 CrossRef CAS; (d) M. Bielawski, D. Aili and B. Olofsson, J. Org. Chem., 2008, 73, 4602–4607 CrossRef CAS PubMed; (e) L. Qin, B. Hu, K. D. Neumann, E. J. Linstad, K. McCauley, J. Veness, J. J. Kempinger and S. G. DiMagno, Eur. J. Org. Chem., 2015, 5919–5924 CrossRef CAS PubMed.
- J. Ratniyom, N. Dechnarong, S. Yotphan and S. Kiatisevi, Eur. J. Org. Chem., 2014, 2014, 1381–1385 CrossRef CAS.
- W. Zhu and D. Ma, J. Org. Chem., 2005, 70, 2696–2700 CrossRef CAS PubMed.
- D. Wang, L. Kuang, Z. Li and K. Ding, Synlett, 2008, 69–72 Search PubMed.
- S. Izquierdo, S. Essafi, I. Del Rosal, P. Vidossich, R. Pleixats, A. Vallribera, G. Ujaque, A. Lledos and A. Shafir, J. Am. Chem. Soc., 2016, 138, 12747–12750 CrossRef CAS PubMed.
- (a) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1981452. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02737e |
| This journal is © The Royal Society of Chemistry 2020 |