
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting radical-pair intersystem crossing for maximizing singlet oxygen quantum yields in pure organic fluorescent photosensitizers†
Xuanhang
Wang
a,
Yucong
Song
b,
Guocui
Pan
a,
Wenkun
Han
a,
Boyu
Wang
a,
Li
Cui
d,
Huili
Ma
c,
Zhongfu
An
 c,
Zhigang
Xie
*b,
Bin
Xu
*a and
Wenjing
Tian
a
c,
Zhigang
Xie
*b,
Bin
Xu
*a and
Wenjing
Tian
a
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Qianjin Street No. 2699, Changchun 130012, China. E-mail: xubin@jlu.edu.cn
bState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, PR China. E-mail: xiez@ciac.ac.cn
cKey Laboratory of Flexible Electronics (KLOFE), Institute of Advanced Materials (IAM), Nanjing Tech University (NanjingTech), 30 South Puzhu Road, Nanjing 211816, China
dDepartment of Neurology, First Hospital of Jilin University, Changchun, 130000, PR China
First published on 2nd September 2020
Abstract
Fluorescent photosensitizers (PSs) often encounter low singlet oxygen (1O2) quantum yields and fluorescence quenching in the aggregated state, mainly involving the intersystem crossing process. Herein, we successfully realize maximizing 1O2 quantum yields of fluorescent PSs through promoting radical-pair intersystem crossing (RP-ISC), which serves as a molecular symmetry-controlling strategy of donor–acceptor (D–A) motifs. The symmetric quadrupolar A–D–A molecule PTP exhibits an excellent 1O2 quantum yield of 97.0% with bright near-infrared fluorescence in the aggregated state. Theoretical and ultrafast spectroscopic studies suggested that the RP-ISC mechanism dominated the formation of the triplet for PTP, where effective charge separation and an ultralow singlet–triplet energy gap (0.01 eV) enhanced the ISC process to maximize 1O2 generation. Furthermore, in vitro and in vivo experiments demonstrated the dual function of PTP as a fluorescent imaging agent and an anti-cancer therapeutic, with promising potential applications in both diagnosis and theranostics.
Introduction
Photodynamic therapy (PDT)1 has attracted significant attention over the past few decades due to its high spatiotemporal precision, controllability, and noninvasive properties for clinical cancer treatment.2 PDT is a photochemical-based therapeutic method that relies on light-activated photosensitizers (PSs) to generate cytotoxic species under irradiation for killing cancer cells.3 It involves intersystem crossing (ISC) from the lowest singlet excited state (S1) of the light-excited PS to its triplet state (Tn), and then vibrational relaxation brings it into the lowest vibrational level (T1), subsequently transferring its energy to produce cytotoxic singlet oxygen (1O2) or other reactive oxygen species (ROS).4 One of the most promising approaches to improve the ISC efficiency of PSs is to incorporate heavy atoms into the molecular structure to enhance the spin–orbit perturbations (SOP), but this method raises concerns over their cytotoxicity.5 Previous studies on the ISC process in charge transfer (CT) excited states showed us that the triplet formation could undergo a radical-pair intersystem crossing (RP-ISC) mechanism in donor–acceptor (D–A) molecules, which required rapid formation of a radical-pair (RP) upon charge separation (CS) and occurred through quantum mechanical mixing of the singlet and triplet RP sublevels under hyperfine coupling effects.6 In addition, the separation of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) attributed to the D–A structure can reduce the electron–electron exchange energy (J values), thus leading to a small singlet–triplet energy gap (ΔEST).7 Therefore, the incorporation of D–A motifs may represent an alternative approach to facilitate the ISC process and enhance the photosensitization of PSs.In addition to the efficient production of singlet oxygen, PSs can also serve as real-time fluorescence imaging agents.8 A fluorescently detectable PS is beneficial for aiding the definition and adjustment of parameters, therapy monitoring, and guidance of surgery or other therapies.9 However, the majority of conventional organic PSs, such as porphyrin and chlorin derivatives, suffer from substantially reduced fluorescence in the biological system due to aggregation-caused quenching of the excited state.10 In contrast, aggregation-induced emission (AIE) luminogens usually exhibit negligible fluorescence in dilute solution but fluoresce strongly in the aggregated state.11 Although significant progress on AIE-active PSs has been witnessed recently,12 developing a high-performance fluorescent PS and obtaining an in-depth understanding of the structure–property relationship, as well as the involved intrinsic photosensitization mechanism, still remain an unsolved challenge. Guidelines are therefore anticipated and will direct the future design of novel PSs with tunable characteristics, such as efficient photosensitization and strong fluorescence, which would warrant applications in advanced PDT.
Herein, we present a molecular symmetry-controlling strategy to fine-tune the excited state dynamics and enhance the ISC process of fluorescent PSs. Two types of PSs containing asymmetric dipolar D–A molecules and symmetric quadrupolar A–D–A molecules were designed and synthesized, where AIE-active tetraphenylethylene (TPE) served as the donor unit, while a strong electron-withdrawing group, dicyanoethylene, served as the acceptor unit. Notably, the symmetric molecule PTP not only exhibits an outstanding 1O2 quantum yield of 97.0%, but also exhibits bright NIR fluorescence with an efficiency of 2.96% in the aggregated state, where the exciton utilization is approximately 100%. The photosensitization capability of symmetric PTP far exceeds that of the asymmetric molecule TP or the widely used chlorin E6 (Ce6). Theoretical and ultrafast spectroscopic investigations revealed that the triplet formation of these PSs underwent the RP-ISC mechanism. More importantly, we found a distinctive symmetry-breaking process of the quadrupolar excited state in the symmetric molecule PTP, which facilitated charge separation to effectively generate RPs. Moreover, an ultralow singlet–triplet energy gap (0.01 eV) was observed in PTP, thereby promoting the high triplet formation by the hyperfine coupling interactions to realize the efficient RP-ISC process.13In vitro and in vivo studies verified the application of PTP as a PDT agent in cancer therapy, which allowed simultaneous imaging and killing of tumors.
Results and discussion
Molecular design and optical properties
Four AIE-active PSs, including the symmetric A–D–A molecules (MTM and PTP) and the asymmetric D–A molecules (TM and TP), were synthesized and characterized (Fig. S23–S33†). For instance, when dissolved in tetrahydrofuran (THF), both TP and PTP exhibited two absorption bands covering the 300–550 nm wavelength region (Fig. 1b). The two bands can be attributed to the locally excited (LE) state and the CT excited state, respectively, where the absorption bands of the LE state of the donor TPE ends at 400 nm (Fig. S2†). Compared to TP, PTP exhibits weaker absorbance in the LE state (∼330 nm) but stronger absorbance in the CT state (∼400 nm). This observation indicates that PTP exhibits more effective intramolecular charge transfer. In addition, we found that the significant bathochromic shift in the emission spectrum of these PSs was associated with the increase of solvent polarity. This solvation effect coincides with the characteristics of the twisted intramolecular charge transfer (TICT) state (Fig. S5†).14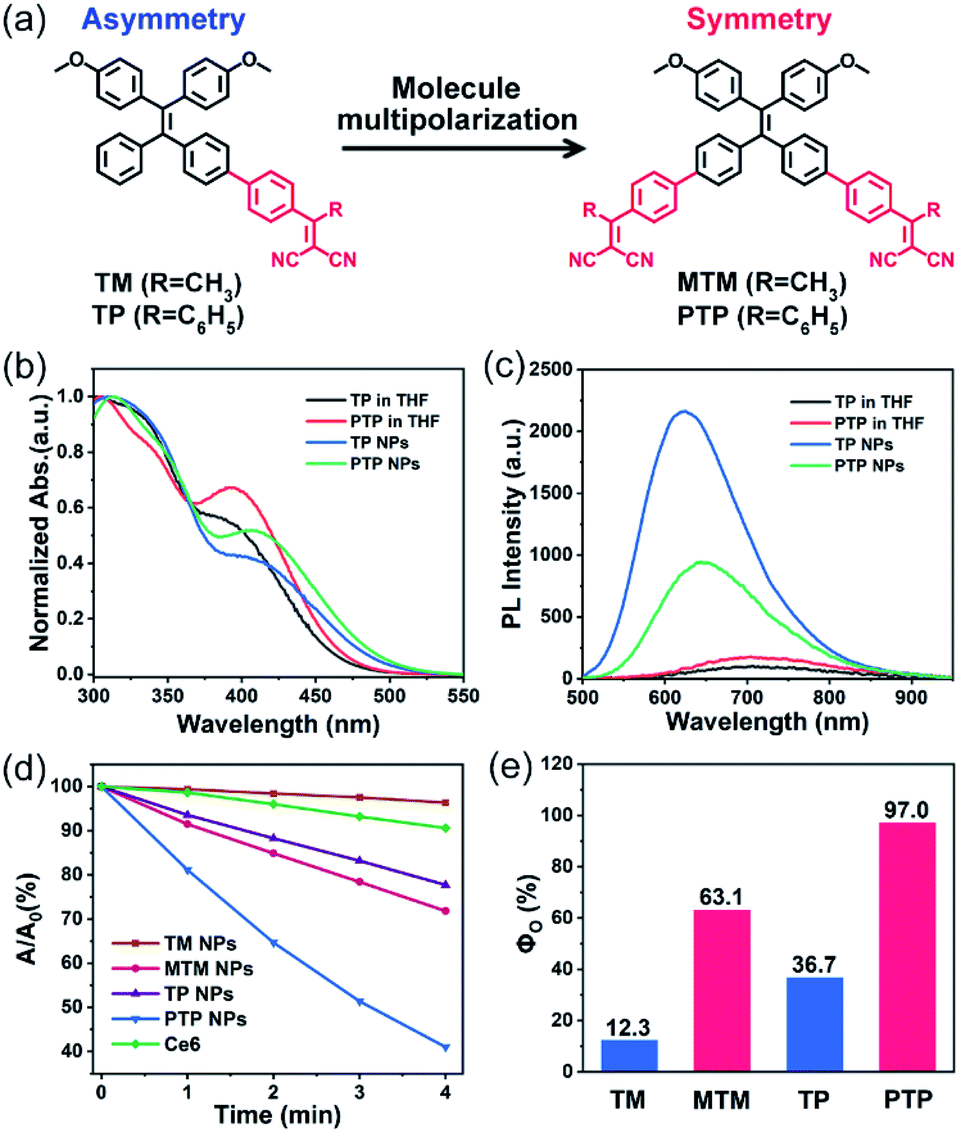 | ||
| Fig. 1 (a) Molecular structures of TM, TP, MTM and PTP. (b) UV-visible absorption spectrum of TP and PTP in THF and NP aqueous solutions. (c) PL spectra of TP and PTP in THF and NP aqueous solutions. [PSs] = 100 μM. (d) The absorbance decomposition rates of ABDA at 400 nm in the presence of different PS NPs under light irradiation (400–800 nm); A0 and A represent the absorbance of ABDA at 400 nm before and after illumination, [PSs] = 10 μM, [ABDA] = 100 μM. (e) Singlet oxygen quantum yields (ΦO) of TM, MTM, TP and PTP NPs. | ||
Both TP and PTP show negligible fluorescence in pure THF because strong intramolecular vibration and rotation inactivate their excited state (Fig. 1c). Upon addition of water to THF, a dramatically enhanced emission is observed (Fig. S3†), which manifests typical AIE characteristics.15 To realize good biological applications in aqueous media, these PSs were encapsulated using an amphiphilic block copolymer, Pluronic F127. The resulting PTP nanoparticles (NPs) are observed to be monodispersed under a transmission electron microscope (TEM) and by dynamic light scattering (DLS) (Fig. S6†). PTP NPs display a near-infrared emission covering the wavelength range of 500–900 nm, with a fluorescence quantum yield (ΦQY) of 2.96%. In comparison with PTP NPs at the same concentration, a brighter emission is observed for TP NPs. These observations suggest that these AIE-active PSs can effectively avoid fluorescence quenching as well as stabilizing the excited state in their aggregation.
Chemical detection of singlet oxygen
To evaluate the 1O2 production of these AIE-active PSs, 9,10-anthracenediylbis(methylene)dimalonic acid (ABDA) was used as a 1O2-trapping agent to detect the generation of 1O2 specially, with an obvious decrease in its absorbance.16 The mixture dissolving ABDA (100 μM) and different PSs (10 μM) was exposed to white light irradiation (400–800 nm) and the changes in the UV-vis absorption spectra of ABDA were recorded using a spectrophotometer at various irradiation times. The evaluation of the 1O2 generation efficiency was first carried out in dilute N,N-dimethylformamide (DMF) solution. But no remarkably decreased absorbance of ABDA was observed among these PSs in the isolated state, even if we extended the total irradiation time to 10 minutes (Fig. S7†). Interestingly, a significant enhancement of 1O2 production was observed once these PSs molecules were aggregated into NPs in aqueous solution (Fig. S8†). While the asymmetric TM and TP NPs showed a moderate decomposition rate towards ABDA under light irradiation, sharply decreased absorbance of ABDA was observed in their symmetric counterparts MTM and PTP NPs (Fig. 1d). We also included the Ce6, one of the most widely used PSs, as a reference in our test. After irradiation for 4 min under the same circumstances, approximately 60% ABDA consumption occurred in PTP NPs, but only 10% ABDA was degraded in Ce6, indicating an excellent photosensitizing ability of PTP NPs. Furthermore, taking ABDA as an indicator and Rose Bengal (RB) as the standard PS, the 1O2 quantum yields of these AIE-active PSs were calculated (Fig. S9†). The high values of the 1O2 quantum yields are up to 63.1% and 97.0% for the symmetric molecules MTM and PTP, whereas the corresponding asymmetric molecules TM and TP show modest values of 12.3% and 36.7%, respectively (Fig. 1e). It should be noted that the total efficiency of 1O2 sensitization and fluorescence of PTP is approximately 100%, suggesting that the excitons can be almost completely utilized. These observations thus demonstrated that the 1O2 production of these PSs could be manipulated and maximized by employing the molecular symmetry of D–A molecules.Intrinsic photophysical mechanism of the fluorescent PSs
Generally, symmetric A–D–A molecules exhibit typical quadrupolar characteristics, whereas asymmetric D–A molecules exhibit dipolar features.17 Encouraged by the significant promotion of 1O2 quantum yields from our experimental results, we carried out a series of theoretical calculations based on time-dependent density functional theory (TD-DFT). To study the structure–property relationship of dipolar and quadrupolar molecules, we select asymmetric TP and symmetric PTP as representative model molecules. The optimized geometries of both TP and PTP show twisted three-dimensional conformations with a large torsion angle between the donor TPE and the acceptor dicyanoethylene subunits (Fig. S13†). Meanwhile, complete separation between the HOMO and LUMO was observed, where the HOMO was dominated by the TPE moiety while the LUMO was distributed in dicyanoethylene subunits (Fig. S14†). The clearly separated HOMO and LUMO often match with small electron exchange energy, resulting in a small singlet–triplet energy gap.18 As expected, the calculated ΔEST values of both asymmetric TP and symmetric PTP are very small (Fig. 2a and c), especially for PTP with an ultralow ΔEST of 0.01 eV, which benefits the ISC process. However, the calculated spin–orbit coupling coefficients (SOCs) of both TP and PTP are very small as well, due to the absence of heavy atoms or aromatic carbonyl induced (n, π*) transition.19 Therefore, we speculate that the triplet formation in TP and PTP is not a result of the spin–orbit intersystem crossing (SO-ISC).20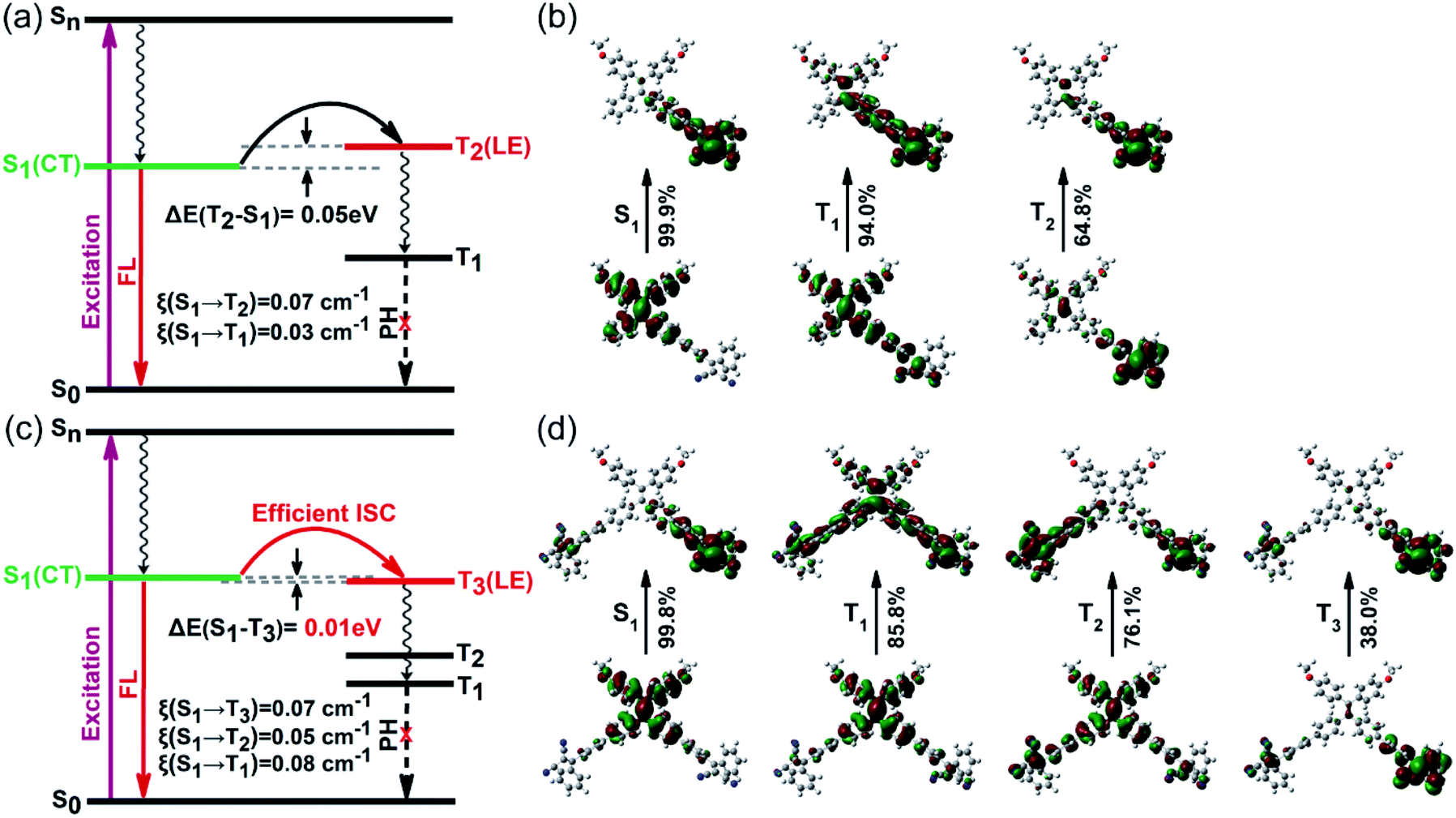 | ||
| Fig. 2 The calculated energy level diagram and the spin–orbit coupling coefficient of (a) TP and (c) PTP. The natural transition orbitals (hole ones at the bottom and electron ones at the top) for (b) TP and (d) PTP. | ||
In contrast, well-separated molecular orbitals are conducive for photo-driven charge transfer reactions to generate RPs (e.g., electron–hole pairs) under strong D–A interaction, which has an important influence on their spin dynamics. To study the dynamic process in the excited state, we further calculated the natural transition orbitals (NTOs) of TP and PTP. Asymmetric TP exhibits a clear dipolar feature of charge transfer in the S1 state, where electrons and holes are completely separated (Fig. 2b). Upon activation, the initial 1LE state (D*–A) will directly transform into the 1RP (D+–A−) by one-step charge transfer. Interestingly, the symmetric PTP exhibited different excited-state dynamics. The asymmetric excitation suggests that the quadrupolar PTP behaves as a dipolar 1RP (A–D+–A−) in the S1 state (Fig. 2d). This excited state characteristic is usually accompanied by the breakup of the symmetry in the quadrupolar state (A−–D+–A−).21 Moreover, two types of NTOs of PTP with near-degenerate energy (S1 ≈ S2) caused by symmetry-breaking were observed, which corresponded to the right acceptor branch and left acceptor branch, respectively (Fig. S15†). Subsequently, fast internal conversion brings the S2 electrons back to the S1 level to generate more 1RPs in contrast with TP. This also helps to explain the stronger CT absorption of PTP in the UV-visible absorption spectrum. These results illustrated that both TP and PTP participated in the generation of RPs, and the formation of triplets might involve the RP-ISC mechanism.
When 1RPs form in the previous step of charge separation in both TP and PTP, RP-ISC may subsequently occur through quantum mechanical mixing of the 1RP and 3RP sublevels. This process is dominated by electron-nuclear hyperfine coupling interactions, followed by spin selective charge recombination (CR) to either the ground state or the locally excited triplet state of either the donor or the acceptor subunits.22 It is worth noting that the high-lying triplet excited states of TP (T2) and PTP (T3) exhibit large overlap extents of electrons and holes, with the feature of a pure locally excited state of either the donor or the acceptor (Fig. 2b, d, S16 and S17†), which perfectly aligns with all circumstances of the RP-ISC process. Yet, for asymmetric TP, the calculated potential energy curve of T2 is above S1 with a large energy gap (Fig. S19a†), indicating that the 1RP needs external energy to overcome the energy barrier for the spin-flip transitions to complete the ISC process.23 Meanwhile, the radiative transition process competes with the RP-ISC process. Consequently, singlet RPs of TP tend to decay back to the ground state with strong emission instead of the RP-ISC process (energy gap law).24 This could be why TP exhibits relatively high fluorescence quantum yield but moderate efficiency for 1O2 generation.
Unlike TP, the donor of PTP exhibits two approximately equal torsion angles with both acceptor branches in the ground state (Fig. S18a†). But in the excited state, the twist angles between the donor and acceptor increase with the charge separation to reduce the energy of the whole system (Fig. S18b†). Notably, the right acceptor branch shows a bigger twisted angle than the left acceptor branch due to the asymmetric excitation of the S1 state, further verifying the symmetry-breaking process of the excited state.25 Such an increased twist angle of the donor–acceptor of PTP in the excited state can effectively reduce the overlap of the HOMO and LUMO, thereby narrowing the singlet–triplet energy gap (ΔEST = 0.01 eV) and providing an efficient channel for the RP-ISC process. Moreover, the intersection of potential energy curves between S1 and T3 was observed in the symmetric molecule PTP (Fig. S19b†), suggesting that energy level degeneracy occurred between S1 and T3. Such near-degeneracy orbitals could effectively boost the coupling of singlets and triplets via hyperfine interactions, thereby promoting the RP-ISC process to realize high formation of triplets,26 accounting for its excellent 1O2 generation efficiency.
Insight into the RP-ISC mechanism based on an ultrafast system
To gain insight into the ISC process in both the asymmetric molecule TP and symmetric molecule PTP, we performed femtosecond-resolved transient absorption (fs-TA) measurements to further study their internal excited-state dynamics. As shown in Fig. 3a, the fs-TA spectra of TP are comprised of excited state absorption (ESA) bands overlapping with stimulated emission (SE) bands. Three excited state components that existed in TP were obtained by a global fitting procedure combined with singular value decomposition (SVD) (Fig. 3c). The first femtosecond component (832.5 fs) is ascribed to an internal conversion process of the highly excited state. After this, the charge separation process occurred immediately and ended at 4.364 ps. Meanwhile, significant spectral changes were observed, in which the SE peaks exhibited a red-shift from 550 nm of the LE state (blue line) to 650 nm of the CT state (red line) and the ESA peaks revealed a blue-shift from 650 nm of the LE state to 520 nm of the CT state. In particular, TP exhibits strong SE bands of the CT state in the three-dimensional (3D) map of the absorption spectra (Fig. 3e), indicating that substantial 1RPs are inactivated by the emission pathway, which is consistent with the calculated high radiation transition rate (kr) (Table S1†). Besides, the ESA peak of the CT state at 520 nm is attributed to dicyanoethylene radical anion species (A−-ESA). This radical anion absorption band was also observed by electrochemical spectroscopy upon bulk electrolysis of the dicyanoethylene at a controlled-potential of −1.5 V (Fig. S11†), providing further spectral evidence of the RP.27 The third picosecond component of TP (213.4 ps) corresponds to the charge recombination process of RPs. | ||
| Fig. 3 Femtosecond transient absorption spectra of (a) TP and (b) PTP in toluene. Evolution-associated difference spectra (EADS) obtained from global fitting for (c) TP and (d) PTP. Three-dimensional map of the femtosecond transient absorption spectra of (e) TP and (f) PTP. Excitation: 400 nm, probe light: 450–780 nm. | ||
Similarly, three excited state components existing in PTP were also obtained by a global fitting procedure (Fig. 3d). However, due to fast vibrational relaxation of the highly excited state, PTP exhibits a shorter internal conversion lifetime (577.9 fs) than TP. The SE peak (∼550 nm) and ESA peak (∼650 nm) of the LE state (blue line) emerged in PTP, originating from the same donor group as in TP. Subsequently, the LE bands disappeared followed by a newly formed ESA peak (∼520 nm) of radical anion species (red line), which corresponded to the charge separation process (5.699 ps) of PTP. Despite massive RPs generated from the effective charge separation process, the disappearance of the SE peak of the CT state in the 3D map of the absorption spectra of PTP (Fig. 3f) suggested that most RPs don't choose the fluorescence channel to decay back to the ground state. In contrast, a rapid charge recombination process (117.8 ps) was observed in PTP, indicating that the efficient RP-ISC instead of CT emission afforded its excellent 1O2 production. Owing to intensive intramolecular vibration and rotation in the single molecule state, the excited state is easily deactivated through strong nonradiative transition, which results in the overall excited state lifetime remaining in the order of picoseconds.
Taken together, the theoretical and experimental data enable us to draw an overall diagram of the excited state dynamics of these symmetric and asymmetric molecules, where RP-ISC plays a key role in the formation of triplets as well as further 1O2 generation (Fig. 4). The LE state (D*–A) of light-activated TP is converted into singlet RP 1(D+–A−) via rapid charge separation with a lifetime of 4.364 ps. Due to the large singlet–triplet energy gap (T2 > S1) of TP, more 1RPs may choose the emission pathway to decay back to the ground state instead of picking the lengthy charge recombination (213.4 ps) to form triplet 3(D–A*). This preference results in a high radiation transition rate but a moderate singlet oxygen quantum yield. In contrast, the quadrupolar structure of PTP can effectively facilitate charge separation with the formation of a quadrupolar excited state 1(A−–D+–A−), followed by a unique symmetry-breaking process to convert into the asymmetric dipolar state 1(A–D+–A−). Exactly opposite to TP, the ultralow singlet–triplet energy gap (0.01 eV) of PTP accelerates spin-flip transitions of 1RPs and enhances the RP-ISC by a rapid charge recombination process (117.8 ps), which weakens the CT emission but boosts the formation of triplet 3(A–D–A*). The resulting high triplet formation gives rise to the maximum 1O2 quantum yield of PTP through further sensitization with triplet oxygen. Otherwise, there are still some singlet excitons which can deactivate through the CT emission pathway owing to the AIE effect, resulting in bright NIR fluorescence in its aggregated state.
 | ||
| Fig. 4 The schematic diagram of the excited state dynamics of dipolar TP and quadrupolar PTP. Dashed lines represent forbidden transition, solid lines indicate allowed transition and wavy lines correspond to the internal conversion (IC) process. | ||
Imaging and PDT in vitro and in vivo
Given its bright NIR fluorescence and excellent 1O2 quantum yield, we explored the potential of PTP NPs in bioimaging and apoptosis towards HeLa cells. The lysosome colocalization experiment of PTP NPs was performed with Lyso-Tracker, a commercial lysosome-specific imaging agent, to track their cellular uptake (Fig. 5b). After 6 h of incubation, not only are PTP NPs internalized into the HeLa cells, but their fluorescence signals also align with the ones from the lysosome (stained with LysoTracker). This observation indicates that PTP NPs may undergo the endosomal–lysosomal pathway of endocytosis. While PTP NPs alone exhibit high cytocompatibility at various concentrations (up to 4 μg mL−1), the light activation of these NPs induces potent cytotoxicity in a dose-dependent manner (Fig. 5c). Similar results are observed for live/dead cell staining, in which the light activation of PTP NPs triggers massive apoptosis in HeLa cells but moderate results occur in TP NPs (Fig. 5d). Moreover, PTP NPs display more effective elevation of intracellular oxidative stress in comparison with TP NPs, as evidenced by the more obvious green fluorescence appearing in PTP NPs treated HeLa cells using the commercial 2′,7′-dichlorofluorescin diacetate (DCFH-DA) as an intracellular 1O2 fluorescence probe (Fig. 5e). Therefore, this light-switchable property would warrant their application in cancer therapy.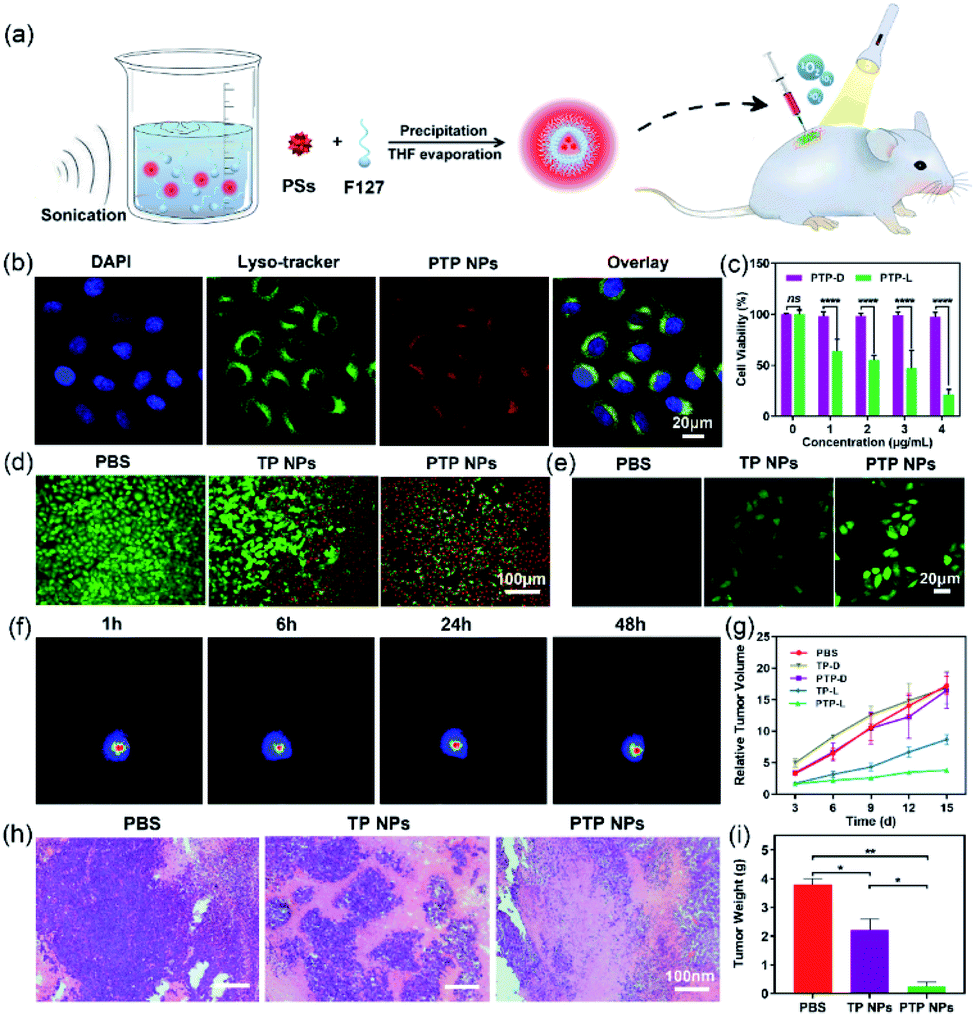 | ||
| Fig. 5 (a) The preparation procedure of nanoparticles and the animal model diagram of photodynamic therapy. (b) Confocal and merged images of HeLa cells stained with DAPI, Lyso-Tracker and PTP NPs, excitation: 488 nm. (c) Cell viability of HeLa cells treated with PTP NPs in different concentrations under light irradiation or dark conditions (15 mW cm−2). (d) Live/dead staining of HeLa cells with calcein-AM (green, 50 μg mL−1) and propidium iodide (red, 100 μg mL−1) in the presence of light irradiation. (e) Detection of intracellular singlet oxygen generation in stained HeLa cells using DCFH-DA (green fluorescence, 10 μM) under light irradiation. (f) Tumor imaging in mice after intravenous injection of PTP NPs at different times. (g) Relative volume of tumors in different treatment groups at different times. (h) H&E staining of tumor sections from different treatment groups after 15 d of treatment. (i) The weight of tumor tissues in different treatment groups on day 15 (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). | ||
We further examine the anti-cancer effect of PTP and TP NPs in tumor-bearing mice. U14 cervical cancer cells were subcutaneously injected into the right leg of BALB/c mice to obtain a solid-tumor model. 7 days after tumor formation, we performed intratumoral injection of PTP and TP NPs and tracked the tumor progression. Under optical imaging, intense fluorescence signals from PTP NPs are observed in the tumor region 48 h post-injection, indicating their good stability in vivo (Fig. 5f). The tumor site was then exposed to light irradiation for 20 min per day to trigger the generation of 1O2 by PTP and TP NPs for cancer-killing. In the absence of light irradiation, the mice treated with PTP and TP NPs develop similar tumor-growth curves to those of the control group (PBS-treated). In contrast, we observed significant inhibition of tumor growth in mice treated with PTP NPs and light irradiation, starting from day 6 (Fig. 5g), whereas the tumor growth in mice treated with TP NPs and light irradiation is slightly inhibited. Even after 2 weeks, PTP NPs in the presence of light effectively limited tumor expansion. When we examined the tumor tissues with H&E staining, substantial cell necrosis was observed in the group treated with PTP NPs and light irradiation while only partial damage was caused in the group treated with TP NPs and light irradiation, and no remarkable damage was observed in the control group (Fig. 5h). Consistently, tumor necrosis also results in dramatically reduced tumor weight (Fig. 5i). These results suggest that the PTP NPs exhibit more efficient photosensitization towards biological tissue oxygen in contrast with TP NPs, which guarantees their excellent anti-cancer behavior for in vivo PDT.
Conclusions
In summary, we have developed a molecular symmetry-controlling strategy to design high-performance fluorescent PSs and demonstrated a systematic and in-depth investigation of the photosensitization mechanism. Our strategy not only successfully solved the matter of photosensitization efficiency roll-off caused by self-quenching at high concentrations of conventional photosensitizers, but also fine-tuned the excited state dynamics to exploit the initial ISC process, affording an excellent 1O2 quantum yield and bright NIR fluorescence in the aggregated state. Furthermore, theoretical and spectroscopic investigations revealed that an RP-ISC mechanism dominated the triplet formation, where the effective charge separation and ultra-small singlet–triplet energy splitting in symmetric A–D–A systems promoted the RP-ISC process. Despite the efficient RP-ISC from the perspective of excited state dynamics being demonstrated in the present molecular models, future in-depth theoretical and physicochemical studies on the impact of molecular symmetry will be beneficial for understanding the nature of the intersystem crossing process and clarifying the validity of the “symmetry-controlling” strategy. Furthermore, the ability to efficiently enhance singlet oxygen generation with strong fluorescence will encourage us to develop high performance fluorescent PSs following the new guidelines, especially the extension of the absorption wavelength and biocompatibility for advanced applications in photodynamic therapy and related imaging.Ethical statement
All animal studies were performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication No. 85-23 Rev. 1985) and were approved by the Committee on Animal Use and Care of Chinese Academy of Sciences.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the Natural Science Foundation of China (21835001, 51773080, 21674041, and 21221063), the Program for Changbaishan Scholars of Jilin Province, and the “Talents Cultivation Program” of Jilin University.Notes and references
- (a) T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889 CrossRef CAS; (b) D. Cui, J. G. Huang, X. Zhen, J. C. Li, Y. Y. Jiang and K. Y. Pu, Angew. Chem., Int. Ed., 2019, 58, 5920 CrossRef CAS.
- (a) S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990 CrossRef CAS; (b) G. R. Jin, R. Y. He, Q. Liu, Y. Q. Dong, M. Lin, W. F. Li and F. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 10634 CrossRef CAS.
- (a) P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250 CrossRef; (b) J. C. Li, D. Cui, J. G. Huang, S. S. He, Z. B. Yang, Y. Zhang, Y. Luo and K. Y. Pu, Angew. Chem., Int. Ed., 2019, 58, 12680 CrossRef CAS.
- (a) W. P. Fan, P. Huang and X. Y. Chen, Chem. Soc. Rev., 2016, 45, 6488 RSC; (b) Y. Z. Shen, A. J. Shuhendler, D. J. Ye, J.-J. Xu and H.-Y. Chen, Chem. Soc. Rev., 2016, 45, 6725 RSC; (c) K. K. Ng and G. Zheng, Chem. Rev., 2015, 115, 11012 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS.
- (a) Z. E. X. Dance, Q. X. Mi, D. W. McCamant, M. J. Ahrens, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. B, 2006, 110, 25163 CrossRef CAS; (b) I. R. Gould, J. A. Boiani, E. B. Gaillard, J. L. Goodman and S. Farid, J. Phys. Chem. A, 2003, 107, 3515 CrossRef CAS.
- (a) S. Y. Lee, T. Yasuda, Y. S. Yang, Q. S. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402 CrossRef CAS; (b) M. Baroncini, G. Bergamini and P. Ceroni, Chem. Commun., 2017, 53, 2081 RSC.
- (a) J. C. Li, J. G. Huang, Y. Lyu, J. S. Huang, Y. Y. Jiang, C. Xie and K. Y. Pu, J. Am. Chem. Soc., 2019, 141, 4073 CrossRef CAS; (b) Q. Y. Li, Y. Li, T. L. Min, J. Y. Gong, L. L. Du, D. L. Phillips, J. K. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, R. T. K. Kwok, C. L. Ho, K. Li, J. G. Wang and B. Z. Tang, Angew. Chem., Int. Ed., 2019, 58, 2 CrossRef.
- (a) J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839 CrossRef CAS; (b) J. C. Li, D. Cui, Y. Y. Jiang, J. G. Huang, P. H. Cheng and K. Y. Pu, Adv. Mater., 2019, 31, 1905091 CrossRef CAS.
- (a) A. B. Ormond and H. S. Freeman, Materials, 2013, 6, 817 CrossRef CAS; (b) M. Taniguchi and J. S. Lindsey, Chem. Rev., 2017, 117, 344 CrossRef CAS.
- (a) J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001,(18), 1740 RSC; (b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS.
- (a) S. D. Xu, W. B. Wu, X. L. Cai, C.-J. Zhang, Y. Y. Yuan, J. Liang, G. X. Feng, P. Manghnani and B. Liu, Chem. Commun., 2017, 53, 8727 RSC; (b) F. Hu, S. D. Xu and B. Liu, Adv. Mater., 2018, 30, 1801350 CrossRef.
- (a) S. M. Sartor, B. G. McCarthy, R. M. Pearson, G. M. Miyake and N. H. Damrauer, J. Am. Chem. Soc., 2018, 140, 4778 CrossRef CAS; (b) Y. Dong, A. A. Sukhanov, J. Z. Zhao, A. Elmali, X. L. Li, B. Dick, A. Karatay and V. K. Voronkova, J. Phys. Chem. C, 2019, 123, 22793 CrossRef CAS.
- J. B. Zhang, B. Xu, J. L. Chen, L. J. Wang and W. J. Tian, J. Phys. Chem. C, 2013, 117, 23117 CrossRef CAS.
- Y. Y. Yuan, G. X. Feng, W. Qin, B. Z. Tang and B. Liu, Chem. Commun., 2014, 50, 8757 RSC.
- (a) S. D. Xu, Y. Y. Yuan, X. L. Cai, C.-J. Zhang, F. Hu, J. Liang, G. X. Zhang, D. Q. Zhang and B. Liu, Chem. Sci., 2015, 6, 5824 RSC; (b) W. B. Wu, D. Mao, F. Hu, S. D. Xu, C. Chen, C.-J. Zhang, X. M. Cheng, Y. Y. Yuan, D. Ding, D. L. Kong and B. Liu, Adv. Mater., 2017, 29, 1700548 CrossRef.
- G. S. He, L.-S. Tan, Q. D. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245 CrossRef CAS.
- T. Chen, L. Zheng, J. Yuan, Z. F. An, R. F. Chen, Y. Tao, H. H. Li, X. J. Xie and W. Huang, Sci. Rep., 2015, 5, 10923 CrossRef.
- H. L. Ma, Q. Peng, Z. F. An, W. Huang and Z. G. Shuai, J. Am. Chem. Soc., 2019, 141, 1010 CrossRef CAS.
- (a) H. Levanon and J. R. Norris, Chem. Rev., 1978, 78, 185 CrossRef CAS; (b) M. C. Thurnauer, J. J. Katz and J. R. Norris, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 3270 CrossRef CAS.
- (a) F. Terenziani, A. Painelli, C. Katan, M. Charlot and M. Blanchard-Desce, J. Am. Chem. Soc., 2006, 128, 15742 CrossRef CAS; (b) B. Dereka, A. Rosspeintner, Z. Q. Li, R. Liska and E. Vauthey, J. Am. Chem. Soc., 2016, 138, 4643 CrossRef CAS.
- (a) M. T. Colvin, A. B. Ricks, A. M. Scott, D. T. Co and M. R. Wasielewski, J. Phys. Chem. A, 2012, 116, 1923 CrossRef CAS; (b) M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282 CrossRef CAS.
- (a) P. Alam, N. L. C. Leung, J. K. Liu, T. S. Cheung, X. P. Zhang, Z. K. He, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, C. C. S. Chan, K. S. Wong, Q. Peng and B. Z. Tang, Adv. Mater., 2020, 2001026 CrossRef CAS; (b) J. Yang, Z. C. Ren, Z. L. Xie, Y. J. Liu, C. Wang, Y. J. Xie, Q. Peng, B. Xu, W. J. Tian, F. Zhang, Z. G. Chi, Q. Q. Li and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 880 CrossRef CAS.
- M. Bixon, J. Jortner, J. Cortes, H. Heitele and M. E. Michel-Beyerle, J. Phys. Chem., 1994, 98, 7289 CrossRef CAS.
- B. Carlotti, E. Benassi, A. Spalletti, C. G. Fortuna, F. Elisei and V. Barone, Phys. Chem. Chem. Phys., 2014, 16, 13984 RSC.
- Z. E. X. Dance, Q. Mi, D. W. McCamant, M. J. Ahrens, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. B, 2006, 110, 25163 CrossRef CAS.
- (a) D. Gosztola, M. P. Niemczyk, W. Svec, A. S. Lukas and M. R. Wasielewski, J. Phys. Chem. A, 2000, 104, 6545 CrossRef CAS; (b) A. M. Lifschitz, R. M. Young, J. Mendez-Arroyo, V. V. Roznyatovskiy, C. M. McGuirk, M. R. Wasielewski and C. A. Mirkin, Chem. Commun., 2014, 50, 6850 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03128c |
| This journal is © The Royal Society of Chemistry 2020 |