
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic, multimodal hydrogel actuators using porphyrin-based visible light photoredox catalysis in a thermoresponsive polymer network†
Faheem
Amir
a,
Xuesong
Li
 a,
Max C.
Gruschka
a,
Nathan D.
Colley
a,
Lei
Li
a,
Ruihan
Li
a,
Houston R.
Linder
b,
Scott A.
Sell
b and
Jonathan C.
Barnes
*a
a,
Max C.
Gruschka
a,
Nathan D.
Colley
a,
Lei
Li
a,
Ruihan
Li
a,
Houston R.
Linder
b,
Scott A.
Sell
b and
Jonathan C.
Barnes
*a
aDepartment of Chemistry, Washington University, One Brookings Drive, St. Louis, MO 63130, USA. E-mail: jcbarnes@wustl.edu
bDepartment of Biomedical Engineering, Saint Louis University, St. Louis, MO 63103, USA
First published on 3rd September 2020
Abstract
Hydrogels that can respond to multiple external stimuli represent the next generation of advanced functional biomaterials. Here, a series of multimodal hydrogels were synthesized that can contract and expand reversibly over several cycles while changing their mechanical properties in response to blue and red light, as well as heat (∼50 °C). The light-responsive behavior was achieved through a photoredox-based mechanism consisting of photoinduced electron transfer from a zinc porphyrin photocatalyst in its excited state to oligoviologen-based macrocrosslinkers, both of which were integrated into the hydrogel polymer network during gel formation. Orthogonal thermoresponsive properties were also realized by introducing N-isopropyl acrylamide (NIPAM) monomer simultaneously with hydroxyethyl acrylate (HEA) in the pre-gel mixture to produce a statistical 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 HEA:NIPAM polymer network. The resultant hydrogel actuators – crosslinked with either a styrenated viologen dimer (2V4+-St) or hexamer (6V12+-St) – were exposed to red or blue light, or heat, for up to 5 h, and their rate of contraction, as well as the corresponding changes in their physical properties (i.e., stiffness, tensile strength, Young's modulus, etc.), were measured. The combined application of blue light and heat to the 6V12+-St-based hydrogels was also demonstrated, resulting in hydrogels with more than two-fold faster contraction kinetics and dramatically enhanced mechanical robustness when fully contracted. We envision that the reported materials and the corresponding methods of remotely manipulating the dynamic hydrogels may serve as a useful blueprint for future adaptive materials used in biomedical applications.
:40 HEA:NIPAM polymer network. The resultant hydrogel actuators – crosslinked with either a styrenated viologen dimer (2V4+-St) or hexamer (6V12+-St) – were exposed to red or blue light, or heat, for up to 5 h, and their rate of contraction, as well as the corresponding changes in their physical properties (i.e., stiffness, tensile strength, Young's modulus, etc.), were measured. The combined application of blue light and heat to the 6V12+-St-based hydrogels was also demonstrated, resulting in hydrogels with more than two-fold faster contraction kinetics and dramatically enhanced mechanical robustness when fully contracted. We envision that the reported materials and the corresponding methods of remotely manipulating the dynamic hydrogels may serve as a useful blueprint for future adaptive materials used in biomedical applications.
Introduction
Stimuli-responsive soft materials1–4 that can sense external environmental cues and change their size, shape, and mechanical properties have found use in a number of applications, such as actuators,5,6 soft robotics,7,8 tissue culture,9–11 and the controlled release of cells, biomolecules and drugs.12–14 Often, the responsivity is programmed into the soft material at the (macro)molecular level prior to formation of a liquid crystal elastomer or gel network for example. Some common external stimuli that have been investigated previously include changes in pH15–17 or temperature,18–20 reduction and oxidation,21–24 magnetic fields,25 or irradiation with light (e.g., UV, visible, near-IR (NIR)),26–37 where the selection of a material's responsive components is typically based on the desired application and preference for a specific external stimulus.Thermally responsive hydrogels in particular possess distinct biomedical advantages since they can be fabricated to mimic soft biological tissues and formulated with a lower critical solution temperature (LCST) that is close to that of a physiological setting (37 °C). One of the most commonly used polymers in thermoresponsive hydrogels is poly(N-isopropyl acrylamide) (PNIPAM)38–40 because at temperatures below its LCST, it is fully hydrated, but at temperatures above the threshold temperature, desolvation is entropically favored and precipitation of the polymer leads to contraction of the overall material. This process may be reversed after the temperature is lowered below the LCST and the polymeric material is re-expanded by swelling in H2O. Building off this temperature-driven phenomenon, many investigations reported in the literature have elected to couple PNIPAM's thermoresponsive process with that of a photothermal one, such as surface plasmon resonance-based heating of gold nanomaterials,35–37 or the irradiation of graphene41,42 and carbon nanotubes43–45 embedded in the gels. In these examples, localized heating induces precipitation of PNIPAM at the site of light irradiation, resulting in controlled contraction of the material in precise areas. Thus, the dependent, photothermal mechanism converts light-based energy into heat and provides spatiotemporal control over actuation and changes in the gel's mechanical properties.
Alternatively, stimuli-responsive soft materials that possess light- and temperature-responsive components operated independently through a decoupled, or orthogonal, mechanism have also been investigated. One such example from Shinbo and co-workers involved the synthesis of a PNIPAM-based hydrogel that incorporated blue light-responsive spirobenzopyran (SP) as part of the gel network.46 Upon irradiation with blue light, the open merocyanine form of SP isomerized to the neutral, ring-closed spiro form, resulting in hydrogel contraction. The authors used this reversible switch at low pH to control the permeability of a hydrogel-based membrane. Similarly, azobenzene and its derivatives have also been used as the UV-visible-active component in dual-responsive (i.e., photo- and thermo-responsive) hydrogels.47,48 In each of these cases though, the photoresponsive component is usually activated by higher energy, lower wavelength visible light. However, for potential biomedical applications, longer wavelengths of light in the near-IR optical window (650–950 nm) are required to penetrate human tissue.49,50 Along these lines, many examples have been reported involving the fabrication of functional soft materials that are programmed to respond to red51–53 and/or NIR light.35–37,41 But, to the best of our knowledge, none of the aforementioned materials and related designs are capable of operating using multiple wavelengths of light, whilst also simultaneously possessing thermoresponsive behavior.
The advent of photoredox catalysis in the fields of synthetic organic chemistry54–59 and polymer chemistry60–65 opened up the possibility to enact complex chemical transformations through photoinduced electron/energy transfer processes. By tuning the photocatalyst66 – commonly a transition metal complex or a metal-free organic dye – different photophysical properties can be accessed, depending on the desired application. Metalloporphyrins, often used as a photosensitizer in photodynamic therapies,67,68 or as a means to harvest photons in dye-sensitized solar cells,69–71 are quickly becoming a versatile tool in the field of photocatalysis.62,72–74 One of the earliest demonstrations of their use in polymer synthesis was described62 by Boyer and co-workers in 2015 when they investigated the multi-wavelength and oxygen-tolerant properties of zinc (Zn)-based tetraphenylporphyrin in the photoinduced electron transfer addition–fragmentation chain transfer (PET-RAFT) polymerization of a range of monomers and trithiocarbonate chain-transfer agents. Yet as versatile as they have proven to be, metalloporphyrins, to the best of our knowledge, have not yet been exploited as photoredox catalysts that can initiate macroscale motion in hydrogel actuators.
Recently, we demonstrated75–77 a novel photoredox-based mechanism for actuating gels (Fig. 1a), whereby a bis(viologen)-based crosslinker, 2V4+-St, was used to establish a network comprising poly(hydroxyethyl acrylate) (poly(HEA)) and a visible-light-absorbing photocatalyst (PC) – i.e., [Ru(bpy)3]Cl2 – soaked into the gel. Irradiation of the gels with blue light in the presence of a sacrificial reductant like triethanolamine (TEOA) induced a dramatic reduction in the gel's volume (contracting anywhere from 40 to 80%) and caused a 2–3 fold increase in its Young's modulus and maximum tensile strength. These changes in size and mechanical properties resulted from the photoreduction of the crosslinker (2V4+ to 2V˙+) inside the gel network, leading to a loss of ions and electrostatic repulsion, as well as the “turning on” of favorable radical spin-pairing interactions between viologen radical cations (Fig. 1a).78–83 Removing the light source and allowing O2 and H2O to diffuse into the contracted gel caused the viologen radical cations to oxidize (2V˙+ to 2V4+) and the gel to re-swell as it absorbed H2O upon expansion.
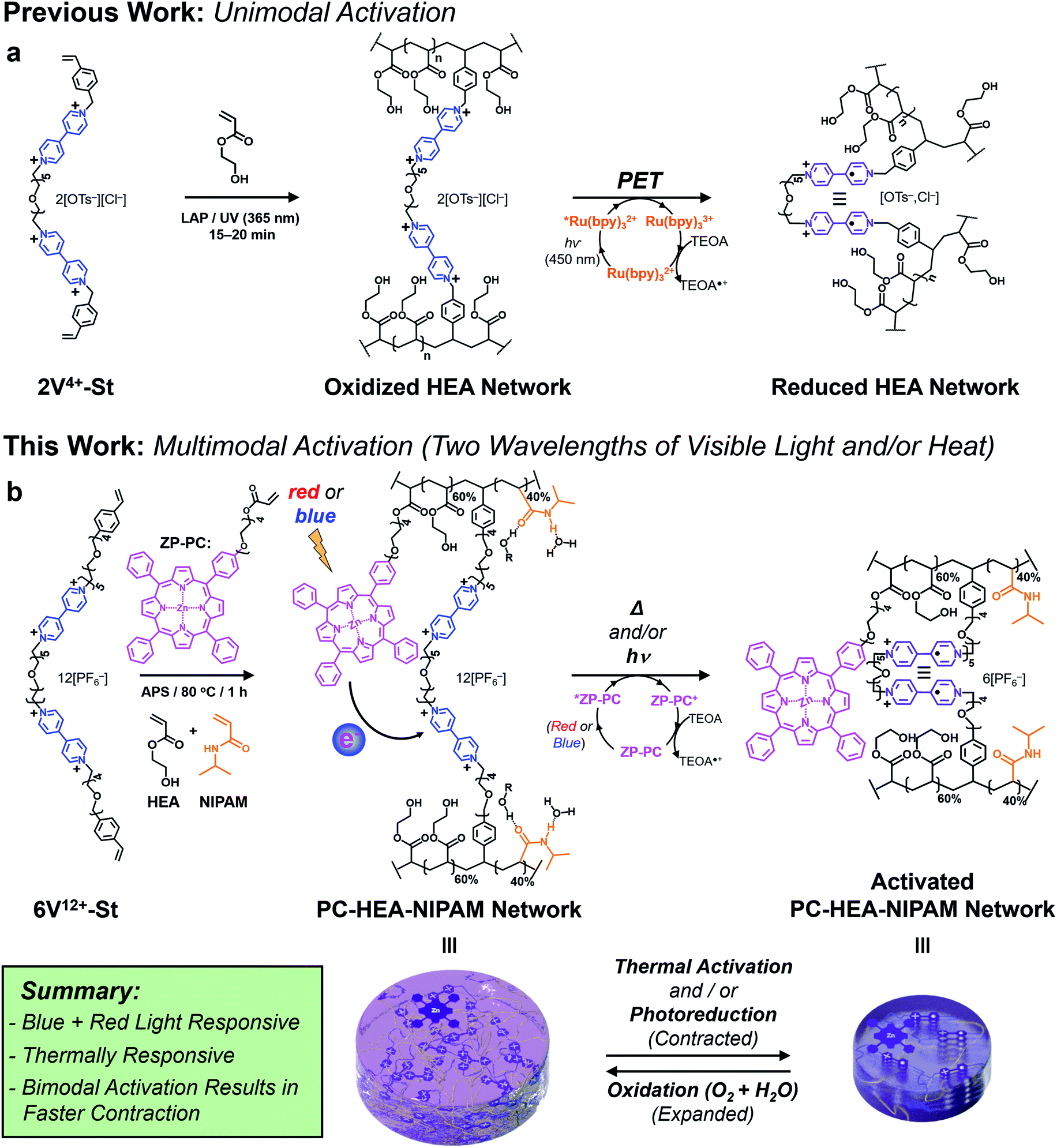 | ||
| Fig. 1 Design and synthesis of viologen-based hydrogel actuators. (a) Previous gel network design composed of HEA monomer that was actuated using a photoinduced electron transfer (PET) process between a blue light-absorbing photocatalyst (Ru(bpy)32+) and a (bis)viologen-based crosslinker. (b) New hydrogel network design composed of 60:40 HEA:NIPAM monomer ratio, a Zn-based porphyrin photocatalyst (ZP-PC), and a six viologen-containing macrocrosslinker. Actuation of the multimodal gel is achieved with blue or red light, heat, or the combination of light and heat (bimodal). | ||
Although we demonstrated that this novel light-based mechanism for actuating gels was completely reversible over many contraction–expansion cycles, it is still not without limitations. Firstly, the ruthenium-based PC was soaked into the gel along with the sacrificial reductant (TEOA). This means the gels needed to be replenished after each contraction cycle by soaking them in a solution consisting of 0.15 mM PC and 3 mM TEOA, allowing for consistent actuation kinetics and performance. Secondly, the oft-used PC in this case ([Ru(bpy)3]Cl2) is commonly photoexcited with blue light (i.e., 450 nm), which falls short for its use in biomedical applications that require longer wavelengths of light.
With an eye towards future potential biomedical applications, we sought to redesign the photoredox platform (Fig. 1b) so a more versatile PC could be employed and integrated into the hydrogel network. Specifically, a mono-functionalized zinc(II) porphyrin (ZP-PC) was prepared with an oligoethylene glycol chain bearing a polymerizable acrylate group. Incorporation of the derivatized PC into the hydrogel network crosslinked with the bis- or hexa-viologen containing macrocrosslinkers (2V4+-St or 6V12+-St, respectively) was screened and optimized to ensure efficient gel actuation performance and kinetics. Additionally, an orthogonal, thermoresponsive mode of activation was built into the stimuli-responsive material by implementing a 60:40 HEA:NIPAM monomer ratio prior to gel formation. Here, we describe (i) the synthesis of functional precursors and the final multi-responsive gel product, (ii) photoredox-based activation of the gel actuators using blue or red light, (iii) gel actuation using only heat, and (iv) enhanced gel actuation kinetics and performance when blue light and heat are used in concert.
Results and discussion
Synthesis and characterization of the viologen-based crosslinkers and photocatalyst
Synthesis of the hexaviologen-based macrocrosslinker (6V12+-St) was performed (Fig. 2a) by using a modified version of our previously reported84 iterative synthetic protocol to make well-defined polyviologens. Specifically, a dibromo-end-capped hexaethylene glycol linker (HEG-Br) was used in this investigation instead of its tosylated equivalent on account of the latter being more susceptible to hydrolysis during workup and storage. Potassium hexafluorophosphate (KPF6) was also included in several of the reaction steps (vide infra) to obtain the final product macrocrosslinker. The purpose of adding the salt was to keep the precursors and intermediate products soluble in MeCN during the reaction, which in turn yielded nearly quantitative conversion for each step. With these protocol changes in hand, the synthesis of 2V2+·2PF6− was performed (Fig. 2a) using a Teflon screw-capped high-pressure flask, in which 1 equiv. of HEG-Br was reacted with 20 equiv. of 4,4′-bipyridine (BIPY) in the presence of 4 equiv. of KPF6, all dissolved in anhydrous acetonitrile (MeCN; 100 mg mL−1) heated to 130 °C and stirred for 16 h. The salt (KPF6) was used to exchange the Br− counteranion with PF6− ions, thus enhancing the solubility of the resultant product (2V2+·2PF6−) in MeCN. After completion, the crude reaction mixture was cooled to room temperature, filtered, and precipitated in PhMe:Et2O (1:2), followed by centrifugation at 4500 rpm for 30 min. Next, the supernatant solution was decanted, the resultant pellet was re-dissolved in MeCN, and the precipitation process was repeated four times to yield 2V2+·2PF6− in 75% yield. The moderate loss in yield may be attributed to the workup procedure, as proton nuclear magnetic resonance (1H NMR) spectroscopy was used to confirm full conversion of HEG-Br in the crude reaction mixture. Similarly, 2V2+·2PF6− was converted to 2V4+·2PF6−·2Br− by stirring it for 16 h with 20 equiv. of HEG-Br in anhydrous MeCN (10 mg mL−1 of 2V2+·2PF6−) heated to 130 °C in a high-pressure flask. However, it is important to note that in this step KPF6 was not used because the terminal HEG-Br chains of 2V4+·2PF6−·2Br− improved its solubility in MeCN. Isolation of the pure product was carried out like the previous step, affording 2V4+·2PF6−·2Br− in 74% yield. Subsequent extension of the chain to a tetraviologen intermediate was carried out by reacting 2V4+·2PF6−·2Br− with 20 equiv. of BIPY in the presence of 4 equiv. of KPF6, all dissolved in anhydrous MeCN (at 10 mg mL−1 of the viologen-based starting material), heated to 130 °C in a high-pressure flask while stirring for 20 h. The product 4V6+·6PF6− was isolated as described in the previous steps, in 70% yield. Further repetition from the previous steps also yielded the hexaviologen precursor 6V10+·10PF6− in 67% yield. In the final reaction step, 6V10+·10PF6− was treated in a high-pressure flask with a mesylated oligoethylene glycol chain attached to styrene (MsO-TEG-St; 35 equiv.) in the presence of KPF6 (3 equiv.) and 4-tert-butylcatechol (6 equiv.) dissolved in anhydrous dimethylformamide (DMF; 75 mg mL−1) heated to 80 °C. After 48 h of stirring, the crude reaction mixture was cooled to room temperature and diluted with MeOH, followed by filtering and precipitating out the semi-pure product in PhMe:Et2O (1:2). The isolated product was then re-dissolved in DMF and precipitated with THF:Et2O (1:2) and this process was repeated four additional times to produce nearly 3 g of 6V12+-St·12PF6− in 66% yield. For reference, the shorter bis(viologen)-based crosslinker (2V2+·2PF6−) was treated with 4-vinylbenzyl chloride (50 equiv.) dissolved in anhydrous DMF (75 mg mL−1 of 2V2+·2PF6−) and heated at 55 °C while stirring for 24 h. The product 2V4+-St·2PF6−·2Br− was isolated by precipitation – in PhMe:Et2O (1:2) – to afford 1.5 g of the crosslinker in 57% yield.
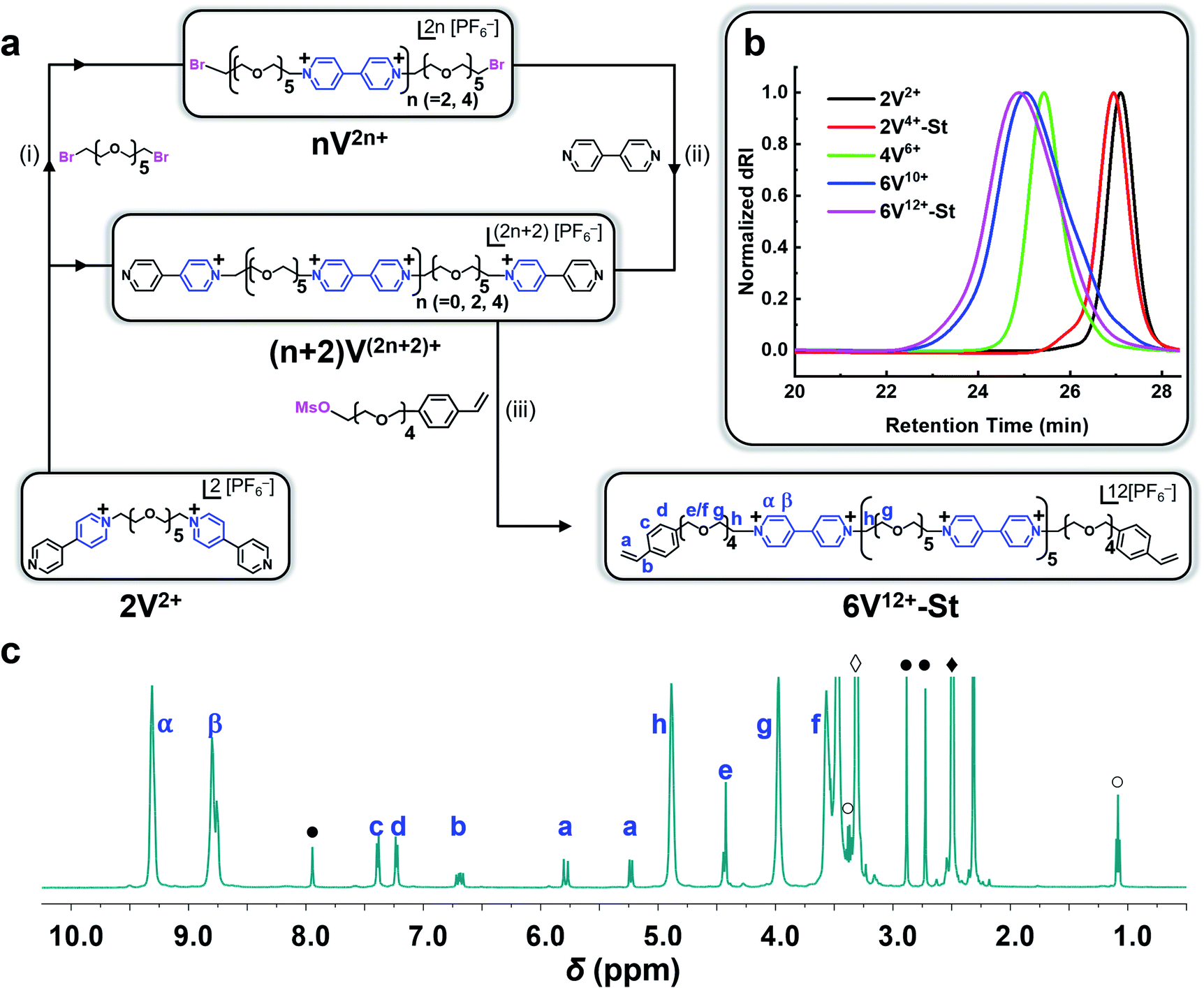 | ||
| Fig. 2 Synthesis of a hexaviologen-based macrocrosslinker 6V12+-St as its PF6− salt. (a) Iterative synthesis: (i) HEG-Br (20.0 equiv.), KPF6 (4.0 equiv.) dissolved in MeCN in a high-pressure flask heated to 130 °C and stirred for 16 h; (ii) BIPY (20 equiv.) dissolved in MeCN in a high-pressure flask heated to 130 °C and stirred for 16 h; (iii) MsO-TEG-St (35 equiv.), KPF6 (3.0 equiv.), 4-tert-butylcatechol (6 equiv.) dissolved in DMF (in a non-high-pressure flask) and heated to 80 °C while stirring for 48 h; (b) GPC traces (H2O with 0.025 M Na2SO4 at 23 °C with 1.0 mL min−1) of the oligoviologen intermediates and final macrocrosslinker (all counterions are HSO4−); (c) 1H NMR data of 6V12+-St (500 MHz, DMSO-d6). The residual solvent peaks are labeled as: DMF (●), DMSO (◆), H2O (◊), and Et2O (○). | ||
Synthesis of the monofunctionalized photocatalyst (ZP-PC, Fig. 1b) was performed by modifying previously reported procedures.85,86 The first step in the synthesis to generate the monohydroxyl free-base tetraphenylporphyrin (TPP-OH) was the most costly in terms of yield (i.e., ∼5%) given that the reaction produces a statistical mixture of mono-, bis-, tris-, and tetra-functionalized TPP. However, isolation of the desired product (TPP-OH) was readily achieved using standard silica gel chromatography using a gradient mobile phase of hexane:CH2Cl2. Functionalization of the singular hydroxyl group with a triethylene glycol linker was carried out under mildly basic conditions (K2CO3) in DMF at 80 °C by reacting TPP-OH with a monotosylated triethylene glycol (TEG-OTs) precursor, resulting in a 55% yield for the free-base porphyrin intermediate TPP-TEG-OH. Next, a polymerizable group was added to the terminus of the TEG chain by treating TPP-TEG-OH with Et3N in refluxing CH2Cl2, followed by the addition of acryloyl chloride and heating the reaction at 60 °C. After an aqueous workup, the product was isolated by silica gel chromatography (CH2Cl2:EtOAc, 9:1) to afford TPP-TEG-Acrylate in 75% yield. The final metalated ZP-PC photocatalyst was obtained by treating TPP-TEG-Acrylate with Zn(OAc)2 (10 equiv.) in CHCl3:MeOH (7:3), stirring overnight at room temperature and in the dark. After another aqueous workup, several hundred milligrams of the final product were isolated in 95% yield. For complete synthetic details and reaction schemes to prepare ZP-PC, see ESI.†
Each compound synthesized in this investigation was characterized by 1H and 13C NMR (Fig. 2c and S1–S9†), and each oligoviologen product underwent additional characterization that included gel permeation chromatography (GPC) (Fig. 2b and S13†) and MALDI-ToF mass spectrometry (see ESI† for mass spectral data). It is important to note that although the smaller molecular weight oligomers – namely, 2V2+, 2V4+-St, and 4V6+ – displayed narrow elution bands by GPC, the hexaviologen macrocrosslinker and its corresponding precursor (6V12+-St and 6V10+, respectively) exhibited considerable peak broadening. However, this has been observed in previous reports by us,75,76 and is expected as more positive charges are built into the higher molecular weight oligomers, thus allowing for increased counterion exchange to occur on the column (mobile phase: H2O with 0.025 M Na2SO4 at 23 °C). To corroborate the purity and unimolecular nature of each oligoviologen intermediate and the final macrocrosslinker, end-group analysis was performed using 1H NMR spectroscopy (Fig. 2c and S9†). In particular, the intermediates terminated by unfunctionalized pyridine rings showed four distinct proton resonances between 8.0–9.4 ppm, which arise from the asymmetry of the α and β protons associated with each pseudo-viologen end group. Conversely, the bromine-end capped precursor sequences contain symmetrical viologen subunits, which leads to only two proton resonances for the α and β protons of each viologen subunit at 8.7 and 9.3 ppm. Similarly, 6V12+-St possesses symmetrical viologen subunits throughout (Fig. 2c), hence two peaks were observed at 8.79 and 9.31 ppm for the α and β protons associated with each end group. Additionally, a distinct singlet arises at 4.43 ppm (labelled as ‘e’ in Fig. 2c), representing the methylene proton next to the styrene whose own aromatic proton resonances appear as doublets at 7.24 and 7.40 ppm (labelled ‘c’ and ‘d’). Moreover, the vinylogous proton resonances of styrene appear at 5.24, 5.79, and 6.70 ppm as doublet, doublet, and doublet of doublets, respectively (labelled ‘a’ and ‘b’ in Fig. 2c). When comparing the relative integrations of the styrenyl methylene and aromatic proton resonances to that of the α and β protons of the viologen subunits (Fig. S9†), the expected ratio (24:4 and 24:8, respectively) was observed for a macrocrosslinker consisting of six viologen subunits. For the 2V4+-St crosslinker, each viologen subunit is asymmetric, hence three proton resonances were observed (Fig. S9†) at 8.73, 9.28, and 9.48 ppm. Additionally, a distinct singlet appears at 5.93 ppm, representing the methylene proton adjacent to the styrene whose aromatic proton resonances show up at 7.58 ppm. Similar to the hexaviologen macrocrosslinker, the vinylogous proton resonances for 2V4+-St appear at 5.33, 5.90, and 6.76 ppm as a doublet, doublet, and doublet of doublets, respectively.
Next, the ability of 2V4+-St and 6V12+-St to undergo intramolecular self-assembly (i.e., folding) in MeCN through a radical–radical spin pairing mechanism involving photoreduced viologens (V˙+⋯V˙+) was verified using ultraviolet-visible-near-infrared (UV-vis-NIR) absorption spectroscopy. This experiment was performed by preparing an N2-purged MeCN solution (0.7 mL) containing 0.25 mM 2V4+-St·2PF6−·2Br−, 0.3 mM ZP-PC, and 3.0 mM TEOA as a sacrificial reductant, and adding it to a cuvette in a N2-filled glovebox, followed by irradiating it with blue light (450 nm) for 60 min. UV-vis-NIR absorption spectra were recorded (Fig. S10–S12†) before and after photoreduction, and a distinct absorption peak in the NIR centered around 850–900 nm was observed for the irradiated sample. This peak is commonly associated with the formation of viologen radical dimers87 and was also observed in our previously reported investigations of oligoethylene glycol-based polyviologens.75,77,84 Moreover, the absence of either the photocatalyst, viologen dimer, or light irradiation resulted in absorption spectra that lacked the diagnostic electronic features at ∼900 nm associated with the pairing of viologen radical cations. The same experiment was repeated for 6V12+-St·12PF6− at the same concentration as the dimer (Fig. S12b†). Again, the NIR absorption peak at 850–900 nm was observed, only at a higher intensity on account of the hexamer containing three times as many viologen subunits. These results in solution support the notion that intramolecular radical-based molecular recognition upon photoirradiation is possible with each crosslinker, however, it is important to note that the data does not entirely rule out the possibility that some intermolecular affinity may also be possible, even under these dilute conditions, and certainly when crosslinked into hydrogels (vide infra).
Fabrication of multimodal hydrogels
Next, a series of hydrogels were synthesized in triplicate using 2V4+-St·2PF6−·2Br− (at 0.365 mol% or 3.5 wt%) or 6V12+-St·12PF6− (at 0.365 mol% or 12.4 wt%) crosslinkers in DMSO and using the thermal initiator ammonium persulfate (APS) to go along with 2-hydroxyethyl acrylate (HEA; 0.58 mol%) and N-isopropylacrylamide (NIPAM) (0.40 mol%) monomers, and ZP-PC. The total mass of all the pre-gel material was 443.5 mg in 1.11 mL of DMSO (Table S1†). The gelation time was 60 min, while the pre-gel solution was contained in a 2 cm diameter rubber septum. Complete conversion of the crosslinker, monomer, and ZP-PC photocatalyst was confirmed using Fourier transform infrared (FTIR) spectroscopy and monitoring the disappearance of corresponding vinyl groups (Fig. S14†). The resultant three-dimensional polymer network was soaked in H2O for at least 48 h to yield swollen hydrogels with an average volume of 9.68 cm3 for 2V4+-St and 11.16 cm3 for 6V12+-St crosslinked gels. The shear modulus (G) was obtained by measuring the storage (G′) and loss (G′′) moduli for each hydrogel. Subsequently, the corresponding swollen hydrogels were lyophilized and their swollen and dry masses were used to calculate the volumetric swelling ratio (Q)88 (Table S3†), crosslinking density89 (0.978 and 0.744 mol m−3 for 2V4+-St and 6V12+-St crosslinked hydrogels, respectively), and molecular weight of the polymer chains in between crosslinking junctions (175 and 118 kDa for the 2V4+-St and 6V12+-St crosslinked hydrogels) (see ESI† for all related equations). From these calculations, it is apparent that by increasing the number of viologen–hexaethylene glycol units in the chain, as well as the overall amount of the crosslinker, the hydrophilicity of the corresponding hydrogel increases. The increased affinity for water thus leads to the increased swelling ratios and lower G and MC values, meaning the corresponding gel becomes inherently softer.Performance of the hydrogel actuators
With the hydrogels synthesized in triplicate, their ability to perform as photo-, thermal-, and bimodal-responsive (photo + thermal) actuators was assessed (Fig. 3). The photoresponsive experiments were performed by soaking the hydrogels in an aqueous 3.0 mM solution of TEOA inside a N2-filled glove box for 24 h. Next, the hydrogels were placed in a clear sealed Petri dish and irradiated simultaneously from above and below (5.5 cm from dish) with blue (450 nm) or red (660 nm) light (see inset pictures in Fig. 3a). For the control experiments, the hydrogels were soaked only in Milli-Q H2O (with no photocatalyst) and irradiated with blue or red light. For the thermoresponsive experiments, one hydrogel each was placed in three 250 mL beakers (containing 125 mL H2O). These beakers were then placed in a pre-heated (50 °C) H2O bath (Fig. S17†), representing the starting point of the experiment. The combined activation using light and heat (i.e., bimodal actuation) was investigated by soaking the hydrogels in 3.0 mM TEOA solution (50 mL) in 125 mL glass bottles inside a N2-filled glove box. After soaking, each of the bottles were sealed with a rubber septum inside the N2-filled glove box and then immediately connected to a N2-line after removing from the glovebox. The bottles were then placed in a pre-heated (50 °C) H2O bath (Fig. S21†) to start the contraction kinetics experiments. For reference, only the 6V12+-St crosslinked hydrogels were evaluated for bimodal actuation. For all actuation methods, the volume was measured (Fig. 3b and c) as the hydrogels contracted over a period of 5 h.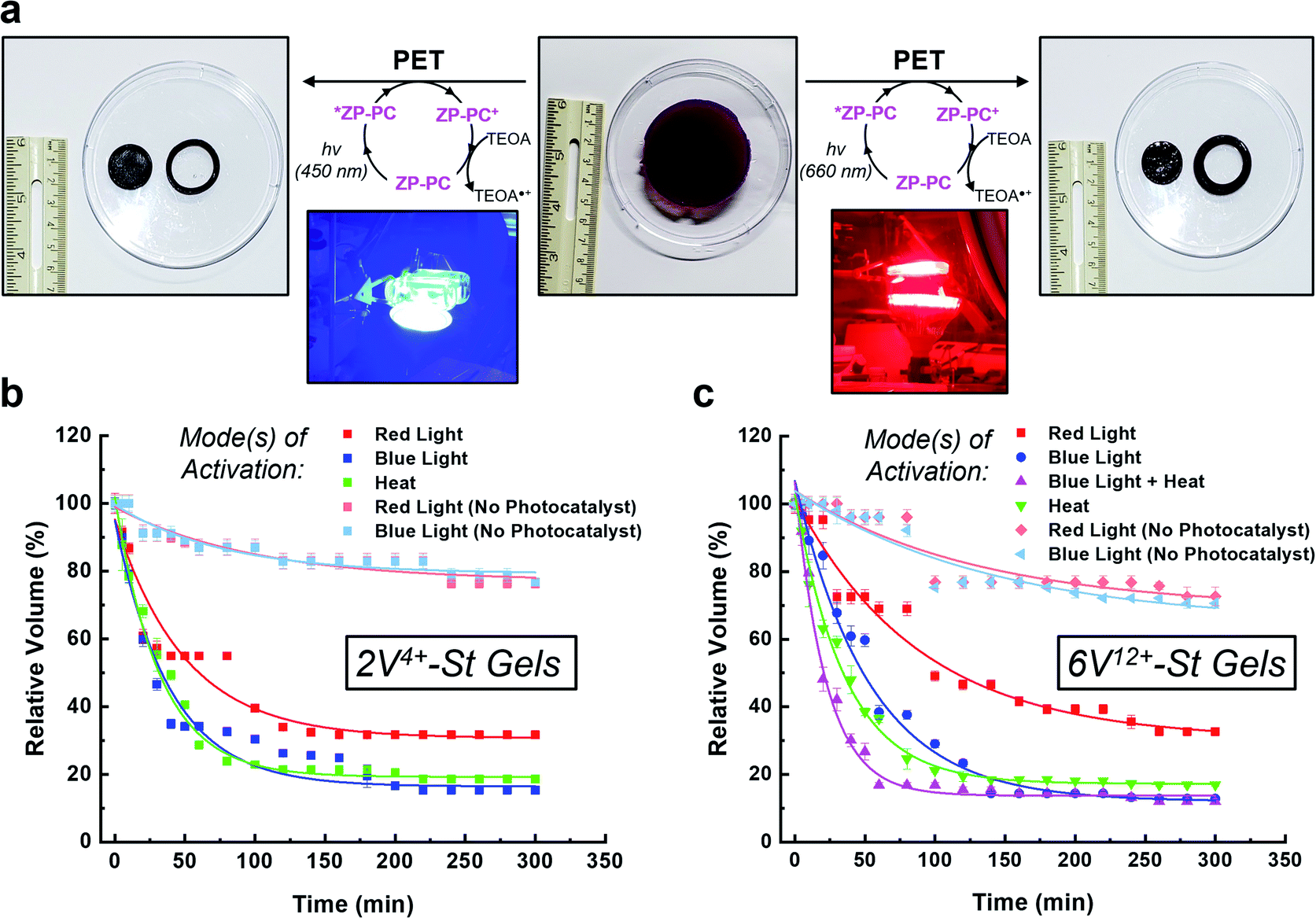 | ||
| Fig. 3 (a) Pictures of a representative swollen hydrogel (middle) were acquired before and after irradiation with either blue (450 nm) or red (660 nm) light (left and right, respectively). The inset pictures show the experimental setup for irradiating each hydrogel from above and below using lamps. Contraction kinetics of hydrogels composed of (b) 2V4+-St or (c) 6V12+-St crosslinkers were investigated by measuring the change in volume of each hydrogel when activated by visible light (red or blue) and/or heat over the course of 5 h. | ||
When the 2V4+-St crosslinked hydrogels (Fig. 3b) were irradiated with red light for 2 h, their volumes decreased down to 34% of their original starting size, and only an additional 2% contraction was observed thereafter from 2 to 5 h (i.e., the final relative volume was 32%). By comparison, irradiating the hydrogels with blue light resulted in volume contraction down to 26% in 2 h, and then down to 15% after another 3 h of irradiation. As a control, two sets of hydrogels possessing no photocatalyst were irradiated with red or blue light. After 2 h of irradiation, both sets of hydrogels contracted to 82% of their original starting volumes, and then down to 76% after an additional 3 h of light irradiation. The loss in volume for the control hydrogels is attributed to mild dehydration that results from the arid glovebox environment and potentially due to some localized heating from the LED light bulbs. Alternatively, when the viologen dimer-based hydrogels were exposed to heat in a H2O bath at 50 °C, they contracted down to 22% of their original volumes within the first 2 h, and then down to 18% after a total of 5 h at 50 °C.
Photoreduction of the 6V12+-St crosslinked hydrogels (Fig. 3c) by red light resulted in contraction down to 46% of the starting volume after 2 h, and to 33% after 5 h. When irradiated with blue light, the hydrogels contracted down to 24% in 2 h and to 13% after 5 h. Additionally, two sets of control hydrogels (with no photocatalyst) were irradiated by red or blue light for 5 h. After the first 2 h, each set contracted to 75% on average relative to their original sizes. Only an additional 5% contraction was observed for the remaining 3 h of irradiation with either wavelength of light, yielding volumes that were 70% of the original starting sizes. The results from the control experiments again indicate that the primary mode of activation is dehydration when no photocatalyst is present and that contraction is greatly accelerated when the photoredox mechanism is the primary mode of activation. Lastly, the hydrogels were exposed to heat in a H2O bath pre-heated to 50 °C, which induced contraction of the hydrogels down to 19% within the first 2 h, and to 17% after 5 h total.
It is important to note that the contraction kinetics between the 2V4+-St and 6V12+-St crosslinked hydrogels were almost alike, regardless of the fact that they were incorporated at equimolar concentrations and the latter possessed four more viologen subunits than the former. We attribute the similar performance to differences in hydrophilicity, swelling ratio, and the crosslinking density (vide supra), which offset any additional radical–radical spin-pairing interactions and favourable loss of electrostatic charge and corresponding counteranions that may be attained for the hexaviologen-based hydrogels.
Next, bimodal activation of the 6V12+-St crosslinked hydrogels using blue light (450 nm) and heat (50 °C) was investigated. The setup for this experiment (Fig. S21†) involved placing one hydrogel in N2-purged H2O inside a glass bottle sealed with a rubber septum. The experiment was started when the glass bottle was placed in a H2O bath pre-heated to 50 °C, while simultaneously being irradiated with blue light emitted from two desk lamps. Within 1 h, the hydrogels contracted on average down to 17% of their original starting volume. After an additional 4 h of irradiation/heating, the hydrogels underwent 5% more contraction, stopping at a relative volume of 12% of the original starting size. Thus, there is a distinct advantage to activating orthogonal mechanisms of actuation – i.e., photoredox and thermoresponsive – inside the hydrogels. The contraction rate more than doubled by reaching 94% of the maximum observed contraction of the multimodal hydrogel within 1 h.
Contraction of the hydrogels described in this investigation are completely reversible. For example, the in situ photoreduction of the viologen subunits of the crosslinker results in contracted hydrogels that appear black in colour. However, once they are placed in a Petri dish containing O2-saturated H2O, the embedded viologens oxidize in 20–60 min and the hydrogels re-swell in a few hours. The same is true for the thermally activated hydrogels once they are removed from the hot H2O bath and allowed to re-swell. To demonstrate the reversibility of the bimodal actuation process, three 6V12+-St crosslinked hydrogels were cycled three times each by shining blue light on them and simultaneously applying heat (50 °C) to induce contraction, followed by turning off the light and removing the hydrogels from the hot plate to induce expansion (re-swelling). The results from these cycling experiments (Fig. 4) demonstrate that it is possible to enact multiple contraction–expansion cycles (i.e., actuation) on the hydrogels using orthogonal modes of activation. Also, the slight changes in volume between each cycle may possibly be explained by counterion exchange that occurs after each contraction step, whereby each viologen subunit loses its unpaired radical electron to molecular O2, thus generating superoxide anion (O2−) and presumably its hydroxide byproduct (OH−) in the process, and therefore potentially changing the volume slightly each cycle.
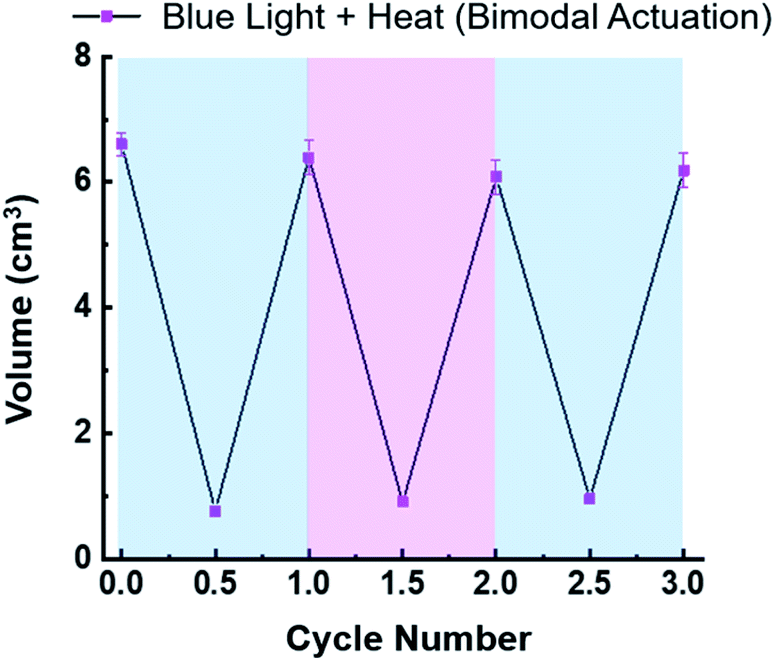 | ||
| Fig. 4 Plot of hydrogel volume change over three actuation cycles, demonstrating the reversibility and cyclability of 6V12+-St crosslinked hydrogel actuators using orthogonal modes of activation (blue light and heat (50 °C)) simultaneously. | ||
Mechanical properties of the hydrogel actuators
The change in stiffness of the hydrogels before and after 5 h of photo-, thermal-, and bimodal-induced contraction was measured using oscillatory shear rheology (Fig. S19 and S20†). The 2V4+-St crosslinked hydrogels contracted by red light, heat, or blue light modes of activation showed (Fig. S19a, b, S20a and b†) storage moduli (G′) of 2.3, 3.7, and 5.2 kPa, respectively. After oxidation and re-swelling in H2O, the G′ value decreased by 1.6–4.3 kPa due to the softening of the respective hydrogels. For comparison, the red and blue light-activated control hydrogels (with no photocatalyst present) underwent only a slight change in storage moduli (∼0.5 kPa): red light-activated hydrogel's storage moduli changed from 1.1 to 0.5 kPa upon oxidation and re-swelling, and the blue light-activated hydrogel's storage moduli changed from 1.2 to 0.7 kPa.Enabling the different modes of activation to contract the 6V12+-St crosslinked hydrogels via red light, heat, blue light, and bimodally (blue light and heat) was also performed to assess any potential changes in the hexaviologen-containing hydrogel's stiffness. Accordingly, oscillatory shear rheology experiments were carried out to measure the corresponding G′ values of each hydrogel (Fig. 5, S19c, d, S20c, d† and Table 1), which were found to be 2.0, 3.5, 4.6, and 5.0 kPa, respectively, for each method of activation. After oxidation and re-swelling of the hydrogels, the G′ values decreased by 1.5–4.3 kPa; a similar outcome as that for the viologen dimer-based hydrogels. Also, the same experimental conditions were applied to the control hydrogels (with no photocatalyst), which resulted in hydrogels exhibiting a G′ value of 1.1 kPa after 5 h of irradiation with red light and 0.8 kPa when oxidized and re-swollen in H2O. Likewise, the blue light-irradiated control hydrogels exhibited a G′ value of 0.8 kPa, and upon oxidation and re-swelling, a lower G′ value of 0.6 kPa was measured. The differences of only 0.3 and 0.2 kPa, respectively, indicate that the larger macroscale changes in the 6V12+-St crosslinked hydrogels are only observed when either the photoredox, thermal, or bimodal modes of activation are employed. In other words, a small amount of dehydration does not yield large changes in hydrogel stiffness.
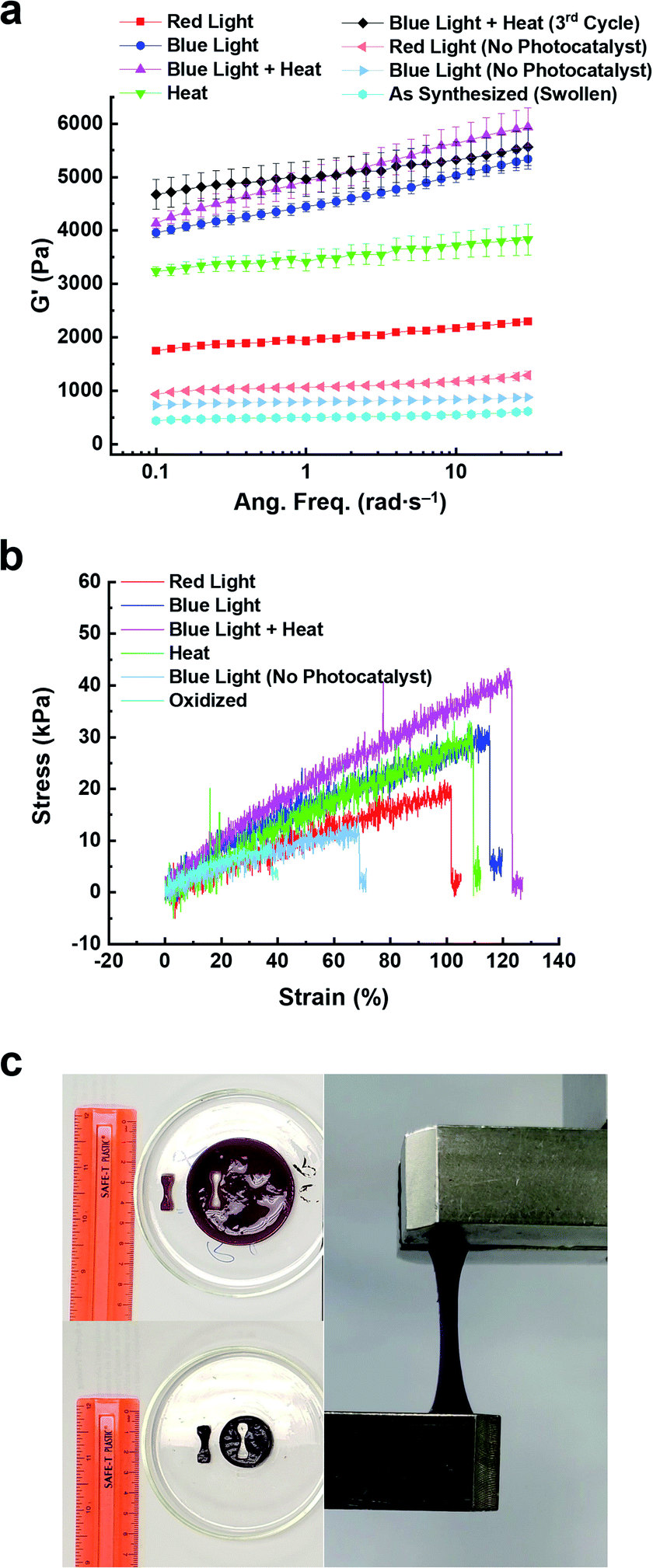 | ||
| Fig. 5 Rheological and tensile data plots for 6V12+-St crosslinked hydrogels that were contracted using different modes of activation. (a) Storage modulus (G′) vs. angular frequency (rad s−1) for hydrogels activated by red or blue light (red square, blue sphere, resp.), heat (50 °C, green triangle), or bimodally using blue light and heat (pink triangle). Data is also shown for control (i.e., no photocatalyst, light blue and red horizontal triangles) and pre-activation hydrogels (light blue sphere). (b) Stress (kPa) versus strain (%) data plots of hydrogels activated with red or blue light (red and blue data traces, resp.), heat (green trace), and bimodally (pink trace). Also shown is data for the control (no photocatalyst, blue-grey trace) and oxidized hydrogels (light blue) (c) Pictures of an as-synthesized hydrogel before (upper left) and after (bottom left) photoreduction, as well as during tensile testing. | ||
| Hydrogel state | E (kPa) | TSb (kPa) | ε B (%) | G′d (Pa) | G′′e (Pa) |
|---|---|---|---|---|---|
| a Young's modulus (E), determined from the slope of the linear elastic region of each stress vs. strain curve. b Tensile Strength (TS), determined from the maximum stress value just before break. c Elongation at break (εB), determined from the maximum percent strain when the sample fractured. d Storage modulus (G′), determined at 10 rad s−1. e Loss modulus (G′′), determined at 10 rad s−1. | |||||
| Oxidized | 14.4 ± 0.4 | 5.5 ± 0.4 | 37.3 ± 1.5 | 494 | 36 |
| Blue light (control) | 17.7 ± 1.2 | 13.2 ± 1.2 | 69.7 ± 3.0 | 787 | 36 |
| Red light | 17.9 ± 0.2 | 18.8 ± 0.6 | 103.8 ± 1.3 | 1953 | 131 |
| Heat | 23.2 ± 1.8 | 22.8 ± 2.7 | 103.2 ± 4.7 | 3465 | 237 |
| Blue light | 23.9 ± 2.2 | 28.3 ± 0.7 | 110.7 ± 2.5 | 4397 | 297 |
| Blue + heat | 33.0 ± 1.9 | 40.3 ± 3.1 | 116.5 ± 4.7 | 4841 | 399 |
To further assess the performance of the 6V12+-St crosslinked hydrogels, the storage moduli was measured after three contraction–expansion cycles were performed using bimodal activation (see Fig. S21† for the experimental setup and Fig. 4, which shows the changes in volume for each cycle). After the third contraction, the hydrogels shrunk down to 14% of their starting volume, and the change in stiffness was quantified by oscillatory shear rheology (Fig. 5a, S19c, d, S20c and d†). The contracted hydrogels exhibited a storage modulus of 5.1 kPa (Fig. 5a, black data trace), and after oxidation and re-swelling in H2O, the G′ value decreased to 0.6 kPa. This experiment proves that the mechanical properties of the hydrogel actuators are also reversible and can be restored to nearly the same level of stiffness that was observed prior to the first contraction–expansion cycle. See ESI† for full rheology data plots (Fig. S19 and S20†), including the data corresponding to the loss moduli (G′′) associated with each set of hydrogels.
Next, the Young's modulus (E), tensile strength (TS), and percent elongation at break (εB) were measured (Fig. 5b, c and Table 1) for the 6V12+-St crosslinked hydrogels before and after 5 h of activation by red or blue light, heat, or the concerted application of blue light and heat (bimodal). First, dog-bone-shaped hydrogels were punched out from larger disc-shaped ones and loaded onto a mechanical testing system (Fig. 5c). Each dog-bone hydrogel was extended at a rate of 5.0 mm min−1 until the sample fractured. The stress (kPa) versus strain (%) curves were recorded (Fig. 5b) during this process. The oxidized, as-synthesized hydrogels (i.e., no applied modes of activation) afforded the lowest values for E, TS, and εB, as expected (see the first row in Table 1). The control hydrogels that were irradiated with blue light, but possessed no photocatalyst, yielded a Young's modulus that was 22.9% higher than the starting hydrogel, whereas the tensile strength increased by 140% and the percent elongation before breaking increased by 86.9% (Table 1, second row). Again, these increases in mechanical parameters for the control hydrogels irradiated with blue light are attributed to some dehydration that occurs as the experiment is carried out in a N2-filled glovebox. By comparison, the hydrogels irradiated with red light for 5 h (Table 1, third row) resulted in a 24.3% increase in the Young's modulus and an increase of 241.8% and 178.3% in the tensile strength and percent elongation at break, respectively. Exposing the hydrogels to heat by placing them in beakers that were lowered into a H2O bath at 50 °C further increased E, TS, and εB by 61.1, 314.5, and 176.7%, respectively (Table 1, fourth row), relative to the oxidized, as-synthesized hydrogels. This trend continued upon assessing the mechanical properties of hydrogels that were irradiated with blue light for 5 h. Specifically, E, TS, and εB all increased by 66.0, 414.5, and 196.8%, respectively, over the same values measured for the starting hydrogels. Lastly, the full, multimodal actuator performance was demonstrated when a set of hydrogels were irradiated with blue light while simultaneously being heated in a H2O bath at 50 °C for 5 h. These hydrogels exhibited increases of 129.2, 632.7, and 212% for E, TS, and εB, respectively. Taken collectively, these tensile experiments reveal how the physical properties of the reported porphyrin–NIPAM-based hydrogel actuators can be enhanced dramatically when multiple modes of activation are employed simultaneously. It is also important to note that the signal noise for these tensile experiments was higher than expected. We attribute this result to the increased NIPAM component and the viologen-based crosslinker, which has a high affinity for H2O.
Conclusions
The design, synthesis, properties, and performance of dynamic, multimodal hydrogel actuators is reported. The hydrogels were synthesized through thermal polymerization of HEA and NIPAM monomers in the presence of custom viologen-based crosslinkers bearing two or six viologen subunits. A zinc-based porphyrin photocatalyst was also polymerized into the hydrogel polymer network, imbuing the soft material with the ability to undergo multi-wavelength, visible-light photoredox catalysis when irradiated by blue or red light. Excitation of the embedded porphyrin resulted in photoinduced electron transfer from the excited-state porphyrin to the oligoviologen-based crosslinkers; a photoreduction process which induced chain folding of the crosslinker and hydrogel contraction down to as low as 15% of the starting volume after 5 h of irradiation with blue light (450 nm). Upon removal of the light source and exposure to O2 and H2O, the hydrogels re-swelled to their original volumes, demonstrating complete reversibility of the photo-based actuation process. Thermal activation of the hydrogel actuators was also demonstrated by heating them for 5 h at 50 °C in H2O, leading to a maximum contraction down to 17% of its original volume. The simultaneous application of blue light and heat to the hydrogels (thus, bimodal activation) doubled the rate of contraction and allowed them to shrink down to 17% of their original volume within the first hour, and down to 12% after a total of 5 h of exposure. Assessing the change in mechanical properties (i.e., stiffness, Young's modulus, tensile strength, etc.) for each reported hydrogel was also performed before and after exposure to one or more of the external stimuli. It was determined that bimodal activation yielded the greatest enhancement of physical properties, where Young's modulus, tensile strength, and percent elongation at break increased by 129.2, 632.7, and 212% relative to that of the starting hydrogel. These results suggest that the physical properties of the dynamic, multimodal hydrogel actuators can be finely tuned over select time periods with either visible light, heat, or the combination therein. We envision that this dynamic hydrogel platform may find application in biomedical research endeavours, such as in 3D/4D cell culture, tissue engineering, and regenerative medicine.Author contributions
J. C. B. conceived the idea for the project, and J. C. B. and F. A. designed the experiments. F. A., X. L., M. G., N. D. C., L. L., and R. L. carried out the synthesis of gel precursors, and F. A. and M. G. synthesized all gels studied in this investigation. F. A. and M. G. performed the gel activation and contraction experiments, as well as the rheological measurements. F. A. and H. R. L. did tensile measurements of the gels at Saint Louis University, supervised by J. C. B. and S. A. S. Finally, J. C. B., F. A., and X. L. co-wrote the manuscript and ESI† documents, and all authors contributed to the refinement of each document.Conflicts of interest
J. C. B. has filed a non-provisional patent application (U.S. 15/939,515).Acknowledgements
We thank the David & Lucile Packard Foundation which supported this research in the form of J. C. B.‘s Packard Fellowship for Science and Engineering. M. G. also acknowledges support from Washington University's Office of Undergraduate Research for two 10 week Summer Undergraduate Research Awards.References
- C. Echeverria, S. N. Fernandes, M. H. Godinho, J. P. Borges and P. I. P. Soares, Gels, 2018, 4, 54 CrossRef.
- S.-J. Jeon, A. W. Hauser and R. C. Hayward, Acc. Chem. Res., 2017, 50, 161–169 CrossRef.
- M. Ding, L. Jing, H. Yang, C. E. Machnicki, X. Fu, K. Li, I. Y. Wong and P. Y. Chen, Materials Today Advances, 2020, 8, 100088 CrossRef.
- M. C. Koetting, J. T. Peters, S. D. Steichen and N. A. Peppas, Mater. Sci. Eng., R, 2015, 93, 1–49 CrossRef.
- L. Ionov, Mater. Today, 2014, 17, 494–503 CrossRef.
- X. Le, W. Lu, J. Zhang and T. Chen, Adv. Sci., 2019, 6, 1801584 CrossRef.
- H. Banerjee, M. Suhail and H. Ren, Biomimetics, 2018, 3, 15 CrossRef.
- M. Wehner, R. L. Truby, D. J. Fitzgerald, B. Mosadegh, G. M. Whitesides, J. A. Lewis and R. J. Wood, Nature, 2016, 536, 451–455 CrossRef CAS.
- A. M. Rosales and K. S. Anseth, Nat. Rev. Mater., 2016, 1, 15012 CrossRef CAS.
- N. Sood, A. Bhardwaj, S. Mehta and A. Mehta, Drug Delivery, 2016, 23, 748–770 CrossRef CAS.
- S. Mantha, S. Pillai, P. Khayambashi, A. Upadhyay, Y. Zhang, O. Tao, H. M. Pham and S. D. Tran, Materials, 2019, 12, 3323 CrossRef CAS.
- B. A. Badeau, M. P. Comerford, C. K. Arakawa, J. A. Shadish and C. A. DeForest, Nat. Chem., 2018, 10, 251–258 CrossRef CAS.
- E. R. Ruskowitz and C. A. DeForest, Nat. Rev. Mater., 2018, 3, 17087 CrossRef CAS.
- C. A. Dreiss, Curr. Opin. Colloid Interface Sci., 2020, 48, 1–17 CrossRef CAS.
- S. K. De, N. R. Aluru, B. Johnson, W. C. Crone, D. J. Beebe and J. Moore, J. Microelectromech. Syst., 2002, 11, 544–555 CrossRef CAS.
- M. Krogsgaard, M. A. Behrens, J. S. Pedersen and H. Birkedal, Biomacromolecules, 2013, 14, 297–301 CrossRef CAS.
- S. H. Park, H. S. Shin and S. N. Park, Carbohydr. Polym., 2018, 200, 341–352 CrossRef CAS.
- L. E. Bromberg and E. S. Ron, Adv. Drug Delivery Rev., 1998, 31, 197–221 CrossRef CAS.
- D. Kuckling, J. Hoffmann, M. Plötner, D. Ferse, K. Kretschmer, H.-J. P. Adler, K.-F. Arndt and R. Reichelt, Polymer, 2003, 44, 4455–4462 CrossRef CAS.
- F.-J. Xu, E.-T. Kang and K.-G. Neoh, Biomaterials, 2006, 27, 2787–2797 CrossRef CAS.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef.
- W. Cheng and Y. Liu, in Biopolymer-Based Composites, ed. S. Jana, S. Maiti and S. Jana, Woodhead Publishing, Cambridge, England, 2017, ch. 2, pp. 31–60 Search PubMed.
- Y. Che, S. Zschoche, F. Obst, D. Appelhans and B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2590–2601 CrossRef CAS.
- F. Li, X. Zhang, S. Hu, Z. Lv, J. Lv, W. Yu, X. Xu and D. Yang, Chem. Mater., 2020, 32, 2156–2165 CrossRef CAS.
- Y. Li, G. Huang, X. Zhang, B. Li, Y. Chen, T. Lu, T. J. Lu and F. Xu, Adv. Funct. Mater., 2013, 23, 660–672 CrossRef CAS.
- L. Li, J. M. Scheiger and P. A. Levkin, Adv. Mater., 2019, 31, 1807333 CrossRef.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS.
- R. Yang, S. Peng, W. Wan and T. C. Hughes, J. Mater. Chem. C, 2014, 2, 9122–9131 RSC.
- Q. Li, G. Fuks, E. Moulin, M. Maaloum, M. Rawiso, I. Kulic, J. T. Foy and N. Giuseppone, Nat. Nanotechnol., 2015, 10, 161–165 CrossRef CAS.
- J. T. Foy, Q. Li, A. Goujon, J.-R. Colard-Itté, G. Fuks, E. Moulin, O. Schiffmann, D. Dattler, D. P. Funeriu and N. Giuseppone, Nat. Nanotechnol., 2017, 12, 540–545 CrossRef CAS.
- M. Hendrikx, J. ter Schiphorst, E. P. A. van Heeswijk, G. Koçer, C. Knie, D. Bléger, S. Hecht, P. Jonkheijm, D. J. Broer and A. P. H. J. Schenning, Small, 2018, 14, 1803274 CrossRef.
- S. Li, G. Han and W. Zhang, Macromolecules, 2018, 51, 4290–4297 CrossRef CAS.
- G. Stoychev, A. Kirillova and L. Ionov, Adv. Opt. Mater., 2019, 7, 1900067 CrossRef.
- C. Li, A. Iscen, L. C. Palmer, G. C. Schatz and S. I. Stupp, J. Am. Chem. Soc., 2020, 142, 8447–8453 CrossRef CAS.
- G. Wu, A. Mikhailovsky, H. A. Khant, C. Fu, W. Chiu and J. A. Zasadzinski, J. Am. Chem. Soc., 2008, 130, 8175–8177 CrossRef CAS.
- M. S. Yavuz, Y. Cheng, J. Chen, C. M. Cobley, Q. Zhang, M. Rycenga, J. Xie, C. Kim, K. H. Song, A. G. Schwartz, L. V. Wang and Y. Xia, Nat. Mater., 2009, 8, 935–939 CrossRef CAS.
- S.-W. Lv, Y. Liu, M. Xie, J. Wang, X.-W. Yan, Z. Li, W.-G. Dong and W.-H. Huang, ACS Nano, 2016, 10, 6201–6210 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- Y. Guan and Y. Zhang, Soft Matter, 2011, 7, 6375–6384 RSC.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A, 1968, 2, 1441–1455 CrossRef CAS.
- E. Wang, M. S. Desai and S.-W. Lee, Nano Lett., 2013, 13, 2826–2830 CrossRef CAS.
- W. Li, J. Wang, J. Ren and X. Qu, Adv. Mater., 2013, 25, 6737–6743 CrossRef CAS.
- X. B. Zhang, C. L. Pint, M. H. Lee, B. E. Schubert, A. Jamshidi, K. Takei, H. Ko, A. Gillies, R. Bardhan, J. J. Urban, M. Wu, R. Fearing and A. Javey, Nano Lett., 2011, 11, 3239–3244 CrossRef CAS.
- L. Chen, M. Weng, W. Zhang, Z. Zhou, Y. Zhou, D. Xia, J. Li, Z. Huang, C. Liu and S. Fan, Nanoscale, 2016, 8, 6877–6883 RSC.
- T. Wang, D. Torres, F. E. Fernandez, C. Wang and N. Sepulveda, Sci. Adv., 2017, 3, 9 Search PubMed.
- K. Sumaru, K. Ohi, T. Takagi, T. Kanamori and T. Shinbo, Langmuir, 2006, 22, 4353–4356 CrossRef CAS.
- J. Wang, Q. Li, S. Yi and X. Chen, Soft Matter, 2017, 13, 6490–6498 RSC.
- Y. Guan, H.-B. Zhao, L.-X. Yu, S.-C. Chen and Y.-Z. Wang, RSC Adv., 2014, 4, 4955–4959 RSC.
- S. L. Jacques, Phys. Med. Biol., 2013, 58, R37–R61 CrossRef.
- H. Zhang, D. Salo, D. M. Kim, S. Komarov, Y.-C. Tai and M. Y. Berezin, J. Biomed. Opt., 2016, 21, 126006 CrossRef.
- Z. Jiang, M. L. Tan, M. Taheri, Q. Yan, T. Tsuzuki, M. G. Gardiner, B. Diggle and L. A. Connal, Angew. Chem., Int. Ed., 2020, 59, 7049–7056 CrossRef CAS.
- Z. Jiang, M. Xu, F. Y. Li and Y. L. Yu, J. Am. Chem. Soc., 2013, 135, 16446–16453 CrossRef CAS.
- D. Wang, M. Wagner, H.-J. Butt and S. Wu, Soft Matter, 2015, 11, 7656–7662 RSC.
- A. Hossain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, eaav9713 CrossRef.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS.
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS.
- X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS.
- J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082 CrossRef CAS.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- H. Abrahamse and M. R. Hamblin, Biochem. J., 2016, 473, 347–364 CrossRef CAS.
- J. Kou, D. Dou and L. Yang, Oncotarget, 2017, 8, 81591–81603 CrossRef.
- T. Higashino and H. Imahori, Dalton Trans., 2015, 44, 448–463 RSC.
- F. Mendizabal, R. Mera-Adasme, W.-H. Xu and D. Sundholm, RSC Adv., 2017, 7, 42677–42684 RSC.
- M. K. Nazeeruddin, R. Humphry-Baker, D. L. Officer, W. M. Campbell, A. K. Burrell and M. Grätzel, Langmuir, 2004, 20, 6514–6517 CrossRef CAS.
- R. Costa e Silva, L. Oliveira da Silva, A. de Andrade Bartolomeu, T. J. Brocksom and K. T. de Oliveira, Beilstein J. Org. Chem., 2020, 16, 917–955 CrossRef.
- A. A. N. de Souza, N. S. Silva, A. V. Müller, A. S. Polo, T. J. Brocksom and K. T. de Oliveira, J. Org. Chem., 2018, 83, 15077–15086 CrossRef CAS.
- K. Rybicka-Jasińska, W. Shan, K. Zawada, K. M. Kadish and D. Gryko, J. Am. Chem. Soc., 2016, 138, 15451–15458 CrossRef.
- F. Amir, K. P. Liles, A. O. Delawder, N. D. Colley, M. S. Palmquist, H. R. Linder, S. A. Sell and J. C. Barnes, ACS Appl. Mater. Interfaces, 2019, 11, 24627–24638 CrossRef CAS.
- A. O. Delawder, A. Natraj, N. D. Colley, T. Saak, A. F. Greene and J. C. Barnes, Supramol. Chem., 2019, 31, 523–531 CrossRef CAS.
- K. P. Liles, A. F. Greene, M. K. Danielson, N. D. Colley, A. Wellen, J. M. Fisher and J. C. Barnes, Macromol. Rapid Commun., 2018, 39, 1700781 CrossRef.
- L. Michaelis and E. S. Hill, J. Gen. Physiol., 1933, 16, 859–873 CrossRef CAS.
- L. Michaelis, Chem. Rev., 1935, 16, 243–286 CrossRef CAS.
- C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49–82 RSC.
- T. M. Bockman and J. K. Kochi, J. Org. Chem., 1990, 55, 4127–4135 CrossRef CAS.
- A. Trabolsi, N. Khashab, A. C. Fahrenbach, D. C. Friedman, M. T. Colvin, K. K. Cotí, D. Benítez, E. Tkatchouk, J.-C. Olsen, M. E. Belowich, R. Carmielli, H. A. Khatib, W. A. Goddard iii, M. R. Wasielewski and J. F. Stoddart, Nat. Chem., 2009, 2, 42 CrossRef.
- A. C. Fahrenbach, J. C. Barnes, D. A. Lanfranchi, H. Li, A. Coskun, J. J. Gassensmith, Z. Liu, D. Benítez, A. Trabolsi, W. A. Goddard, M. Elhabiri and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 3061–3072 CrossRef CAS.
- A. F. Greene, M. K. Danielson, A. O. Delawder, K. P. Liles, X. Li, A. Natraj, A. Wellen and J. C. Barnes, Chem. Mater., 2017, 29, 9498–9508 CrossRef CAS.
- B. Fu, H.-C. Yu, J.-W. Huang, P. Zhao, J. Liu and L.-N. Ji, J. Mol. Catal. A: Chem., 2009, 298, 74–80 CrossRef CAS.
- Y. Chu, N. Corrigan, C. Wu, C. Boyer and J. Xu, ACS Sustainable Chem. Eng., 2018, 6, 15245–15253 CrossRef CAS.
- M. R. Geraskina, A. S. Dutton, M. J. Juetten, S. A. Wood and A. H. Winter, Angew. Chem., Int. Ed., 2017, 56, 9435–9439 CrossRef CAS.
- S. J. Bryant, T. T. Chowdhury, D. A. Lee, D. L. Bader and K. S. Anseth, Ann. Biomed. Eng., 2004, 32, 407–417 CrossRef.
- C. A. DeForest and K. S. Anseth, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 421–444 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic protocols, purification and characterization data for all compounds and polymers, and mechanical testing data. See DOI: 10.1039/d0sc04287k |
| This journal is © The Royal Society of Chemistry 2020 |