
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactions of an anionic chelate phosphane/borata-alkene ligand with [Rh(nbd)Cl]2, [Rh(CO)2Cl]2 and [Ir(cod)Cl]2†‡
Kohei
Watanabe§
 a,
Atsushi
Ueno
a,
Xin
Tao
a,
Karel
Škoch
a,
Xiaoming
Jie
a,
Sergei
Vagin
b,
Bernhard
Rieger
b,
Constantin G.
Daniliuc
a,
Matthias C.
Letzel
a,
Gerald
Kehr
a and
Gerhard
Erker
*a
a,
Atsushi
Ueno
a,
Xin
Tao
a,
Karel
Škoch
a,
Xiaoming
Jie
a,
Sergei
Vagin
b,
Bernhard
Rieger
b,
Constantin G.
Daniliuc
a,
Matthias C.
Letzel
a,
Gerald
Kehr
a and
Gerhard
Erker
*a
aOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: erker@uni-muenster.de
bWacker-Lehrstuhl für Makromolekulare Chemie, Fakultät für Chemie, Technische Universität München, Lichtenbergstraße 4, 85747 Garching bei München, Germany
First published on 19th June 2020
Abstract
Borata-alkenes can serve as anionic olefin equivalent ligands in transition metal chemistry. A chelate ligand of this type is described and used for metal coordination. Deprotonation of the Mes2P(CH2)2B(C6F5)2 frustrated Lewis pair in the α-CH[B] position gave the methylene-bridged phosphane/borata-alkene anion. It reacted with the [Rh(nbd)Cl] or [Rh(CO)2Cl] dimers to give the respective neutral chelate [P/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B][Rh] complexes. The reaction of the [P/CB]− anion with [Ir(cod)Cl]2 proceeded similarly, only that the complex underwent a subsequent oxidative addition reaction at the mesityl substituent. Both the resulting Ir(III)hydride complex 15 and the P/borata-alkene Rh system 12 were used as hydrogenation catalysts. The [P/CB(C6F5)2]Rh(nbd) complex 12 served as a catalyst for arylacetylene polymerization.
B][Rh] complexes. The reaction of the [P/CB]− anion with [Ir(cod)Cl]2 proceeded similarly, only that the complex underwent a subsequent oxidative addition reaction at the mesityl substituent. Both the resulting Ir(III)hydride complex 15 and the P/borata-alkene Rh system 12 were used as hydrogenation catalysts. The [P/CB(C6F5)2]Rh(nbd) complex 12 served as a catalyst for arylacetylene polymerization.
Introduction
Carbanions in the α-position to boryl groups show a conjugative interaction with the adjacent Lewis acid. Such systems can be described as borata-alkenes. Borata-alkenes derived from some alkyldiarylboranes had previously been prepared.1 Typically, short CB bond lengths around 1.45 Å were found in these systems. In addition, a variety of related boryl-carbanion ↔ borata-alkene systems were in situ generated and employed as reagents e.g. in borata-Wittig olefination chemistry.2 These reactions are the formal boron analogues of the conventional phosphorus ylide derived Wittig olefination reaction of organic carbonyl compounds.3
It was recently shown that the presence of the strongly electron-withdrawing –B(C6F5)2 group resulted in a markedly increased α-CH acidity in the respective boranes. A DFT study had revealed that e.g. H3C–B(C6F5)2 showed a pKa-value comparable to that of cyclopentadiene.4 According to this study the H3C–B(C6F5)2 borane must be considered >10 pKa values more C–H acidic than the related H3C-BMes2 borane. Consequently, R–H2C–B(C6F5)2 systems were easily deprotonated to give the corresponding [R–HCB(C6F5)2]− borata-alkene systems. Several of such systems were isolated as their Li+ salts. Some were used in borata-Wittig olefination reactions.5
Neutral bora-alkene compounds had previously been used as ligands6 and there are reports about the use of borata-benzenes in organometallic chemistry.7 There are a few examples of η3-borata-allyl metal complexes and related systems known.8 Piers et al. had prepared the borata-alkene tantalocene complex 4 (Scheme 1)9 and emphasized the relation of the anionic η2-[H2CB(C6F5)2]− ligand with the neutral η2-olefin analogues. The Piers group developed some follow-up chemistry of complex 4.9 C. Martin et al. have just recently described a conceptually related borata-phenanthrene gold complex.10,11
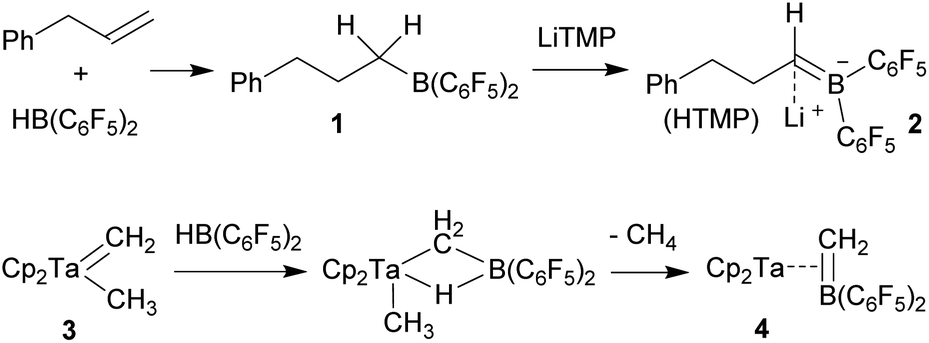 | ||
| Scheme 1 Formation of borata-alkene derivatives containing the B(C6F5)2 unit. | ||
Formal substitution of a hydrogen atom of the borata-alkene CH2– terminus by a Mes2P–CH2-substituent now gave an anionic [P/CB] system that served as a chelate ligand in Rh and Ir coordination chemistry.12 The preparation of first examples of this class of compounds and some uses are described in this account.
Results and discussion
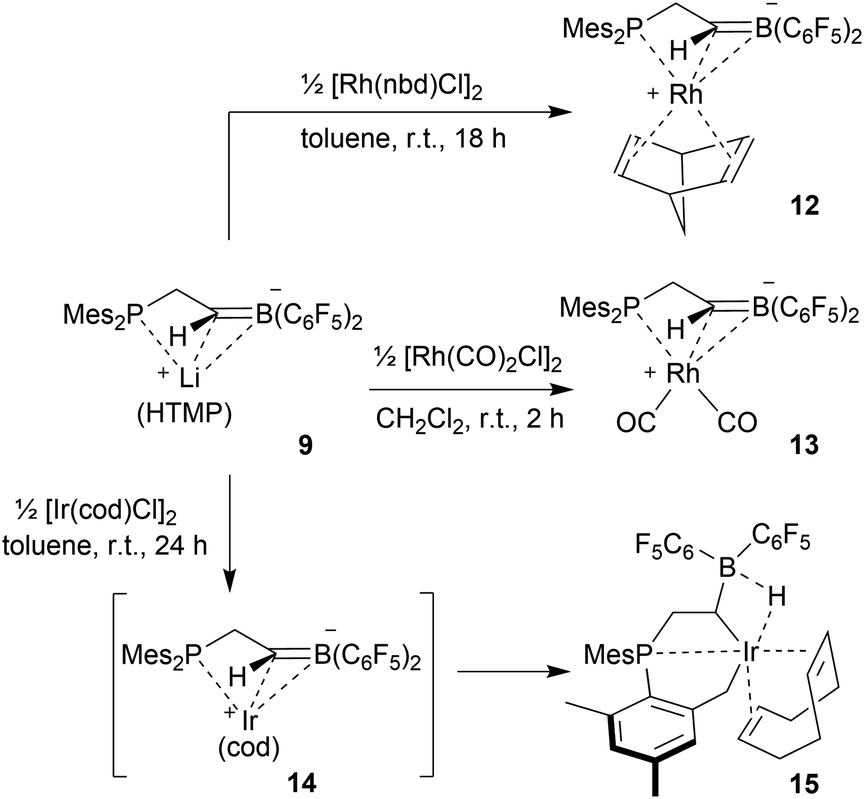
Development of the chelate phosphane/borata-alkene ligand system
We started our phosphane/borata-alkene chelate ligand synthesis from the ethylene-bridged frustrated P/B Lewis pair (FLP) 7.13 This was obtained from the hydroboration reaction of Mes2P-vinyl (5) with Piers' borane [HB(C6F5)2] (6)14 as we had previously reported.15We first attempted deprotonation of 7 at the α-position to the boron atom by treatment with LDA (r.t., pentane, 16 h). Compound 7 is α-CH acidic, but it is also an active boron Lewis acid that is able to abstract hydride from amines in the α-position to nitrogen with iminium salt formation.16 We found that a variant of the latter reaction is favoured in this system. Hydride abstraction from an isopropyl substituent of the LDA reagent by the borane Lewis acid functional group of 7 generated the respective imine. This is found as a component in the product 8 that we isolated from the reaction mixture as a white solid in 67% yield (Scheme 2). Compound 8 was characterized by an X-ray crystal structure analysis (Fig. 1). It shows the intact Mes2PCH2CH2B(C6F5)2 backbone. The boron atom shows a pseudotetrahedral coordination geometry (ΣB1CCC 333.2°); it has hydride attached. The lithium cation shows contacts to the [B]-H moiety, the phosphorus atom and one ortho-C6F5 fluorine atom. The Li cation has the newly formed imine moiety N-coordinated. In solution compound 8 shows a 11B NMR [B]-H doublet at δ −18.4 with a 1JBH ∼70 Hz coupling constant and a 7Li NMR (C6D6) signal at δ 1.4. The –NCMe2 imine 13C NMR resonance occurs at δ 173.5.
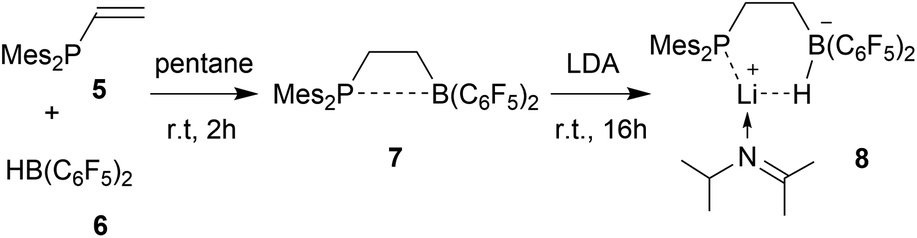 | ||
| Scheme 2 Reaction of the FLP 7 with LDA. | ||
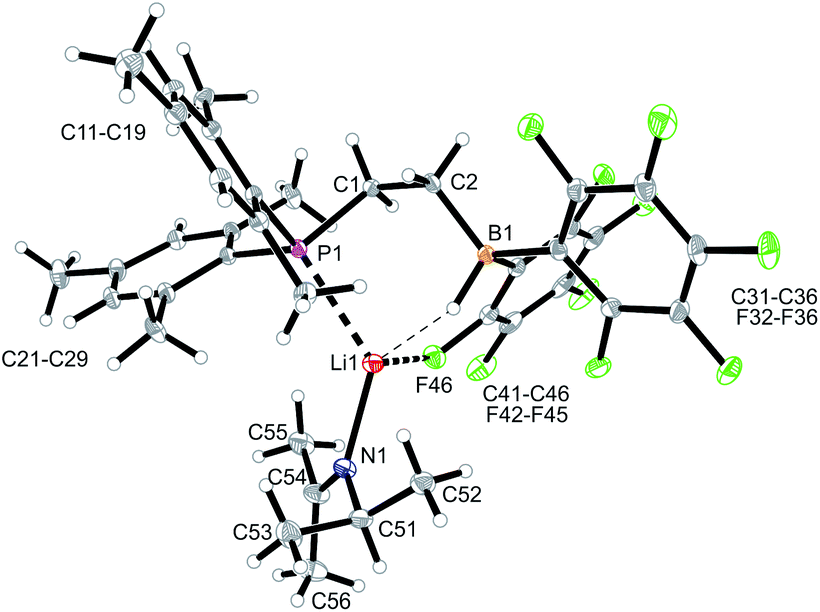 | ||
| Fig. 1 Molecular structure of compound 8 (thermal ellipsoids at 15% probability). Selected bond lengths (Å) and angles (°): P1–Li1 2.536(6) F46–Li1, 2.051(7), N1–C51 1.475(5), N1–C54 1.297(5), C2–B1–C31 115.8(3), C2–B1–C41 108.3(3), C31–B1–C41 109.1(3). | ||
In order to avoid the unwanted N–CH hydride abstraction we reacted the FLP 7 with the LiTMP reagent, a base that has no CH groups α to nitrogen. The reaction was carried out with the in situ generated FLP 7. Treatment with LiTMP in pentane for 16 h at room temperature followed by workup gave the methylene-linked phosphane/borata-alkene product 9 (Scheme 3), that we isolated as a white solid in 78% yield.
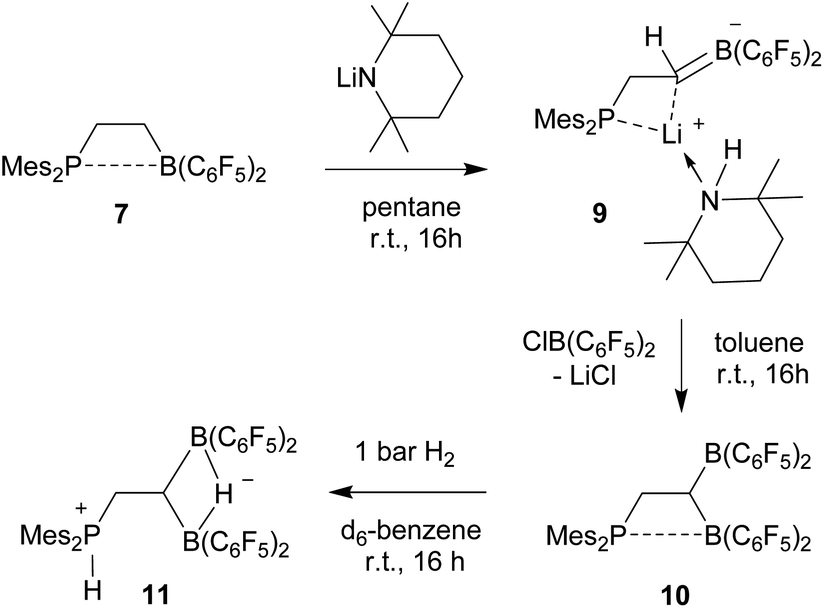 | ||
| Scheme 3 Formation and borylation reaction of the borata-alkene 9. | ||
Compound 9 was characterized by C,H,N elemental analysis, by spectroscopy and by X-ray diffraction. The X-ray crystal structure analysis confirmed the formation of the borata-alkene functionality. It shows the typical short B1–C2 linkage of 1.441(3)Å, which is much shorter than the adjacent boron-aryl bonds (B1–C31: 1.607(3)Å, B1–C41: 1.602(3)Å). The boron coordination geometry in compound 9 is trigonal-planar (ΣB1CCC 360.0°). The B1–C2–C1 angle amounts to 126.4(2)°. The lithium ion in 9 shows contacts to the borata-alkene unit as well as to the phosphorus atom and one ortho-C6F5 fluorine atom. The lithium atom Li+ also has the HTMP amine ligand bonded to it that had been formed in the deprotonation process (Fig. 2).
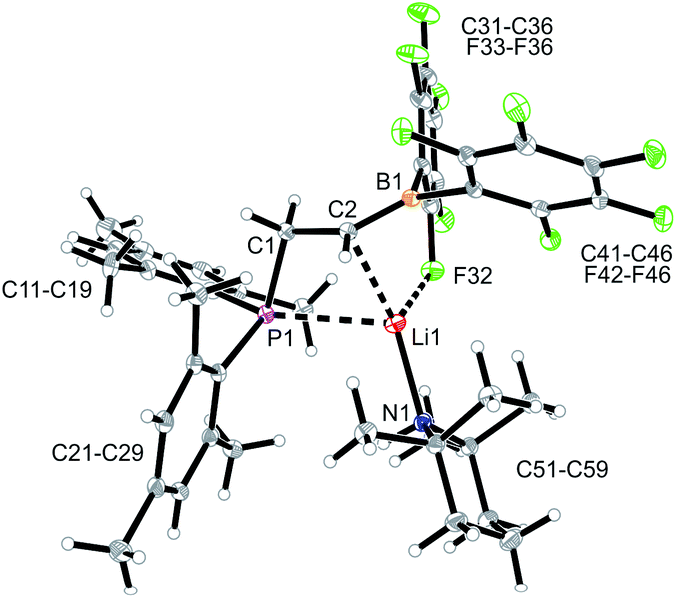 | ||
| Fig. 2 A view of the molecular structure of the phosphane/borata-alkene system 9 (thermal ellipsoids at 15% probability). Selected bond lengths (Å) and angles (°): B1–C2 1.441(3), B1–C31 1.607(3), B1–C41 1.602(3), B1–Li1 2.654(5), C1–P1 1.855(2), C2–Li1 2.367(4), F32–Li1 2.076(4), B1–C2–Li1 84.6 (2), B1–C2–C1 126.4(2), B1–Li1–C2 32.7(1), C2–B1–Li1 62.7(2), C2–B1–C31 120.7(2), C2–B1–C41 123.0(2), C31–B1–C41 116.3(2). | ||
In solution (THF-d8) compound 9 features a typical borata-alkene 11B NMR signal at δ 18.6. The 31P NMR signal is at δ −20.6 and the –CH2–CH backbone shows 1H NMR resonances at δ 4.21 (BCH; 13C: δ 106.7 (br)) and δ 3.36 (CH2; 13C: δ 33.5, 1JPC = 16.0 Hz). The 19F NMR spectrum of compound 9 shows two sets of o,p,m-resonances of the CB(C6F5)2 moiety (E and Z to the alkyl group at the adjacent sp2-hybridized borata-alkene carbon atom C2).
We briefly investigated the nucleophilic property of the borata-alkene unit in compound 9. For that purpose, we reacted it with the ClB(C6F5)2 reagent.14,17 The reaction (in toluene, r.t., 16 h) resulted in a substitution reaction at boron to give the P/B/B compound 10 (isolated as a yellow solid in 52% yield). It was characterized by C,H-elemental analysis, by spectroscopy and by its reaction with dihydrogen (see below). Compound 10 is a typical intramolecular FLP, showing a P–B interaction with one boron atom and having the other one free. However, the temperature dependent 19F NMR spectrum showed exchange between the pair of B(C6F5)2 groups at e.g. 299 K. Only at low temperature (e.g. 203 K) we observed a set of three broad 19F NMR resonances of a free trigonal planar B(C6F5)2 unit and a set of ten separate signals [four ortho, two para and four meta] of the rotationally hindered P⋯B(C6F5)2 group. The 31P NMR (299 K) signal of compound 10 is at δ 16.3 and the –CH2–CH backbone shows 1H NMR features at δ 3.55 and δ 4.30, respectively (13C: δ 29.2, 38.1 (br)).
Compound 10 reacted rapidly with dihydrogen under mild conditions (d6-benzene, r.t., 16 h, 1 bar H2) to give the phosphonium/hydridoborate dihydrogen splitting product 11 (isolated as a solid in 71% yield). The X-ray crystal structure analysis (Fig. 3) showed the presence of the phosphonium unit (ΣP1CCC 339.7°) and the newly formed hydride-bridged bis-borane moiety. In solution (CD2Cl2) the phosphonium [P]-H unit showed up at δ 7.58 (1H NMR) and δ −3.7 (31P, 1JPH ∼ 480 Hz), respectively. We recorded a broadened 11B NMR signal at δ −18.1 with a corresponding broad 1H NMR [B](μ-H) feature at δ 5.45. The 19F NMR spectrum of compound 11 shows two equal-intensity sets of o,p,m-C6F5 signals of the pair of B(C6F5)2 groups and we observed the 1H/13C NMR signals of the –CH2–CH backbone at δ 2.84/27.8 (PCH2) and δ 1.80/7.6(br)(BCH), respectively.
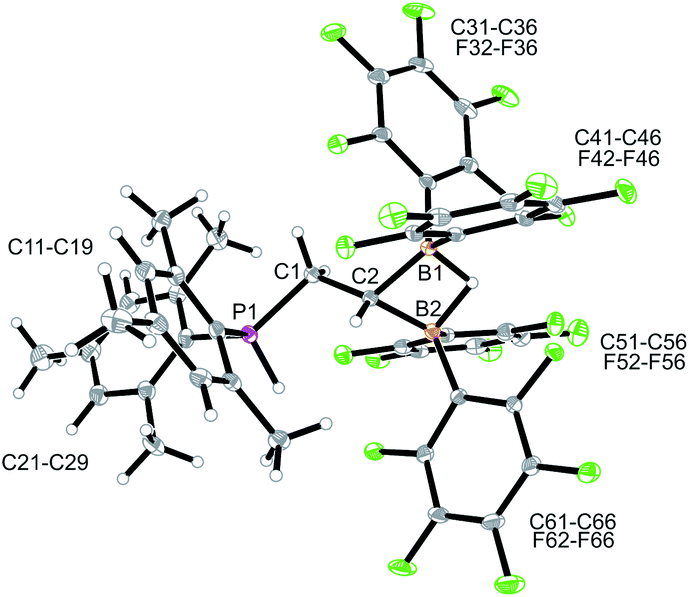 | ||
| Fig. 3 Molecular structure of the P/B/B dihydrogen splitting product 11 (thermal ellipsoids at 30% probability). Selected bond lengths (Å) and angles (°): B1–C2 1.601(3), B2–C2 1.601(3), C1–C2 1.524(3), C1–P1–C11 107.3(1), C1–P1–C21 121.1(1), C11–P1–C21 111.3(1). | ||
Synthesis and characterization of the P/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) B chelate metal complexes
B chelate metal complexes
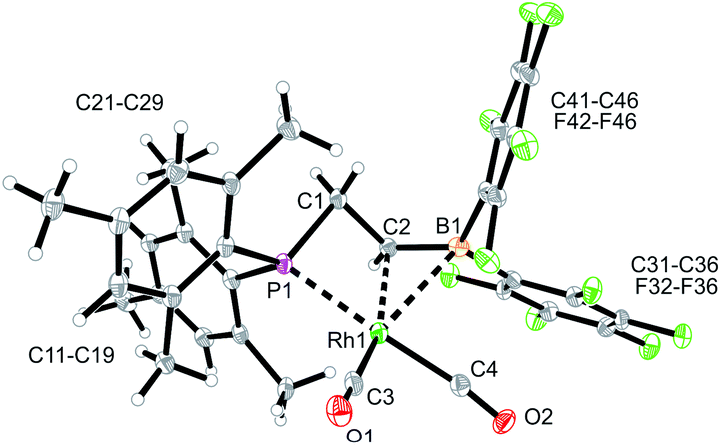
We used the methylene-bridged phosphane/borata-alkene anion of the lithium salt 9 as a chelate ligand in Rh chemistry. For that purpose, we treated the (norbornadiene)RhCl dimer with the prefabricated borata-alkene reagent 9 for 18 h in toluene solution at room temperature. Workup then gave the respective neutral chelate phosphane/borata-alkene(norbornadiene)Rh complex 12 in >60% yield (Scheme 4). Suitable crystals for the X-ray crystal structure analysis were obtained from slow diffusion of pentane into a saturated solution in dichloromethane at −30 °C (Fig. 4). Compound 12 shows a distorted square-planar coordination geometry at rhodium. The P/CB system serves as a chelate ligand. It is unsymmetrically η2-coordinated through both backbone atoms of the borata-alkene moiety and κP-bonded to the attached phosphanyl group. As the P/CB ligand is mono-anionic, the resulting Rh complex is neutral. The C2–B1 bond is only marginally elongated, it is still within the typical CB distance of borata-alkene examples1,5 (Table 1). The metal center has both olefinic π-systems of the norbornadiene ligand bonded through its endo-face. Both olefinic units are oriented perpendicular to the mean coordination plane of the transition metal center. In complex 12 the phosphane donor exhibits a stronger structural trans-effect18 than the borata-alkene ligand as judged from the respective Rh–C (olefin) bond lengths [trans: Rh1–C54: 2.214(2) Å, Rh1–C55: 2.213(2) Å; cis: Rh1–C51: 2.172(2) Å, Rh1–C52: 2.162(2) Å, see Fig. 4]
 | ||
| Scheme 4 Preparation of Rh and Ir complexes from the anion 9. | ||
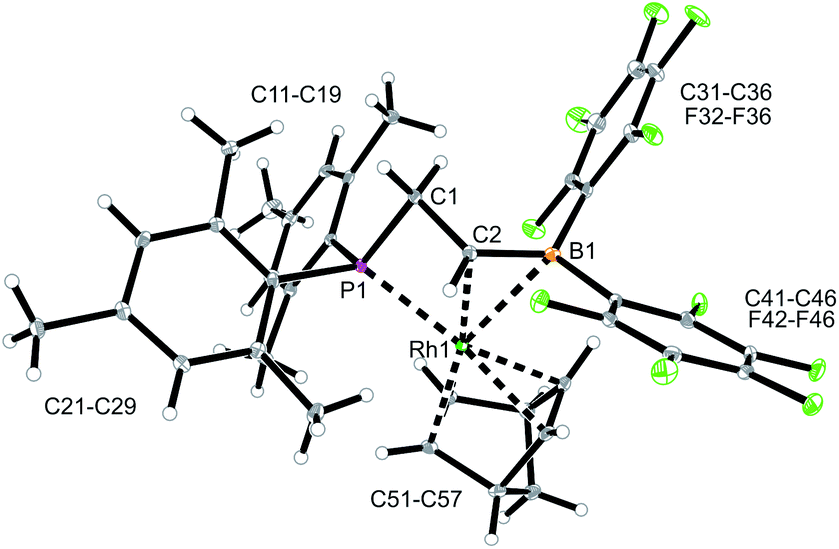 | ||
| Fig. 4 A view of the molecular structure of the chelate phosphane/borata-alkene Rh complex 12 (thermal ellipsoids at 30% probability). | ||
| 12 (Rh) | 15 (Ir)b | |
|---|---|---|
| a Bond lengths in Å, angles in °. b Two independent molecules, values are given for molecule A. | ||
| M–C2 | 2.270(2) | 2.166(4) |
| M–B1 | 2.611(2) | 2.463(5) |
| M–P1 | 2.346(1) | 2.328(1) |
| C2–B1 | 1.476(3) | 1.545(6) |
| M–C51 | 2.172(2) | 2.243(4) |
| M–C52 | 2.162(2) | 2.256(4) |
| B1–C2–C1 | 124.2(2) | 129.4(3) |
| C1–P1–M | 88.6(1) | 88.4(1) |
| ΣB1CCC | 357.2 | 346.2 |
In solution, complex 12 shows a 31P NMR signal (CD2Cl2) at δ −89.0 with a 1JRhP ∼ 120 Hz coupling constant. This changed only marginally when the spectrum of 12 was recorded in d8-THF solution. Compound 12 shows a 11B NMR signal at δ 24.3, a value that is similar to that of the uncomplexed borata-alkene anion 9 (see above).5 The 19F NMR spectrum of 12 shows two sets of o,p,m-C6F5 signals for the pair of pentafluorophenyl substituents at boron. We observed the 1H NMR signals of the chelate ligand backbone at δ 4.14/3.90 (PCH2) and δ 3.68 (BCH–), respectively (corresponding 13C NMR signals at δ 42.9 and 59.8(br)), and there are the 1H/13C NMR signals of the coordinated norbornadiene ligand at rhodium (see the ESI‡ for details).
The reaction of the P/borata-alkene lithium salt 9 with the chloro(dicarbonyl)Rh dimer was carried out at r.t (in dichloromethane, 2 h). Workup involving extraction with pentane and crystallization gave the neutral chelate [P/borata-alkene]Rh(CO)2 complex 13 as a yellow crystalline solid in 46% yield. The X-ray crystal structure analysis (Fig. 5) showed a distorted square planar coordination geometry around Rh. The phosphane (P1–Rh1: 2.335(1) Å) and the borata-alkene moiety of the chelate ligand are both bonded to rhodium (Rh1–C2: 2.251(4) Å, Rh1–B1: 2.590(5) Å). The B1–C2 linkage is found in the typical borata-alkene range at 1.476(7) Å. Again, the phosphane exerts a stronger trans effect than the CB unit [Rh1–C4 (CO trans to P1): 1.915(5) Å, Rh1–C3 (CO cis to P1): 1.868(5) Å]. Compound 13 shows strong IR CO bands at ν = 2069 and 1997 cm−1.19 In CD2Cl2 solution it shows a 11B NMR signal at δ 27.3, i.e. in the typical borata-alkene range. The 31P NMR resonance was located at δ −105.0 with a 1JRhP = 88.5 Hz coupling constant. The borata-alkene unit in complex 13 shows 19F NMR signals of a pair of inequivalent C6F5 substituents at boron.
 | ||
| Fig. 5 A view of the molecular structure of the chelate P/borata-alkene dicarbonyl Rh complex 13 (thermal ellipsoids at 30% probability). | ||
The reaction between the borata-alkene reagent 9 and the iridium(cyclooctadiene)chloride dimer was carried out similarly (toluene, 24 h, r.t.). It gave a slightly different outcome. We assume that initially a (P/borata-alkene)Ir(cod) complex 14 was generated, analogous to the formation of the Rh system 12. However, it was apparently not persistent under the prevailing reaction conditions but underwent intramolecular C–H bond activation20 at an ortho-methyl group of a mesityl substituent at phosphorus to give the oxidative addition product 15 (Scheme 4). It was isolated in 44% yield. Complex 15 was characterized spectroscopically and by X-ray diffraction (single crystals were obtained by crystallization from pentane at −30 °C).
The X-ray crystal structure analysis of complex 15 revealed that the iridium atom has undergone oxidative addition at a mesityl group at phosphorus, with formation of a new benzylic –CH2–Ir–H moiety (Fig. 6). The resulting Ir-hydride shows a contact to the boron atom. We note that the C2–B1 linkage in 15, consequently, is much longer than in 9 or 12, it corresponds to a short boron-carbon σ-bond. The Ir–C2 linkage is rather short (Table 1). The hydride is bridging between Ir and B [independent molecule A: Ir1A-H01 1.64(4) Å, H01–B1A 1.56(4) Å; molecule B: Ir1B–H02 1.59(4) Å, H02–B1B 1.50(4) Å] (Fig. 6).
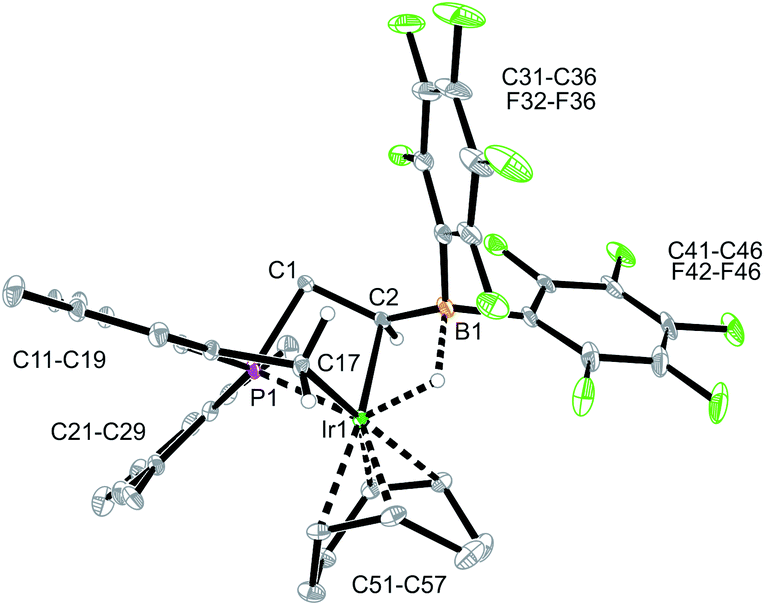 | ||
| Fig. 6 A projection of the molecular structure of the Iridium complex 15 (thermal ellipsoids at 30% probability). | ||
In solution (CD2Cl2) the iridium complex 15 shows four olefinic 1H NMR signals of the coordinated cyclooctadiene ligand. It also features four arene CH 1H NMR signals of the mesitylene and the CH-activated Mes substituents at phosphorus. Complex 15 shows a broadened 11B NMR resonance at δ −17.5. The 31P NMR signal is observed at δ −104.0. It shows coupling to the Ir–H moiety (2JPH ∼ 70 Hz).21 Consequently, the Ir-hydride signal shows up at δ −10.4 with ca. 70 Hz coupling to phosphorus (for additional details see the ESI‡).
Catalytic reactions
Our study has shown that the methylene linked phosphane/borata-alkene anion of the salt 9 served well as a chelate ligand in Rh coordination chemistry. It is likely that the Ir(III) complex 15 was actually formed by an oxidative addition reaction at a mesityl methyl group at the stage of the analogous intermediate 14. We carried out some preliminary investigation toward the use of the new chelate phosphane/borata-alkene complexes in catalysis. For this reason, we performed two sets of catalytic reactions using either of the complexes 12 and 15. We first turned to alkene and alkyne hydrogenation catalysis.22 Exposure of complex 15 to dihydrogen (1.0 bar, r.t.) revealed the stoichiometric formation of cyclooctane, the reduction product of the cod ligand of the Ir complex 15. Consequently, we employed compound 15 as a catalyst in our hydrogenation experiments. The hydrogenation of styrene is a typical example. With both 1 or 0.5 mol% of 15 quantitative hydrogenation to ethylbenzene was achieved (Scheme 5); with 0.1 mol% catalyst still a ca. 50% conversion was obtained. The catalytic hydrogenation sequence starting from complex 15 may possibly involve the not directly observed equilibration with its likely synthetic precursor 14, the Ir(cod) analogue of the Rh complex 12 (see above).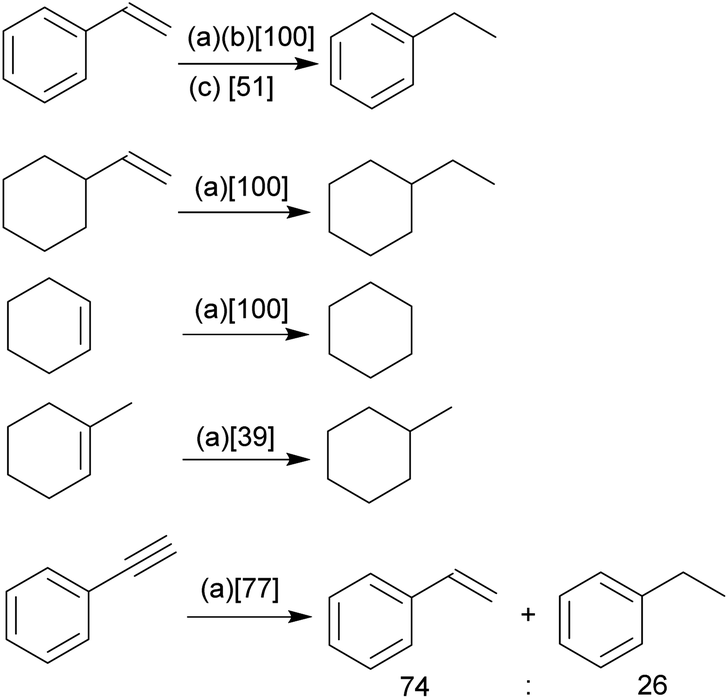 | ||
| Scheme 5 Catalytic hydrogenation of unsaturated substrates using an Ir catalyst derived from 15 under our standard conditions {1.0 bar H2, d6-benzene, r.t., 16 h, (a): 1 mol% catalyst, (b): 0.5 mol%, (c): 0.1 mol%; [% conversion achieved]}. | ||
Quantitative alkene hydrogenation was found at the 15 derived catalyst system with 1 mol% of vinylcyclohexane or cyclohexene, as well. The more sterically encumbered 1-methylcyclohexene substrate gave only a 39% conversion under these conditions and phenylacetylene eventually furnished a ca. 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of styrene and ethylbenzene with a combined conversion of 77% after 16 h.
:1 mixture of styrene and ethylbenzene with a combined conversion of 77% after 16 h.
Styrene was quantitatively hydrogenated to ethylbenzene with 0.5 mol% of the Rh catalyst 12 under our standard conditions (Scheme 6). With 0.1 mol% a ca. 50% conversion was obtained, similar as with the Wilkinson catalyst under these conditions. Cyclohexene was hydrogenated at the catalyst system 12 (0.1 mol%, 34% conversion). The bulkier 1-methylcyclohexene was not hydrogenated at the Rh catalyst system 12 under our typical conditions.
 | ||
| Scheme 6 Catalytic hydrogenation of alkenes with Rh complexes: comparison of the reaction with complex 12 and the Wilkinson catalyst (1 bar H2, r.t., d6-benzene, 16 h).23 | ||
So far we assume a conventional pathway of dihydrogen activation at the metal centre in the complexes 12 or 15, but we presently cannot rule out an alternative “FLP-like” metal/borane dihydrogen splitting reaction.24
A variety of Rh catalysts are able to polymerize arylacetylenes and so does the phosphane/borata-alkene complex 12.25 The phenylacetylene polymerization reaction by the neutral system 12 was carried out in the non-polar solvent benzene or in ethereal solution (diethylether or tetrahydrofuran). We carried out the phenylacetylene polymerization at room temperature for a duration between 30 min (in ether) or 2 h (in benzene). With decreasing catalyst amounts (0.1 mol%, 0.05 mol%) an almost quantitative amount of polyphenylacetylene was isolated from the reaction in benzene. Even with 0.025 mol% as well as 0.01 mol% of the catalyst poly(phenylacetylene) was isolated, albeit in lower yields (45%, 28%). The obtained polymer was similar in appearance (yellow to orange solids) as the poly(phenylacetylene) obtained by Noyori et al. at the remotely related neutral [(Ph3P)n(nbd)Rh-CCPh] (n: 1 or 2) derived catalysts, so we assume it has a similar structure.25a We also polymerized p-fluorophenylacetylene and p-methoxyphenylacetylene at the catalyst system 12 (0.1 mol%) and isolated the respective polyacetylenes in close to quantitative yields (Scheme 7).26
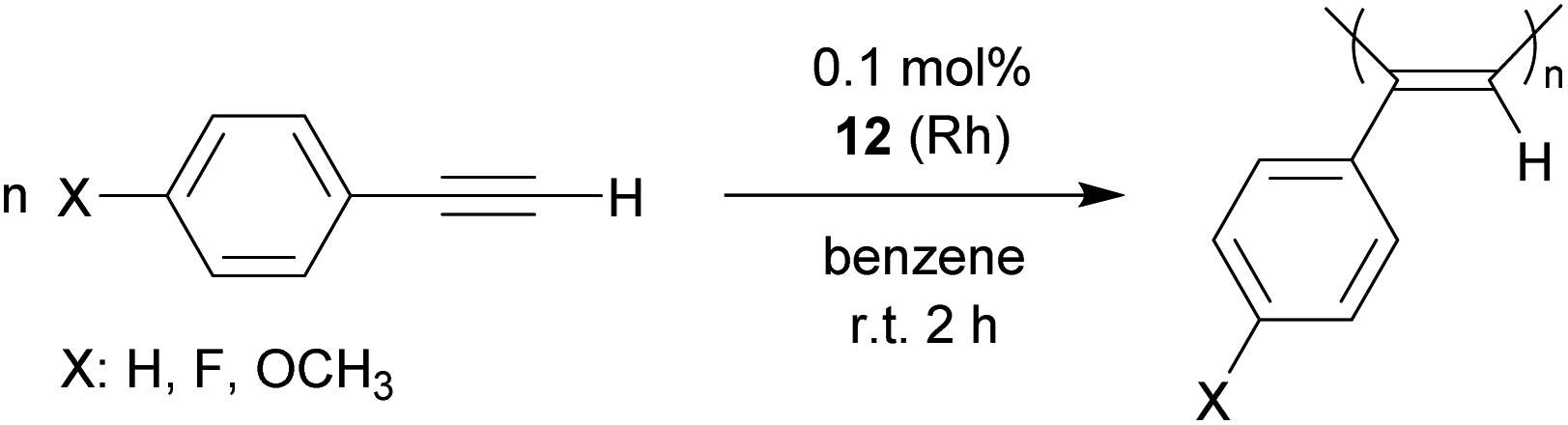 | ||
| Scheme 7 Polymerization of arylacetylenes. | ||
Each of the arylacetylene polymers shows a single set of 1H NMR signals, which indicates its origin from a stereo- and regioselective polymerization process25a (see the ESI‡ for details). The poly(p-anisylacetylene) sample was characterized by MALDI-TOF mass spectrometry, which showed the regular sequence of signals separated by the mass of the respective monomer unit of 132 (depicted in the ESI‡). The molecular weights of the polyacetylene samples were determined by GPC. Under our typical conditions, the polymerization reactions in benzene or THF furnished polymers of somewhat lower molecular weight than in ether. The latter reaction produced higher molecular weight polyacetylenes.27 The samples contained varying amounts of insoluble material (potentially very high molecular weight polymer). For the sizable soluble fraction of the poly(p-anisylacetylene) sample obtained in ether with the Rh complex 12 derived catalyst we found a molecular weight of Mn ≥ 100000. The respective poly(phenylacetylene) sample had an about twice as high Mn, and the poly(p-fluorophenylacetylene) had the highest measured Mn in the series of >400000 (Table 2). In all cases rather large polydispersities of close to 3 were found (see the ESI‡ for further details).
| X | Yield (%) | M n | PD | |
|---|---|---|---|---|
| a Conditions: solvent Et2O (5 mL), compound 12 (0.015 mmol = 2 mol%), monomer phenylacetylenes (0.75 mmol). b Soluble fraction measured (see the ESI for details), molecular weights determined by GPC, rel. to polystyrene standards. | ||||
| 1 | MeO | 86 | 114320 | 2.98 |
| 2 | H | 99 | 240006 | 2.69 |
| 3 | F | 97 | 444085 | 2.84 |
Conclusions
Our study has shown that the seminal study published by Piers et al. on the use of a borata-alkene as a π-ligand equivalent to ethene at an early transition metal can be substantially extended. In the Piers' system the H2CB(C6F5)2− ligand was generated by a typical organometallic reaction pathway within the coordination sphere of the metal (in that case at tantalum). Since we had found about the vastly increased α-CH acidity of the B(C6F5)2 boranes4 an improved and potentially more general pathway to κ2C,B-borata-alkene complexes has become evident: deprotonation5 of the respective suitably substituted [P]–CH2–CH2–B(C6F5)2 borane gave the borata-alkene in an independent initial step. Our syntheses of the methylene-bridged chelate phosphane/borata-alkene Rh and Ir complexes serve as examples of this development. The complexes are readily prepared, although the Ir system undergoes a subsequent rearrangement reaction. This new approach will probably allow for some variation on the ligand side, and it may open pathways to choosing variations on the metal side. The P/CB ligands in the here reported complexes do not interfere with catalytic features in our examples. To us this indicates that the readily available borata-alkenes might see useful applications as polar alkene ligand analogues in organometallic and coordination chemistry as well as in catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft is gratefully acknowledged. KW thanks the Uehara memorial foundation for a postdoctoral fellowship. AU thanks the JSPS for a postdoctoral stipend.Notes and references
- (a) R. A. Bartlett and P. P. Power, Organometallics, 1986, 5, 1916 CrossRef CAS; (b) M. M. Olmstead, P. P. Power and K. J. Weese, J. Am. Chem. Soc., 1987, 109, 2541 CrossRef CAS; (c) M. Pilz, J. Allwohn, R. Hunold, M. Massa and A. Berndt, Angew. Chem., Int. Ed. Engl., 1988, 27, 1370 CrossRef; (d) J. Yu, G. Kehr, C. G. Daniliuc and G. Erker, Eur. J. Inorg. Chem., 2013, 3312 CrossRef CAS; (e) J. D. Hoefelmeyer, S. Solé and F. P. Gabbaï, Dalton Trans., 2004, 1254 RSC.
- (a) G. Zweifel and H. Arzoumanian, Tetrahedron Lett., 1966, 7, 2535 CrossRef; (b) R. Kow and M. W. Rathke, J. Am. Chem. Soc., 1973, 95, 2715 CrossRef CAS; (c) B. G. Ramsey and L. M. Isabelle, J. Org. Chem., 1981, 46, 179 CrossRef CAS; (d) H. Klusik and A. Berndt, Angew. Chem., Int. Ed. Engl., 1983, 22, 877 CrossRef; (e) A. Höfner, B. Ziegler, W. Massa and A. Berndt, Angew. Chem., Int. Ed. Engl., 1989, 28, 186 CrossRef; (f) R. Hunold, M. Pilz, J. Allwohn, M. Stadler, W. Massa, P. v. R. Schleyer and A. Berndt, Angew. Chem., Int. Ed. Engl., 1989, 28, 781 CrossRef; (g) M. Pilz, J. Allwohn, W. Massa and A. Berndt, Angew. Chem., Int. Ed. Engl., 1990, 29, 399 CrossRef; (h) P. Willershausen, C. Kybart, N. Stamatis, W. Massa, M. Bühl, P. v. R. Schleyer and A. Berndt, Angew. Chem., Int. Ed. Engl., 1992, 31, 1238 CrossRef; (i) R. Littger, H. Nöth, M. Thomann and M. Wagner, Angew. Chem., Int. Ed. Engl., 1993, 32, 295 CrossRef; (j) C. Balzereit, C. Kybart, H.-J. Winkler, W. Massa and A. Berndt, Angew. Chem., Int. Ed. Engl., 1994, 33, 1487 CrossRef; (k) U. Kawashima, N. Yamashita and R. Okazaki, J. Am. Chem. Soc., 1995, 117, 6142 CrossRef; (l) J. J. Eisch, Adv. Organomet. Chem., 1996, 39, 355 CrossRef CAS; (m) R. Littger, H. Nöth and M. Suter, Eur. J. Inorg. Chem., 2000, 1571 CrossRef CAS; (n) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877 CrossRef CAS PubMed; (o) T. Tomioka, Y. Takahashi, T. G. Vaughan and T. Yanase, Org. Lett., 2010, 12, 2171 CrossRef CAS PubMed; (p) T. Tomioka, R. Sankranti, T. G. Vaughan, T. Maejima and T. Yanase, J. Org. Chem., 2011, 76, 8053 CrossRef CAS PubMed.
- (a) B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 4863 CrossRef; (b) Modern Carbonyl Olefination, ed. T. Takeda, Wiley-VCH, Weinheim, 2005 Search PubMed.
- P. Moquist, G.-Q. Chen, C. Mück-Lichtenfeld, K. Bussmann, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Sci., 2015, 6, 816 RSC.
- (a) S. Kohrt, S. Dachwitz, C. G. Daniliuc, G. Kehr and G. Erker, Dalton Trans., 2015, 44, 21032 RSC; (b) T. Wang, S. Kohrt, C. G. Daniliuc, G. Kehr and G. Erker, Org. Biomol. Chem., 2017, 15, 6223 RSC.
- (a) S. Helm and H. Nöth, Angew. Chem., Int. Ed. Engl., 1988, 27, 1331 CrossRef; (b) S. Channareddy, G. Linti and H. Nöth, Angew. Chem., Int. Ed. Engl., 1990, 29, 199 CrossRef.
- (a) G. C. Bazan, G. Rodriguez, A. J. Ashe III, S. Al-Ahmad and C. Müller, J. Am. Chem. Soc., 1996, 118, 2291 CrossRef CAS; (b) G. C. Bazan, G. Rodriguez, A. J. Ashe III, S. Al-Ahmad and J. W. Kampf, Organometallics, 1997, 16, 2492 CrossRef CAS; (c) A. J. Ashe III, S. Al-Ahmad, X. G. Fang and J. W. Kampf, Organometallics, 1998, 17, 3883 CrossRef; (d) A. J. Ashe III, S. Al-Ahmad and X. G. Fang, J. Organomet. Chem., 1999, 581, 92 CrossRef; (e) J. S. Rogers, X. H. Bu and G. C. Bazan, J. Am. Chem. Soc., 2000, 122, 730 CrossRef CAS.
- (a) F. Jiang, P. J. Shapiro, F. Fahs and B. Twamley, Angew. Chem., Int. Ed., 2003, 42, 2651 CrossRef CAS PubMed; (b) D. J. H. Emslie, L. E. Harrington, H. A. Jenkins, C. M. Robertson and J. F. Britten, Organometallics, 2008, 27, 5317 CrossRef CAS; (c) K. B. Kolpin and D. J. H. Emslie, Angew. Chem., Int. Ed., 2010, 49, 2716 CrossRef CAS PubMed; (d) X. Zhao, E. Otten, D. Song and D. W. Stephan, Chem.–Eur. J., 2010, 16, 2040 CrossRef CAS PubMed; (e) D. J. H. Emslie, B. E. Cowie and K. B. Kolpin, Dalton Trans., 2012, 41, 1101 RSC.
- (a) K. S. Cook, W. E. Piers, T. K. Woo and R. McDonald, Organometallics, 2001, 20, 3927 CrossRef CAS; (b) K. S. Cook, W. E. Piers, P. G. Hayes and M. Parvez, Organometallics, 2002, 21, 2422 CrossRef CAS; (c) K. S. Cook, W. E. Piers and R. McDonald, J. Am. Chem. Soc., 2002, 124, 5411 CrossRef CAS PubMed.
- For a very recent conceptually related study see: T. A. Bartholome, A. Kaur, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie. 202002125.
- See for a comparison: A. Amgoune, S. Ladeira, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2012, 134, 6560 CrossRef CAS PubMed.
- There are a number of metal complexes of the neutral ambiphilic P/B systems known, see e.g. (a) S. Bontemps, G. Bouhadir, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 12056 CrossRef CAS PubMed; (b) S. Bontemps, M. Sircoglou, G. Bouhadir, H. Puschmann, J. A. K. Howard, P. W. Dyer, K. Miqueu and D. Bourissou, Chem.–Eur. J., 2008, 14, 731 CrossRef CAS PubMed; (c) G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2016, 45, 1065 RSC.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef CAS PubMed.
- (a) D. J. Parks, R. E. von H. Spence and W. E. Piers, Angew. Chem., Int. Ed. Engl., 1995, 34, 809 CrossRef CAS; (b) D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492 CrossRef CAS; (c) M. Hoshi, K. Shirakawa and M. Okimoto, Tetrahedron Lett., 2007, 48, 8475 CrossRef CAS; (d) A. Schnurr, K. Samigullin, J. M. Breunig, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2011, 30, 2838 CrossRef CAS; (e) J. Zhang, S. Park and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 13757 CrossRef CAS PubMed.
- P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072 RSC.
- (a) C. Höltke, G. Erker, G. Kehr, R. Fröhlich and O. Kataeva, Eur. J. Inorg. Chem., 2002, 2789 CrossRef; (b) N. Millot, C. C. Santini, B. Fenet and J. Marie, Eur. J. Inorg. Chem., 2002, 3328 CrossRef CAS; (c) F. Focante, P. Mercandelli, A. Sironi and L. Resconi, Coord. Chem. Rev., 2006, 250, 170 CrossRef CAS; (d) S. Schwendemann, R. Fröhlich, G. Kehr and G. Erker, Chem. Sci., 2011, 2, 1842 RSC; (e) M. Shang, J. Z. Chan, M. Cao, Y. Chang, Q. Wang, B. Cook, S. Torker and M. Wasa, J. Am. Chem. Soc., 2018, 140, 10593 CrossRef CAS.
- (a) R. D. Chambers and T. Chivers, J. Chem. Soc., 1965, 3933 RSC; (b) W. E. Piers and R. E. H. Spence, Acta Crystallogr., 1995, C51, 1688 CAS; (c) M. Bochmann, S. J. Lancaster and O. B. Robinson, J. Chem. Soc., Chem. Commun., 1995, 2081 RSC; (d) W. E. Piers, Adv. Organomet. Chem., 2004, 52, 1 CrossRef; (e) A. Ueno, J. Li, C. G. Daniliuc, G. Kehr and G. Erker, Chem.–Eur. J., 2018, 24, 10044 CrossRef CAS PubMed.
- A. Pitcock, R. E. Richards and L. M. Venanzi, J. Chem. Soc., 1966, 1707 RSC.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457 CrossRef CAS PubMed.
- (a) T. Yano, Y. Moroe, M. Yamashita and K. Nozaki, Chem. Lett., 2008, 37, 1300 CrossRef CAS; (b) M. Yamashita, Y. Moroe, T. Yano and K. Nozaki, Inorg. Chim. Acta, 2011, 369, 15 CrossRef CAS.
- (a) B. Punji, T. J. Emge and A. S. Goldman, Organometallics, 2010, 29, 2702 CrossRef CAS; (b) S. Kundu, J. Choi, D. Y. Wang, Y. Choliy, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2013, 135, 5127 CrossRef CAS PubMed; (c) M. Rimoldi and A. Mezzetti, Inorg. Chem., 2014, 53, 11974 CrossRef CAS PubMed; (d) D. A. Laviska, T. Zhou, A. Kumar, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Organometallics, 2016, 35, 1613 CrossRef CAS.
- (a) R. Crabtree, Acc. Chem. Res., 1979, 12, 331 CrossRef CAS; (b) A. Pfaltz, J. Blankenstein, R. Hilgraf, E. Hörmann, S. McIntyre, F. Menges, M. Schönleber, S. P. Smidt, B. Wüstenberg and N. Zimmermann, Adv. Synth. Catal., 2003, 345, 33 CrossRef CAS; (c) T. L. Church and P. G. Andersson, Coord. Chem. Rev., 2008, 252, 513 CrossRef CAS; (d) J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130 CrossRef CAS PubMed.
- J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A, 1966, 1711 RSC.
- See for a comparison: W. H. Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 5080 CrossRef CAS PubMed.
- (a) Y. Kishimoto, P. Eckerle, T. Miyatake, M. Kainosho, A. Ono, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1999, 121, 12035 CrossRef CAS; (b) I. Saeed, M. Shiotsuki and T. Masuda, Macromolecules, 2006, 39, 8977 CrossRef CAS; (c) N. Onishi, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2009, 42, 4071 CrossRef CAS; (d) M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, E. Vispe, F. J. Lahoz and L. A. Oro, Macromolecules, 2009, 42, 8146 CrossRef; (e) N. I. Nikishkin, J. Huskens and W. Verboom, Polymer, 2013, 54, 3175 CrossRef CAS; (f) J. Sedláček and H. Balcar, J. Macromol. Sci., Part C: Polym. Rev., 2017, 57, 31 Search PubMed.
- (a) S. B. T. Nguyen, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1993, 115, 9858 CrossRef CAS; (b) H. H. Fox, M. O. Wolf, R. O'Dell, B. L. Lin, R. R. Schrock and M. S. Wrighton, J. Am. Chem. Soc., 1994, 116, 2827 CrossRef CAS; (c) H. Nishide, Adv. Mater., 1995, 7, 937 CrossRef CAS; (d) O. A. Scherman and R. H. Grubbs, Synth. Met., 2001, 124, 431 CrossRef CAS; (e) O. A. Scherman, I. M. Rutenberg and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 8515 CrossRef CAS PubMed; (f) S. Karabulut, Polym. J., 2009, 41, 629 CrossRef CAS.
- (a) J. Chen, K. K.-L. Cheuk and B. Tang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1153 CrossRef CAS; (b) M. Shiotsuki, N. Onishi, F. Sanda and T. Masuda, Polym. J., 2011, 43, 51 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Wolfgang Kirmse on the occasion of his 90th birthday. |
| ‡ Electronic supplementary information (ESI) available: Additional experimental details, further spectral and crystallographic data. CCDC deposition numbers are 1960302–1960306 and 2008240. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02223c |
| § Current address: Graduate School of Pharmaceutical Sciences, University of Tokyo, Hongo 7-3-1, Bunkyo-ku, Tokyo, Japan. |
| This journal is © The Royal Society of Chemistry 2020 |