
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective α,δ-hydrocarboxylation of conjugated dienes utilizing CO2 and electrosynthesis†
Ahmed M.
Sheta
 ab,
Mohammad A.
Mashaly
b,
Samy B.
Said
b,
Saad S.
Elmorsy
c,
Andrei V.
Malkov
a and
Benjamin R.
Buckley
*a
ab,
Mohammad A.
Mashaly
b,
Samy B.
Said
b,
Saad S.
Elmorsy
c,
Andrei V.
Malkov
a and
Benjamin R.
Buckley
*a
aDepartment of Chemistry Loughborough University, Ashby Road, Loughborough, Leicestershire LE11 3TU, UK. E-mail: b.r.buckley@lboro.ac.uk
bDepartment of Chemistry, Damietta University, Damietta El-Gadeeda City, Kafr Saad, Damietta Governorate 34511, Egypt
cDepartment of Chemistry, Mansoura University, 25 El Gomhouria St, Dakahlia Governorate 35516, Egypt
First published on 20th July 2020
Abstract
To date the majority of diene carboxylation processes afford the α,δ-dicarboxylated product, the selective mono-carboxylation of dienes is a significant challenge and the major product reported under transition metal catalysis arises from carboxylation at the α-carbon. Herein we report a new electrosynthetic approach, that does not rely on a sacrificial electrode, the reported method allows unprecedented direct access to carboxylic acids derived from dienes at the δ-position. In addition, the α,δ-dicarboxylic acid or the α,δ-reduced alkene can be easily accessed by simple modification of the reaction conditions.
Introduction
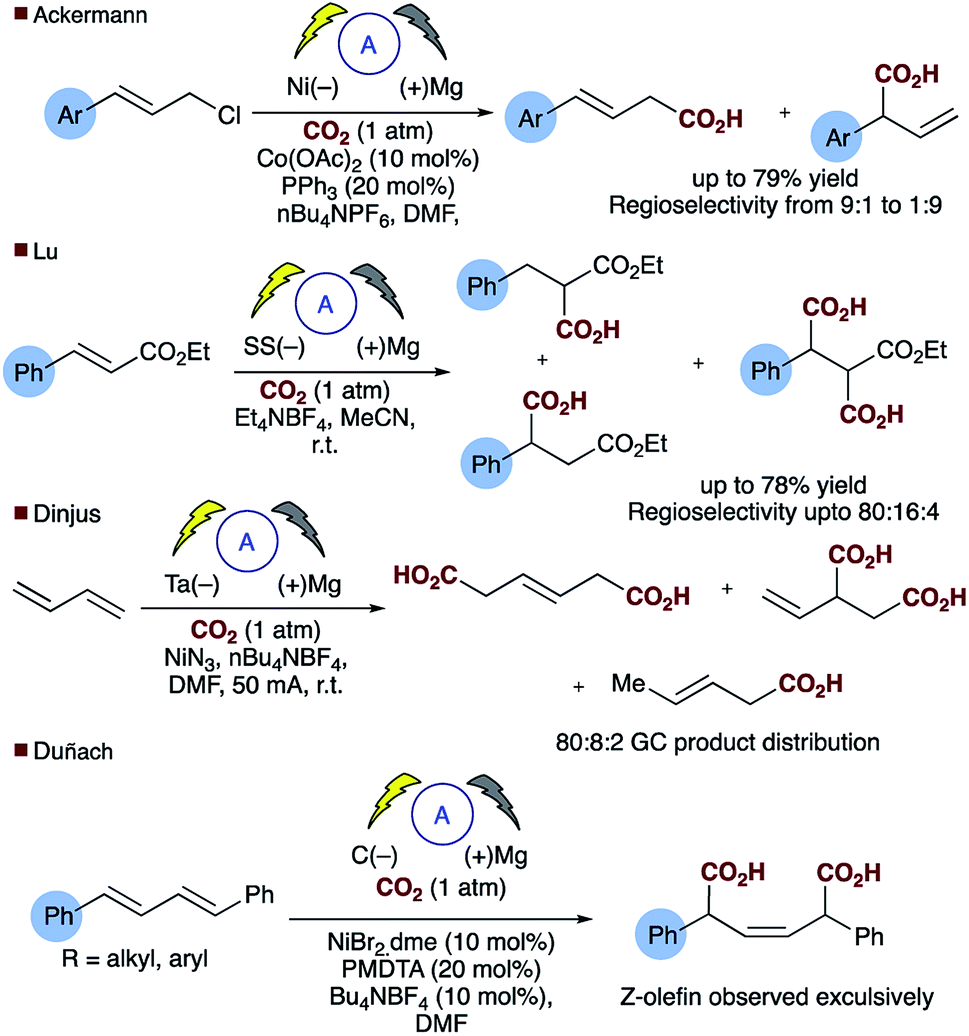
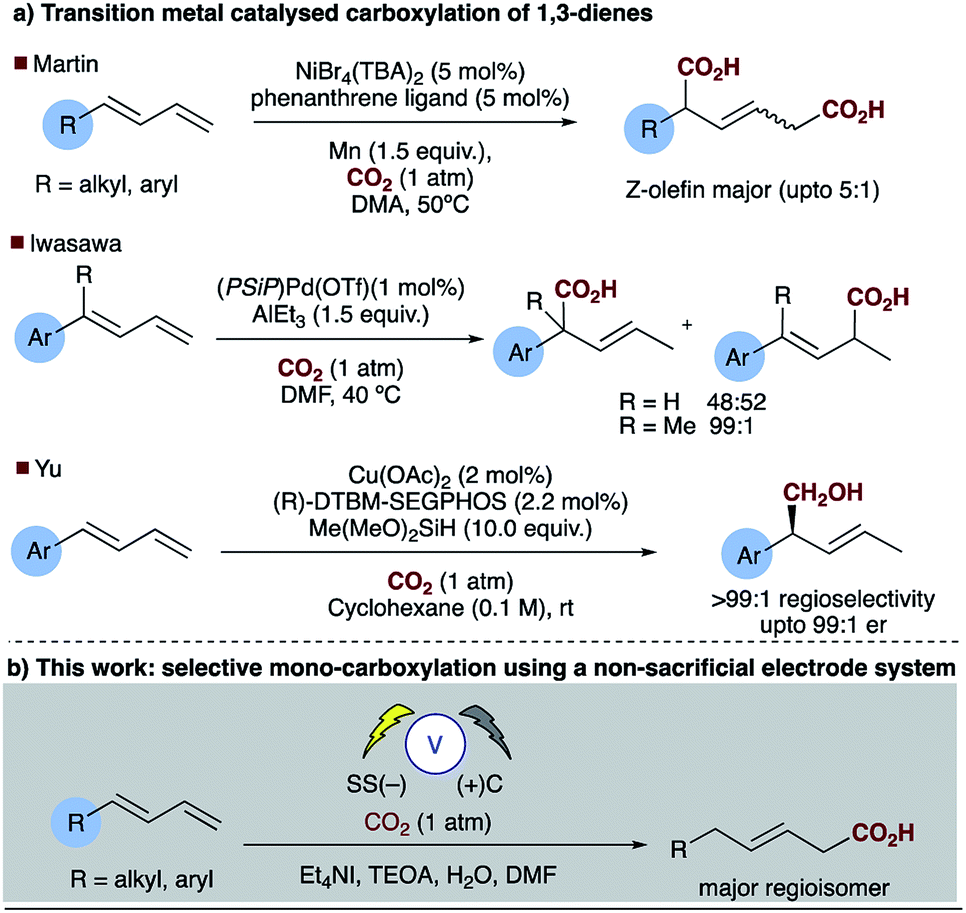
Direct carboxylation of low value olefin feedstocks utilising carbon dioxide is regarded as a “dream reaction” owing to the desire to utilise this low value, high volume waste gas.1–3 However, selectivity in these, and related carboxylation reactions remains a significant challenge (see for example Scheme 1). Electrochemical approaches to carboxylation have been known for some time and recently Ackermann and others have examined the electro–reductive transition metal carboxylation of allyl chlorides with a sacrificial electrode system and up to 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regioselectivity being observed.4,5 Lu has described a sacrificial magnesium anode system in which the carboxylation of α,β-unsaturated esters leads to a mixture of mono- and di-carboxylic acids.6 Electrochemical approaches to α,δ-dicarboxylic acids from dienes have been known for some time, early reports in the patent literature by Loveland describe the electrolytic production of acyclic carboxylic acids from olefins.7 However, several drawbacks to the system, such as the use of a mercury electrode, prompted further research in this area.8 Dinjus reported the transition metal assisted electrocarboxylation of 1,3-butadiene to afford predominantly the straight chain dicarboxylic acid, again utilising a sacrificial magnesium anode.9 Duñach and Perichon later described a sacrificial magnesium/nickel catalysed system with exclusive dicarboxylic acid formation and good Z-olefin selectivity.10 Aside from the electrochemical literature Mori originally reported the stoichiometric nickel dicarboxylation of 1,3-dienes and more recently Martin has described a catalytic approach to the α,δ-dicarboxylation of diene feedstocks.11,12 However, hydrocarboxylation of 1,3-dienes have scarcely been reported, perhaps due to the inherent selectivity issues associated with the site of carboxylation. Some selectivity has been observed for mono-carboxylation at the α-carbon by Iwasawa but this tends to depend on the substrate employed (Scheme 2a).13 Yu has reported a particularly impressive catalytic asymmetric approach to α-hydroxymethylation of 1,3-dienes utilising CO2.14 Only one report of selective δ-carboxylation, reported by Walther and Schonecker in the early 1990's, utilizing stoichiometric nickel complexes on a steroidal substrate is known.15
:1 regioselectivity being observed.4,5 Lu has described a sacrificial magnesium anode system in which the carboxylation of α,β-unsaturated esters leads to a mixture of mono- and di-carboxylic acids.6 Electrochemical approaches to α,δ-dicarboxylic acids from dienes have been known for some time, early reports in the patent literature by Loveland describe the electrolytic production of acyclic carboxylic acids from olefins.7 However, several drawbacks to the system, such as the use of a mercury electrode, prompted further research in this area.8 Dinjus reported the transition metal assisted electrocarboxylation of 1,3-butadiene to afford predominantly the straight chain dicarboxylic acid, again utilising a sacrificial magnesium anode.9 Duñach and Perichon later described a sacrificial magnesium/nickel catalysed system with exclusive dicarboxylic acid formation and good Z-olefin selectivity.10 Aside from the electrochemical literature Mori originally reported the stoichiometric nickel dicarboxylation of 1,3-dienes and more recently Martin has described a catalytic approach to the α,δ-dicarboxylation of diene feedstocks.11,12 However, hydrocarboxylation of 1,3-dienes have scarcely been reported, perhaps due to the inherent selectivity issues associated with the site of carboxylation. Some selectivity has been observed for mono-carboxylation at the α-carbon by Iwasawa but this tends to depend on the substrate employed (Scheme 2a).13 Yu has reported a particularly impressive catalytic asymmetric approach to α-hydroxymethylation of 1,3-dienes utilising CO2.14 Only one report of selective δ-carboxylation, reported by Walther and Schonecker in the early 1990's, utilizing stoichiometric nickel complexes on a steroidal substrate is known.15
 | ||
| Scheme 1 Representative attempts at selective electrocarboxylation. | ||
 | ||
| Scheme 2 Attempts at selective carboxylation of 1,3-dienes, (a) transition metal catalysed and (b) this work. | ||
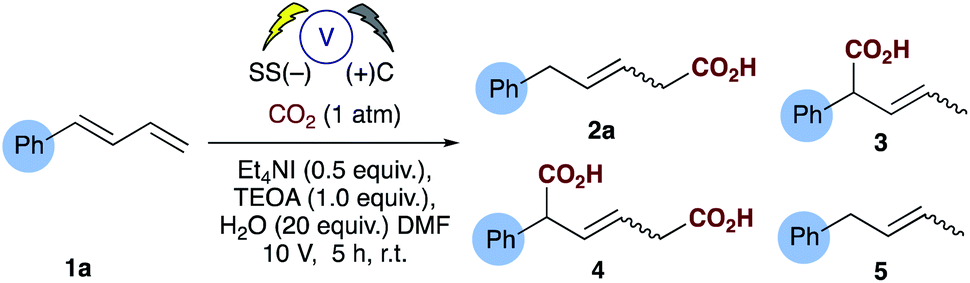
The electrosynthesis of carboxylates from dienes, like those from alkenes readily rely on the use of a sacrificial electrode to enable successful carboxylation and in some cases regiocontrol. As we and others have expressed previously from a practical and sustainability point of view, a non-sacrificial metal system would naturally be more desirable.16,17 We have recently reported the selective hydrocarboxylation of substituted aromatic alkenes utilising electrosynthesis and carbon dioxide.16 This novel carbon–carbon bond forming process provides unprecedented access to all carbon quaternary centres through the carboxylation of β,β-substituted olefins. Unlike the majority of previously reported transition metal catalysed approaches this chemistry selectively affords the β-carboxylated product. Herein, we present a new practical electrosynthetic approach to highly regioselective 1,4-hydrocarboxylation of dienes (Scheme 2b, Table 1, entries 1 and 2).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Deviation from standard conditions | Ratiob | Conv. 1ac (%) | |||
| 2a | 3 | 4 | 5 | |||
|
a General conditions: CO2 (1 atm), stainless steel cathode, carbon anode, Et4NI (0.5 equiv.), TEOA (1.0 equiv.), DMF, single compartment cell, 10 V (60–100 mA), 5 h rt.
b Ratios determined by GC-MS analysis, all products displayed approximately a 3:1 E:Z ratio.
c Conversion evaluated by GC-MS analysis, numbers in parenthesis indicate isolated yield of 2a.
|
||||||
| 1 | None | 9 | 1 | 0.5 | 0.7 | 99 (57) |
| 2 | No TEOA | 9 | 1 | 0.5 | 0.7 | 99 |
| 3 | C (−), C (+), no H2O | 1.7 | 1 | 1 | 1 | 99 |
| 4 | Ni (−), C (+), no H2O | 0 | 0 | 0 | 1 | 80 |
| 5 | Cu (−), C (+), no H2O | 3.8 | 1 | 1 | 2 | 99 |
| 6 | SS (−), C (+), no H2O | 9 | 1 | 1 | 1 | 99 |
| 7 | SS (−), SS (+), no H2O | 3.1 | 1 | 1 | 0 | 83 |
| 8 | H2O (5.0 equiv.) | 9 | 1 | 0.82 | 0.7 | 99 |
| 9 | H2O (10.0 equiv.) | 9 | 1 | 0.74 | 0.7 | 99 |
| 10 | No TEOA or H2O | 0 | 0 | 1 | 0 | 99 |
Results and discussion
Investigations commenced by utilising our previously reported conditions for alkene hydrocarboxylation, using carbon cathode and anodes (Table 1, entry 3). Disappointingly this resulted in the formation of almost a statistical mixture of the δ-monocarboxylated alkene 2a, the α-monocarboxylated alkene 3, the α,δ-dicarboxylated alkene 4 and the α,δ-reduced alkene 5. Initially we screened a range of electrode pairs; Ni/C resulted in selective formation of 5 with no sign of any carboxylation by GC/MS analysis (Table 1, entry 4).18 Utilising Cu/C couple resulted in higher selectivity towards the δ-monocarboxylated alkene 2a but significant amounts of the reduced diene 5 were observed. Switching to stainless steel (SS) and carbon provided a significant improvement in the selectivity towards 2a (Table 1, entry 6) and further optimization through the addition of water (Table 1, entries 8–10) provided our standard conditions in which the side products 3, 4 and 5 have been significantly reduced (Table 1, entry 1). Interestingly the use of stainless steel as cathode and anode eliminated the reduced product 5 but showed no improvement in selectivity towards 2a (Table 1, entry 7). In the absence of TEOA we observed little change in the selectivity of the reaction, but more unidentifiable by-products (by GCMS) were observed (unlike the corresponding reaction carried out with alkenes16), however, in the absence of a suitable proton source (e.g. both TEOA and water) the dicarboxylic acid 4 was the sole product in up to 50% unoptimised isolated yield (Table 1, entry 10).With the optimized conditions in hand, initial examination of the scope of the reaction was carried out with a variety of aryl and aliphatic dienes (1a–m, Table 2), good selectivity of the 1,4-hydrocarboxylated products were obtained and, in some cases (2f–h), the δ-substituted carboxylic acid was obtained exclusively. The thiophene substrate 1g resulted in exclusive formation of 2g. Interestingly substitution of the diene in the 4-position with a methyl group 1h also afforded exclusive formation of the 4-substituted acid 2h. The symmetrical diene 1f afforded 2f in good yield, however, for reasons which are unclear at present, its isomer 1i did not incorporate carbon dioxide and the corresponding reduced alkene 5i was the only detectable product by GC-MS analysis. Anthracene 1j proved to be a good substrate and the corresponding mono-carboxylate 2j was isolated in 65% yield. We then turned our attention to several challenging aliphatic dienes: for reasons which are unclear 1k and 1l did not incorporate CO2 under our optimised conditions.
|
a General conditions: CO2 (1 atm), stainless steel cathode, carbon anode, Et4NI (0.5 equiv.), TEOA (1.0 equiv.), DMF, single compartment cell, 10 V (60–100 mA), 5 h rt. Products were isolated as the corresponding methyl esters and 2/3 ratios determined after hydrogenation of the double bond. Products 2/3a–e and 2/3g & h displayed approximately a 3:1 E:Z ratio.
b Ratios determined by GC-MS analysis.
c Isolated as the alkene.
d The reduced diene was the major product by GC-MS analysis.
e Conditions from Table 1, entry 6 employed.
|
|---|

|
In order to form low yields of 2l we had to employ the non-aqueous conditions from Table 1 (entry 6). However, cyclohexadiene 1m did react under our optimised conditions to afford a mixture of alkenes from direct and conjugate addition in excellent yield (80%).
An examination of the aryl substituent (1a–e) reveals that selectivity of the hydrocarboxylation (2vs.3) is in some way related to the electron density of the aryl ring. Electron withdrawing groups (4-F, CF3) resulted in a slightly poorer ratio when compared to more electron rich aryl substituents (4-H, Me, OMe). Initial Hammett correlation using σ, or the modified σ+ or σ− values revealed no obvious correlation. However, when we plotted the regioselectivity of (2a–d/3a–d) over the modified Swain–Lupton parameters (σS–L)19 a correlation (r2 = 0.992) was observed (Fig. 1).20 Addition of the intermediate radical anion to CO2 is accompanied by decrease in negative charge. Electronic effects of aryl substituents should influence benzylic radical to a lesser extent compared to benzylic anion. Therefore, a modest slope of the line (ρ = −0.55) with F (field-inductive constant) dominant over R (resonance constant) appears to be in line with the preferential formation of linear isomer 2. The 4-F substituted 1,3-diene 1c produces 2/3 with lower regioselectivity vs. 4-H, OMe and CF3 due to its increased ability to inductively destabilise the formation of positive character at the benzylic position. In addition, this provides further evidence that in these particular cases reduction of the diene occurs in preference to that of CO2, thus route b shown in Scheme 3a likely dominates.
 | ||
| Fig. 1 Swain–Lupton correlation of regioselectivity of carboxylation for the 4-aryl substituted 1,3-dienes (2/3). | ||
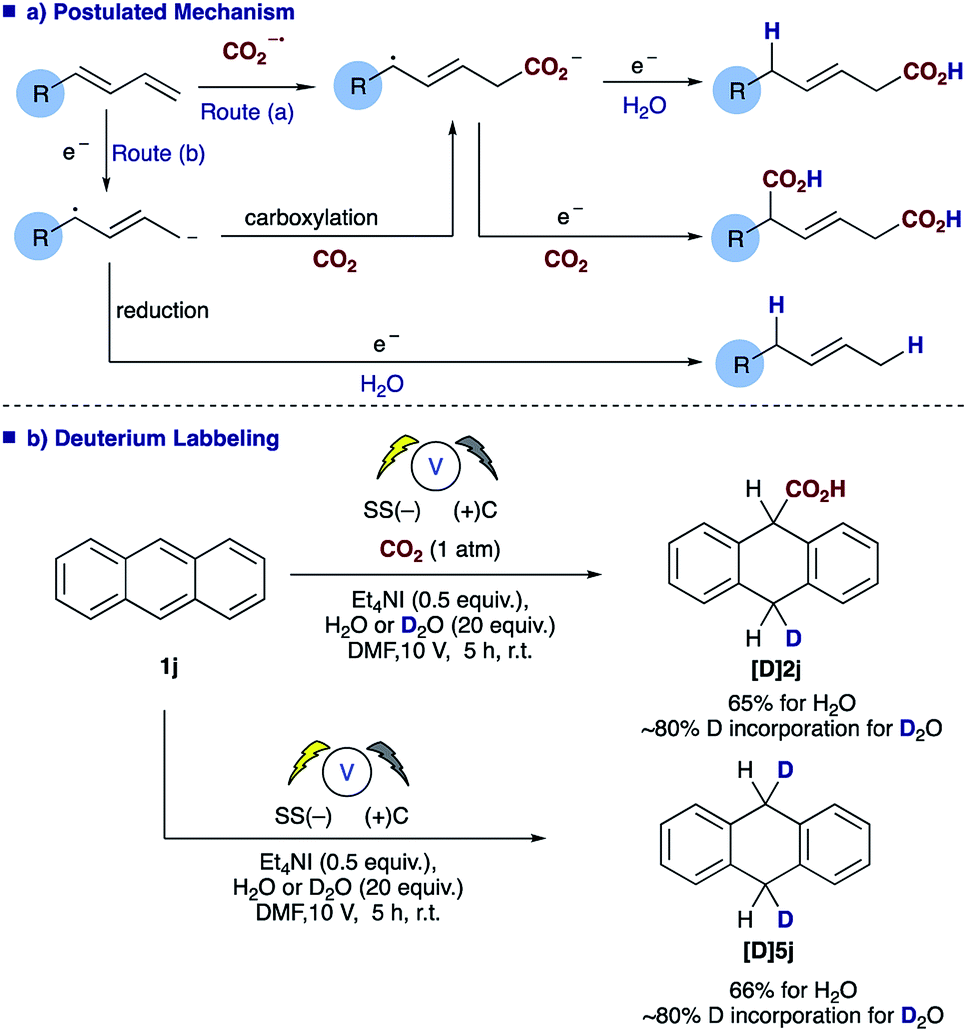 | ||
| Scheme 3 Postulated mechanism (a) and deuterium labelling studies (b). | ||
The role of water in the reaction was confirmed through deuterium labelling studies of anthracene 1j employing D2O which resulted in formation of [D]2j or in the absence of CO2[D]5j both with ∼80% D incorporation (Scheme 3b). This leads us to propose the mechanism highlighted [Scheme 3a, route (b)] in which electron transfer to the diene proceeds to form the adsorbed radical anion of the diene, subsequent carboxylation and further electron transfer and protonation from water affords the final α,δ-monocarboxylated product. The reaction at the counter electrode has been eloquently described by Chang and co-workers in their study of deuteration reactions employing electrosynthesis and D2O.21 In the absence of water the dicarboxylated product dominates and in the absence of CO2 the α,δ-reduction of the diene occurs (Fig. 2).
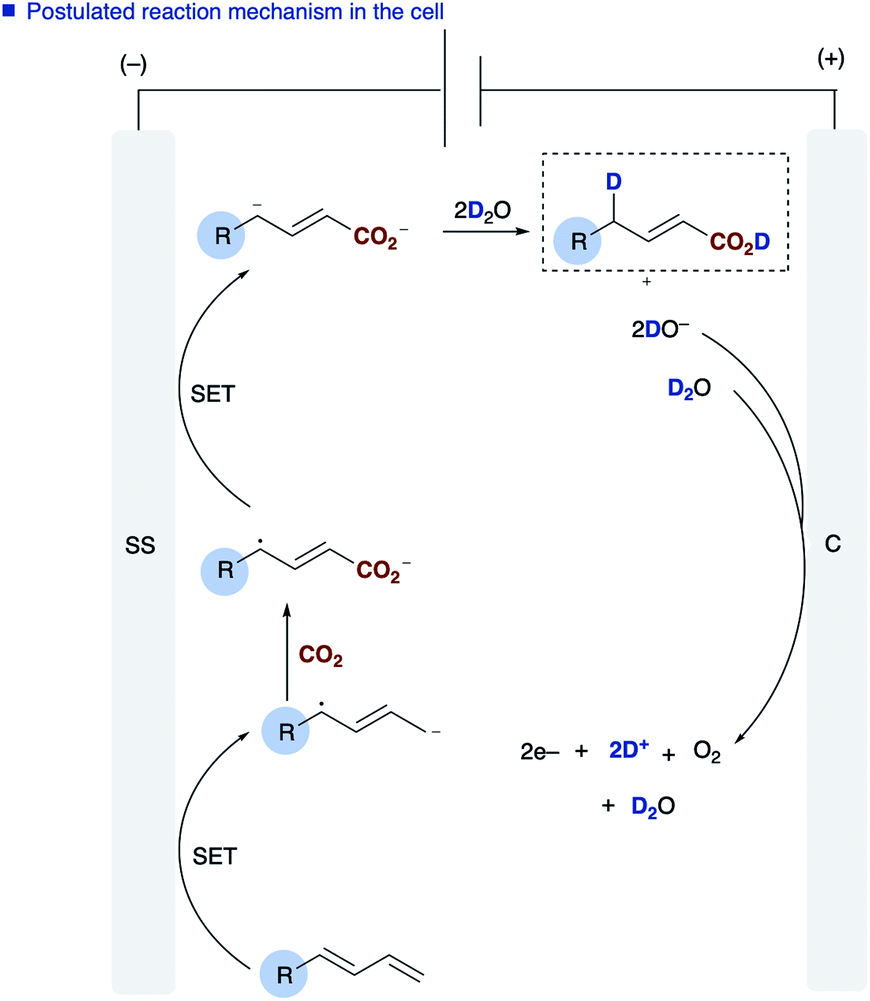 | ||
| Fig. 2 Overall processes at both the anode and cathode. | ||
Probing the substrates a little further revealed that selective carboxylation of non-conjugated alkenes, such as 6, is possible under the reaction conditions affording the α,β-hydrocarboxylated product 7 (Scheme 4a), thus demonstrating that conjugation of the diene is essential for successful carboxylation to occur, in a similar fashion to the reported styrene hydrocarboxylation.16 The methodology could also be extended to trienes such as 8 with the α,ϕ-hydrocarboxylated product 9a predominating when using the conditions employed for dienes in Table 2 (Scheme 4b). In addition, depending on the reaction conditions employed one can choose the product distribution required (Scheme 4c); addition of H2O to reaction provides the α,δ-monocarboxylated product; removal of any proton source affords the solely the α,δ-dicarboxylated product and switching the electrode system results in α,δ-reduction of the diene.
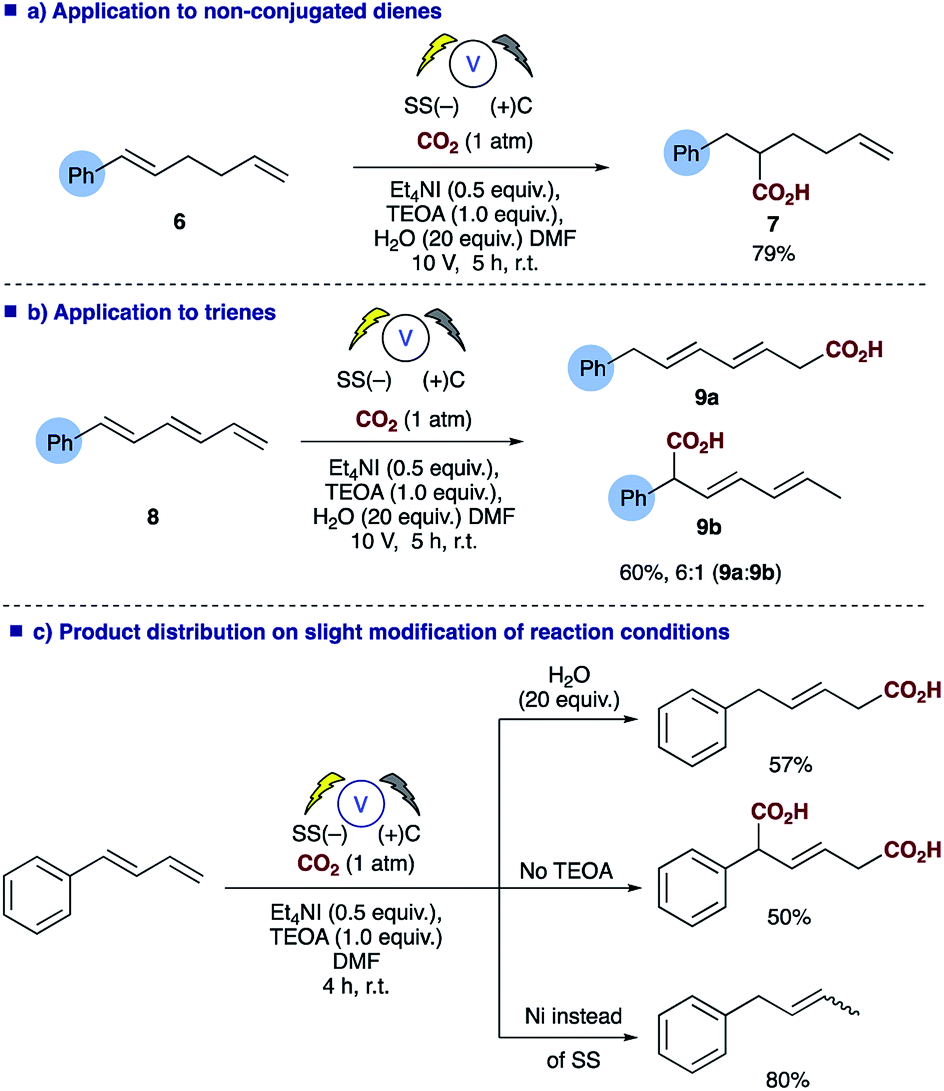 | ||
| Scheme 4 Summary of the versatility of the electrosynthetic conditions to selectively afford 3 different products. | ||
Conclusions
In summary, a highly regioselective hydrocarboxylation process that enables the direct formation of carboxylic acids from dienes giving access to α,δ-hydrocarboxylation products has been reported. A wide variety of substrates have been tolerated under these electrosynthetic conditions; Thus, this approach is complimentary to the current literature in which α-addition and dicraboxylation dominates. Preliminary mechanistic studies suggest that field inductive effects are dominant over resonance effects in the transition state. Thus lower regioselectivties of 2vs.3 are observed when the 4-substituent is inductively withdrawing. The current process goes beyond these state-of-the-art systems enabling the selective mono-carboxylation of 1,3-dienes, non-conjugated dienes and trienes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Egyptian cultural affairs and missions sectors for a visiting fellowship to AMS, the EPSRC (EP/P030599/1) and Loughborough University for additional funding. Anas Alkayal, and Volodymr Tabas for initial substrate screening and Sophie Ezeilo for assistance in the preparation of compounds 6 and 7.References
- (a) J.-B. Peng, H.-Q. Geng and X.-F. Wu, Chem, 2019, 5, 526–552 CrossRef CAS; (b) D. U. Nielsen, X.-M. Hu, K. Daasbjerg and T. Skrydstrup, Nat. Catal., 2018, 1, 244–254 CrossRef CAS; (c) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed; (d) H. Alper, J. Organomet. Chem., 1986, 300, 1–6 CrossRef CAS.
- The corresponding carbonylation process is well established and recently an attractive route to adipic acid esters has been reported using CO: J. Yang, J. Liu, H. Neumann, R. Franke, R. Jackstell and M. Beller, Science, 2019, 366, 1514–1517 CrossRef CAS PubMed.
- (a) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (b) S.-S. Yan, Q. Fu, L. L. Liao, G.-Q. Sun, J.-H. Ye, L. Gong, Y.-Z. Bo-Xue and D.-G. Yu, Coord. Chem. Rev., 2018, 374, 439–463 CrossRef CAS; (c) Y. Cao, X. He, N. Wang, H.-R. Li and L.-N. He, Chin. J. Chem., 2018, 36, 644–659 CrossRef CAS; (d) Z. Zhang, L. Gong, X.-Y. Zhou, S.-S. Yan, J. Li and D.-G. Yu, Acta Chim. Sin., 2019, 77, 783–793 CrossRef.
- N. W. J. Ang, J. C. A. de Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202003218.
- (a) L.-X. Wu, Y.-G. Zhao, Y.-B. Guan, H. Wang, Y.-C. Lan, H. Wang and J.-X. Lu, RSC Adv., 2019, 9, 32628–32633 RSC; (b) M. J. Medeiros, C. Pintaric, S. Olivero and E. Dunach, Electrochim. Acta, 2011, 56, 4384–4389 CrossRef CAS; (c) S. Torii, H. Tanaka, T. Hamatani, K. Morisaki, A. Jutand, F. Pfluger and J.-F. Fauvarque, Chem. Lett., 1986, 15, 169–172 CrossRef; (d) O. Sock, M. Troupel and J. Perichon, Tetrahedron Lett., 1985, 26, 1509–1512 CrossRef CAS; (e) J.-C. Folest, J.-M. Duprilot, J. Perichon, Y. Robin and J. Devynck, Tetrahedron Lett., 1985, 26, 2633–2636 CrossRef CAS; (f) Y. Sasaki, Y. Inoue and H. Hashimoto, J. Chem. Soc., Chem. Commun., 1976, 605–606 RSC.
- H. Wang, Y.-F. Du, M.-Y. Lin, Z. Kai and J.-X. Lu, Chin. J. Chem., 2008, 26, 1745–1748 CrossRef CAS.
- J. W. Loveland, Electrolytic production of acyclic carboxylic acids from hydrocarbons, US Pat., 3032489, May 1, 1962.
- (a) W. J. M. van Tilborg and C. J. Smit, Recl. Trav. Chim. Pays-Bas, 2010, 100, 437–438 CrossRef; (b) C.-H. Li, G.-Q. Yuan, X.-C. Ji, X.-J. Wang, J.-S. Ye and H.-F. Jiang, Electrochim. Acta, 2011, 56, 1529–1534 CrossRef CAS; (c) R. Matthessen, J. Fransaer, K. Binnemans and D. E. De Vos, RSC Adv., 2013, 3, 4634–4642 RSC; (d) R. Matthessen, J. Fransaer, K. Binnemans and D. E. De Vos, ChemElectroChem, 2014, 2, 73–76 CrossRef.
- J. Bringmann and E. Dinjus, Appl. Organomet. Chem., 2001, 15, 135–140 CrossRef CAS.
- S. Derien, J. C. Clinet, E. Dunãch and J. Perichon, Tetrahedron, 1992, 48, 5235–5248 CrossRef CAS.
- M. Takimoto and M. Mori, J. Am. Chem. Soc., 2001, 123, 2895–2896 CrossRef CAS PubMed.
- A. Tortajada, R. Ninokata and R. Martin, J. Am. Chem. Soc., 2018, 140, 2050–2053 CrossRef CAS PubMed.
- J. Takaya, K. Sasano and N. Iwasawa, Org. Lett., 2011, 13, 1698–1701 CrossRef CAS PubMed.
- (a) Y.-Y. Gui, N. Hu, X.-W. Chen, L. L. Liao, T. Ju, J.-H. Ye, Z. Zhang, J. Li and D.-G. Yu, J. Am. Chem. Soc., 2017, 139, 17011–17014 CrossRef CAS PubMed; (b) X.-W. Chen, L. Zhu, Y.-Y. Gui, K. Jing, Y.-X. Jiang, Z.-Y. Bo, Y. Lan, J. Li and D.-G. Yu, J. Am. Chem. Soc., 2019, 141, 18825–18835 CrossRef CAS PubMed.
- G. Bäunlich, D. Walther, H. Eibisch and B. Schönecker, J. Organomet. Chem., 1993, 453, 295–298 CrossRef.
- A. Alkayal, V. Tabas, S. Montanaro, I. A. Wright, A. V. Malkov and B. R. Buckley, J. Am. Chem. Soc., 2020, 142, 1780–1785 CrossRef CAS PubMed.
- (a) R. Matthessen, J. Fransaer, K. Binnemans and D. E. De Vos, Beilstein J. Org. Chem., 2014, 10, 2484–2500 CrossRef PubMed; (b) H. Senboku and A. Katayama, Current Opinion in Green and Sustainable Chemistry, 2017, 3, 50–54 CrossRef.
- All of which displayed a pair of E/Z-isomers about the alkene bond.
- C. H. A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef.
- J. Lehmann and G. C. Lloyd-Jones, Tetrahedron, 1995, 51, 8863–8874 CrossRef CAS , For a note of caution when applying simple correlation see: C. G. Swain, S. H. Unger, N. R. Rosenquist and M. S. Swain, J. Am. Chem. Soc., 1983, 105, 492–502 CrossRef , in which they quote “….one should avoid the pernicious practice of simple linear correlation vs. F alone…….Such simple correlation is defensible only if the correlation with F or R alone is good (simple correlation coefficient > 0.965).
- X. Liu, R. Liu, J. Qiu, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202005765.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound data and 1H, 13C and 19F NMR data. See DOI: 10.1039/d0sc03148h |
| This journal is © The Royal Society of Chemistry 2020 |