
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoscale battery cathode materials induce DNA damage in bacteria†
Tian A.
Qiu‡
 a,
Valeria
Guidolin§
b,
Khoi Nguyen L.
Hoang§
c,
Thomas
Pho§
c,
Andrea
Carra'
b,
Peter W.
Villalta
b,
Jiayi
He
a,
Xiaoxiao
Yao
a,
Robert J.
Hamers
d,
Silvia
Balbo
b,
Z. Vivian
Feng
*c and
Christy L.
Haynes
*a
a,
Valeria
Guidolin§
b,
Khoi Nguyen L.
Hoang§
c,
Thomas
Pho§
c,
Andrea
Carra'
b,
Peter W.
Villalta
b,
Jiayi
He
a,
Xiaoxiao
Yao
a,
Robert J.
Hamers
d,
Silvia
Balbo
b,
Z. Vivian
Feng
*c and
Christy L.
Haynes
*a
aDepartment of Chemistry, University of Minnesota, 207 Pleasant St SE, Minneapolis, MN 55455, USA. E-mail: chaynes@umn.edu
bMasonic Cancer Center, University of Minnesota, 2231 6th Street SE, Minneapolis, MN 55455, USA
cChemistry Department, Augsburg University, 2211 Riverside Ave, Minneapolis, MN 55454, USA. E-mail: feng@augsburg.edu
dDepartment of Chemistry, University of Wisconsin, 1101 University Avenue, Madison, WI 53706, USA
First published on 21st September 2020
Abstract
The increasing use of nanoscale lithium nickel manganese cobalt oxide (LixNiyMnzCo1−y−zO2, NMC) as a cathode material in lithium-ion batteries poses risk to the environment. Learning toxicity mechanisms on molecular levels is critical to promote proactive risk assessment of these complex nanomaterials and inform their sustainable development. We focused on DNA damage as a toxicity mechanism and profiled in depth chemical and biological changes linked to DNA damage in two environmentally relevant bacteria upon nano-NMC exposure. DNA damage occurred in both bacteria, characterized by double-strand breakage and increased levels of many putative chemical modifications on bacterial DNA bases related to direct oxidative stress and lipid peroxidation, measured by cutting-edge DNA adductomic techniques. Chemical probes indicated elevated intracellular reactive oxygen species and transition metal ions, in agreement with DNA adductomics and gene expression analysis. By integrating multi-dimensional datasets from chemical and biological measurements, we present rich mechanistic insights on nano-NMC-induced DNA damage in bacteria, providing targets for biomarkers in the risk assessment of reactive materials that may be extrapolated to other nano–bio interactions.
Introduction
Lithium-ion batteries (LIB) have become a critical high-density energy storage solution in the last few decades. One key component determining the capacity of LIBs is the cathode material.1 While LiCoO2 is widely used in portable electronics, nanostructured lithium nickel manganese cobalt oxide (LixNiyMnzCo1−y−zO2, NMC) materials are being rapidly scaled up for use in electric vehicles because this modified composition yields high energy storage capacity with lower raw material cost and good stability, critical factors for electric vehicle batteries.2,3While the high energy density makes LIBs suitable for large-scale applications as energy materials, the low cost of NMC materials reduces economic incentives for recycling compared with LiCoO2 or lead–acid batteries.4 Currently, most LIBs end up in landfills or being incinerated as municipal solid waste, with the potential to produce undesirable leachate from buried solid waste or metal-rich ash.5 In one report, standardized leaching tests and life-cycle impact assessments showed potential environmental and human health impacts of LIBs through metallic leachates.6 The leachates contain transition metal ions, which are shown to substantially reduce the biodiversity within microbial communities.7,8 Considering the projected increase in LIB production and consumption, the amount of LIB waste is expected to increase. Therefore, developing adequate strategies to assess the biological impact of this new class of energy material in a proactive manner is essential to inform sustainable development and production of battery cathode materials.
Commercially available cathode materials typically consist of micron-sized fused clusters of smaller primary particles. Nanoscale primary particles are become more widely adapted because shorter paths for Li ion diffusion and greater surface area for electrical contact can yield improved performance.9 In addition, we previously reported that cycling of batteries caused stress-induced fracture of micro-sized commercial NMC, generating sheet-like nanostructures.10 However, the increased surface area-to-volume ratio inherent in nanoscale materials also results in increased toxicity compared to microscale NMC, as we have recently demonstrated.11 Our lab and others also showed that nanoscale NMC inhibits bacterial growth and impacts survival and reproduction of Daphnia magna, partially or completely due to the dissolution of toxic nickel and cobalt ions.10–14 More recently, Mitchell, et al. showed that bacteria Shewanella oneidensis rapidly developed resistance to nano-NMC after chronic exposure, indicating the potential effect of NMC on the genetic materials in biological organisms.15 Such discoveries, along with the fact that nickel and cobalt ions are known to induce genotoxicity,16,17 indicate that DNA damage is a likely potential toxicity mechanism of nano-NMC in bacteria.
Nanoparticles (NPs) are known to induce genotoxicity in various cell lines and organisms across the food web.18,19 Mechanisms of genotoxicity induced by NPs include DNA methylation variations,20 oxidative DNA adduct formation21 and DNA strand breakage,22 and can lead to mutagenesis.23 A lack of evidence for nano-NMC entering bacterial cells suggests that indirect mechanisms of genotoxicity through the generation of reactive oxygen species (ROS) and ion dissolution are likely to be more relevant.12 Since transition metal ions are known to cause oxidative stress via Fenton reactions,24 we postulated that nano-NMC can cause DNA damage through oxidative stress, related to dissolved nickel and cobalt ions. As the aim of this work is to understand environmental risk of nano-NMC, two ecologically relevant but distinct bacterial species were studied. Shewanella oneidensis MR-1 (S. oneidensis) is a Gram-negative bacterium with unique heavy metal-reducing capability in the environment.25 The Gram-positive bacterium Bacillus subtilis SB491 (B. subtilis) is commonly found in soil and on plant undergrowth and is widely used for nanotoxicity studies.26 While plenty of previous work has revealed the role of ROS and metal ions in nano-metal/metal oxide toxicity to microorganisms (Table S1†), the goal of this study is to focus on DNA damage, a potentially universal toxicity mechanism, and reveal relevant molecular details of nano-bacteria interactions that can be potentially extrapolated to a wide range of organisms.
A sensitive and rapid tool to assess DNA damage is the Comet assay; this method has been applied widely to study strand breakage induced by NPs.14,27 Therefore, we characterized bacterial DNA damage upon nano-NMC exposure using an adapted Comet assay for bacterial DNA double strand breakage.28 To further investigate the chemistry of DNA damage, we used a high-resolution mass spectrometry-based DNA adductomics approach to characterize chemical modification of DNA bases upon nano-NMC exposure.29 DNA adducts include redox reactions or fusion of molecular species on DNA molecules and have been informative for the risk assessment of carcinogens.30,31 Previous studies have used DNA adduct formation in bacteria Salmonella typhimurium32 and Escherichia coli33 to study mechanisms of mutagenesis of chemicals in risk assessment. However, the limited number of investigation on DNA adducts in nanotoxicology has focused on detecting a single DNA modification, 8-hydroxy-2′-deoxyguanosine (8-oxo-dG, 8-OHdG), an adduct indicative of direct oxidative stress.34,35 Yet, oxidative stress can result in a variety of DNA modifications including those from lipid peroxidation. Recently developed DNA adductomic approaches provide the possibility to detect a wider variety of DNA modifications and reveal molecular mechanisms of genotoxicity.30 DNA adductomics has been applied to investigate modes of genotoxicity in mice upon iron oxide NP exposure36 and in a soil bacterium.37 In this work, we used state-of-the-art DNA adductomic techniques based on the use of high resolution accurate mass (HRAM) mass spectrometry to detect a large variety of DNA modifications, providing information on the chemical nature of the cellular responses upon nano-NMC exposure.
We combined DNA adductomic results with data from toxicity, intracellular ROS, metal ion uptake, and gene expression to construct an understanding of the molecular mechanisms of nano-NMC-induced DNA damage with unprecedented details, pushing beyond the phenomenological assessment of nanomaterial–bacteria interactions. We chose the specific composition LiNi1/3Mn1/3Co1/3O2 as this is the most stable stoichiometry within the NMC family. We found that nano-NMC induced DNA damage in both bacteria, characterized by double-strand breakage and increased levels of putative endogenous DNA adducts related to direct oxidative stress and lipid peroxidation; such DNA damage was accompanied by changes in metal homeostasis and increased levels of intracellular ROS. Since DNA damage is a potentially universal toxicity mechanism, the molecular details we reveal in this study provide insight on how we may extrapolate the results and apply this multidimensional mechanistic approach to other organisms for the discovery of useful biomarkers and the environmental risk assessment of reactive nanomaterials.
Results
Nano-NMC caused dose-dependent bacterial growth inhibition and viability decrease over time
Nano-NMC was synthesized using a method we described previously;11 this method yields nano-sheets approximately 50–70 nm in diameter and 3–5 nm thick. Representative SEM images are shown in the ESI.† Previous work showed that nano-NMC exposure inhibited bacterial growth in a dose-dependent manner mainly due to dissolution of toxic nickel and cobalt ions from NMC.11,14 We initiated NMC exposure at the mid-log phase of bacterial growth as bacterial cells actively replicate during exponential growth; thus, the impact on DNA during the log phase potentially has a high probability to lead to noticeable consequences. After spiking NMC during exponential growth, a dose-dependent growth inhibition was observed for both bacteria species (Fig. 1 and S1†). A growth-based viability (GBV) assay was performed at the time point when the bacterial population was about to enter stationary phase (8 hour for S. oneidensis and 5 hour for B. subtilis) to measure bacterial viability compared to controls,38 as optical density does not effectively differentiate live and dead cells. Results showed that for S. oneidensis, an 1-hour short exposure slightly inhibited growth and compromised viability, while an 8 hour exposure caused significantly decrease in both normalized optical density and viability in a dose-dependent manner, indicating that NMC toxicity is time-dependent. Interestingly, compared to control, while the optical density at 8 hour of 50 mg L−1 NMC post-exposure indicated 50% viable cells, the viability assay showed only 10% viable cells. This discrepancy showed that not all cells accounted for by optical density measurements are viable, and it is important to assess toxicity with different endpoints. For B. subtilis, it was intriguing to observe that, besides the expected dose-dependent behavior, the 5 mg L−1 treatment to B. subtilis resulted in almost doubled viability compared to control, while the optical density readings were not different (Fig. 1(c) and (d)). Hormesis, a biphasic response of organism responding to potentially toxic agents, has been widely reported in different organisms from bacteria to yeast to invertebrates.39 The stimulatory effect of As(III) at low dose and inhibitory effect at high dose on the metabolic growth of B. subtilis has been previously reported.40 It is possible that the low dose of NMC (5 mg L−1) induced hormesis effects in B. subtilis and stimulated cell division without increasing cell volume, resulting in increased viability yet the same optical density compared to the control. In fact, we found that the increased viability at 5 mg L−1 is mainly due to the dissolved manganese ions (Fig. S2†), which is less relevant to our hypothesis related to nickel and cobalt ions. In general, both growth inhibition and viability assays indicate that 5 mg L−1 NMC either has no effect or is stimulatory while 50 mg L−1 is highly toxic for both bacteria species.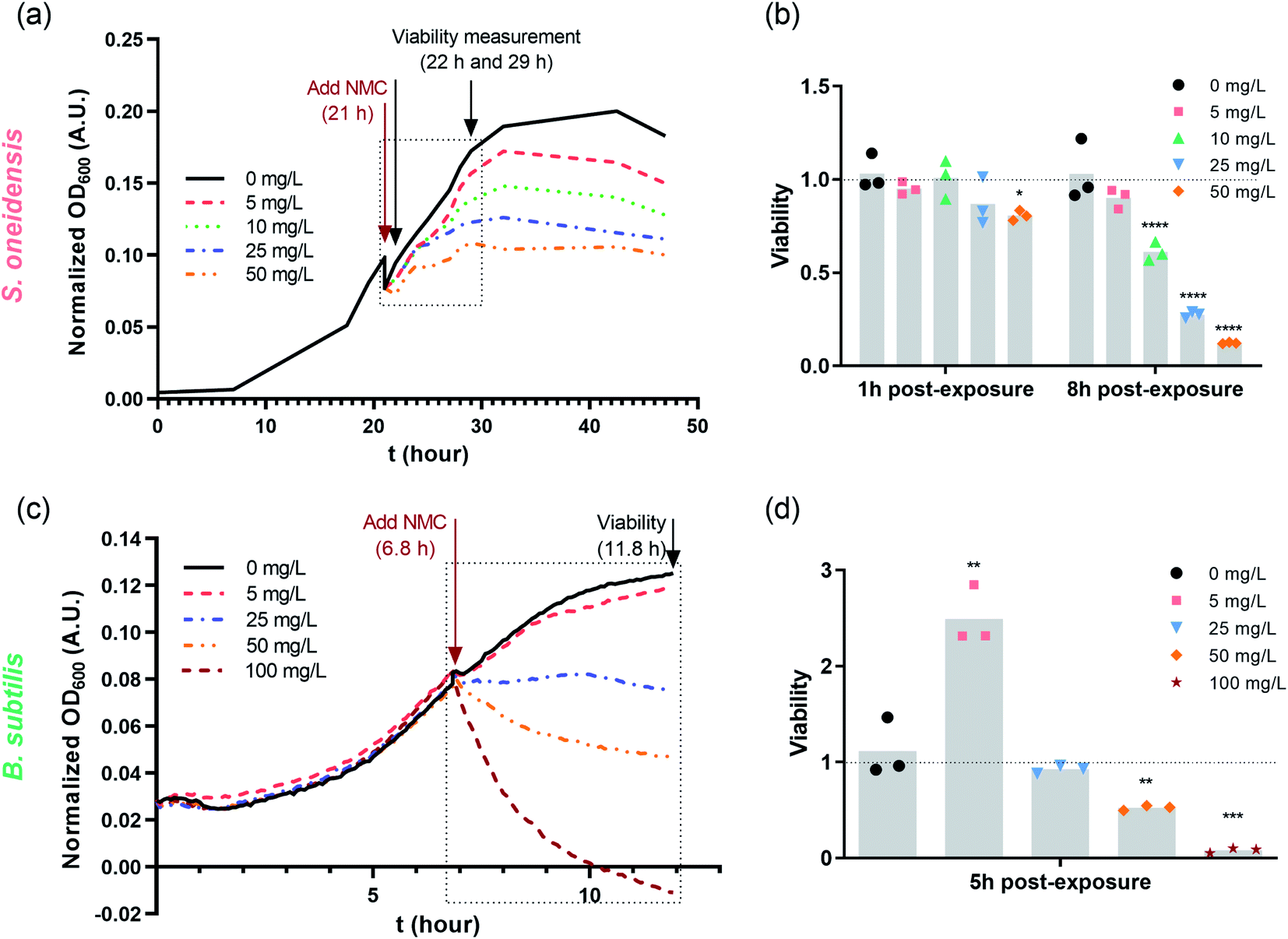 | ||
| Fig. 1 Dosing NMC at mid-log phase of (a) S. oneidensis MR-1 and (c) B. subtilis growth. The time points marked with “Add NMC” indicate the time points when NMC suspension or blank media was added to make the final concentration of 0, 5, 10, 25 and 50 mg L−1 nano-NMC. (a) and (c) show the normalized average optical density over time before and after adding NMC (n = 3, error bar not shown for clarity. Plots with error bars and magnified insets can be found in Fig. S1†); (b) and (d) show viability of nano-NMC-treated bacterial population compared to control (0 mg L−1) at different time points post-exposure (n = 3). Different concentrations are represented by different colors. Individual data points are shown. Asterisks denote statistically significant difference compared to control (* for p < 0.05, ** for p < 0.01, *** for p < 0.001 and **** for p < 0.0001, ANOVA followed by Dunnett's multiple comparisons test performed on log2-transformed viability).38 | ||
Nano-NMC resulted in DNA double strand breakage
The Comet assay is commonly used to characterize DNA damage by measuring DNA strand breakage due to its simplicity and sensitivity, and it has been used previously to assess NP genotoxicity.41 Upon strand breakage, supercoiled DNA relaxes, resulting in “halo”-like tails for mammalian cells or “strand”-like tails for bacterial cells of stained DNA upon electrophoresis.28,42 Generally, it is believed that alkaline conditions (pH > 12.3) mostly measure single-strand breakage while neutral conditions (pH < 10) measure double-strand breakage.43,44 Since double-strand breakage is more lethal than single-strand breakage which can be quickly repaired, we used a neutral Comet assay to assess DNA double-strand breakage in bacterial cells, and the tail lengths were used to characterize the extent of damage.28,45We treated cells with 5 or 50 mg L−1 nano-NMC at the mid-log phase and collected bacterial cells 8 hours (S. oneidensis) or 5 hours (B. subtilis) post-exposure, matching the conditions of toxicity measurements. Fig. 2(a) shows typical microscopic images of bacterial DNA “tails”. To quantify DNA double-strand breakage, the lengths of the stretched DNA upon electrophoresis were measured (Fig. 2(b) and (c)). The figures show that, compared to control groups, longer DNA tails were observed in both bacteria exposed to NMC, indicating DNA double-strand breakage. While B. subtilis DNA tail lengths showed a dose-dependent response, it is interesting that there was an increased but no dose-dependent DNA damage over 8-hour exposure in S. oneidensis.
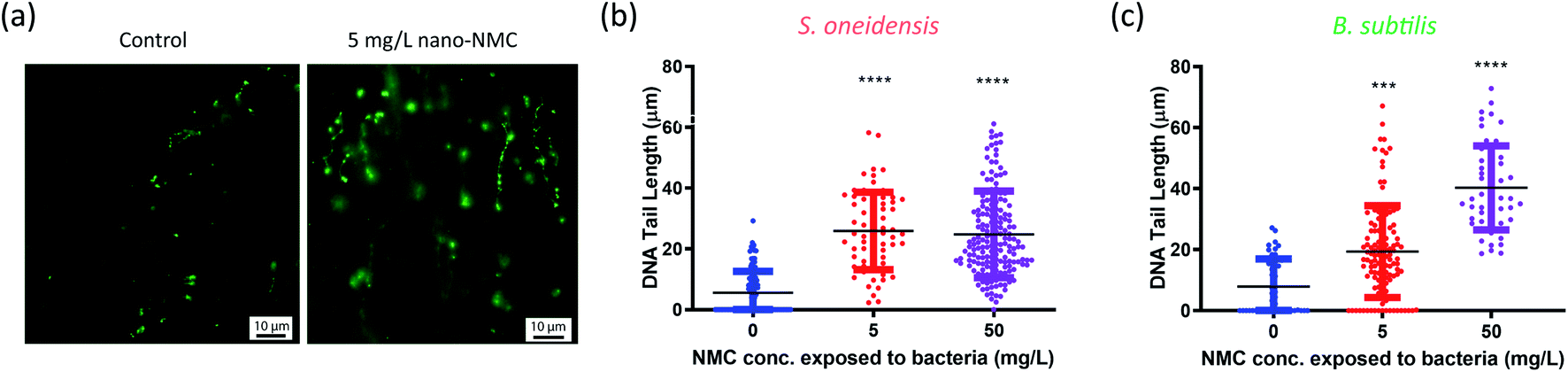 | ||
| Fig. 2 Comet assay results show DNA double strand breakage in nano-NMC-treated bacterial cells. (a) Representative fluorescence images of the Comet assay on B. subtilis. (b and c) Tail length measured after single-cell electrophoresis of S. oneidensis MR-1 and B. subtilis SB491, respectively. Asterisks denote statistically significant differences compared to the nanoparticle-free control group (*** for p < 0.001 and **** for p < 0.0001, Kruskal–Wallis test followed by Dunn's multiple comparisons test). | ||
High-resolution DNA adductomics revealed a wide range of alterations in DNA adduct patterns
To explore the potential mechanisms of DNA damage, a high-resolution accurate mass (HRAM) mass spectrometry DNA adductomic approach was used to interrogate the chemical modifications on bacterial DNA upon NMC exposure. This approach is based on the empirical observation that, once the DNA is hydrolyzed, all the deoxyribonucleosides show a common neutral loss of the deoxyribose moiety upon fragmentation in the mass spectrometer. For this reason, the instrument monitors the neutral loss of the deoxyribose (dR = 116.0474 m/z ± 0.0006 m/z) to trigger MS3 fragmentation of possible DNA adducts.29,30 This approach leads to a highly specific method for the detection of DNA adducts and the generation of structural information allowing for their identification. Specifically, the MS2 and MS3 spectra provide structural information for characterization of the modifications and identification of the nucleobases (i.e. C, T, A, or G) which were modified. An example of HRAM mass spectrometry detecting DNA adducts is shown in Fig. S3.† This powerful high-resolution DNA adductomics method has never been used to characterize covalent modifications of bacterial DNA, and a preliminary experiment was carried out by incubating extracted bacterial DNA with nanoscale NMC, along with naked calf-thymus DNA as a positive control, to validate the method. We successfully detected a collection of DNA modifications on bacterial DNA using this approach. We then exposed log-phase bacteria to sub-lethal dosage (5 mg L−1 NMC for 8 or 5 hours). After exposure, DNA was extracted, hydrolyzed, purified and analyzed as described in the Experimental procedures section.Table 1 and 2 present an overview of DNA adducts that either appeared in all triplicate samples of NMC-exposed bacteria or showed a statistically significant change in adduct levels (semi-quantified via signal intensity of the precursor ion) in NMC-exposed bacteria compared to the control. Fig. 3 shows semi-quantified levels of putative DNA adducts, selected via more stringent criteria of (1) statistically significant increase in exposed samples compared to control, (2) appeared in all three exposed samples, and (3) a fold change larger than 10 if there is any zero value in control group. These selected m/z values in Tables 1, 2 and Fig. 3 refer to putative DNA adducts whose level changed upon NMC exposure; other putative DNA adducts that did not fit the criteria (a.k.a. no level change upon exposure) are not presented herein. A full list of all putative DNA adducts detected in all 12 samples can be found in SI_AdductInfo.xlsx.† It should be noted that all structures presented here are putative structures consistent with the accurate mass and fragmentation detected by tandem MS; however, without isotopically labeled standards, these structures cannot be confirmed.
| [M + H]+ | Fold change | Putative name | Origin | Category |
|---|---|---|---|---|
| 229.0931 | 12 | dIZ | Degradation 8-oxo-dG | Oxidative stress |
| 244.0928 | Only in exposed | 5-OH-dC | Direct or indirect oxidation via ROS | Oxidative stress |
| 260.0877 | Only in exposed | 5,6-OH-dC | Direct or indirect oxidation via ROS | Oxidative stress |
| 270.1197 | Only in exposed | FAPY-A | Direct or indirect oxidation via ROS | Oxidative stress |
| 284.0989 | 19 | 8-oxo-dG | Direct or indirect oxidation via ROS | Oxidative stress |
| 286.1146 | 6 | FAPY-dG | Direct or indirect oxidation via ROS | Oxidative stress |
| 300.0939 | 5 | Spiroiminodihydantoin | Direct or indirect oxidation via ROS | Oxidative stress |
| 310.1146 | 110 | N 6 -hydroxyacetyl-dA | Direct or indirect oxidation via ROS | Oxidative stress |
| 382.1721 | 42 | N 2 -dimethyldioxane-dG | Lipid peroxidation | Lipid peroxidation |
| 436.2191 | Only in exposed | DDE-I-dA | Lipid peroxidation | Lipid peroxidation |
| 299.1238 | 66 | Acr-dT_I | Acrolein | Lipid peroxidation |
| 340.1252 | 165 | CE-dG | Glycation | Glycation |
| 308.0989 | 337 | 2-oxo-1,N2-εdG | Etheno | Alkylation |
| [M + H]+ | Fold change | Putative name | Origin | Category |
|---|---|---|---|---|
| a Two isomers were found for this m/z at different retention times. | ||||
| 229.0931 | 62 | dIZ | Degradation of 8-oxo-dG | Oxidative stress |
| 244.0928 | 519 | 5-OH-dC | Direct or Indirect Oxidation via ROS | Oxidative stress |
| 260.0877 |
15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 234 234
|
5,6-OH-dC | Direct or Indirect Oxidation via ROS | Oxidative stress |
| 262.1034 | 4581 | 5,6-H-5,6-OH-dC | Direct or Indirect Oxidation via ROS | Oxidative stress |
| 284.0989 | 204 | 8-oxo-dG | Direct or Indirect Oxidation via ROS | Oxidative stress |
| 286.1146 | Only in exposed | FAPY-dG | Direct or Indirect Oxidation via ROS | Oxidative stress |
| 297.0942 | Only in exposed | Nitro-dA | Direct or Indirect Oxidation via ROS | Oxidative stress |
| 310.1146 | Only in exposed | N 6 -hydroxyacetyl-dA | Direct or Indirect Oxidation via ROS | Oxidative stress |
| 252.0979 | Only in exposed | 3,N2-εdC | Lipid peroxidation | Lipid peroxidation |
| 382.1721 | Only in exposed | N 2 -dimethyldioxane-dG | Lipid peroxidation | Lipid peroxidation |
| 306.1197 | Only in exposed | M1dA | Malonaldehyde | Lipid peroxidation |
| 386.1459 | Only in exposed | M2AA-dA | Malonaldehyde | Lipid peroxidation |
| 402.1408 | Only in exposed | M2AA-dG_I | Malonaldehyde | Lipid peroxidation |
| 338.1459 | Only in exposed | CRO-dG | Crotonaldehyde | Lipid peroxidation |
| 299.1238 | Only in exposed | Acr-dT_I | Acrolein | Lipid peroxidation |
| 308.1353 | Only in exposed | Acr-dA_I | Acrolein | Lipid peroxidation |
| 591.2270 | 943 | dG-ACR-dG | Acrolein | Lipid peroxidation |
| 300.1190 | 18 | Carboxy-dC | Glycation | Glycation |
| 324.1302 | 52/Only in exposeda | Carboxy-dA | Glycation | Glycation |
| 370.1357 | Only in exposed | CHPG-dG | Glycation | Glycation |
| 266.1248 | 7 | N 6-methyl-dA | Alkylating agent | Alkylation |
| 296.1353 | 27 | N 2-ethyl-dG | Alkylating agent | Alkylation |
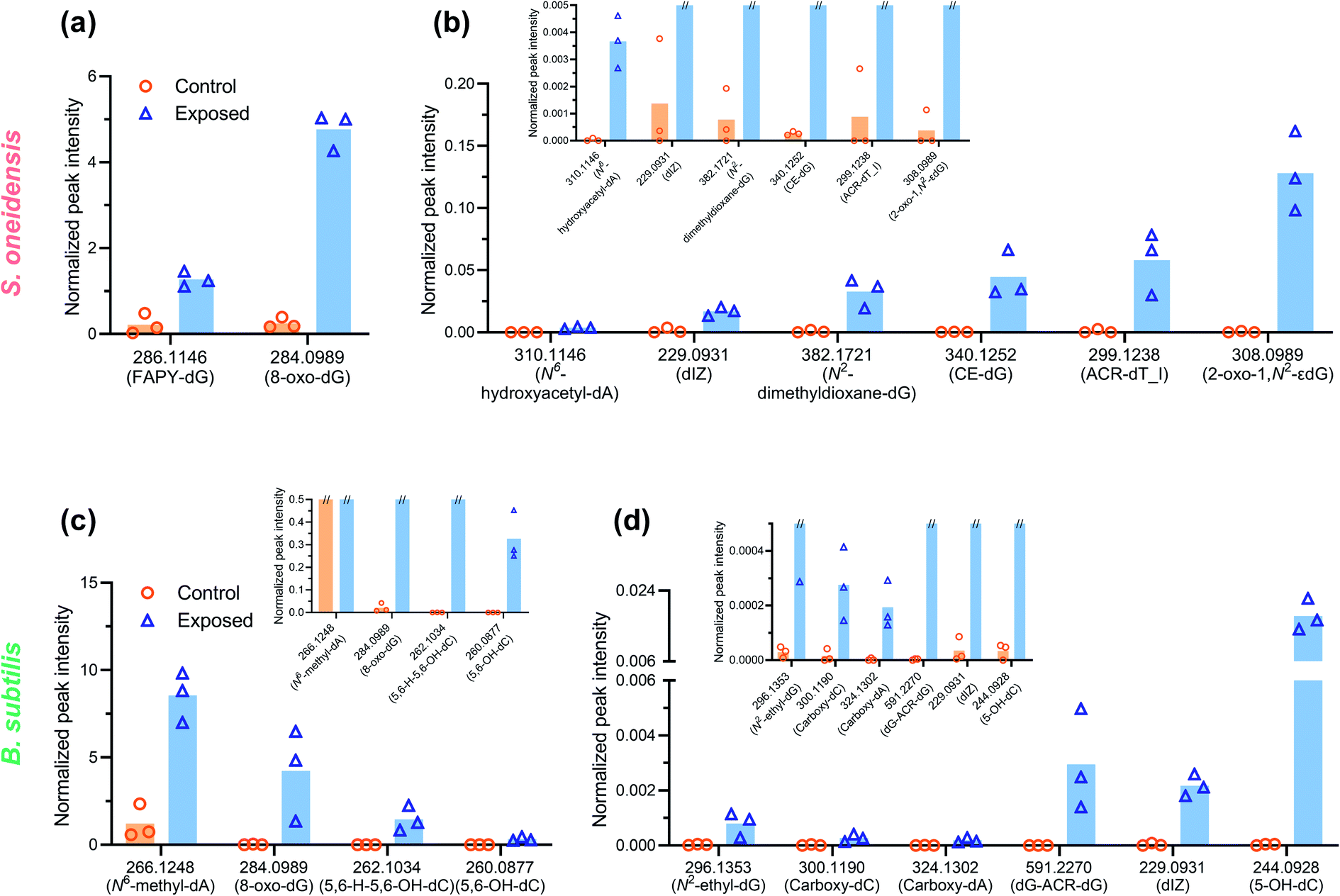 | ||
| Fig. 3 DNA adducts with increased levels (n = 3, p < 0.05 from individual two-tailed t-test) in the NMC-exposed samples (blue open triangle) compared to negative control (orange open circle). Signal intensity was normalized by the dG content in extracted DNA and peak area of internal standard. Magnified subsets show the level and variation of control samples (mark of “//” at the top of a column indicates that specific value exceeds the Y-axis maximum of that subset). Putative structures and names are reported, as structural identification can only be confirmed by comparison with synthetic standards. Normalized peak intensity was calculated as described in the Experimental procedures section. Note that the normalized intensity of two different adducts cannot be directly compared due to different ionization efficiencies of various structures. | ||
Overall, a variety of putative DNA adducts were detected in the DNA of bacterial cells, and NMC caused changes in the level and profile of putative DNA adducts. In the DNA of both NMC-exposed S. oneidensis and B. subtilis cells, we found the appearance and increased ion signal levels of the m/z values that are consistent with adducts that are known to result from oxidative stress, lipid peroxidation, glycation, and alkylation. A striking feature of the data in the tables is that the general categories of modifications are the same between two species despite significant biological differences. Comparing responses from two bacterial species, increased level or appearance of m/z values corresponding to putative adducts from oxidative stress (m/z 229, 244, 260, 284, 286, 310) and lipid peroxidation (m/z 382, 299) were found as shared features between the two bacteria (Tables 1 and 2). It is not surprising as oxidative stress and lipid peroxidation are two general mechanisms of genotoxicity. However, NMC-exposed B. subtilis showed a much higher variety of putative DNA adducts related to lipid peroxidation. In addition, fold changes are much higher in NMC-exposed B. subtilis cells compared to S. oneidensis.
Fluorometric assays detected an increase of intracellular ROS and metal ions
ROS generation and Ni(II)/Co(II) ion uptake were hypothesized to be two biochemical processes related to observed DNA damage. We used two fluorescent dyes, 2′,7′-dichlorodihydrofluorescein diacetate (DCFH2-DA, also known as 2′,7′-dichlorofluorescin diacetate) and Newport Green DCF diacetate (NPG-Ac), to measure the level of abiotic/intracellular ROS and metal ion uptake upon nano-NMC exposure, respectively. Utilizing the spontaneous hydrolysis of DCFH2-DA in aqueous medium, we first examined abiotic ROS production induced by NMC (Fig. S4†) in bacterial growth medium. Results showed that compared to the fast increase of fluorescence due to H2O2, nano-NMC induced slower but steady increase in fluorescence signal in a dose-dependent manner. This indicates that, unlike the direct oxidation by H2O2, nano-NMC might induce oxidation in an indirect manner, possibly through generation of ROS via reactions between aqueous media and NMC surfaces or released metal ions.11 Using the same dye, DCFH2-DA, we measured intracellular ROS upon short NMC exposure (15 minute) in both bacteria (Fig. 4(a) and (c)). Measurements of intracellular ROS were repeated multiple times with biological replicates, as described in the Experimental procedures section, and the average of each experimental run was plotted using different shapes. In S. oneidensis, most of the experimental runs showed that at all doses, nano-NMC can cause an increased level of intracellular ROS compared to control, while the general dose-dependent trend was not clear. It is possible that at higher concentrations, NMC started to kill bacterial cells during exposure, resulting in overall reduced fluorescence intensity. In B. subtilis, 25 mg L−1 nano-NMC appeared to cause higher levels of ROS compared to 5 mg L−1 in all experimental runs, while 5 mg L−1 also caused slight increase in intracellular ROS but not statistically significant compared to the control.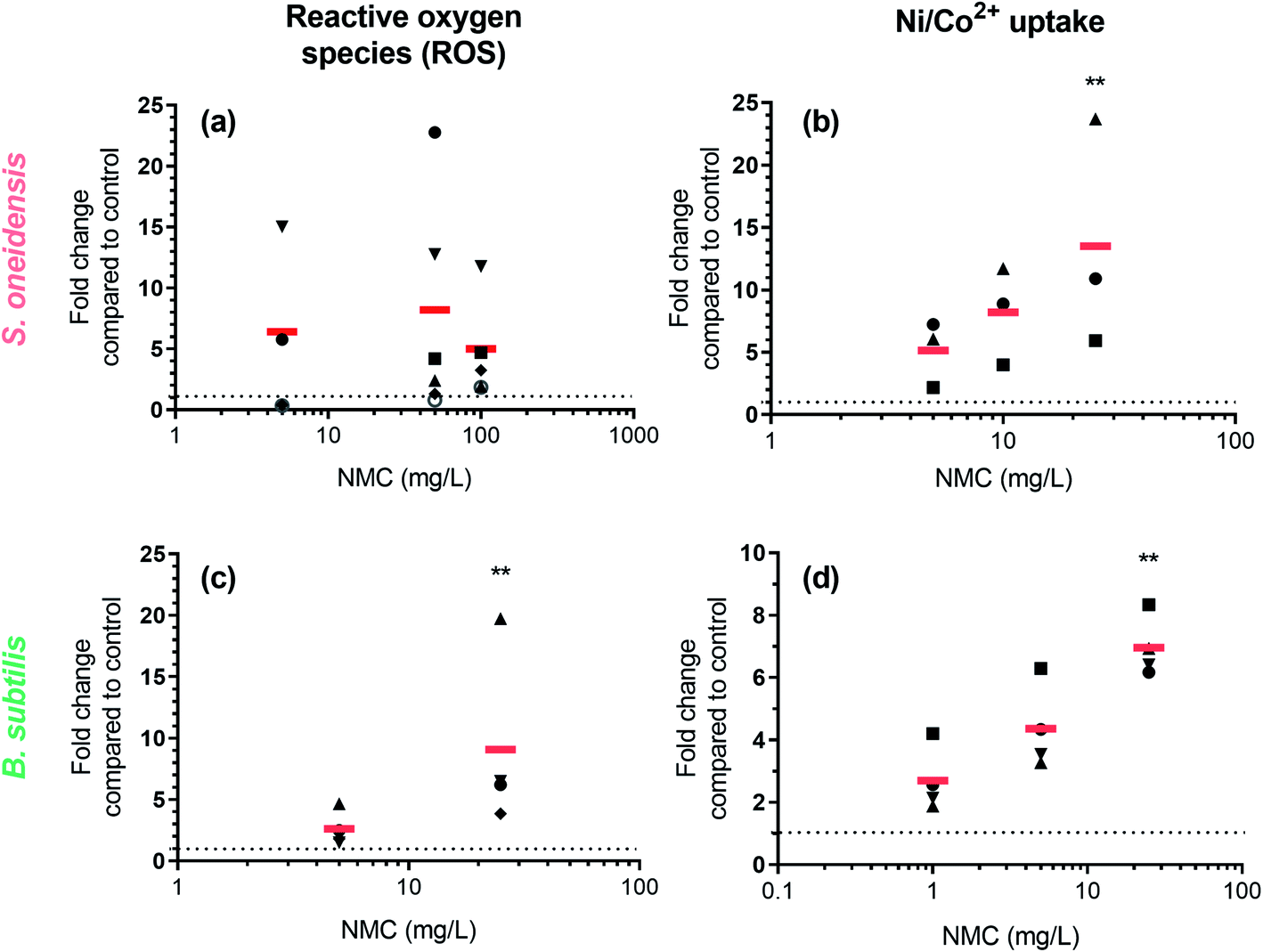 | ||
| Fig. 4 Increase in fluorescence intensity in dye-loaded cells shows increased level of intracellular ROS and nickel/cobalt ions. Left panels: Fluorescence intensity of DCFH2-DA-loaded (a) S. oneidensis and (c) B. subtilis cells upon NMC exposure. Right panels: Fluorescence intensity of NPG-Ac-loaded (b) S. oneidensis and (d) B. subtilis cells upon NMC exposure. In all figures, each data point represents the average of 2–3 biological replicates in one experimental run, and the same shape denotes data points from the same experimental run. The red bar represents the average of all experimental runs. Asterisks denote statistically significant difference of average (red bar) compared to control (dashed line) (Dunn's multiple comparison, alpha = 0.05). | ||
Newport Green DCF diacetate (NPG-Ac) is a cell-permeable dye sensitive to many transition metal ions, including Zn(II), Ni(II), Co(II), and Fe(II), but not Mn(II) or Fe(III).46 As it was found earlier that the toxicity of NMC to both S. oneidensis and B. subtilis is due to released nickel and cobalt, but not manganese, ions,11,14 the NPG-Ac dye is a great choice to measure metal ion uptake into bacterial cells. In fact, we have already applied this dye to measure the uptake of cobalt in S. oneidensis upon exposure to nanoscale multiphase lithiated cobalt phosphate.47 To validate the dye, we exposed bacterial cells pre-loaded with NPG-Ac to both Ni(II) and Co(II) ions (Fig. S5†). A clear dose-dependent trend in fluorescence intensity increase was observed. The response induced by intracellular Ni(II) is about 1 order of magnitude higher than Co(II) in general, possibly because the probe is more sensitive to Ni(II),46 bacterial cells uptake more Ni(II) than Co(II), or a mixed effect. Upon probe validation, we exposed NMC to bacterial cells pre-loaded with the NPG-Ac dye. As expected, results showed a dose-dependent increase in fluorescence intensity, indicating cellular uptake of nickel and cobalt ions released from NMC (Fig. 4(b) and (d)). It should be noted that NPG-Ac dye is not able to differentiate between Ni(II) and Co(II) ions, nor is it quantitative.
Expression levels of DNA repair/metabolism related genes changed upon NMC exposure
Gene expression was used to further understand the mechanism of DNA damage. We performed two different kinds of exposures: 8-hour (S. oneidensis) or 5-hour (B. subtilis) long exposure at the low dose of 5 mg L−1 NMC and 1 hour short exposure at the high dose of 50 mg L−1, as bacterial population remains ≥100% viability at these exposure scenarios according to growth and viability measurements. Table 3 lists selected genes and their related functions. Fig. 5(a) shows a heatmap of selected gene expression level changes. For details of selected genes, see SI_GeneInfo.xlsx.† Results show that despite the shorter exposure time, 50 mg L−1 NMC elicited more drastic response in both bacteria with mostly up-regulations compared to 5 mg L−1 with longer exposure time, indicating the shock of high dosage to cells. S. oneidensis responded to low and high dosages with different patterns in up/down-regulation, while B. subtilis showed similar patterns at two dosages with differences in expression levels. We noticed several shared gene expression features in the two bacteria responding to NMC, such as ruvC/recU, ruvA, and uvrA, related to DNA repair mechanisms. Looking closely at the gene expression levels and their statistical significance compared to control (Fig. 5(b) and (c)), we confirmed that 50 mg L−1 NMC induced many statistically significant changes in gene expression levels in both bacteria compared to 5 mg L−1, and these changes involved genes in all categories except for sporulation. Several oxidative stress-related genes, including katB, trxC, ahpC and gst, were all up-regulated upon 50 mg L−1 exposure to S. oneidensis. Interestingly, aphC was significantly down-regulated in B. subtilis upon high-dosage exposure, opposite to the trend in S. oneidensis. While two metal ion-related genes in B. subtilis did not show changes in expression levels, we found significant changes in levels of genes related to nickel sensing and transportation (hypB, nicT and hupE) in S. oneidensis.| S. oneidensis | B. subtilis | ||
|---|---|---|---|
| DNA repair | Base excision repair (BER) | mutM | mutM |
| Nucleotide excision repair (NER) | uvrA | uvrA | |
| Mismatch repair (MMR) | mutS | ||
| Error-prone DNA repair | umuC | ||
| dinB | yqjH | ||
| Recombinational repair | recA | recA | |
| Recombinational repair (dsDNA) | recB | addB | |
| Recombinational repair (gap repair) | recJ | recJ | |
| recF | |||
| Recombinational repair (Holliday junction removal; double-strand breakage) | recG | ||
| ruvA | ruvA | ||
| ruvC | recU | ||
| yqgF | |||
| General DNA stress response | lexA | lexA | |
| DNA replication | polA | ||
| Metal homeostasis | Divalent metal transport | corA | |
| Nickel uptake | hupE | ||
| nicT | |||
| Nickel chaperone | hypB | ||
| Manganese-containing enzyme | ydbD | ||
| Divalent metal efflux | czcD | ||
| Oxidative stress | Antioxidant enzyme | gst | |
| katB | |||
| sodB | |||
| ahpC | ahpC | ||
| trxC | |||
| Transcriptional regulator | oxyR | ||
| Cell machinery | Ribosomal RNA | 16S | |
| Protease | radA | radA | |
| RNA polymerase | rpoA | ||
| Fermentative lactate dehydrogenase | ldhA | ||
| Arsenate regulator | arsR | ||
| Sporulation | Sporulation | spoVK | |
| spo0A |
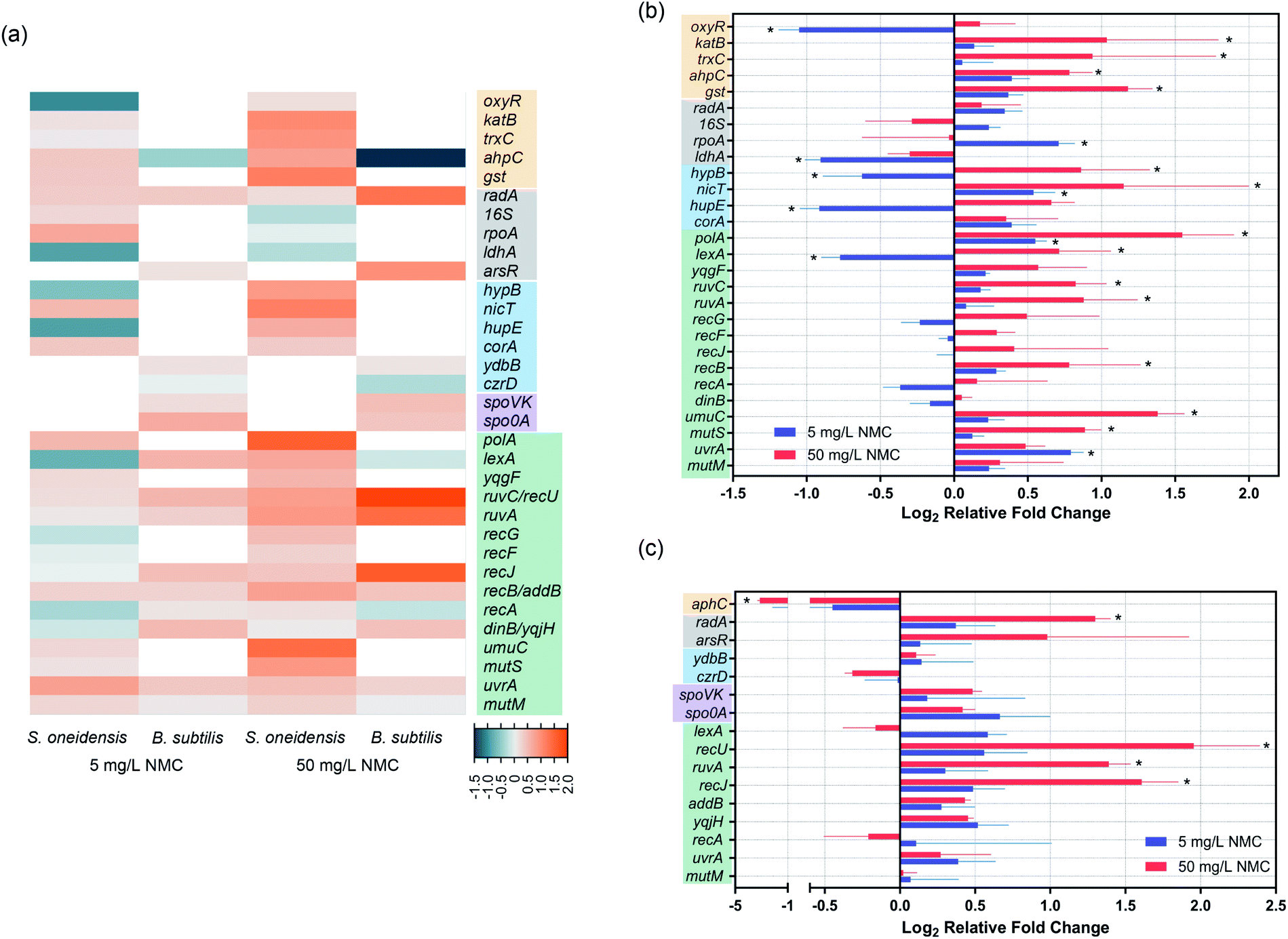 | ||
| Fig. 5 Gene expression results show regulation of genes related to oxidative stress, metal uptake, DNA repair and metabolism in S. oneidensis MR-1 and B. subtilis SB491. (a) A heatmap of all tested genes. White blocks stand for genes not tested. Color codes of heatmap are indicated by the legends at the bottom right corner: teal for down-regulation, orange for up-regulation, and light grey for zero fold change. Different color-shaded gene names denote categories, including oxidative stress (yellow), basic cell machinery (grey, housekeeping), metal ion homeostasis (blue), sporulation (purple, only for B. subtilis), and DNA repair/metabolism (green). Details of gene expression fold and statistical significance are shown for (b) S. oneidensis and (c) B. subtilis. Asterisks denote statistically significant difference compared to corresponding controls (multiple t-tests with p value correction, p < 0.05. n = 5 for S. oneidensis and n = 4 for B. subtilis). Error bars denote standard deviation. | ||
In terms of gene expression level changes related to pathways in DNA repair and general stress response, low dosage of NMC did not cause significant change in either bacteria except for down-regulation of lexA and up-regulation of polA and uvrA in S. oneidensis. High dosage of NMC, although for a much shorter time, induced up-regulation of many DNA repair-related genes in both bacteria, including polA, lexA, ruvC, ruvA, recB, umuC and mutS in S. oneidensis and recU, ruvA and recJ in B. subtilis. Pathways affected included DNA replication, general DNA stress response, DNA recombinational repair, error-prone DNA repair, DNA mismatch repair in S. oneidensis and DNA recombinational repair in B. subtilis.
Discussion
Dissolution to produce metal ions from metal oxide nanomaterials is one major source of NP toxicity.48 We showed previously that nickel(II) and cobalt(II) ions were primarily responsible for the inhibition of bacterial growth upon exposure to NMC.11,14 Since transition metal ions are known to be carcinogens and can cause DNA damage, we hypothesized the NMC toxicity mechanism to be DNA damage related to cobalt and nickel ions.To test this hypothesis, we applied the same assays to two distinct bacterial species, Gram-negative Shewanella oneidensis MR-1 and Gram-positive Bacillus subtilis SB491. Results from the Comet assay clearly show DNA double-strand breakage in both bacterial species upon sub-lethal (5 mg L−1 for 8 or 5 hours) and lethal (50 mg L−1 for 8 or 5 hours) NMC exposure (Fig. 2), and metal ion-sensing dye confirmed increased levels of intracellular metal ions after 30 min NMC exposure (Fig. 4). Changes in the expression levels of genes related to metal homeostasis in S. oneidensis further support fluorescence assays (Fig. 5). In S. oneidensis, it is clear that a long exposure at low dose (8 hour, 5 mg L−1) and a short “shock” at high dose (1 hour, 50 mg L−1), although showing no change in bacterial viability (Fig. 1), make differences in the cells' metal homeostasis genes. Bacterial HupE proteins are widespread among bacterial species and reported to potentially be nickel uptake transporters.49 The product of the hypB gene in Escherichia coli is essential for nickel insertion into [NiFe]hydrogenases.50 Both hupE and hypB genes were up-regulated at high dosage of NMC and down-regulated at low dosage. It is possible that cells respond to the drastic increase of nickel ions due to the high NMC dose in a short period of time by increasing uptake and incorporation of nickel, but then decrease expression of these genes to maintain a reasonable intracellular nickel concentration over time while NMC outside cells continues to release metal ions. Interestingly, nicT, a putative nickel transporter,51 showed up-regulation upon low dosage exposure, while the function of this gene is not well-understood. No genes related to nickel homeostasis in B. subtilis was tested.
We confirmed the presence of oxidative stress with fluorescence assays, gene expression, and DNA adductomics. DCFH2-DA is a fluorescence indicator for several ROS species, including superoxide anion, hydroxyl radical, nitric oxide, and hydrogen peroxide. We detected increased levels of fluorescence in cells upon 15 min NMC exposure in both bacteria (Fig. 4). Gene expression results regarding oxidative stress-related genes showed that S. oneidensis cells respond to high dosage of NMC via up-regulation of many antioxidant enzymes, including KatB for hydrogen peroxide, TrxC for thio-disulfide reduction, AhpC for primary and alkyl hydroperoxides, and Gst for general oxidative and xenobiotic stress, indicating the existence of oxidative stress (Fig. 5(b)). The transcriptional regulator OxyR was down-regulated at the high dose; this is interesting because the activation of OxyR is post-translational. Nevertheless, it is possible that oxyR gene was down-regulated to indirectly increase the expression of its downstream antioxidant genes such as peroxidase and alkyl hydroperoxide reductase.52 Interestingly, the ahpC gene expression in B. subtilis was largely decreased upon high dosage of NMC, opposite to the trend observed in S. oneidensis (Fig. 5(c)). Gene expression is a dynamic process, and the cellular responses may change over time after exposure.53 Although such down-regulation of aphC gene is unexpected, with the fluorescence assay clearly showing intracellular ROS and DNA adductomics showing evidence of oxidative stress, we believe that B. subtilis also experienced oxidative stress while reacting to NMC in a different way. The oxidative stress can be attributed to the uptake of transition metal ions followed by Fenton reactions, which generates ROS species in cells. However, it should be noted that a previous report from Lloyd, et al. showed that when incubated with circular DNA and hydrogen peroxide, Ni(II) and Co(II) mainly induced single-strand breakage instead of the double-strand breakage as we observed in this study.54 This indicates that the Fenton reaction is likely not the only mechanism causing oxidative stress in cells. Another possible source of oxidative stress is the generation of ROS from the nano-NMC surface, particularly from high oxidation state transition metals (+III or +IV) oxidizing water when they are dissolved into the aqueous medium. High oxidation state transition metal compounds are known as water oxidation catalysts and can generate ROS as intermediates when reacting with water.55,56 As NMC does not enter bacterial cells, ROS generated from nano-NMC surface may directly interact with bacterial cell envelope and cause lipid peroxidation, as indicated by DNA adductomics. Instead of only looking at 8-oxo-dG, DNA adductomics using high-resolution LC-tandem MS allows us to identify and semi-quantify a large collection of putative DNA base modifications. We identified many putative DNA adducts related to oxidative stress rather than simply 8-oxo-dG, the most widely used DNA modification to indicate DNA oxidative stress (Tables 1 and 2). Previous reports showed that Ni(II) and Co(II) with the presence of H2O2 induced very little increase in 8-oxo-dG level in cell-free in vitro experiments with DNA.54,57 In contrast, we found an increased level of putative 8-oxo-dG in the DNA of both NMC-exposed bacteria (Fig. 3). This indicates that the mechanisms causing oxidative stress by NMC in cells are more complex than in vitro Fenton reactions with DNA alone. Here, the results from our adductomic analyses showed that oxidative stress can result in many forms of putative oxidative stress-related DNA adducts other than 8-oxo-dG, such as putative adducts consistent with the oxidation of dC, nitrogen-containing ROS adducts, and lipid peroxidation adducts such as malonaldehyde adducts (Tables 1 and 2). DNA adductomic results further indicate that the specific mechanisms regarding oxidative stress in two bacteria are different, despite the similar response shown in fluorescence assays. Overall, B. subtilis shows more drastic response in adduct level changes than S. oneidensis. In B. subtilis, much larger fold changes of putative DNA adducts related to direct oxidation of DNA bases are observed compared to in S. oneidensis, indicating ROS plays a big role upon NMC exposure to B. subtilis. The up-regulation of antioxidant enzymes likely helped to mitigate the oxidative stress on S. oneidensis.
Interestingly, increase in lipid peroxidation-related DNA adducts is observed in both bacteria while B. subtilis putative adducts show higher diversity and levels compared to S. oneidensis. Lipid peroxidation has been thought not to be a primary mechanism for oxidative stress in bacterial species due to the lack of polyunsaturated fatty acids in bacteria.58 Yet, our DNA adductomic results showed the presence and increased levels of lipid peroxidation-related DNA adducts in both bacteria. S. oneidensis is found to produce polyolefinic hydrocarbons,59 and polyunsaturated fatty acids were found in B. subtilis with mass spectrometry, although in a very small fraction.60 Additionally, monounsaturated fatty acids commonly exist in bacterial species and can be a source of lipid peroxidation as well.61 This is the first report suggesting the existence of lipid peroxidation in bacteria with chemical evidence, although again, structural confirmation of putative lipid peroxidation DNA adduct can only be confirmed by comparison with synthetic standards in future work. The differences in the profiles of lipid peroxidation-related adducts in two bacteria are possibly due to the different cell envelop structure of Gram-negative and -positive bacteria and the differences between the lipid compositions of the two bacterial species.
Finally, we explored the potential mechanisms of DNA damage by examining gene expression profiles of selected DNA repair and metabolism-related genes. Among all monitored genes, both bacteria show up-regulation of ruvC/recU and ruvA gene expression upon high dosage of NMC. RuvABC complex in E. coli and RuvAB/RecU in B. subtilis are responsible for the Holliday junction resolution at the replication fork.62,63 Holliday junction formation and resolution represent one of the major repair pathways for DNA double-strand breakage,64 in agreement with Comet assay results. In addition to Holliday junction resolvases, other enzymes involved in recombinational repair are also regulated, such as recB in S. oneidensis and recJ in B. subtilis.62,65 Surprisingly, none of the exposures induced expression level changes in mutM, a gene participating in repairing 8-oxo-dG,66 at the time points chosen, which did not correlate to the increased level of putative 8-oxo-dG in either bacterial population. Since there are other two enzymes (MutY and MutT) for repairing 8-oxo-dG, further investigations are needed to evaluate these loci as potential biomarkers. In fact, expression level of a mutT homolog was found to inversely correlate to level of 8-oxo-dG in fetal mice.67 NMC exposure at high dosage also triggers other DNA repair pathways in S. oneidensis. Genes related to mismatch repair (mutS) and error-prone DNA repair (umuC) were found to be up-regulated upon high dosage of NMC in S. oneidensis, and these expression level changes both related to lipid peroxidation adducts. MutS was found to bind to exocyclic adducts derived from malondialdehyde produced via lipid peroxidation and trigger adduct removal by mismatch repair.68 UmuC, a Y-family DNA polymerase, can perform error-prone DNA repair by bypassing bulky DNA adducts, and the expression of the umu operon can be induced by lipid peroxidation products.69,70 This result suggests that increased level of error-prone DNA polymerase is related to the appearance and increase of lipid peroxidation DNA adducts and thus may serve as a biomarker for lipid peroxidation. In general, we find agreement in terms of cellular response between results from gene expression and DNA adductomics.
Conclusion
Cellular response to complex nanomaterials such as nano-NMC, nanoscale lithium nickel manganese cobalt oxide (nano LixNiyMnzCo1−y−zO2), can be much more complicated than nanomaterials of relatively simple or unreactive compositions with one or two modes of toxic actions. With many factors taking actions simultaneously, it remains a challenge to deconvolute the toxicity mechanism of complex nanomaterials. In this work, focusing on DNA damage, we combined conventional toxicology tools, chemical indicators, biological methodology, and cutting-edge analytical methods to reveal the toxicity mechanism of nano-NMC toxicity to two bacterial species and revealed unprecedented molecular details. Two environmentally relevant bacterial species, S. oneidensis and B. subtilis were used to interrogate the bacterial response to nano-NMC. We found nano-NMC induced DNA damage characterized by double-strand breakage and increased levels of putative endogenous DNA adducts. We found in good agreement, different approaches point to the same toxicity mechanisms, including direct oxidative stress, lipid peroxidation, and changes in metal homeostasis; these results also allow us to propose some genes and/or DNA adducts as potential biomarkers for future research on the toxicity of nano-complex metal oxides, such as genes for recombinational repair (e.g. ruvC/recU and ruvA) and several putative DNA adducts that increased upon exposure in both bacteria. Comparing two bacterial species, B. subtilis displayed a broader range of DNA adduct changes related to direct oxidative stress and lipid peroxidation, while S. oneidensis showed less changes in DNA adduct profiles. The comparison indicates that under the same exposure, B. subtilis may experience a much higher level of oxidative stress compared to S. oneidensis. Holliday junction resolution was found to be a shared mechanism in both bacteria, agreeing with the double-strand breakage observed in Comet assay, while S. oneidensis specifically exhibited DNA repair mechanisms related to nucleotide excision repair, mismatch repair, and error-prone DNA repair. The unprecedented molecular details revealed by our study can be a basis of future research on molecular mechanisms of DNA damage in nanotoxicology and can be potentially extrapolated to understand the impact of complex metal oxides to other organisms. Evaluation of DNA damage, a potentially universal toxicity mechanism, should be considered for the risk assessment of complex metal oxides of similar categories and other reactive nanomaterials.Experimental procedures
Bacterial culture
Shewanella oneidensis MR-1 BAA-1096 (S. oneidensis) was purchased from ATCC. S. oneidensis was plated from a frozen stock onto an 15% w/w LB agar plate and incubated at 30–32 °C overnight, and the colonies were used for subsequent bacteria experiments. Bacillus subtilis SB-491 (B. subtilis) was purchased from the Bacillus Genetic Stock Center (Columbus, OH). Frozen bacterial stock was inoculated on a lysogeny broth (LB) agar plate at 37 °C overnight, and the colonies were used. A sterile minimal medium (MM) was used for bacterial growth (0.68 g NaCl, 0.3 g KCl, 0.285 g MgCl2·6H2O, 0.3975 g Na2SO4, 0.15 g NH4Cl, 2.383 g HEPES, 0.0125 g Na2HPO4 and 0.0056 g CaCl2 per liter, pH 7.2–7.3) supplied with 100 mM sodium lactate for S. oneidensis and 10 mM dextrose for B. subtilis. Details of medium preparation can be found in the ESI.†Bacterial growth inhibition and viability test upon NMC exposure
Bacteria colonies from an agar plate incubated overnight were inoculated into 10 mL fresh LB broth and harvested after 4–5 hours at mid- or late-log phase by centrifugation at 2000 × g for 10 minutes. The pellet was re-suspended in fresh MM with lactate or dextrose, and the OD600 of the cell suspension was measured using a Spectronic 20 Spectrophotometer for S. oneidensis and a visible spectrometer (SpectroVis® Plus, Vernier Software and Technology) for B. subtilis. All other OD readings later were done by transferring 200 μL into a 96-well plate and reading absorbance at 600 nm on a Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek, VT) for S. oneidensis and on a Synergy Mx Microplate Reader (BioTek, VT) for B. subtilis.Comet assay for DNA strand breakage
Single cell gel electrophoresis on NMC-treated S. oneidensis was conducted following published protocols with minor changes.45,71 Bacterial cells at mid-log phase were exposed to 5 mg L−1 and 50 mg L−1 NMC for 8 or 5 hours for S. oneidensis or B. subtilis, respectively. Ten microliters of the NMC-treated cell suspension were mixed in 100 μL of 0.5% low-melting agarose (LMA) solution. Forty μL of the suspension was pipetted onto a Comet assay microscope slide (Travigen®) and spread evenly in a well. Once solidified at 4 °C, a 0.5% lysozyme-LMA layer was placed on top of the gel and solidified. The assembled slide was then incubated for 30 minutes at 30 °C for S. oneidensis and 37 °C for B. subtilis. The slide was then sequentially immersed in a lysing solution (2.5 M NaCl, 100.0 mM EDTA, 10.0 mM Tris–HCl, 1% sodium N-lauryl sarcosine, 0.6% Triton® X-100, pH 10.0) for 1 hour in the dark, and an enzyme digestion solution (2.5 M NaCl, 10.0 mM EDTA, 10.0 mM Tris–HCl, and 0.5 mg mL−1 proteinase K, pH 7.4) at 37 °C for 2 hours. Electrophoresis was performed in an electrophoresis buffer (sodium acetate and Tris at pH 9.0) at 12 V for 30 minutes while being chilled. The slide was then washed and dehydrated with 1.0 M ammonium acetate in ethanol, then absolute ethanol, and was left at room temperature to dry in the dark. The samples were then rehydrated in freshly prepared 5% DMSO in 10 mM NaH2PO4 solution and stained with 50.0 μL of 1.0 μM YOYO-1 in 5% DMSO. After air-drying in the dark for 5 minutes, the stack of microgel was imaged with a fluorescent microscope (100×, Ex/Em = 491/509 nm). ImageJ was used to analyze the DNA tail lengths.NMC exposure and DNA extraction for DNA adductomics
The exposure was done similar to that in the bacterial growth assay. Experiments were done in triplicates. For S. oneidensis, 21.9 mL bacterial suspension was used per replicate and 135 mL was used for B. subtilis. Bacterial growth was monitored before and after exposure. After 8 or 5 hours of exposure for S. oneidensis and B. subtilis, respectively, cell pellets were collected by centrifugation at 4000 × g for 20 minutes and stored at −80 °C until being thawed for DNA extraction. DNA extraction of S. oneidensis was done using Gentra Puregene Yeast/Bacteria Kit (Qiagen) and that of B. subtilis was extracted using a DNeasy PowerLyzer Microbial Kit (Qiagen). Both protocols were optimized to be suitable for the adductomics study. See ESI† for details of cell lysis and DNA extraction.Sample preparation for DNA adductomics
Extracted DNA was first incubated with 30 mg of NaBH3CN to stabilize possibly formed Schiff bases. After desalting, a two-step DNA digestion was performed to digest DNA into single nucleosides using DNase (from E. coli, Aldrich), phosphodiesterase-1 (PDE-1) (from Crotalus adamanteus, Aldrich) and alkaline phosphatase (ALP) (from Pichia pastoris, Aldrich). All the enzymes were purified by using a double filtration membrane Amicon Ultra (0.5 mL, cutoff 10 kDa) prior to use. The collected nucleosides were analyzed using an HPLC Ultimate 3000 equipped with a reversed phase column, Luna C18 (250 × 0.5 mm, 5 μm, 100 Å) and the amount of dG in each sample was quantified for data normalization later in the DNA adduct analysis. Finally, hydrophobic reversed phase fraction collection was done to exclude fractions of unmodified deoxyribonucleosides (dC, dG, dT, dA) and enrich analytes of interest (DNA adducts) for the following LC/MS3 measurement. An HPLC (Ultimate 3000, Thermo Scientific, Waltham, MA) equipped with a C18-Column (4.6 × 250 mm, 100 Å, 5 μm Luna-Phenomenex, Torrace, CA) was used for fraction collection, using as mobile phase H2O (A) and MeOH (B). For analysis of DNA extracted from nano-NMC exposed cells, fractions between four unmodified nucleoside peaks were collected. Collected fractions from one sample were combined, dried in a SpeedVac concentrator, and stored at −20 °C until LC/MS3 adductomic analysis. Two isotopic standards, 15N-N6-methyldA and 15N-N2-ethyldG, were added during DNA digestion and sample concentration for the purpose of quality control. Details of sample preparation can be found in the ESI.†LC/MS3 adductomic analysis
The dried DNA samples were reconstituted in 20 μL of LC-MS water (LCMS grade, Fluka) and then analyzed with a nanoflow UPLC system (Ultimate 3000 RSLCnano, Thermo Scientific, Waltham, MA) coupled to a hybrid Orbitrap mass detector (Fusion Tribrid, Thermo Scientific, Waltham, MA). The LC system was interfaced to the MS-detector using a nanoflow ESI ion source (Nanoflex, Thermo Scientific, Waltham, MA) operating in positive ion mode. The MS-analyses consist of three detection events: full scan, targeted data dependent MS2-acquisition (dd-MS2) and a neutral loss MS3-data acquisition (NL-MS3). An inclusion list of 128 DNA adducts (https://drive.google.com/open?id=14r9mA8NlL908piFCLA5yP-BZAsilxUl7) was used to trigger MS2 scan events. MS2 fragmentation was performed on full scan ions which were within 5 ppm of the inclusion list masses. MS3 data acquisition was performed on MS2 product ions which were different from their parent ions by the mass of the deoxyribose moiety (−dR; 116.0474 ± 0.0006 m/z, 5 ppm). Details of the injection, LC and mass spectrometry configuration can be found in ESI.†Adductomic data analysis and statistics
Data on putative DNA adducts were extracted from spectra acquired from LC/MS3 using Xcalibur Qual Browser. Generally, three levels of evidence were acquired to confirm a putative DNA adduct: the presence of clear m/z values of the precursor ([M + H]+) in full scan, the detection of a fragment after neutral loss in the precursor ion ([MH-dR]+) in MS2, neutral loss triggering of MS3, and the presence of specific fragments in MS3 that are signatures for the fragment of [MH-dR]+ detected in MS2. Additionally, the retention time was checked to exclude inconsistencies between the size of the adduct and unreasonable retention times (e.g. before 5 minutes) in our chromatographic conditions. If not all three levels of MS (full scan/MS2/MS3) were found, the putative DNA adduct was regarded as not detected and recorded as peak area equal to zero. For semi-quantification of a putative DNA adduct in a specific sample, the extracted ion chromatogram area m/z of interest in full scan with a mass range of 5 ppm was used, and the intensity was further normalized using (1) amount of dG determined in that specific sample and (2) extracted ion chromatogram area of the internal standard, 15N5-N2-ethyl-dG. The normalization is shown in the equation below: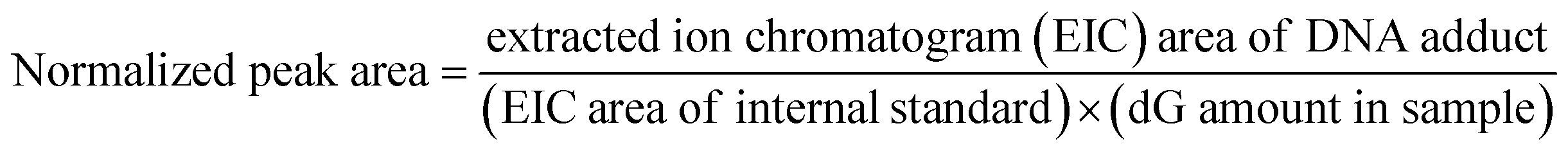
Normalized peak areas of all putative DNA adducts found in DNA extracted from NMC-exposed or control bacterial samples are listed in SI_AdductInfo.xlsx.† For the interpretation of semi-quantification data, if the m/z of a putative adduct was detected in all three NMC-exposed replicates but in none of the control replicates, this putative adduct was defined as present in the “Only in exposed”. For those adducts present in both exposed and non-exposed samples, the fold change in the area of the peak of the precursor ion detected in the full scan was evaluated as follows: the fold change was defined as the average of normalized peak area of NMC-exposed replicates divided by the average of that of control replicates; replicates with peak area zero was also included in calculating average. Individual two-tailed t-tests were performed to compare the normalized peak areas between NMC-exposed and control replicates of the same bacterium (α = 0.05, n = 3); replicates with peak area zero were also included in the t-tests. Comparisons of p < 0.05 were regarded as statistically significant difference between NMC-exposed and control averages.
Fluorescence assays on intracellular ROS and metal ions
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 5 minutes in a microcentrifuge. Pellets were washed with equal volume of MM with lactate or dextrose twice and finally re-suspended into 200 μL of MM with lactate (S. oneidensis) or 250 μL of MM with dextrose (B. subtilis). The resulting suspensions were transferred to a 96-well assay plate to read both optical density at 600 nm and fluorescence at Ex/Em = 493/522 nm (DCF) or 505/535 nm (NPG) on a Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek, VT) for S. oneidensis and on a Synergy Mx Microplate Reader (BioTek, VT) for B. subtilis.
000 × g for 5 minutes in a microcentrifuge. Pellets were washed with equal volume of MM with lactate or dextrose twice and finally re-suspended into 200 μL of MM with lactate (S. oneidensis) or 250 μL of MM with dextrose (B. subtilis). The resulting suspensions were transferred to a 96-well assay plate to read both optical density at 600 nm and fluorescence at Ex/Em = 493/522 nm (DCF) or 505/535 nm (NPG) on a Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek, VT) for S. oneidensis and on a Synergy Mx Microplate Reader (BioTek, VT) for B. subtilis.
Fluorescence assays on both intracellular ROS and metal ion uptake were repeated several times with different concentrations. For data points within one experimental run, background fluorescence of media only was subtracted from each raw fluorescence reading. Nano-NMC did not cause changes in fluorescence reading. After background subtraction, each background-subtracted fluorescence reading was divided by the average of control group readings; the resulting ratio was called “fold change compared to control”. The average fold change value for biological replicates in each experimental run was used to represent that experimental run, used for statistics and plotted in Fig. 4.
RNA extraction, reverse transcription and real-time quantitative PCR (qPCR)
A Direct-zol™ RNA MiniPrep kit (Zymo Research) was used to extract RNA from both S. oneidensis and B. subtilis cells. See ESI† for detailed protocol for cell lysis and RNA extraction. RNA characterization was done using a NanoDrop™ One/OneC Microvolume UV-Vis Spectrophotometer using Nucleic Acid 260/280 Ratio. The total isolated RNA samples were then preserved in at −80 °C until reverse transcription.Total purified RNA was reverse transcribed into complementary deoxyribonucleic acid (cDNA) following Invitrogen's protocols and the iCycler base module of an iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad). cDNA was synthesized using normalized samples of 100 ng of total RNA template. RNA templates were firstly incubated with random primers (Invitrogen, 48190-011) and dNTP mix (Invitrogen, 18427013) at 65 °C for 5 min following a chilling step on ice for 1 min. SuperScript III reverse transcriptase (Invitrogen, 18080-044), dithiothreitol (Invitrogen), and RNaseOUT™ recombinant ribonuclease inhibitors (Invitrogen, 10777019) were then added into samples following incubation steps of 25 °C for 5 minutes, 50 °C for 60 minutes, and 70 °C for 15 minutes for random primer extension. Synthesized cDNA was then stored at −20 °C before qPCR reactions.
A list of genes of interest can be found in Table 3 and SI_GeneInfo.xlsx in ESI.† For qPCR, an iQ5 real-time PCR detection system (Bio-Rad Laboratories) was used with SYBR Green for the fluorescent intercalating dye (iTaq™ Universal SYBR® Green Supermix, Bio-Rad). Each qPCR reaction containing cDNA, primers and fluorescence dye was carried out in 96-Well PCR plate following manufacturer's protocols from Bio-Rad. The reactions started at 95 °C for 10 minutes for DNA denaturing, then 40-times of real-time PCR cycles of amplification (15 s at 95 °C, followed by 30 s at 60 °C). Fluorescence of the SYBR Green was then detected at the end of each of the PCR cycle. All samples were done with technical duplicates.
Statistics (except for adductomics)
Author contributions
T. A. Q., C. L. H., S. B. and Z. V. F. conceived the idea of this manuscript and designed the experiments. R. J. H. designed, provided, and characterized the nanomaterials. T. A. Q. performed experiments on S. oneidensis toxicity, fluorescence assays, gene expression and various data analysis. V. G. performed DNA sample preparation, high-resolution mass spectrometry and data analysis and A. C. and P. W. V. contributed to the optimization of adductomics workflow. T. A. Q., T. P. and Z. V. F. optimized DNA extraction protocol and extracted DNA for adductomics. K. N. L. H. performed experiments on B. subtilis toxicity and fluorescence assays. T. P. conducted the Comet assay for both bacteria. Z. V. F., T. P. and K. N. L. H. performed gene expression on B. subtilis. J. H. and X. Y. contributed to DNA extraction and sample preparation for adductomics. T. A. Q. took the lead in writing the manuscript and V. G., K. N. L. H., T. P. and R. J. H. contributed their parts. All authors contributed to the editing of the manuscript. C. L. H., Z. V. F. and S. B. supervised the project.Data availability
The data that support the findings of this study are available from the authors on reasonable request, see author contributions for specific data sets.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by National Science Foundation under the Center for Sustainable Nanotechnology, CHE-2001611. The CSN is part of the Centers for Chemical Innovation Program. T. A. Q. thanks the support from Student Seed Grant from the Center for Sustainable Nanotechnology. Mass spectrometry was carried out in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, University of Minnesota, funded in part by Cancer Center Support Grant CA-77598. We thank Dr Mimi N. Hang at the University of Wisconsin, Madison for synthesizing and characterizing the starting nano-NMC materials. We thank Dr Yuya Hayashi for access to the NORMA-Gene workbook in qPCR data analysis.References
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 Search PubMed.
- I. Belharouak, Y. K. Sun, J. Liu and K. Amine, J. Power Sources, 2003, 123, 247–252 Search PubMed.
- Z. Wu, S. Ji, J. Zheng, Z. Hu, S. Xiao, Y. Wei, Z. Zhuo, Y. Lin, W. Yang, K. Xu, K. Amine and F. Pan, Nano Lett., 2015, 15, 5590–5596 Search PubMed.
- T. Dutta, K. H. Kim, A. Deep, J. E. Szulejko, K. Vellingiri, S. Kumar, E. E. Kwon and S. T. Yun, Renewable Sustainable Energy Rev., 2018, 82, 3694–3704 Search PubMed.
- K. M. Winslow, S. J. Laux and T. G. Townsend, Resour., Conserv. Recycl., 2018, 129, 263–277 Search PubMed.
- D. H. P. Kang, M. Chen and O. A. Ogunseitan, Environ. Sci. Technol., 2013, 47, 5495–5503 Search PubMed.
- J. Gans, M. Wolinsky and J. Dunbar, Science, 2005, 309, 1387–1390 Search PubMed.
- R.-A. Sandaa, V. Torsvik, Ø. Enger, F. L. Daae, T. Castberg and D. Hahn, FEMS Microbiol. Ecol., 1999, 30, 237–251 Search PubMed.
- H. K. Song, K. T. Lee, M. G. Kim, L. F. Nazar and J. Cho, Adv. Funct. Mater., 2010, 20, 3818–3834 Search PubMed.
- M. N. Hang, I. L. Gunsolus, H. Wayland, E. S. Melby, A. C. Mensch, K. R. Hurley, J. A. Pedersen, C. L. Haynes and R. J. Hamers, Chem. Mater., 2016, 28, 1092–1100 Search PubMed.
- M. N. Hang, N. V. Hudson-Smith, P. L. Clement, Y. Zhang, C. Wang, C. L. Haynes and R. J. Hamers, ACS Appl. Nano Mater., 2018, 1, 1721–1730 Search PubMed.
- I. L. Gunsolus, M. N. Hang, N. V. Hudson-Smith, J. T. Buchman, J. W. Bennett, D. Conroy, S. E. Mason, R. J. Hamers and C. L. Haynes, Environ. Sci.: Nano, 2017, 4, 636–646 Search PubMed.
- J. Bozich, M. Hang, R. Hamers and R. Klaper, Environ. Toxicol. Chem., 2017, 36, 2493–2502 Search PubMed.
- Z. V. Feng, B. R. Miller, T. G. Linn, T. Pho, K. N. L. Hoang, M. N. Hang, S. L. Mitchell, R. T. Hernandez, E. E. Carlson and R. J. Hamers, Environ. Sci.: Nano, 2019, 6, 305–314 Search PubMed.
- S. L. Mitchell, N. V. Hudson-Smith, M. S. Cahill, B. N. Reynolds, S. D. Frand, C. M. Green, C. Wang, M. N. Hang, R. T. Hernandez, R. J. Hamers, Z. V. Feng, C. L. Haynes and E. E. Carlson, Chem. Sci., 2019, 10, 9768–9781 Search PubMed.
- K. Salnikow and A. Zhitkovich, Chem. Res. Toxicol., 2008, 21, 28–44 Search PubMed.
- D. Kirkland, T. Brock, H. Haddouk, V. Hargeaves, M. Lloyd, S. Mc Garry, R. Proudlock, S. Sarlang, K. Sewald, G. Sire, A. Sokolowski and C. Ziemann, Regul. Toxicol. Pharmacol., 2015, 73, 311–338 Search PubMed.
- M. Ghosh, M. Bandyopadhyay and A. Mukherjee, Chemosphere, 2010, 81, 1253–1262 Search PubMed.
- K. S. Butler, D. J. Peeler, B. J. Casey, B. J. Dair and R. K. Elespuru, Mutagenesis, 2015, 30, 577–591 Search PubMed.
- J. Zhang, Y. Chen, S. Liu, Z. Zhang, M. Xu, J. Ma, Y. Xu and M. Gao, Adv. Mater., 2016, 29, 1604580 Search PubMed.
- K. Bhattacharya, M. Davoren, J. Boertz, R. P. Schins, E. Hoffmann and E. Dopp, Part. Fibre Toxicol., 2009, 6, 17 Search PubMed.
- Z. O. Kyjovska, N. R. Jacobsen, A. T. Saber, S. Bengtson, P. Jackson, H. Wallin and U. Vogel, Environ. Mol. Mutagen., 2015, 56, 41–49 Search PubMed.
- N. R. Jacobsen, P. A. White, J. Gingerich, P. Moller, A. T. Saber, G. R. Douglas, U. Vogel and H. Wallin, Environ. Mol. Mutagen., 2011, 52, 331–337 Search PubMed.
- D. Galaris and A. Evangelou, Crit. Rev. Oncol. Hematol., 2002, 42, 93–103 Search PubMed.
- H. H. Hau and J. A. Gralnick, Annu. Rev. Microbiol., 2007, 61, 237–258 Search PubMed.
- R. W. Lewis, P. M. Bertsch and D. H. McNear, Nanotoxicology, 2019, 13, 392–428 Search PubMed.
- A. Azqueta and M. Dusinska, Front. Genet., 2015, 6, 1–4 Search PubMed.
- N. P. Singh, R. E. Stephens, H. Singh and H. Lai, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1999, 429, 159–168 Search PubMed.
- S. Balbo, S. S. Hecht, P. Upadhyaya and P. W. Villalta, Anal. Chem., 2014, 86, 1744–1752 Search PubMed.
- S. Balbo, R. J. Turesky and P. W. Villalta, Chem. Res. Toxicol., 2014, 27, 356–366 Search PubMed.
- J. A. Swenberg, K. Lu, B. C. Moeller, L. Gao, P. B. Upton, J. Nakamura and T. B. Starr, Toxicol. Sci., 2011, 120, 130–145 Search PubMed.
- D. T. Beranek, G. L. White, R. H. Heflich and F. A. Beland, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 5175–5178 Search PubMed.
- L. A. VanderVeen, M. F. Hashim, Y. Shyr and L. J. Marnett, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14247–14252 Search PubMed.
- I. Papageorgiou, C. Brown, R. Schins, S. Singh, R. Newson, S. Davis, J. Fisher, E. Ingham and C. P. Case, Biomaterials, 2007, 28, 2946–2958 Search PubMed.
- O. Bar-Ilan, C. C. Chuang, D. J. Schwahn, S. Yang, S. Joshi, J. A. Pedersen, R. J. Hamers, R. E. Peterson and W. Heideman, Environ. Sci. Technol., 2013, 47, 4726–4733 Search PubMed.
- K. Ishino, T. Kato, M. Kato, T. Shibata, M. Watanabe, K. Wakabayashi, H. Nakagama and Y. Totsuka, Int. J. Mol. Sci., 2015, 16, 3474–3492 Search PubMed.
- R. A. Kanaly, R. Micheletto, T. Matsuda, Y. Utsuno, Y. Ozeki and N. Hamamura, MicrobiologyOpen, 2015, 4, 841–856 Search PubMed.
- T. A. Qiu, T. H. T. Nguyen, N. V. Hudson-Smith, P. L. Clement, D.-C. Forester, H. Frew, M. N. Hang, C. J. Murphy, R. J. Hamers, Z. Vivian Feng and C. L. Haynes, Anal. Chem., 2017, 89, 2057–2064 Search PubMed.
- A. R. D. Stebbing, Sci. Total Environ., 1982, 22, 213–234 Search PubMed.
- Y. Chen, J. Yao, F. Wang, Y. Zhou, H. Chen, N. Gai, H. Chen, K. Chen, T. Maskow, B. Ceccanti, P. Trebse and G. Zaray, Curr. Microbiol., 2008, 57, 258–263 Search PubMed.
- H. L. Karlsson, S. Di Bucchianico, A. R. Collins and M. Dusinska, Environ. Mol. Mutagen., 2015, 56, 82–96 Search PubMed.
- A. R. Collins, V. L. Dobson, M. Dušinská, G. Kennedy and R. Štětina, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1997, 375, 183–193 Search PubMed.
- D. W. Fairbairn, P. L. Olive and K. L. O'Neill, Mutat. Res., Rev. Genet. Toxicol., 1995, 339, 37–59 Search PubMed.
- M. Wojewódzka, I. Buraczewska and M. Kruszewski, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2002, 518, 9–20 Search PubMed.
- D. Solanky and S. E. Haydel, J. Microbiol. Methods, 2012, 91, 257–261 Search PubMed.
- J. Zhao, B. A. Bertoglioa, M. J. Devinney Jr, K. E. Dineley and A. R. Kay, Anal. Biochem., 2009, 384, 34–41 Search PubMed.
- P. L. Clement, J. E. Kuether, J. R. Borgatta, J. T. Buchman, M. S. Cahill, T. A. Qiu, R. J. Hamers, Z. V. Feng and C. L. Haynes, Chem. Res. Toxicol., 2020, 33(3), 806–816 Search PubMed.
- A. B. Djurišić, Y. H. Leung, A. M. C. Ng, X. Y. Xu, P. K. H. Lee, N. Degger and R. S. S. Wu, Small, 2015, 11, 26–44 Search PubMed.
- T. Eitinger, J. Suhr, L. Moore and J. A. C. Smith, BioMetals, 2005, 18, 399–405 Search PubMed.
- T. Maier, A. Jacobi, M. Sauter and A. Bock, J. Bacteriol., 1993, 175, 630–635 Search PubMed.
- D. Agranoff and S. Krishna, Front. Biosci., 2004, 9, 2996–3006 Search PubMed.
- P. J. Pomposiello and B. Demple, Trends Biotechnol., 2001, 19, 109–114 Search PubMed.
- Y. Cui, E. S. Melby, A. C. Mensch, E. D. Laudadio, M. N. Hang, A. Dohnalkova, D. Hu, R. J. Hamers and G. Orr, Nano Lett., 2019, 19, 1990–1997 Search PubMed.
- D. R. Lloyd, P. L. Carmichael and D. H. Phillips, Chem. Res. Toxicol., 1998, 11, 420–427 Search PubMed.
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 Search PubMed.
- O. Diaz-Morales, D. Ferrus-Suspedra and M. T. M. Koper, Chem. Sci., 2016, 7, 2639–2645 Search PubMed.
- D. R. Lloyd and D. H. Phillips, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1999, 424, 23–36 Search PubMed.
- J. A. Imlay, Curr. Opin. Microbiol., 2015, 24, 124–131 Search PubMed.
- D. J. Sukovich, J. L. Seffernick, J. E. Richman, K. A. Hunt, J. A. Gralnick and L. P. Wackett, Appl. Environ. Microbiol., 2010, 76, 3842–3849 Search PubMed.
- J. Gidden, J. Denson, R. Liyanage, D. M. Ivey and J. O. Lay Jr, Int. J. Mass Spectrom., 2009, 283, 178–184 Search PubMed.
- H. Yin, L. Xu and N. A. Porter, Chem. Rev., 2011, 111, 5944–5972 Search PubMed.
- A. Kuzminov, Microbiol. Mol. Biol. Rev., 1999, 63, 751–813 Search PubMed.
- H. Sanchez, D. Kidane, P. Reed, F. A. Curtis, M. C. Cozar, P. L. Graumann, G. J. Sharples and J. C. Alonso, Genetics, 2005, 171, 873–883 Search PubMed.
- T. Helleday, J. Lo, D. C. van Gent and B. P. Engelward, DNA Repair, 2007, 6, 923–935 Search PubMed.
- E. P. C. Rocha, E. Cornet and B. Michel, PLoS Genet., 2005, 1, e15 Search PubMed.
- T. Tajiri, H. Maki and M. Sekiguchi, Mutat. Res., DNA Repair, 1995, 336, 257–267 Search PubMed.
- K. Bialkowski, A. Bialkowska, L. M. Anderson and K. S. Kasprzak, Free Radical Biol. Med., 1999, 27, 90–94 Search PubMed.
- K. A. Johnson, M. L. Mierzwa, S. P. Fink and L. J. Marnett, J. Biol. Chem., 1999, 274, 27112–27118 Search PubMed.
- W. Yang, Curr. Opin. Struct. Biol., 2003, 13, 23–30 Search PubMed.
- M. Benamira and L. J. Marnett, Mutat. Res., DNA Repair, 1992, 293, 1–10 Search PubMed.
- P. L. Olive and J. P. Banáth, Nat. Protoc., 2006, 1, 23–29 Search PubMed.
- L.-H. Heckmann, P. B. Sørensen, P. H. Krogh and J. G. Sørensen, BMC Bioinf., 2011, 12, 250 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02987d |
| ‡ Current address: Beckman Institute, University of Illinois, Urbana-Champaign, 405 N Mathews Ave, Urbana, IL 61801, USA. |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |