DOI:
10.1039/D0SC04478D
(Edge Article)
Chem. Sci., 2020,
11, 11259-11265
Cyclodepsipeptide alveolaride C: total synthesis and structural assignment†
Received
15th August 2020
, Accepted 17th September 2020
First published on 21st September 2020
Abstract
First stereoselective total synthesis of naturally occurring bioactive cyclodepsipeptide alveolaride C has been achieved using a convergent approach. This synthetic study enabled us to establish unambiguously the stereochemistry of three unassigned chiral centres embedded in the nonpeptidic segment as well as revised the stereochemistry of the proposed β-phenylalanine counterpart of the molecule. The key strategic features of this synthesis include Sharpless asymmetric dihydroxylation for installing the vicinal diol moiety, Julia–Kocienski olefination for constructing the aliphatic side chain, the Shiina protocol for intermolecular esterification, amide coupling and macrolactamization for the ring formation.
Introduction
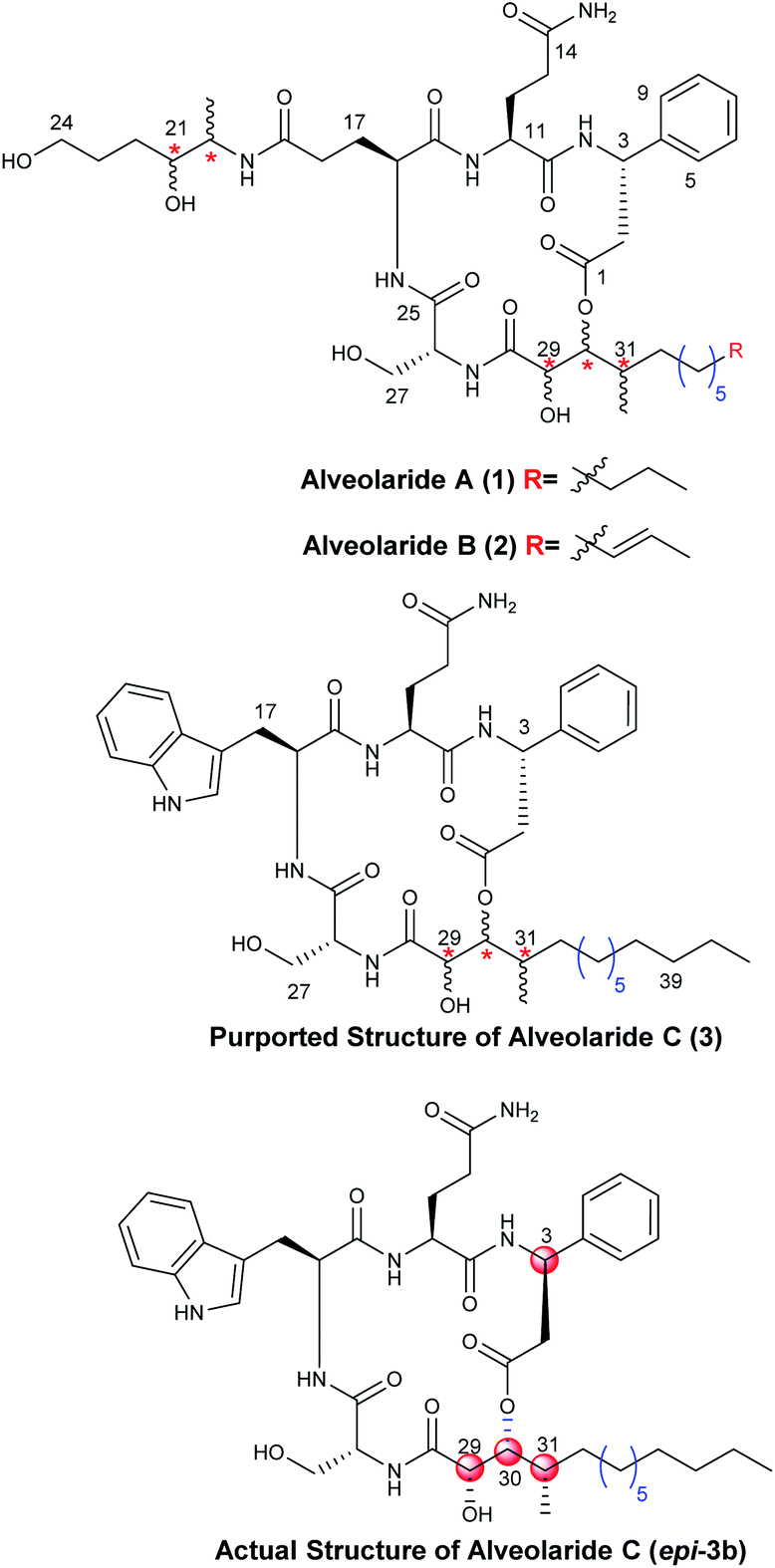
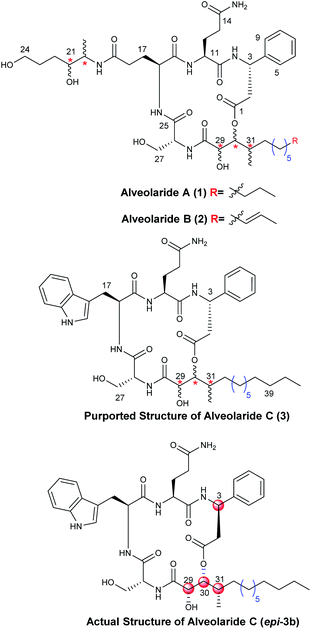
Microorganisms are known as the rich sources of structurally novel secondary metabolites. Many of them possess a broad range of biological activities.1 Chemical synthesis of these metabolites is, thus, crucial for their unambiguous structural confirmation and to ensure adequate supply for exploring their biological importance.2 During the search for naturally occurring environmentally benign pesticides from microorganisms Dow Agro Sciences, in 2018, first identified and isolated a family of novel cyclodepsipeptide alveolarides A–C (1–3, Fig. 1) from the culture broth of Microascus alveolaris strain PF1466.3 Invitro inhibition studies against different harmful plant pathogens Pyricularia oryzae, Zymoseptoria tritici, Ustilago maydis, Puccinia triticina, and Phakopsora pachyrhizi revealed that these cyclodepsipeptides exhibited excellent to moderate activities.3 The structures of these molecules were determined partially using NMR, mass spectroscopic analysis and chemical modification techniques. Architecturally, alveolarides are 17-membered macrocycles bearing rarely found 2,3-dihydroxy-4-methyltetradecanoic acid (DHMTDA) as the nonpeptide unit in which the configurations of all three stereocentres remained undetermined. The DHMTDA segment of alveolaride B was found to be slightly different from that of alveolarides A and C in which an olefin between C39–C40 is present. D-β-Phenylalanine, L-glutamine and D-serine are the residues common to all the members of this family of cyclodepsipeptides. An L-Glutamic acid derivative bearing two additional unassigned stereocentres in alveolarides A & B and L-tryptophan in alveolaride C were observed as the other amino acid counterparts, respectively.3 The chemical structure of alveolaride C was proposed based on the similarities in the 1H and 13C NMR data of alveolarides A and B but no degradation study was performed due to the scarcity of the compound. Our continual interest in synthesis of natural products and their structural assignment4 prompted us to envisage initially the total synthesis of structurally unique alveolaride C (3). The establishment of the stereochemistry of the unassigned centres in the common nonpeptide segment of this family of molecules is very crucial and essential to synthesize the other members including the more active alveolaride A. There are three undetermined stereocentres in the molecule providing the possibility of eight configurational isomers among which one could be expected to be the actual structure of alveolaride C. To narrow down these possibilities, we depended on the reported NMR spectral data of the DHMTDA counterpart of alveolaride C.3 Two diastereomers of the purported structure of alveolaride C (3) having the stereochemistry 29-(R)/30-(S)/31-(R) and 29-(S)/30-(R)/31-(S) were synthesized initially as the specific rotation value of DHMTDA was not reported.3 Comparison of their NMR data with those of the isolated alveolaride C (3) revealed that the latter isomer was a better match with some discrepancies originating from the β-phenylalanine counterpart. Herein, we described a convergent and flexible route for the first stereoselective total synthesis of the actual structure of alveolaride C (epi-3b, Fig. 1) which enabled us to assign the undetermined stereocentres (C-29, C-30, and C-31) successfully and also revise the proposed configuration of the β-phenylalanine (C-3) counterpart of the molecule.
 |
| | Fig. 1 Chemical structures of alveolarides. | |
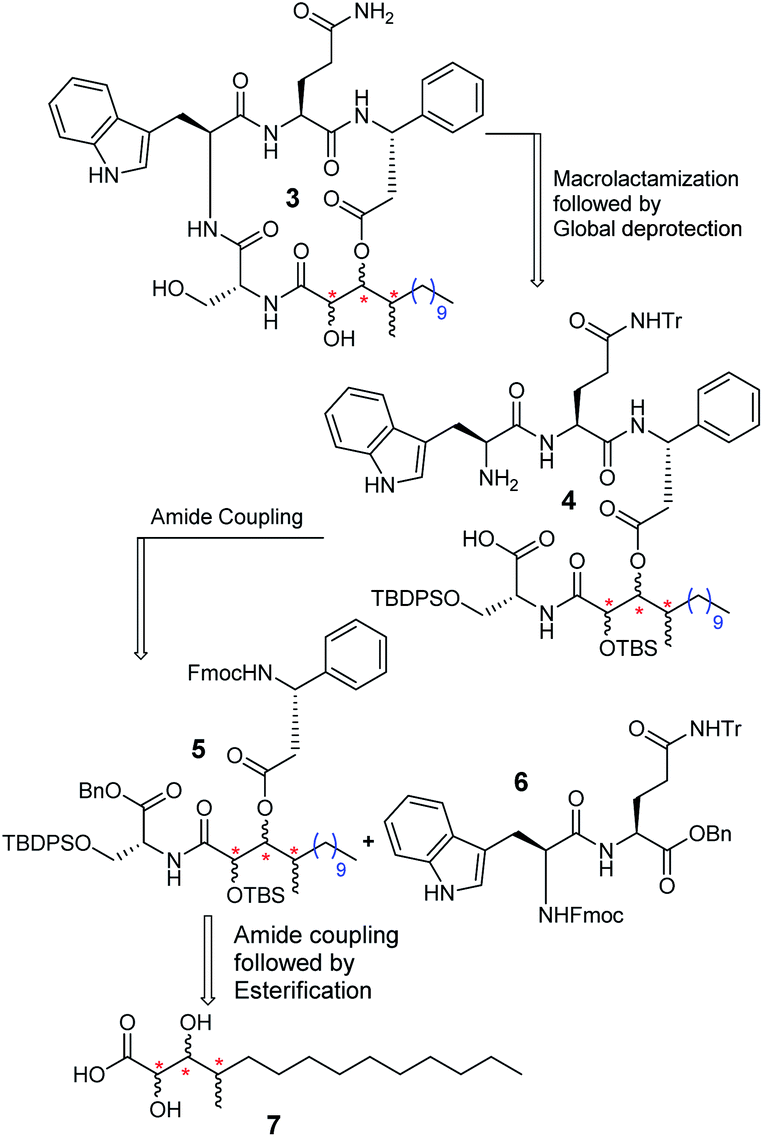
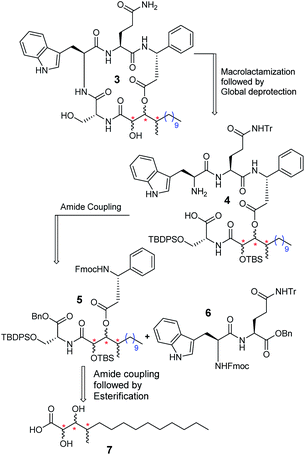
The retrosynthetic analysis of alveolaride C (3) is delineated in Scheme 1. There are a few possible sites in the molecule that are suitable for macrocyclization. We relied on the macrolactamization approach and planned to synthesize the target molecule from precursor 4 which further could be prepared from suitably protected compounds 5 and 6 by amide coupling. Compound 5 could be synthesized from acid 7 by amide coupling with the D-serine residue followed by intermolecular esterification with the N-protected D-β-phenylalanine counterpart.
 |
| | Scheme 1 Retrosynthesis of alveolaride C (3). | |
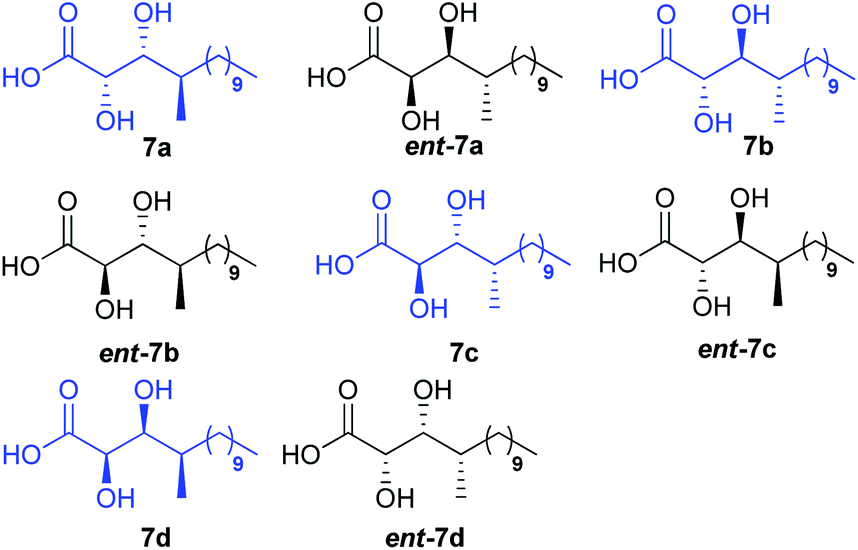
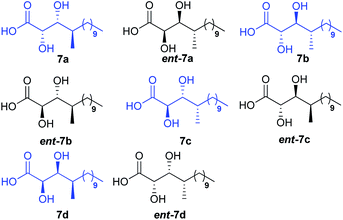
There are three stereocentres in the DHMTDA counterpart of alveolaride C which remained unassigned during its discovery. Thus, it could be conceived that the structure of DHMTDA would match with one out of eight possible stereoisomers [7(a–d) and ent-7(a–d), Fig. 2]. To solve this structural riddle, we initially planned to synthesize four of them 7(a–d) with an intension to compare their spectroscopic data with the reported data of the DHMTDA segment. The initial trial of the isolation group to confirm the absolute configurations of these stereocentres using Mosher's ester method was not successful as the required ester did not form.3
 |
| | Fig. 2 Possible stereoisomers of 2,3-dihydroxy-4-methyl-tetradecanoic (DHMTDA) [7(a–d) and ent-7(a–d)]. | |
Results and discussion
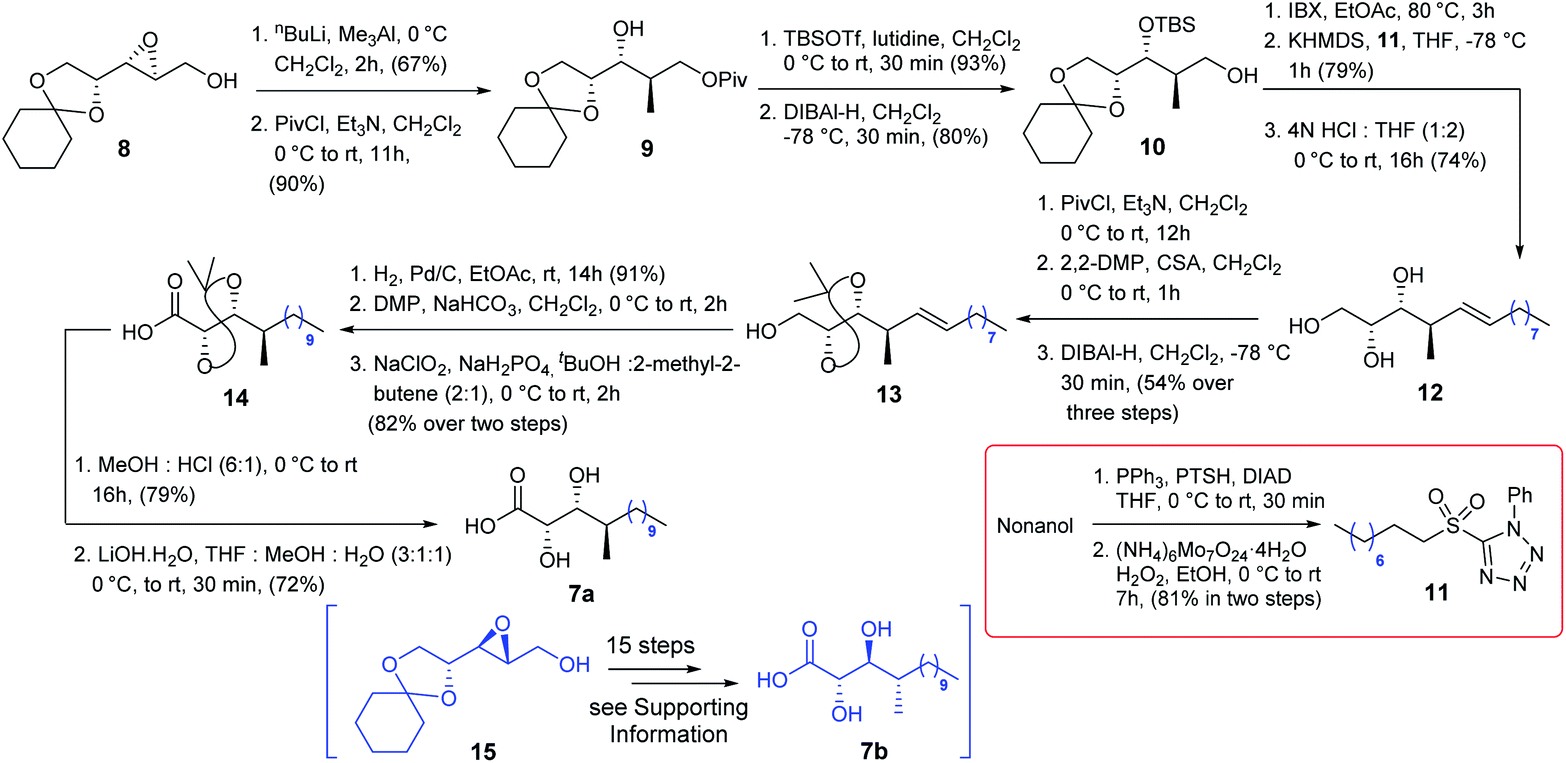
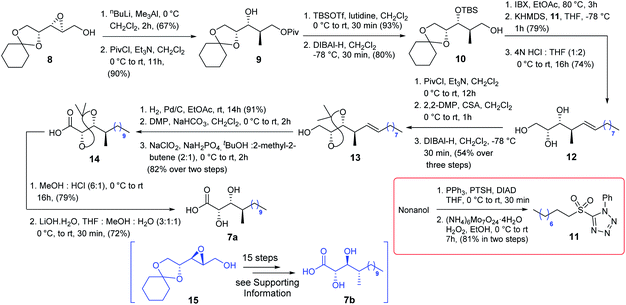
The synthesis of acid 7a commenced with the known epoxide 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5a (Scheme 2) which was treated with nBuLi/Me3Al6 followed by treatment with NaIO4 to obtain the corresponding 1,3-diol which was further reacted with PivCl/Et3N to obtain compound 9. The secondary hydroxy centre of compound 9 was protected as TBS ether and subsequently treated with DIBAL-H to access alcohol 10 which was oxidized further using IBX. The resultant aldehyde was then subjected to Julia–Kocienski olefination7 with sulfone 11, prepared from nonanol in two steps (Scheme 2), in the presence of KHMDS to produce the corresponding olefin with complete E-selectivity and treated further with 4N HCl/THF to obtain triol 12. Next, triol 12 was treated with PivCl/Et3N followed by 2,2-DMP/CSA and finally with DIBAL-H to yield compound 13 efficiently. It was then hydrogenated and subsequently oxidized to acid 14 following a two-step protocol, and DMP oxidation followed by Pinnick oxidation. Many conditions (HCl/THF, AcOH/H2O, HF/Py, CSA/MeOH, and HCl/MeOH) at different temperatures were screened to deprotect the acetonide of acid 14. CSA/MeOH for 23 h as well as HCl/MeOH for 16 h provided the corresponding acetonide deprotected product in satisfactory yield. However, the acid functionality concomitantly transformed into its corresponding methyl ester which was later hydrolyzed using LiOH·H2O to acid 7a.
5a (Scheme 2) which was treated with nBuLi/Me3Al6 followed by treatment with NaIO4 to obtain the corresponding 1,3-diol which was further reacted with PivCl/Et3N to obtain compound 9. The secondary hydroxy centre of compound 9 was protected as TBS ether and subsequently treated with DIBAL-H to access alcohol 10 which was oxidized further using IBX. The resultant aldehyde was then subjected to Julia–Kocienski olefination7 with sulfone 11, prepared from nonanol in two steps (Scheme 2), in the presence of KHMDS to produce the corresponding olefin with complete E-selectivity and treated further with 4N HCl/THF to obtain triol 12. Next, triol 12 was treated with PivCl/Et3N followed by 2,2-DMP/CSA and finally with DIBAL-H to yield compound 13 efficiently. It was then hydrogenated and subsequently oxidized to acid 14 following a two-step protocol, and DMP oxidation followed by Pinnick oxidation. Many conditions (HCl/THF, AcOH/H2O, HF/Py, CSA/MeOH, and HCl/MeOH) at different temperatures were screened to deprotect the acetonide of acid 14. CSA/MeOH for 23 h as well as HCl/MeOH for 16 h provided the corresponding acetonide deprotected product in satisfactory yield. However, the acid functionality concomitantly transformed into its corresponding methyl ester which was later hydrolyzed using LiOH·H2O to acid 7a.
 |
| | Scheme 2 Synthesis of acids 7a and 7b. | |
Acid 7b was also synthesized from the known compound 155 following exactly the same chemistry of acid 7a [see Scheme S2 (page-S28) in the ESI†].
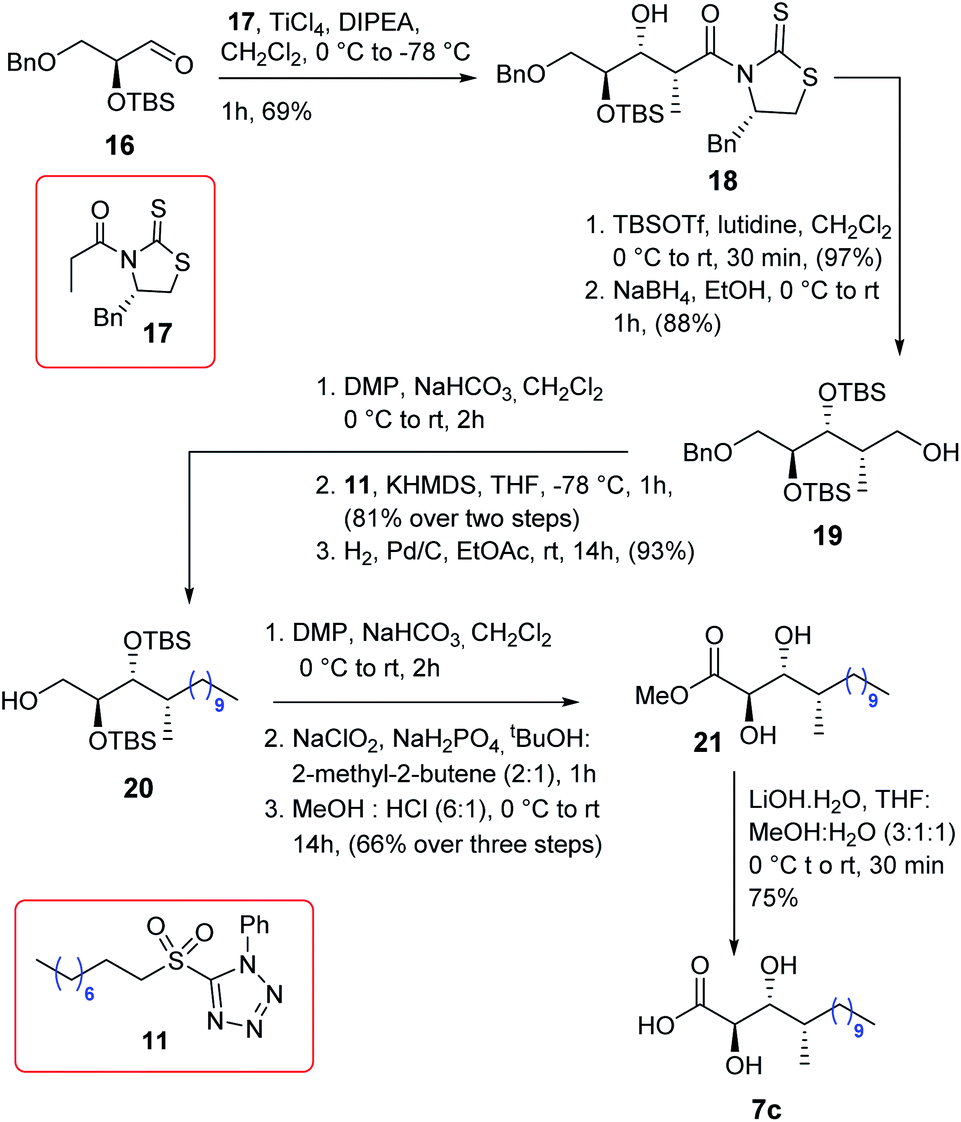
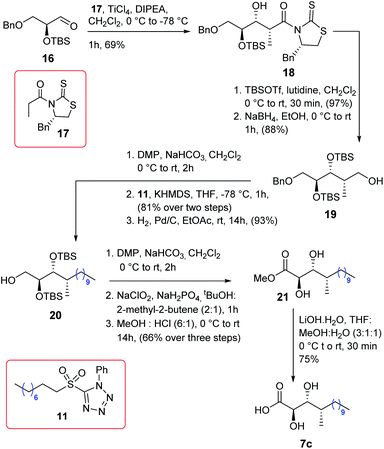
The synthesis of acid 7c is described in Scheme 3. The known aldehyde 168 was subjected to Crimmins aldol9 using the thiazolidinethione 179a in the presence of TiCl4/DIPEA to obtain compound 18 as a single isomer which was further treated using TBSOTf/2,6-lutidine followed by NaBH4/EtOH to alcohol 19. Next, alcohol 19 was oxidized to the corresponding aldehyde using DMP and subsequently subjected to Julia–Kocienski olefination7 using sulfone 11 in the presence of KHMDS to yield the corresponding olefin with complete E-selectivity. The resultant olefin was hydrogenated further to provide compound 20 which was then oxidized to the corresponding acid using DMP following Pinnick oxidations. The acid was treated further with HCl/MeOH to yield compound 21 which finally was hydrolyzed to obtain acid 7c in good overall yield.
 |
| | Scheme 3 Synthesis of acid 7c. | |
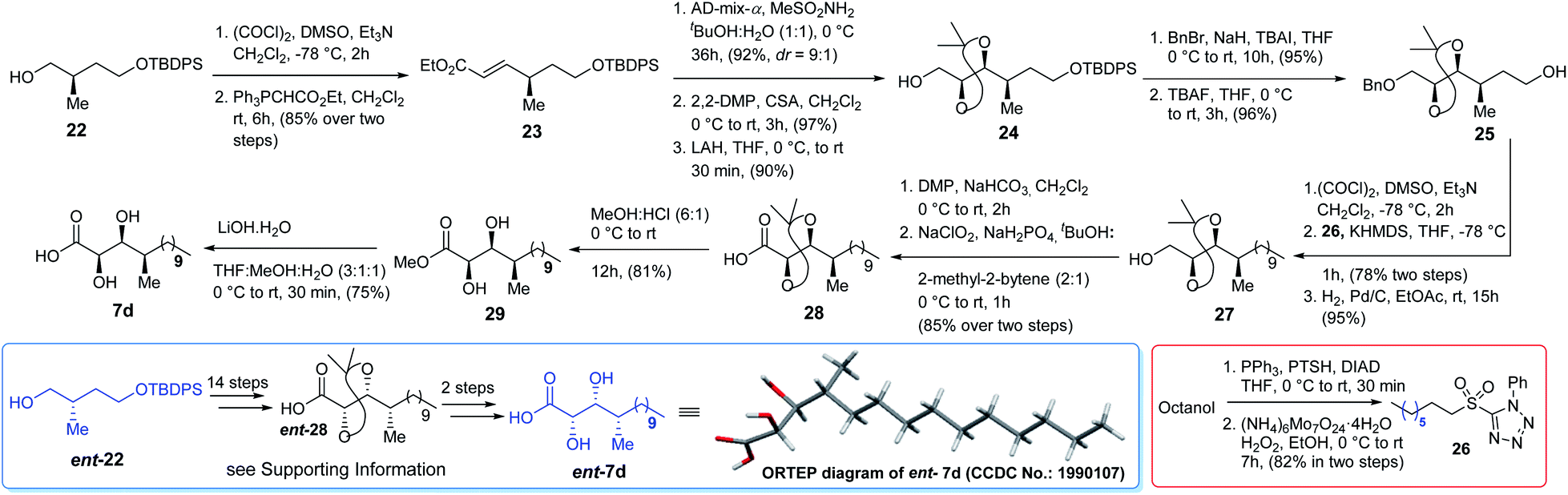
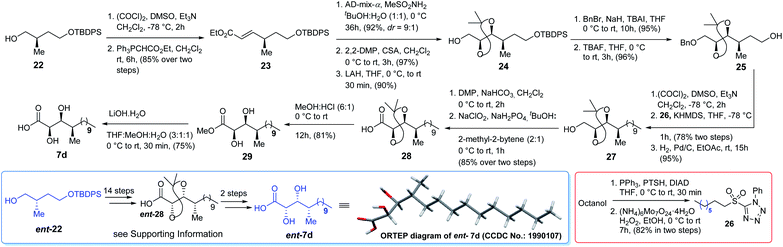
Initially we planned to introduce the syn C-2 and C-3 hydroxy centre of acid 7d adopting the same approach as for the synthesis of acid 7c using auxiliary ent-17 but the required isomer was obtained as a minor product. Moreover, the use of glycolate aldol to install the same chiral centres of acid 7d from compound 2210 was also not fruitful. Thus, compound 22 (Scheme 4) was oxidized using Swern conditions and the resultant aldehyde was subjected to Wittig olefination using Ph3PCHCO2Et to obtain olefin 23 exclusively. Next, olefin 23 was subjected to Sharpless asymmetric dihydroxylation11 using AD-mix-α/MeSO2NH2 to obtain the corresponding diol (d:r = 9:1) which was further treated with 2,2-DMP followed by LAH to alcohol 24. It was then benzylated and treated with TBAF to obtain alcohol 25 which was oxidized further. The resultant aldehyde was subjected to Julia–Kocienski olefination7 using sulfone 26, prepared from octanol in two steps (Scheme 4), to access the corresponding E-olefin exclusively. The resultant olefin finally was hydrogenated to obtain alcohol 27 which was oxidized further to acid 28 in two steps. Treatment of acid 28 with HCl/MeOH provided methyl ester 29 which was hydrolyzed to acid 7d. Comparison of both 1H and 13C NMR data [see Tables ST1 to ST4 (page-S3 to S6) in the ESI†] with the reported data3 revealed that acid 7d was the best match among the four isomers synthesized. The optical rotation of the DHMTDA unit was not reported by the isolation group. Thus, one needs to consider the possibility of its enantiomer ent-7d also as the absolute configurations of those centres were not known. Thus, ent-7d (Scheme 4) was also synthesized from the known compound ent-2210 following exactly an identical approach to acid 7d [see Scheme S5 (page-S49) in the ESI†]. Fortunately, acid ent-7d crystallized and the structure was confirmed unambiguously using X-ray crystallographic analysis [see Fig. SF1 (page-S7) and Table ST5 (page-S7) in the ESI†]. We planned initially to synthesize both the macrocycles bearing acid 7d and ent-7d as the DHMTDA unit to compare their NMR data with the reported data of the isolated alveolaride C.
 |
| | Scheme 4 Synthesis of acids 7d and ent-7d. | |
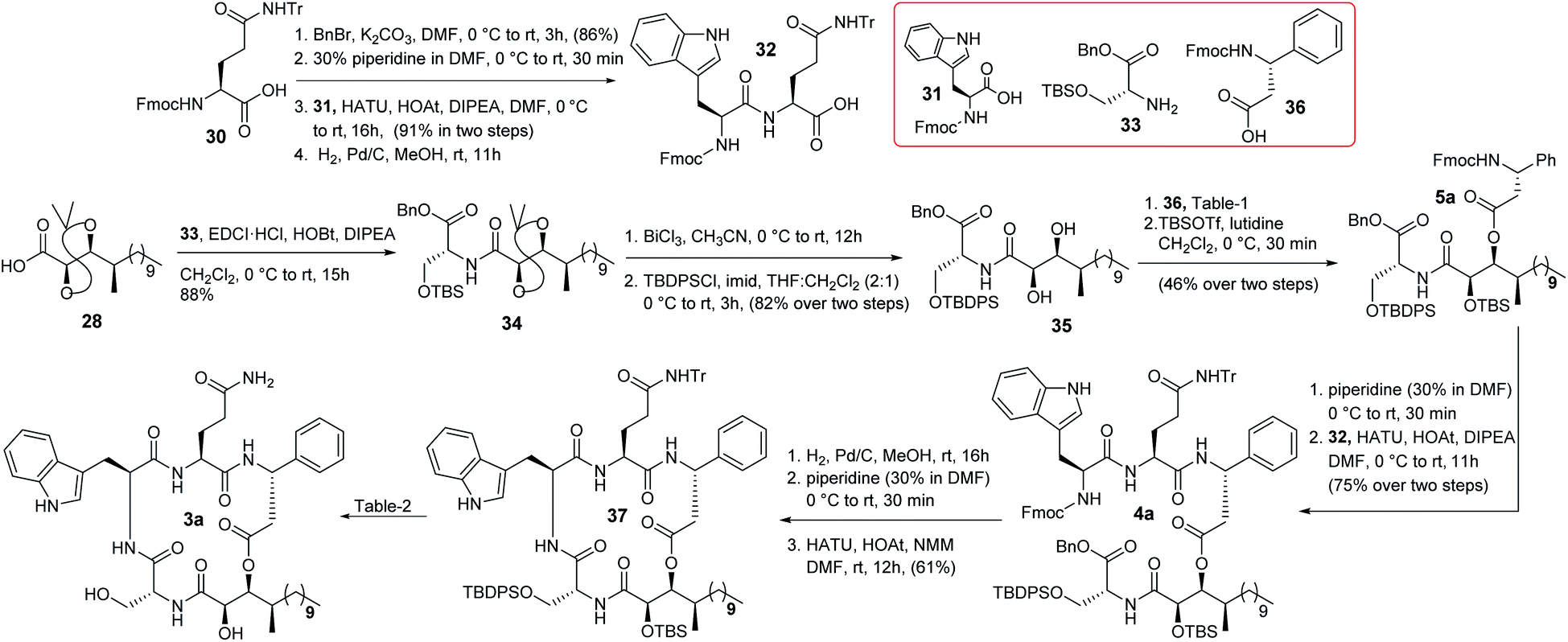
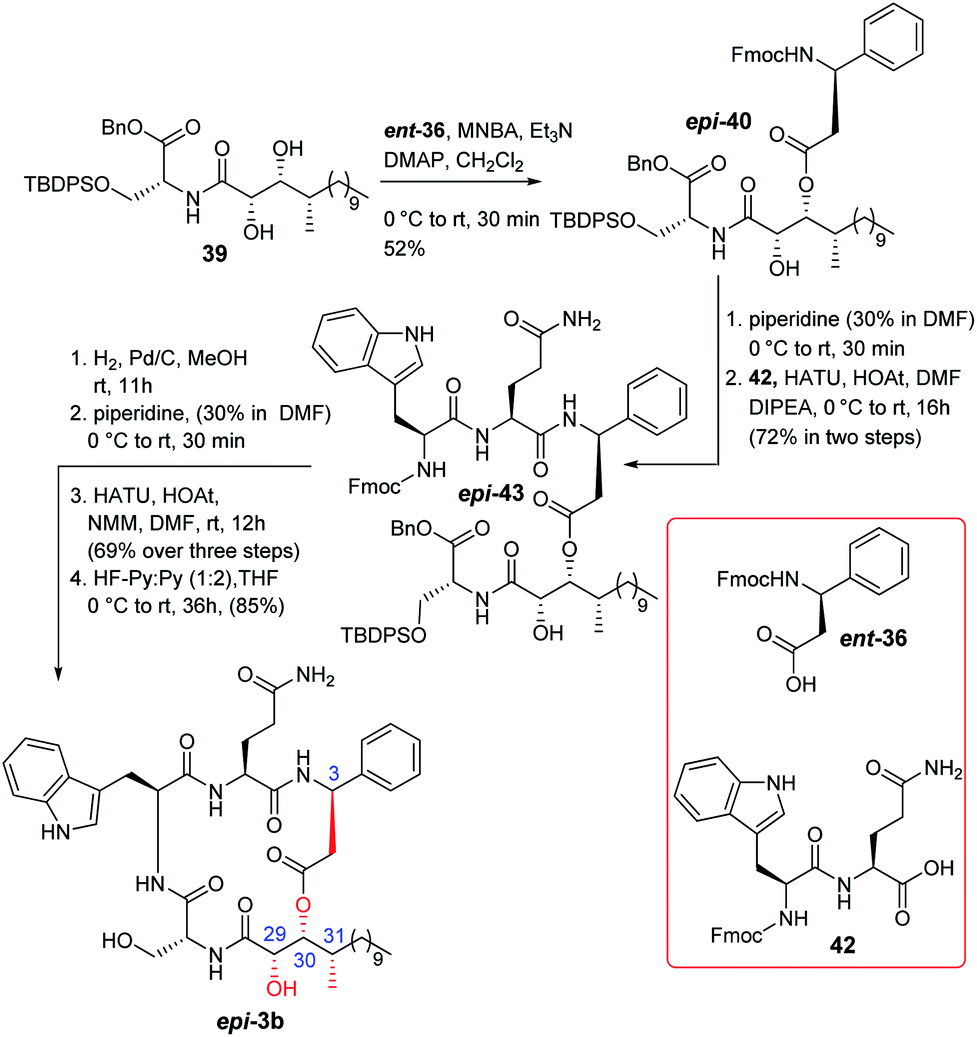
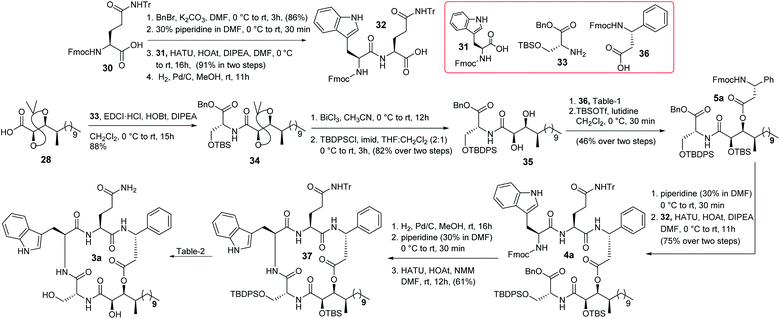
The synthesis of compound 3a is depicted in Scheme 5. N-protected L-glutamine was treated with BnBr/K2CO3 followed by 30% piperidine to obtain the corresponding Fmoc deprotected product that was further coupled with N-protected L-tryptophan in the presence of HATU/HOAt/DIPEA12 to obtain the corresponding amide which was hydrogenated to produce the required dipeptide unit 32. On the other hand, the previously synthesized compound 28 was coupled with the known D-serine counterpart 3313 to access compound 34 which was then treated with BiCl3 to yield the corresponding acetonide and TBS deprotected triol. The primary hydroxy group of the corresponding triol was protected as TBDPS ether to obtain diol 35. Next, different conditions for the selective esterification of diol 35 with protected D-β-phenylalanine (36)14 were screened (Table 1). The Yamaguchi conditions were not tested because the isolation group was unable to obtain the required Mosher's ester under these conditions.3 EDCI based esterification15a,b was found to be ineffective (entry 1). However, the Shiina esterification protocol15c produced (entry 2) the corresponding C-3 esterified product in moderate yield (52%), which was protected further as TBS ether to obtain compound 5a. The change of solvent polarity in the Shiina protocol did not offer any encouraging result (entry 3 & 4). It was observed that the esterification reaction was highly sensitive to moisture and impurity. It is noteworthy that we were unable to detect the formation of any C-2 or bis(C-2/C-3) esterified products during this esterification reaction likely due to the engagement of the C-2 hydroxy group in strong hydrogen bonding16 with the adjacent amide carbonyl. The formation of compound 5a was confirmed by detailed 2D NMR analysis [see Fig. SF2 (page-S8), COSY (page-S110) and HSQC (page-S110) in the ESI†]. Next, compound 5a was treated with 30% piperidine/DMF and the resultant compound was coupled with dipeptide 32 to furnish compound 4a in good overall yield. Hydrogenation followed by treatment with 30% piperidine/DMF resulted in the corresponding precursor of macrolactamization from compound 4a in good yield. Quite a few reaction conditions were screened at this stage for macrocyclization. EDCI/DIPEA/DMF did not function whereas PyBOP/DIPEA/DMF,17a HATU/HOAt/DIPEA/DMF17b and HATU/HOAt/NMM/DMF17c provided the cyclized product 37 in 38%, 51% and 61% yield, respectively. Next, a detailed optimization (Table 2) for the global deprotection was performed. It was observed that compound 37 decomposed completely in the presence of CSA/MeOH (entry 1), HCl/THF (entry 2), AcOH/H2O (entry 3) and TBAF/THF (entry 4). TBAF/AcOH (1:1) (entry 5), HF-Py (entry 6), and HF-Py/Py (1:2) (entry 7) provided only the corresponding desilylated product at different extents. HF-Py/Py was found to be the best desilylation condition compared to others in our case. It was observed that 15% TFA in CH2Cl2 (entry 8) deprotected only the trityl of compound 37 where the silyl ethers remained unaffected. Thus, a two-step protocol was adopted for the global deprotection where compound 37 was first subjected to trityl deprotection (entry 8) and subsequently silyl deprotection (entry 7) to access compound 3a in good overall yield (68% in two steps).
 |
| | Scheme 5 Completion of total synthesis of compound 3a. | |
Table 1 Optimization for esterification
| Entry |
Conditions |
Yield (%) |
| 1 |
EDCI, HCl, CH2Cl2, 0 °C to rt, 6 h |
No reaction |
| 2 |
MNBA, Et3N, DMAP, CH2Cl2, 0 °C to rt, 30 min |
52 |
| 3 |
MNBA, Et3N, DMAP, DMF, 0 °C to rt, 30 min |
Unidentified product |
| 4 |
MNBA, Et3N, DMAP, toluene, 0 °C to rt, 30 min |
47 |
Table 2 Optimization for global deprotection
| Entry |
Conditions |
Result |
| 1 |
CSA in MeOH, 0 °C, 2 h |
Decomposition |
| 2 |
HCl (2N):THF (1:2), 0 °C, 1 h |
Decomposition |
| 3 |
AcOH:H2O (8:2), 0 °C, 4 h |
Decomposition |
| 4 |
TBAF in THF, 0 °C, 1 h |
Decomposition |
| 5 |
TBAF:AcOH (1:1), in THF, 0 °C to rt, 24 h |
Partial deprotection of TBDPS and TBS |
| 6 |
HF-Py in THF, 0 °C to rt, 24 h |
Partial deprotection TBDPS and TBS with some unidentified spot |
| 7 |
HF-Py:Py (1:2) in THF, 0 °C to rt, 36 h |
Only silyls were deprotected |
| 8 |
15% TFA in DCM, 0 °C to rt, 30 min |
Only trityl was deprotected |
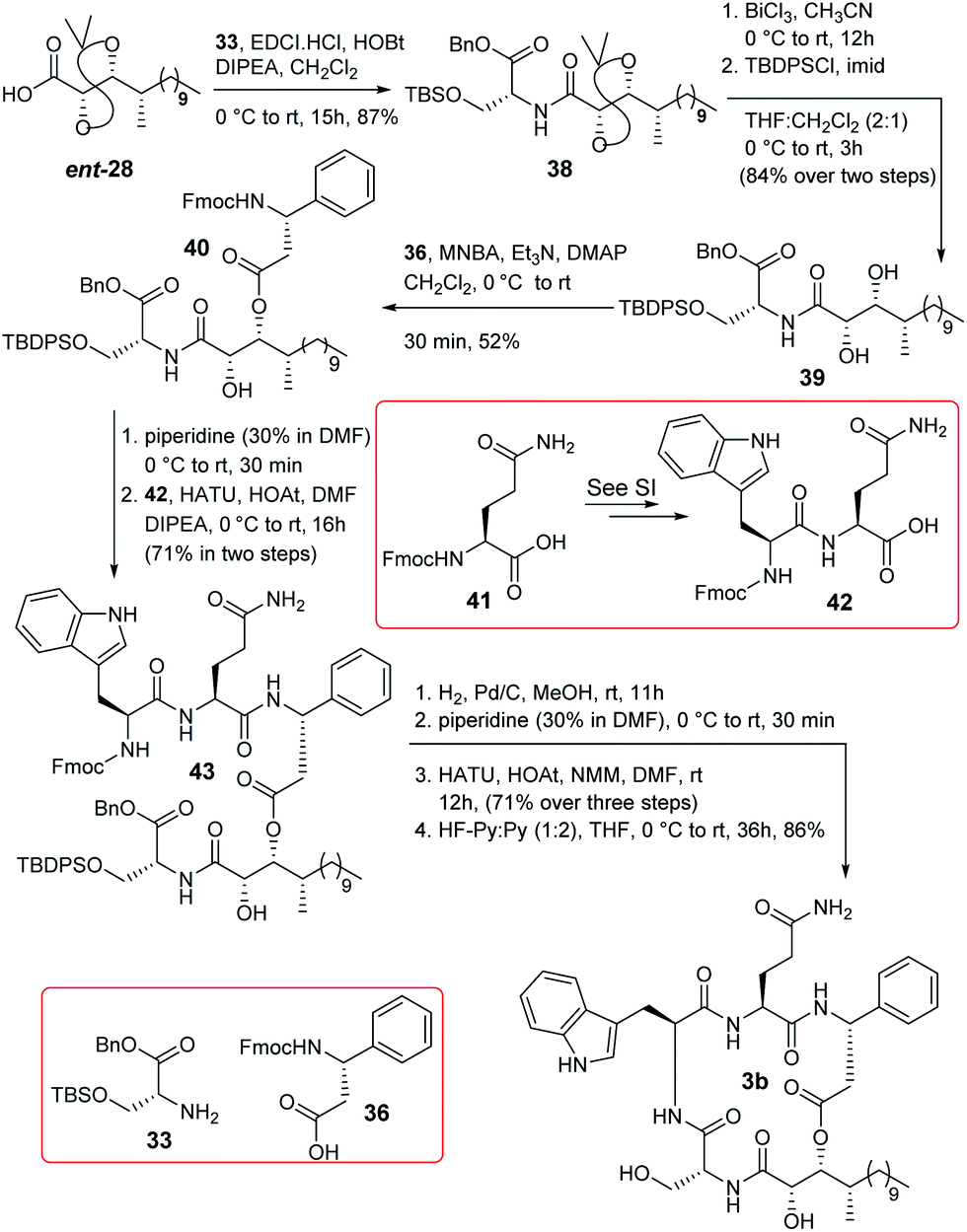
Some changes in the protection group strategy with respect to the route of compound 3a were adopted during the synthesis of compound 3b (Scheme 6) as the global deprotection of compound 37 was challenging. ent-28 was coupled with compound 33 to obtain compound 38 which was then converted to compound 39 by functional group manipulation. Compound 39 was then esterified with acid 36 following the Shiina protocol to obtain compound 40. Next, the Fmoc group of compound 40 was deprotected and the resultant compound was coupled with dipeptide 42 to access compound 43. It is noteworthy that dipeptide 42 was prepared directly from Fmoc protected L-glutamine (41) [see Scheme S8 (page-S65) in the ESI†] to avoid the trityl deprotection step in the final stage of the synthesis. Next, compound 43 was hydrogenated and subsequently reacted with piperidine to furnish the corresponding precursor of macrolactamization. Cyclization using HATU/HOAt/NMM resulted in the corresponding product which finally was treated with HF-Py/Py (1:2) in THF to obtain compound 3b in good overall yield.
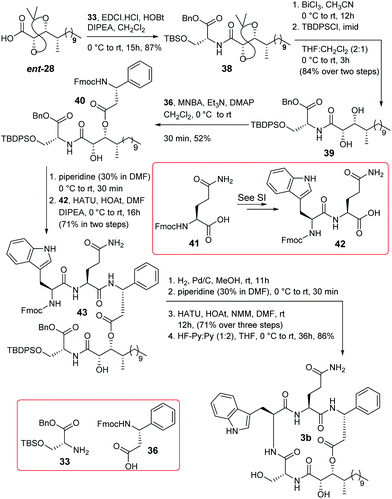 |
| | Scheme 6 Completion of total synthesis of compound 3b. | |
The 1H and 13C NMR data of both compounds 3a and 3b were compared with the reported data of the isolated natural product [see Tables ST6 (pages S9 and S10), and ST7 (pages S11 and S12), Fig. SF3 (page-S15), SF4 (page-S15), SF5 (page-S16) and SF6 (page-S16) in the ESI†]. Major mismatches were observed for compound 3a in both 1H and 13C NMR data whereas the 1H NMR data of compound 3b seemed promising even though some anomalies were observed in its 13C NMR data. However, few noticeable discrepancies in the 1H NMR data of compound 3b were observed for the protons near the β-phenylalanine and DHMTDA counterparts. The H-2 of β-phenylalanine was reported at 2.87 and 3.15 ppm whereas the same protons for the synthesized compound 3b were observed at 2.83 ppm. Moreover, anomalies in H-3 and aromatic protons (H-5, H-6, H-8 and H-9) of the phenyl ring of β-phenylalanine were also observed where the difference in the chemical shift was more than 0.3 ppm. The difference of ∼0.2 ppm in the chemical shift was also noted for H-29, H-30 and H-31. These observations clearly indicated that the stereochemistry of either C-3 or C-29 or C-30 or C-31 or all might not be accurate. However, the stereochemistry of all the centres (C-29, C-30, and C-31) of the DHMTDA counterpart was confirmed by NMR and X-ray crystallographic analysis but the absolute stereochemistry of amino acids of alveolaride C (3) was proposed by comparing the NMR data with those of alveolaride A (1) without performing degradation chemistry due to the lack of compound.3 Therefore, we decided to synthesize the C-3 epimer of compound 3b.
The completion of total synthesis of the actual structure of alveolaride C (epi-3b) is shown in Scheme 7. The chemistry adopted was exactly the same as that of compound 3b. Compound 39 was esterified with Fmoc protected L-β-phenylalanine (ent-36)14 to access compound epi-40 which was then coupled with dipeptide 42 to obtain compound epi-43. It was then hydrogenated and subjected further to Fmoc deprotection. The resultant compound was then cyclized to obtain the corresponding macrocyclic compound. Finally, the TBDPS ether was deprotected to provide compound epi-3b in good overall yield (59% over four steps). The 1H and 13C NMR data were recorded. We were quite delighted to observe that the NMR and optical rotation {observed [α]25D = −15.8 (c 0.21, MeOH); reported [α]25D = −10.2 (c 0.12, MeOH)} data of our synthesized compound epi-3b were in accordance [see comparison Table ST8 (page-S13, S-14) and Fig. SF7 (page-S16) in the ESI†] with the reported data of the isolated natural product which clearly established the actual structure of alveolaride C. It is noteworthy that the 13C signals of C-6 and C-8 of the phenyl ring of β-phenyl alanine were reported at δ 126.4 ppm whereas the same carbons of very similar alveolarides A (1) and B (2) were reported at δ 128.2 ppm by the isolation group. We observed the 13C NMR signals of those carbons at δ 128.27 ppm for the synthesized alveolaride C (epi-3b).18
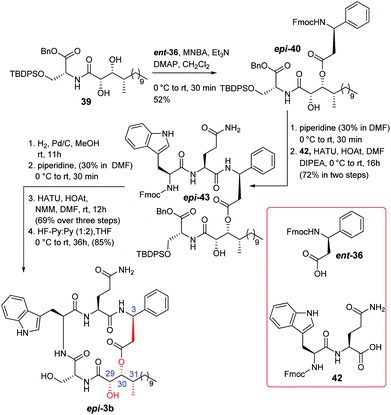 |
| | Scheme 7 Completion of total synthesis of the actual structure of alveolaride C (epi-3b). | |
Conclusions
In summary, a flexible and convergent route for the first total synthesis of structurally attractive alveolaride C was accomplished. The target natural product was prepared in 22 linear steps with 6.05% overall yield from the known compound ent-22. The structural riddle of alveolaride C was solved successfully. The absolute stereochemistry of three undetermined centres on the DHMTDA segment was established unambiguously as 29-(S), 30-(R) and 31-(S). The stereochemistry of β-phenylalanine was revised from S to R. This synthetic study will be immensely useful for establishing the structure of other members of the alveolaride family. The synthesis of other members of this family is under progress in our laboratory which will be reported in due course.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
S. S. and D. P. thank University Grants Commission and the Council of Scientific and Industrial Research, New Delhi for their research fellowship, respectively. The financial support from SERB, Department of Science and Technology (Project no. CRG/2019/001664) and the Council of Scientific and Industrial Research India (Project no. 02(0294)/17/EMR-II), to carry out this work is gratefully acknowledged.
Notes and references
-
(a) T. S. Bugni and C. M. Ireland, Nat. Prod. Rep., 2004, 21, 143–163 Search PubMed;
(b) J. W. Blunt, B. R. Copp, W. P. Hu, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2008, 25, 35–94 Search PubMed;
(c) J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2012, 29, 144–222 Search PubMed;
(d) J. F. Imhoff, Mar. Drugs, 2016, 14, 19 Search PubMed;
(e) B. Schulz, C. Boyle, S. Draeger, A. K. Römmert and K. Krohn, Mycol. Res., 2002, 106, 996–1004 Search PubMed.
-
(a) K. C. Nicolaou and S. Snyder, Angew. Chem., Int. Ed., 2005, 44, 1012–1044 Search PubMed;
(b) P. D. Brown and A. L. Lawrence, Nat. Prod. Rep., 2017, 34, 1193–1202 Search PubMed;
(c) R. M. Wilson and S. D. J. Danishefsky, Org. Chem., 2006, 71, 8329–8351 Search PubMed;
(d) G. M. Cragg, P. G. Grothaus and D. J. Newman, Chem. Rev., 2009, 109, 3012–3043 Search PubMed;
(e) A. Penesyan, S. Kjelleberg and S. Egan, Mar. Drugs, 2010, 8, 438 Search PubMed.
- S. Fotso, P. Graupner, Q. Xiong, J. R. Gilbert, D. Hahn, C. A. Adame, G. Davis and K. Sumiyoshi, J. Nat. Prod., 2018, 81, 10–15 Search PubMed.
-
(a) J. Mondal, R. Sarkar, P. Sen and R. K. Goswami, Org. Lett., 2020, 22, 1188–1192 Search PubMed;
(b) D. Saha, S. Guchhait and R. K. Goswami, Org. Lett., 2020, 22, 745–749 Search PubMed.
-
(a) W. R. Roush, M. A. Adam, A. E. Walts and D. J. Harris, J. Am. Chem. Soc., 1986, 108, 3422–3434 Search PubMed;
(b) Y. J. Kim, M. Ichikawa and Y. Ichikawa, J. Org. Chem., 2000, 65, 2599–2602 Search PubMed.
- B. Ganganna, P. Srihari and J. S. Yadav, Tetrahedron Lett., 2017, 58, 2685–2689 Search PubMed.
- P. R. Blakemore, W. J. Cole, P. J. Kocienski and A. Morley, Synlett, 1998, 1998, 26–28 Search PubMed.
-
(a) M. Kusakabe, H. Kato and F. Sato, Chem. Lett., 1987, 16, 2163–2166 Search PubMed;
(b) A. Whitehead, J. P. McParland and P. R. Hanson, Org. Lett., 2006, 8, 5025–5028 Search PubMed.
-
(a) A. Venkatesham and K. Nagaiah, Tetrahedron: Asymmetry, 2012, 23, 1186–1197 Search PubMed;
(b) M. T. Crimmins, B. W. King, E. A. Tabet and K. Chaudhary, J. Org. Chem., 2001, 66, 894–902 Search PubMed.
- A. F. Salit, C. Meyer and J. Cossy, Synlett, 2007, 2007, 934–938 Search PubMed.
-
(a) E. J. Corey, A. G. Perez and M. C. Noe, J. Am. Chem. Soc., 1995, 117, 10805–10816 Search PubMed;
(b) H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 Search PubMed.
- J. Liu, W. Chen, Y. Xu, S. Ren, W. Zhang and Y. Li, Bioorg. Med. Chem., 2015, 23, 1963–1974 Search PubMed.
- L. Zbigniew Pianowski and N. Winssinger, Chem. Commun., 2007, 3820–3822 Search PubMed.
- A. Müller, C. Vogt and N. Sewald, Synthesis, 1997, 1997, 837–841 Search PubMed.
-
(a) O. Revu and K. R. Prasad, J. Org. Chem., 2017, 82, 438–460 Search PubMed;
(b) Z. Gu and A. Zakarian, Angew. Chem., Int. Ed., 2010, 49, 9702–9705 Search PubMed;
(c) I. Shiina, R. Ibuka and M. A. Kubota, Chem. Lett., 2002, 286–287 Search PubMed.
- The crystal structure of ent-7d revealed a possible hydrogen bonding between the C-2 hydroxy and carbonyl oxygen of the acid moiety. A similar interaction could be expected between the C-2 hydroxy and carbonyl oxygen of the amide of compound 35.
-
(a) N. K. Lim, X. Linghu, N. Wong, H. Zhang and C. G. Sowell, Org. Lett., 2019, 21, 147–151 Search PubMed;
(b) E. Y. Melikhova, R. D. C. Pullin, C. Winter and T. J. Donohoe, Angew. Chem., Int. Ed., 2016, 55, 9753–9757 Search PubMed;
(c) N. K. Lim, X. Linghu, N. Wong, H. Zhang, C. G. Sowell and F. Gosselin, Org. Lett., 2019, 21, 147–151 Search PubMed.
- This anomaly is most likely due to typographic mistakes. No spectra of alveolaride C were made available by the isolation group.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1990107. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04478d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*