
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deoxygenative α-alkylation and α-arylation of 1,2-dicarbonyls†
Shengfei
Jin
 ,
Hang T.
Dang
,
Graham C.
Haug
,
Viet D.
Nguyen
,
Hadi D.
Arman
and
Oleg V.
Larionov
*
,
Hang T.
Dang
,
Graham C.
Haug
,
Viet D.
Nguyen
,
Hadi D.
Arman
and
Oleg V.
Larionov
*
Department of Chemistry, The University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249, USA. E-mail: oleg.larionov@utsa.edu
First published on 1st July 2020
Abstract
Construction of C–C bonds at the α-carbon is a challenging but synthetically indispensable approach to α-branched carbonyl motifs that are widely represented among drugs, natural products, and synthetic intermediates. Here, we describe a simple approach to generation of boron enolates in the absence of strong bases that allows for introduction of both α-alkyl and α-aryl groups in a reaction of readily accessible 1,2-dicarbonyls and organoboranes. Obviation of unselective, strongly basic and nucleophilic reagents permits carrying out the reaction in the presence of electrophiles that intercept the intermediate boron enolates, resulting in two new α-C–C bonds in a tricomponent process.
Introduction
Chemists have long relied on reactions that allow for generation of unstable and reactive intermediates but forcing conditions, as well as strong and unselective reagents are often required to achieve the desired transformations. Much of the recent progress in synthetic methodology has been fueled by the necessity to access transient reactive intermediates in a rapid and modular manner and in the absence of unselective reagents.1Enolates are among the most common and synthetically versatile intermediates used for α-functionalization of carbonyls. Despite their broad utility, the most common method of generation of enolates remains deprotonation with strong bases, e.g., organolithiums, lithium amides and alkoxides that are incompatible with many base-sensitive functional groups.2 Moreover, the deprotonation approach is inherently synthetically inefficient, since it requires prior assembly of the entire carbon skeleton of the carbonyl precursor. Given the central role of enolates in organic synthesis, reactions that allow for modular generation of enolates, e.g., by carbon–carbon bond-forming reactions under neutral conditions and in the absence of strong bases are highly sought after but have remained elusive.
Since their introduction by Mukaiyama in 1973,3 boron enolates have become the preferred class of carbon-centered nucleophiles for many chemo- and stereoselective carbon–carbon bond-forming reactions, due to the presence of the inherently Lewis acidic boryl group that enables more selective reactions with a broader range of electrophiles.2,4 Consequently, there has been an upsurge of interest in new approaches to their generation, exemplified by geminal diboronate-based methods recently developed by Pattison, Liu and Chirik rooted in the earlier studies by Mukaiyama and Matteson (Scheme 1).5
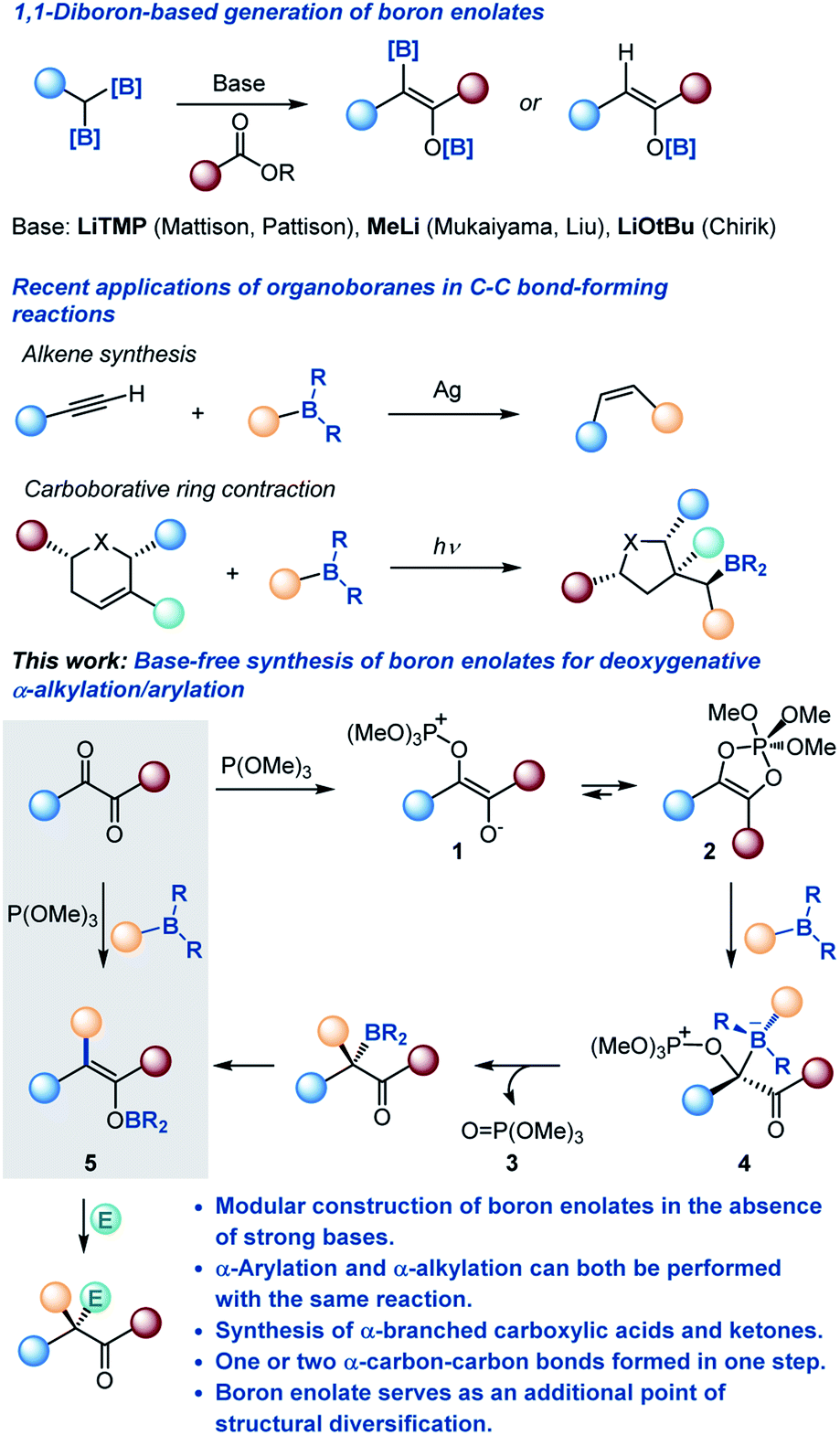 | ||
| Scheme 1 Boron enolates and the deoxygenative α-alkylation/arylation of 1,2-dicarbonyls. | ||
Organoboranes have long served as broadly useful synthetic linchpins,6 and the recent examples of carbon–carbon bond-forming reactions, for instance, Lalic's Z-alkene synthesis7 and the carboborative ring contraction,8 showcase the untapped reactivity of organoboranes. The versatility of organoboranes coupled with the ease of their preparation by well-established routes, e.g., hydroboration, ensures their continued presence at the forefront of organic synthesis.9
In a parallel development, there has been an increased realization of the expanding synthetic potential of 1,2-dicarbonyl compounds, stemming from the proximity of the two reactive carbonyl groups and ready synthetic accessibility of structurally diverse 1,2-dicarbonyls.10
Merging these two classes of versatile synthetic intermediates, we hypothesized that an addition of an organoborane to 1,2-dicarbonyl-phosphite adducts 1 and 2 (ref. 11) would be followed by a 1,2-metallate shift that is accompanied by the departure of phosphate 3 from the zwitterionic intermediate 4 by analogy with the Matteson reaction that has recently been adopted to a variety of valuable synthetic transformations.12 Subsequent 1,3-borotropic shift would result in the formation of boron enolate 5. Notably, construction of the boron enolate would be achieved in the absence of strong bases or strongly Lewis acidic boron reagents (e.g., boryl triflates and halides) and in a modular manner, i.e., with concomitant formation of a carbon–carbon bond in the α-position. Furthermore, the modularity of the reaction combined with the neutral reaction conditions and the weakly Lewis acidic character of the boryloxy group were expected to enable a streamlined synthesis of highly α-branched carboxylic acids and other carbonyls by engaging a range of electrophiles in a tricomponent cascade process.
α-Branched carbonyl compounds, in particular carboxylates, are among the most synthetically valuable and medicinally important structural motifs, whose applications range from non-steroidal anti-inflammatory drugs (NSAIDs)13 to versatile reagents for late stage functionalization enabled by resurgent decarboxylative methodologies.14 A number of synthetic approaches to α-branched carbonyls have recently emerged, including transition metal-catalyzed α-arylation of enolates,15 carbonylation16 and electrophilic α-alkylation.17 However, many challenges remain unsolved, including the lack a unified synthetic platform that can enable both α-arylation and α-alkylation under essentially neutral conditions and in the absence of non-chemoselective or expensive reagents and catalysts. Thus, further work is needed to develop such a synthetic platform for production of structurally diverse α-branched carbonyls by a conceptually distinct aryl–Cα and alkyl–Cα bond forming process that encompasses both α-arylation and α-alkylation.
Results and discussion
We chose triethylborane as a simple and readily available boron reagent for our initial synthetic scope studies, based on our prior experience8 and cognizant that successful development of the reaction with BEt3 would enable utilization of other organoboron sources, including the widely used organo-9-borabicyclo[3.3.1]nonanes (BBN). We found that the reaction of ester 6 with triethylborane and trimethyl phosphite proceeded cleanly at 50 °C in THF, affording α-branched carboxylate 7 in good yield (Scheme 2). Importantly, the more readily available and inexpensive P(OMe)3 can be used, although P(NMe2)3 is also a suitable PIII reagent.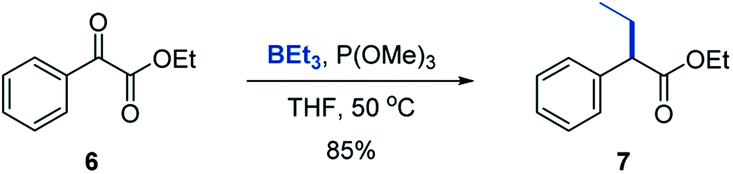 | ||
| Scheme 2 Deoxygenative α-alkylation/arylation of ester 6. Reaction conditions: ester 6 (0.2 mmol), BEt3 (0.3 mmol), P(OMe)3 (0.24 mmol), THF (2 mL), 50 °C. | ||
We then proceeded with the evaluation of the scope of the deoxygenative alkylation reaction (Scheme 3). Halo, alkyl, methoxy and thio-substituted aromatic keto esters were suitable substrates (8–18). The electron-withdrawing cyano, and the medicinally relevant trifluoromethyl and trifluoromethoxy groups were also tolerated (19–21). Polycyclic keto esters were readily converted to the corresponding α-alkyl-branched esters in good yields (22–27). Similarly, heterocyclic keto esters successfully participated in the reaction, including substrates containing pyridine, thiophene, dihydrobenzofuran, indole, carbazole, dibenzothiophene and xanthene cores (28–37). Interestingly, vinylogous substitution in the aromatic ring was also observed for the indole keto ester (33). A reaction with N-methylisatin afforded 3-alkylated indolone 38. Aliphatic keto esters reacted equally well and produced the corresponding 2-alkyl branched esters 39–42 in good yields. 1,2-Diketones were evaluated next with P(NMe2)3 as the optimal PIII reagent, likely due to the increased nucleophilicity imparted by the more basic PIII reagent,18 and a range of α-branched ketones 43–49 were prepared, further highlighting the scope of the reaction. Esters 22, 23, and 35 were converted to corresponding acids, whose structures were confirmed by X-ray crystallographic analysis.
 | ||
| Scheme 3 Scope of 1,2-dicarbonyl compounds. Reaction conditions: 1,2-dicarbonyl (0.1–0.2 mmol), BEt3 (1.5–2 equiv.), P(OMe)3 (1.2–2 equiv.) THF (1–4 mL), 50 °C. a80 °C. bP(NMe2)3 was used. | ||
The reaction can be further extended to other organoboranes (Scheme 4). α-Arylation of carboxylic acids and other carbonyl compounds remains a long-standing synthetic challenge.15 Significantly, facile arylation can be achieved, providing a metal-free access to α-mono- and diarylated esters and ketones (50–55). Other alkylboranes can be used as well (56–62), including those derived from alkenes by hydroboration (58–62). Importantly, 9-BBN-derived organoboranes that can be easily prepared by hydroboration of alkenes and other methods19 proved to be suitable reagents for the transfer of alkyl (63–65), aryl (66, 67), vinyl (68) and heteroaryl groups (69–71).
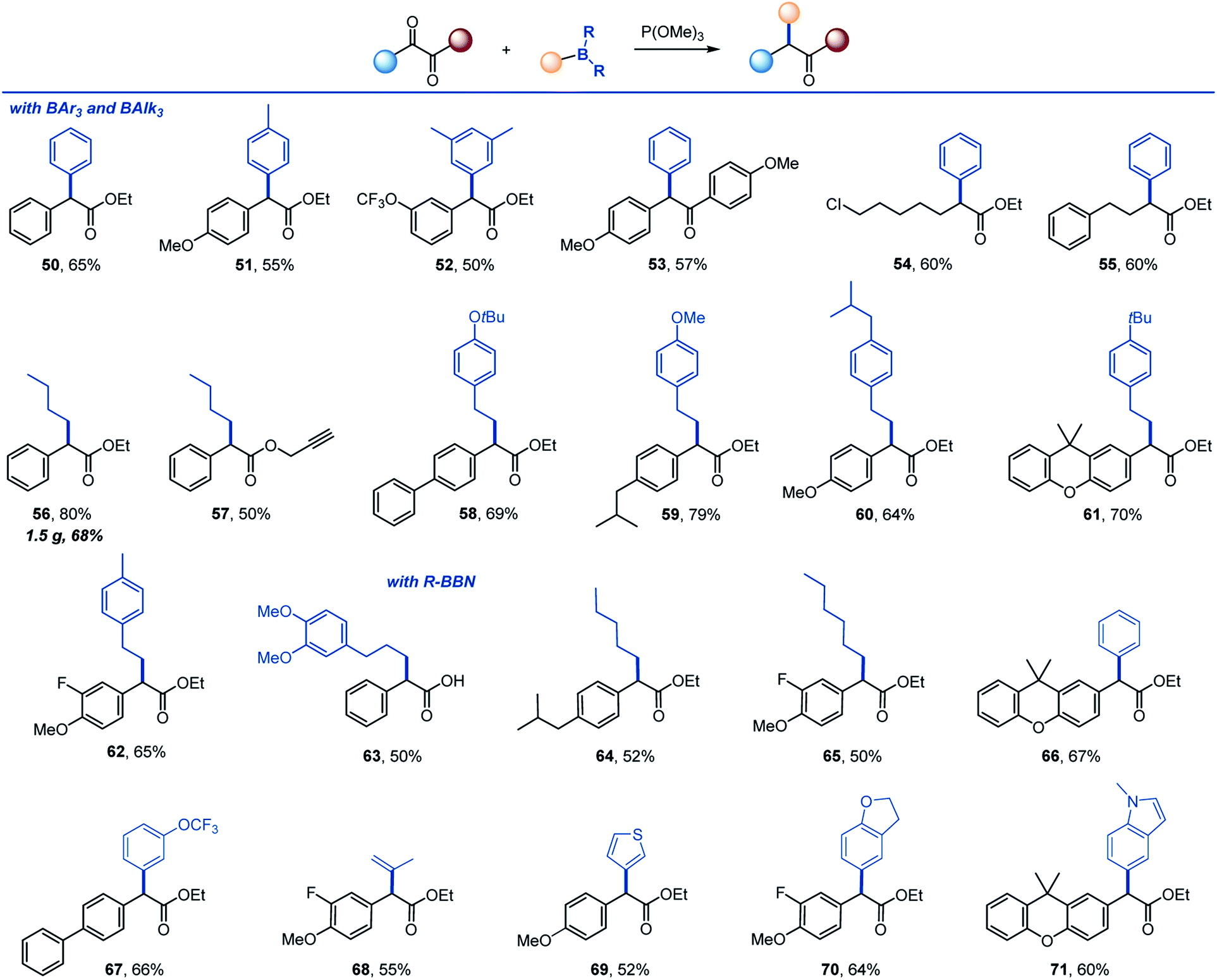 | ||
| Scheme 4 Scope of organoboron reagents. Reaction conditions: see footnote for Scheme 2. | ||
Given the importance of α-arylpropionic acids as NSAIDs, we evaluated trimethylborane in the synthesis of ibuprofen, naproxen and flurbiprofen (72–74, Scheme 5). In all three cases, NSAIDs 72–74 were readily accessed in two synthetic operations from the corresponding precursors 75–77, pointing to the potential of the deoxygenative alkylation reaction in drug discovery applications.
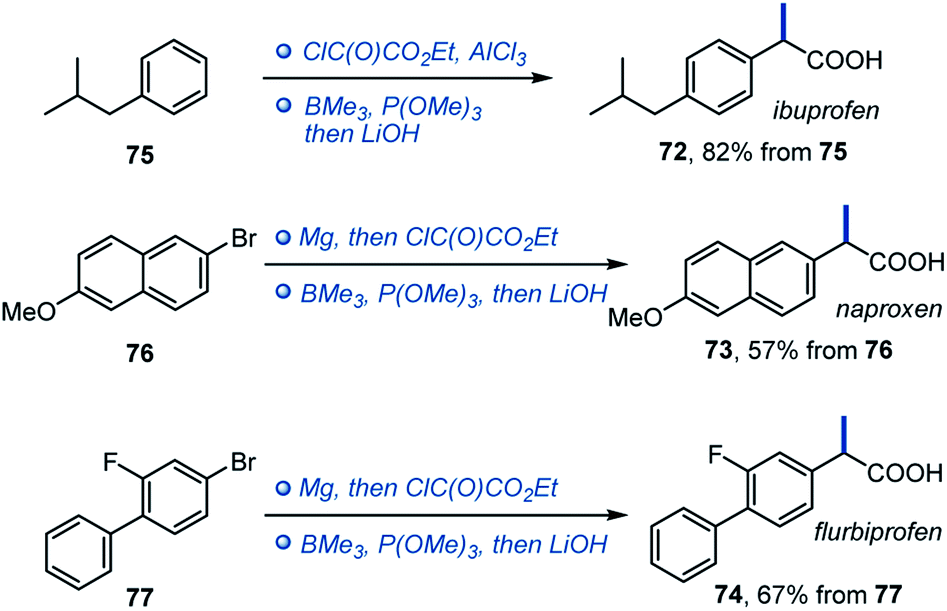 | ||
| Scheme 5 Synthesis of NSAIDs 72–74 with trimethylborane. | ||
A remarkable feature of the reaction is that it produces reactive boron enolates under neutral conditions in the absence of strong bases that are typically required for generation of enolates. We, therefore, next explored the feasibility of carrying out the reaction directly in the presence of electrophiles. This combination would enable a tricomponent coupling process that, due to the mildness of the reaction conditions and the enhanced reactivity of boron enolates, could engage a range of electrophiles, including both very reactive (e.g., proton sources) and typically unreactive ones (e.g., nitriles and ketones). Indeed, the reaction could be successfully carried out in the presence of deuterium oxide, and the corresponding α-deuterated ester 78 was isolated in 72% yield with 92% deuterium incorporation (Scheme 6). This result suggests that the method may be useful in the context of deuterated drug discovery as a means of improving metabolic stability.20
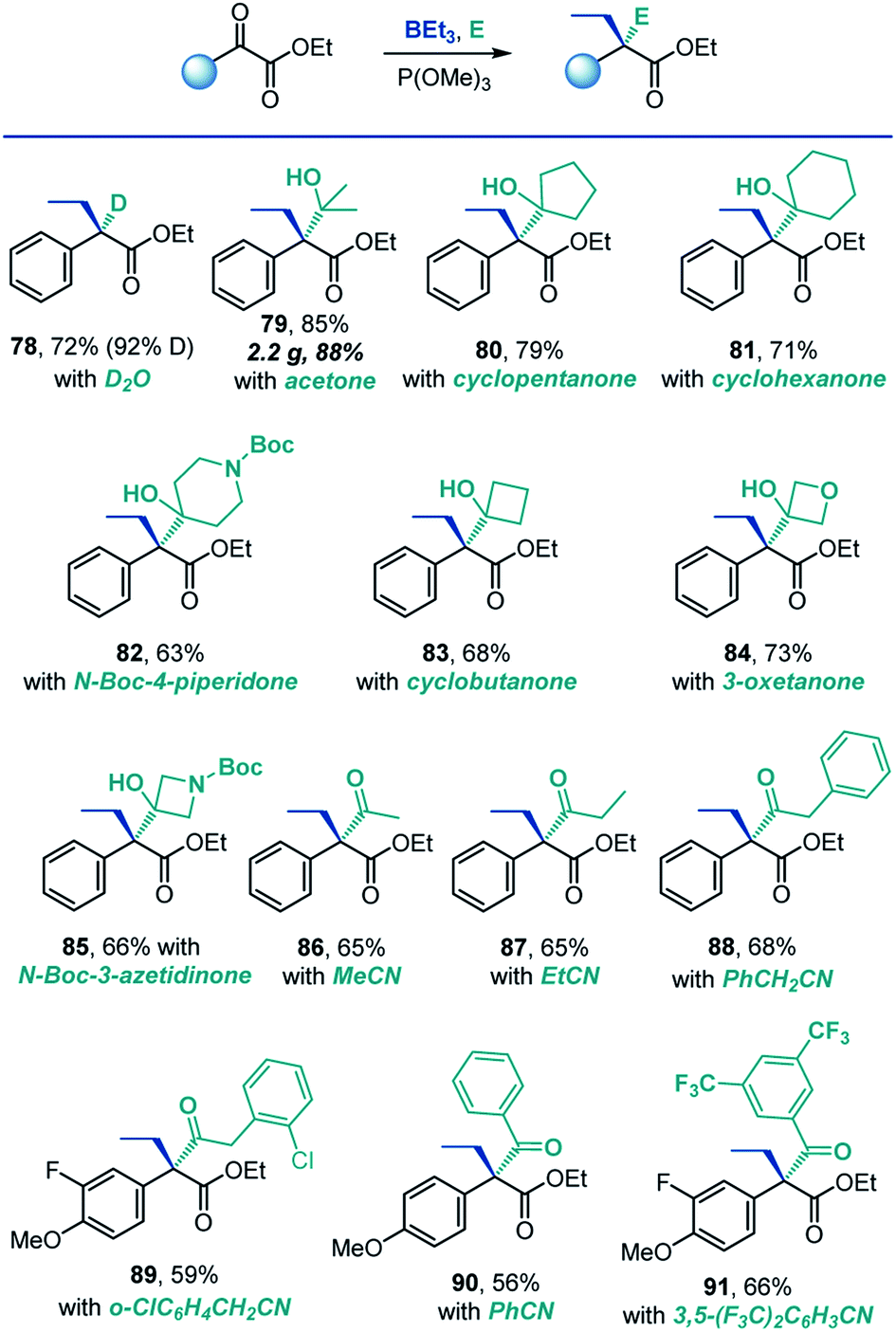 | ||
| Scheme 6 Scope of the tricomponent reaction. Reaction conditions: see footnote for Scheme 2, in the presence of electrophile. | ||
Ketones are typically challenging electrophiles for enolate trapping, because of their facile enolization, lower reactivity and reversibility of the addition reaction.21 Interestingly, a variety of ketones were compatible with the deoxygenative α-alkylation reaction, and the corresponding sterically congested addition products 79–85 featuring two adjacent quaternary carbons were readily formed in good yields.
Intermolecular C-acylation of enolates with nitriles is a potentially synthetically attractive approach to β-dicarbonyl compounds. However, examples of such reactions are rare.22 Significantly, both aliphatic and aromatic nitriles reacted smoothly in a tricomponent manner under the conditions of the deoxygenative α-alkylation (86–91, Scheme 6), presumably due to the Lewis acidic character of the boryl group of the intermediate boron enolate, leading to concomitant construction of two C–C bonds and an all-carbon quaternary α-position in one step.
The deoxygenative α-alkylation can be further leveraged for construction of all-carbon quaternary α-positions in carboxylic acids by the Ireland-Claisen rearrangement of the intermediate boron enolates,23e.g., 92 (Scheme 7). The Ireland-Claisen-enhanced deoxygenative α-alkylation reaction enables installation of allylic α-side chains (93), including the sterically encumbered β,β-dimethylallyl group in the α-branched acid 94. Expansion of the scope to propargylic and allenylic substrates provides straightforward access to carboxylic acids bearing α-allenyl and α-dienyl groups (95 and 96). The deoxygenative α-alkylation can also be combined with the Pd-catalyzed allylation (Scheme 7, 97), indicating that the intermediate boron enolates can serve as a platform for C–C bond-forming functionalization by means of transition metal catalysis. Furthermore, the deoxygenative coupling reaction can be readily carried out on gram scale (8, 56, 79).
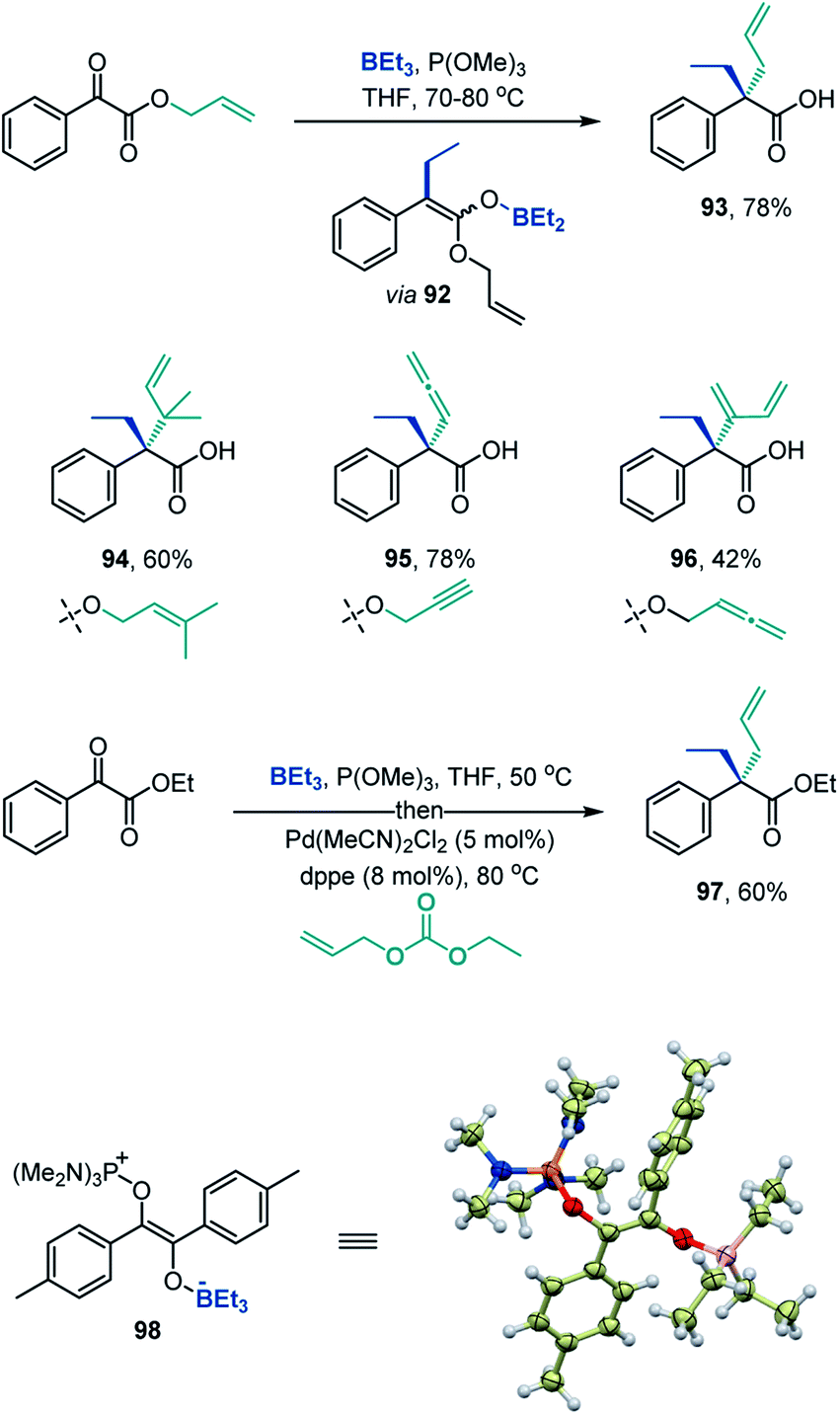 | ||
| Scheme 7 Construction of quaternary all-carbon α-positions in carboxylic acids and the structure of intermediate 98. | ||
Kinetic and computational studies were further performed to clarify the reaction mechanism. We found that TEMPO had no detrimental effect on the reaction performance, indicating that the reaction does not proceed by a radical pathway.
Furthermore, organoboranes are known to form adducts with phosphites,24 and this process was expected to take place under the conditions of the deoxygenative α-alkylation/α-arylation. Indeed, formation of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct was observed by NMR spectroscopy with Kassoc = 1.62 × 103 M−1 for the association of triethylborane with trimethyl phosphite.
:1 adduct was observed by NMR spectroscopy with Kassoc = 1.62 × 103 M−1 for the association of triethylborane with trimethyl phosphite.
Additionally, variable time normalization analysis25 revealed that the deoxygenative α-alkylation was zero order in triethylborane, and first order in α-keto ester and trimethyl phosphite (Fig. 1A–C). The intermediacy of boron enolates was supported by kinetic 11B NMR spectroscopy studies that confirmed formation of the dialkylboryloxy group (δ = 53.1 ppm)2 with concomitant consumption of triethylborane (Fig. 1D). Moreover, zwitterionic adduct 98 crystallized from a P(NMe2)3-mediated reaction of the corresponding 1,2-diketone with triethylborane and was characterized by X-ray crystallography (Scheme 7), confirming participation of adducts 2 in the process. However, an adduct analogous to 98 was not isolated or observed with ester 6 and triethyl phosphite, indicating that it is less stable for these reactants.
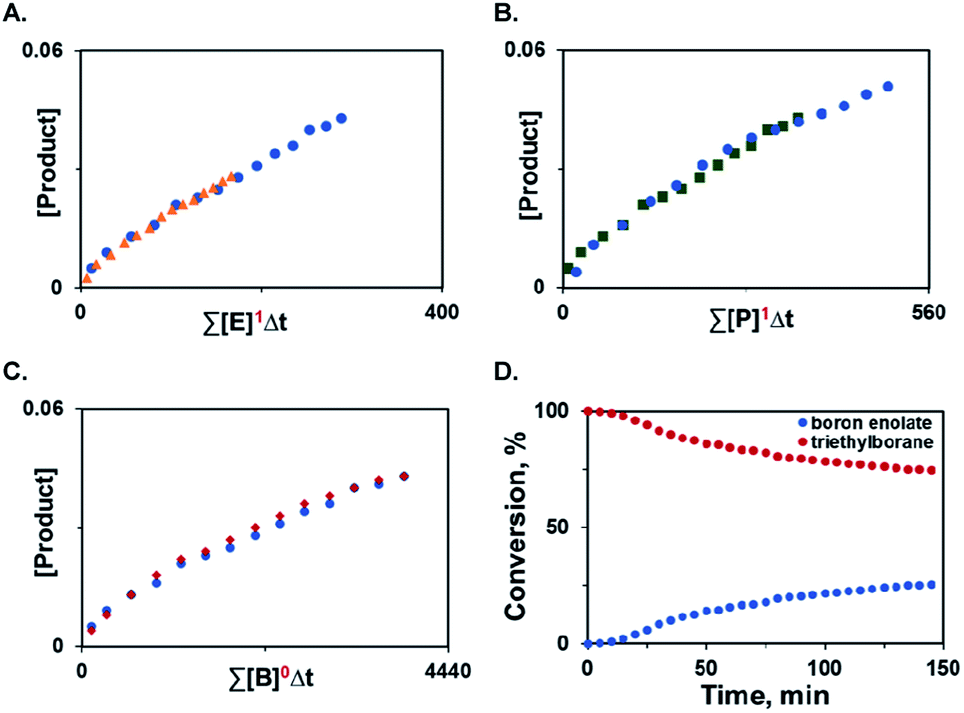 | ||
| Fig. 1 Kinetic studies of the deoxygenative α-alkylation and boron enolate formation. (A) Order in α-keto ester. (B) Order in trimethyl phosphite. (C) Order in triethylborane. (D) Time course of triethylborane consumption and boron enolate formation during the reaction of ester 6 with triethylborane and trimethyl phosphite. | ||
A DFT computational study provided further insights into the mechanism of the reaction that were consistent with the experimental data (Fig. 2). We found that the addition of phosphite to the carbon of the keto group (99) that is followed by a 1,2-phospha-Brook migration of the phosphite group to the oxygen with subsequent ring closure en route to adduct 100 was kinetically accessible (TS-A and TS-B) with an overall barrier of 22.9 kcal mol−1, in excellent agreement with the experimental value (22.7 kcal mol−1). Alternative mechanisms of formation of adduct 100 (e.g., a direct addition of phosphite to the O atom of the carbonyl or a [4 + 1] cheletropic cycloaddition of phosphite to ester 6) were also considered, but were found to proceed over prohibitively high barriers (ΔG≠ > 40 kcal mol−1, see ESI†). The addition of the organoborane to cyclic ester–phosphite adduct 100 further produced zwitterionic intermediate 101 that underwent a Matteson-type displacement viaTS-C en route to α-boryl ester 102. Subsequent low-barrier (TS-D) exergonic 1,3-borotropic shift produces E- and Z-isomers of boron enolate 103, with the E-enolate being the more stable isomer.
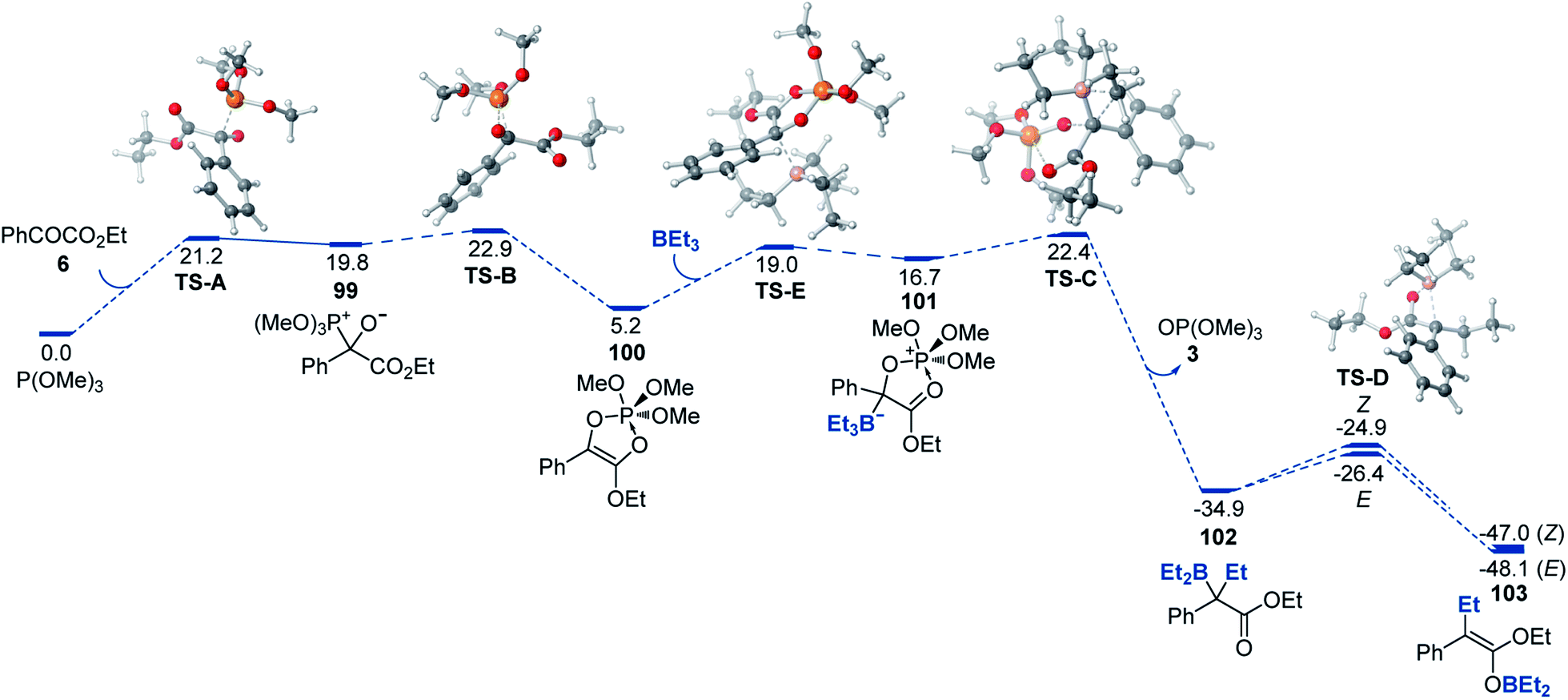 | ||
| Fig. 2 Computed Gibbs free energy profile of the deoxygenative α-alkylation reaction, ΔG, kcal mol−1. | ||
Although the computed difference between TS-B and the second highest barrier (TS-C) is relatively small, we note that formation of adduct 101 as a rate-limiting step (i.e., higher TS-B) is consistent with the experimentally observed zero order in organoborane and first order in both ester 6 and phosphite. The alternative pathway that proceeds via the acyclic adduct of type 1 was also investigated but was found to proceed over higher barriers and with the Matteson-type displacement as the rate-limiting step (see ESI†), thus being inconsistent with the experimental data. Taken together, our experimental and computational studies support the proposed mechanism of the modular boron enolate synthesis from organoboranes and 1,2-dicarbonyl-phosphite adducts.
Conclusions
In conclusion, we have developed a modular construction of boron enolates in the absence of strong bases that enables installation of both α-alkyl and α-aryl groups in a reaction of readily accessible 1,2-dicarbonyls with organoboranes (easily derived by hydroboration from alkenes or from other precursors). The reaction produces α-branched carboxylates and ketones that are valuable synthetic intermediates and medicinally important motifs. The intermediacy of boron enolates enables a tricomponent coupling process with a range of electrophiles, including the typically unreactive nitriles and ketones, resulting in the construction of two α-C–C bonds. The utility and scope of the reaction are demonstrated with profen-type NSAIDs and a variety of substituted carbocyclic and heterocyclic carbonyls that can be further diversified by means of one-pot rearrangements and a palladium-catalyzed allylation of the intermediate boron enolates. The mechanism of the reaction was investigated experimentally and computationally, revealing the central role of 1,2-dicarbonyl-phosphite adducts and a 1,2-metallate shift in enabling the deoxygenative C–C bond-forming process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Welch Foundation (AX-1788), the NSF (CHE-1455061), and NIGMS (GM134371) is gratefully acknowledged. The NMR and X-ray crystallography facilities were supported by the NSF (CHE-1625963 and CHE-1920057). The authors acknowledge the Texas Advanced Computing Center (TACC) at UT Austin for providing computational resources.Notes and references
- (a) M. S. Singh, Reactive Intermediates in Organic Chemistry: Structure, Mechanism, and Reactions, Wiley, Hoboken, 2014 Search PubMed; (b) R. A. Shenvi, D. P. O'Malley and P. S. Baran, Acc. Chem. Res., 2009, 42, 530–541 CrossRef CAS PubMed; (c) N. A. Afagh and A. K. Yudin, Angew. Chem., Int. Ed., 2010, 49, 262–310 CrossRef CAS.
- J. Zabicky, The Chemistry of Metal Enolates; Patai Series: The Chemistry of Functional Groups, John Wiley & Sons, Chichester, UK, 2009 Search PubMed.
- K. Inomata, M. Muraki and T. Mukaiyama, Bull. Chem. Soc. Jpn., 1973, 46, 1807–1810 CrossRef CAS.
- (a) S. B. J. Kan, K. K. -H. Ng and I. Paterson, Angew. Chem., Int. Ed., 2013, 52, 9097–9108 CrossRef CAS PubMed; (b) J. -i. Matsuo and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 9109–9118 CrossRef CAS PubMed; (c) Z. He, A. Zajdlik and A. K. Yudin, Dalton Trans., 2014, 43, 11434–11451 RSC.
- (a) T. Mukaiyama, M. Murakami, T. Oriyama and M. A. Yamaguchi, Chem. Lett., 1981, 10, 1193–1196 CrossRef; (b) D. S. Matteson and R. J. Moody, Organometallics, 1982, 1, 20–28 CrossRef CAS; (c) C. E. Iacono, T. C. Stephens, T. S. Rajan and G. Pattison, J. Am. Chem. Soc., 2018, 140, 2036–2040 CrossRef CAS PubMed; (d) W. Sun, L. Wang, C. Xia and C. Liu, Angew. Chem., Int. Ed., 2018, 57, 5501–5505 CrossRef CAS PubMed; (e) B. Lee and P. J. Chirik, J. Am. Chem. Soc., 2020, 142, 2429–2437 CrossRef CAS PubMed.
- (a) D. S. Matteson, Stereodirected Synthesis with Organoboranes, Springer, Heidelberg, 1995 CrossRef; (b) P. V. Ramachandran and H. C. Brown, Organoboranes for Syntheses, ACS, Washington, DC, 2001 CrossRef.
- M. T. Lee, M. B. Goodstein and G. Lalic, J. Am. Chem. Soc., 2019, 141, 17086–17091 CrossRef CAS.
- (a) S. Jin, V. T. Nguyen, H. T. Dang, D. P. Nguyen, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2017, 139, 11365–11368 CrossRef CAS PubMed; (b) V. A. Roytman, S. Jin, V. T. Nguyen, V. D. Nguyen, G. C. Haug, O. V. Larionov and D. A. Singleton, J. Am. Chem. Soc., 2020, 142, 85–88 CrossRef CAS PubMed.
- (a) J. R. Gonzalez, A. Z. Gonzalez and J. A. Soderquist, J. Am. Chem. Soc., 2009, 131, 9924–9925 CrossRef CAS; (b) Z. Lu, A. Wilsily and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 8154–8157 CrossRef CAS; (c) S. L. Zultanski and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 15362–15364 CrossRef CAS PubMed; (d) M. Chen and W. R. Roush, J. Am. Chem. Soc., 2012, 134, 3925–3931 CrossRef CAS; (e) S. N. Kessler, M. Neuburger and H. A. Wegner, J. Am. Chem. Soc., 2012, 134, 17885–17888 CrossRef CAS PubMed; (f) J. Kister, D. H. Ess and W. R. Roush, Org. Lett., 2013, 15, 5436–5439 CrossRef CAS PubMed; (g) C. G. Watson, A. Balanta, T. G. Elford, S. Essafi, J. N. Harvey and V. K. Aggarwal, J. Am. Chem. Soc., 2014, 136, 17370–17373 CrossRef CAS PubMed; (h) A. M. Bergmann, A. M. Oldham, W. You and M. K. Brown, Chem. Commun., 2018, 54, 5381–5384 RSC; (i) C. L. Hugelshofer, V. Palani and R. Sarpong, Org. Lett., 2018, 20, 2649–2653 CrossRef CAS PubMed; (j) N. Y. Kuznetsov, R. M. Tikhov, T. V. Strelkova and Y. N. Bubnov, Org. Lett., 2018, 20, 3549–3552 CrossRef CAS; (k) N. R. Lee, R. T. H. Linstadt, D. J. Gloisten, F. Gallou and B. H. Lipshutz, Org. Lett., 2018, 20, 2902–2905 CrossRef CAS PubMed; (l) K. M. Baker, D. L. Baca, S. Plunkett, M. E. Daneker and M. P. Watson, Org. Lett., 2019, 21, 9738–9741 CrossRef CAS PubMed; (m) C. Law, Y. Meng, S. M. Koo and J. P. Morken, Angew. Chem., Int. Ed., 2019, 58, 6654–6658 CrossRef CAS PubMed.
- (a) P. Fang, M. Li and H. Ge, J. Am. Chem. Soc., 2010, 132, 11898–11899 CrossRef CAS PubMed; (b) L. Chu, J. M. Lipshultz and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 7929–7933 CrossRef CAS PubMed; (c) S. Yang, H. Tan, W. Ji, X. Zhang, P. Li and L. Wang, Adv. Synth. Catal., 2017, 359, 443–453 CrossRef CAS; (d) Q. Q. Wang, K. Xu, Y. Y. Jiang, Y. G. Liu, B. G. Sun and C. C. Zeng, Org. Lett., 2017, 19, 5517–5520 CrossRef CAS PubMed; (e) T. Karasawa, R. Oriez, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2018, 140, 12290–12295 CrossRef CAS PubMed; (f) T. Morack, C. Mück-Lichtenfeld and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 1208–1212 CrossRef CAS PubMed; (g) Y. H. Wang, J. S. Tian, P. W. Tan, Q. Cao, X. X. Zhang, Z. Y. Cao, F. Zhou, X. Wang and J. Zhou, Angew. Chem., Int. Ed., 2020, 59, 1634–1643 CrossRef CAS PubMed; (h) E. Voutyritsa and C. G. Kokotos, Angew. Chem., Int. Ed., 2020, 59, 1735–1741 CrossRef CAS PubMed.
- (a) V. A. Kukhtin, Dokl. Akad. Nauk SSSR, 1958, 121, 466–469 CAS; (b) F. Ramirez, R. B. Mitra and N. B. Desai, J. Am. Chem. Soc., 1960, 82, 5763–5764 CrossRef CAS; (c) F. Ramirez, Pure Appl. Chem., 1964, 9, 337–370 CAS; (d) F. Ramirez, Acc. Chem. Res., 1968, 1, 168–174 CrossRef CAS . For a review, see: ; (e) Y. Liu, F. Sun and Z. He, Tetrahedron Lett., 2018, 59, 4136–4148 CrossRef CAS.
- (a) D. S. Matteson and R. W. H. Mah, J. Am. Chem. Soc., 1963, 85, 2599–2603 CrossRef CAS; (b) D. S. Matteson and D. Majumdar, J. Am. Chem. Soc., 1980, 102, 7588–7590 CrossRef CAS . For reviews, see: ; (c) V. K. Aggarwal, G. Y. Fang, X. Ginesta, D. M. Howells and M. Zaja, Pure Appl. Chem., 2006, 78, 215–229 CAS; (d) S. P. Thomas, R. M. French, V. Jheengut and V. Aggarwal, Chem. Rec., 2009, 9, 24–39 CrossRef CAS PubMed; (e) C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC.
- C. Lamberth and J. Dinges, Bioactive Carboxylic Compound Classes, Wiley-VCH, Weinheim, 2016 Search PubMed.
- (a) L. J. Allen, P. J. Cabrera, M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 5607–5610 CrossRef CAS PubMed; (b) T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. E. Novak, J. Z. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 260–265 CrossRef CAS PubMed; (c) K. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016–5019 CrossRef CAS PubMed; (d) C. R. Jamison and L. E. Overman, Acc. Chem. Res., 2016, 49, 1578–1586 CrossRef CAS PubMed; (e) S. Jin, G. C. Haug, V. T. Nguyen, C. Flores-Hansen, H. D. Arman and O. V. Larionov, ACS Catal., 2019, 9, 9764–9774 CrossRef CAS; (f) J. Xiang, M. Shang, Y. Kawamata, H. Lundberg, S. H. Reisberg, M. Chen, P. Mykhailiuk, G. Beutner, M. R. Collins, A. Davies, M. Del Bel, G. M. Gallego, J. E. Spangler, J. Starr, S. Yang, D. G. Blackmond and P. S. Baran, Nature, 2019, 573, 398–402 CrossRef CAS PubMed; (g) V. D. Nguyen, V. T. Nguyen, G. C. Haug, N. T. H. Vuong, H. T. Dang, H. D. Arman and O. V. Larionov, Angew. Chem., Int. Ed., 2020, 59, 7921–7927 CrossRef CAS PubMed.
- (a) X. Liu and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 5182–5191 CrossRef CAS PubMed; (b) T. Hama, D. A. Culkin and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 4976–4985 CrossRef CAS PubMed; (c) Z. T. He and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 11749–11753 CrossRef CAS PubMed . For a review, see: ; (d) Y.-J. Hao, X.-S. Hu, Y. Zhou, J. Zhou and J.-S. Yu, ACS Catal., 2020, 10, 955–993 CrossRef CAS.
- (a) L. Kollár, Modern Carbonylation Methods, Wiley-VCH, Weinheim, 2008 CrossRef; (b) Y. Wang, Q. Qian, J. Zhang, B. B. A. Bediako, Z. Wang, H. Liu and B. Han, Nat. Commun., 2019, 10, 5395–5402 CrossRef PubMed.
- (a) L. Guo, X. Ma, H. Fang, X. Jia and Z. Huang, Angew. Chem., Int. Ed., 2015, 54, 4023–4027 CrossRef CAS PubMed; (b) A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- H. R. Hudson, in The Chemistry of Organophosphorus Compounds, ed. F. R. Hartley, Wiley, Chichester, 1990, vol. 1 Search PubMed.
- R. S. Dhillon, Hydroboration and Organic Synthesis: 9-Borabicyclo[3.3.1]nonane (9-BBN), Springer-Verlag, Berlin, 2007 Search PubMed.
- (a) T. G. Gant, J. Med. Chem., 2014, 57, 3595–3611 CrossRef CAS; (b) T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276–5297 CrossRef CAS PubMed.
- (a) H. Zong, H. Huang, J. Liu, G. Bian and L. Song, J. Org. Chem., 2012, 77, 4645–4652 CrossRef CAS PubMed; (b) Organometallics in Synthesis, ed. M. Schlosser, John Wiley & Sons, Inc., Hoboken, 2013 Search PubMed.
- (a) M. Hayashi, S. Bachman, S. Hashimoto, C. C. Eichman and B. M. Stoltz, J. Am. Chem. Soc., 2016, 138, 8997–9000 CrossRef CAS PubMed; (b) C. C. Chong, B. Rao and R. Kinjo, ACS Catal., 2017, 7, 5814–5819 CrossRef CAS.
- E. J. Corey and D. H. Lee, J. Am. Chem. Soc., 1991, 113, 4026–4028 CrossRef CAS.
- J. G. Verkade, R. W. King and C. W. Heitsch, Inorg. Chem., 1964, 3, 884–889 CrossRef CAS.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 16084–16087 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1988922, 1988923, 1988924, 1993289. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03118f |
| This journal is © The Royal Society of Chemistry 2020 |