
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformation control through concurrent N–H⋯S and N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C hydrogen bonding and hyperconjugation effects†
C hydrogen bonding and hyperconjugation effects†
Zeynab
Imani‡
 a,
Venkateswara Rao
Mundlapati‡
b,
Gildas
Goldsztejn§
b,
Valérie
Brenner
b,
Eric
Gloaguen
b,
Régis
Guillot
a,
Jean-Pierre
Baltaze
a,
Katia
Le Barbu-Debus
c,
Sylvie
Robin
ad,
Anne
Zehnacker
c,
Michel
Mons
*b and
David J.
Aitken
*a
a,
Venkateswara Rao
Mundlapati‡
b,
Gildas
Goldsztejn§
b,
Valérie
Brenner
b,
Eric
Gloaguen
b,
Régis
Guillot
a,
Jean-Pierre
Baltaze
a,
Katia
Le Barbu-Debus
c,
Sylvie
Robin
ad,
Anne
Zehnacker
c,
Michel
Mons
*b and
David J.
Aitken
*a
aInstitut de Chimie Moléculaire et des Matériaux d’Orsay (ICMMO), CNRS, Université Paris-Saclay, 91405 Orsay, France. E-mail: david.aitken@u-psud.fr
bLaboratoire Interactions, Dynamiques et Lasers (LIDYL), CEA, CNRS, Université Paris-Saclay, 91191 Gif-sur-Yvette, France. E-mail: michel.mons@cea.fr
cInstitut des Sciences Moléculaires d'Orsay (ISMO), CNRS, Université Paris-Saclay, 91405 Orsay, France
dFaculté de Pharmacie, Université de Paris, 75006 Paris, France
First published on 11th August 2020
Abstract
In addition to the classical N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C non-covalent interaction, less conventional types of hydrogen bonding, such as N–H⋯S, may play a key role in determining the molecular structure. In this work, using theoretical calculations in combination with spectroscopic analysis in both gas phase and solution phase, we demonstrate that both these H-bonding modes exist simultaneously in low-energy conformers of capped derivatives of Attc, a thietane α-amino acid. 6-Membered ring inter-residue N–H⋯S interactions (C6γ), assisted by hyperconjugation between the thietane ring and the backbone, combine with 5-membered ring intra-residue backbone N–H⋯OC interactions (C5) to provide a C5–C6γ feature that stabilizes a planar geometry in the monomer unit. Two contiguous C5–C6γ features in the planar dimer implicate an unprecedented three-centre H-bond of the type CO⋯H(N)⋯SR2, while the trimer adopts two C5–C6γ features separated by a Ramachandran α-type backbone configuration. These low-energy conformers are fully characterized in the gas phase and support is presented for their existence in solution state.
C non-covalent interaction, less conventional types of hydrogen bonding, such as N–H⋯S, may play a key role in determining the molecular structure. In this work, using theoretical calculations in combination with spectroscopic analysis in both gas phase and solution phase, we demonstrate that both these H-bonding modes exist simultaneously in low-energy conformers of capped derivatives of Attc, a thietane α-amino acid. 6-Membered ring inter-residue N–H⋯S interactions (C6γ), assisted by hyperconjugation between the thietane ring and the backbone, combine with 5-membered ring intra-residue backbone N–H⋯OC interactions (C5) to provide a C5–C6γ feature that stabilizes a planar geometry in the monomer unit. Two contiguous C5–C6γ features in the planar dimer implicate an unprecedented three-centre H-bond of the type CO⋯H(N)⋯SR2, while the trimer adopts two C5–C6γ features separated by a Ramachandran α-type backbone configuration. These low-energy conformers are fully characterized in the gas phase and support is presented for their existence in solution state.
Introduction
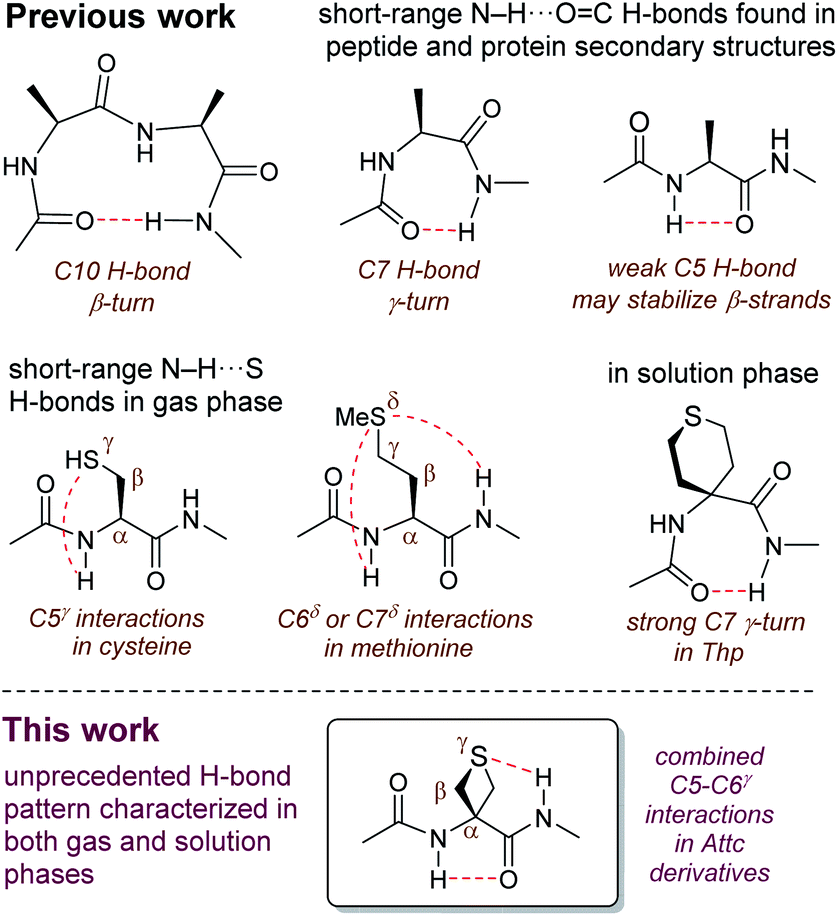
The hydrogen bond (H-bond) plays a supremely important role in determining the structure and the behaviour of countless molecules of all sizes.1 Chemists have harnessed hydrogen bonding as a tool, with applications ranging from selective synthesis,2 to crystal engineering3,4 to the design of self-organizing molecular and supramolecular architectures.5,6 Nature makes use of hydrogen bonding, along with other types of non-covalent interactions, to define and control not only the shapes of biomolecules but also the specific interactions between them.7–9 The most commonly observed (and the best studied) type of H-bond in proteins is the N–H⋯OC interaction between backbone amide functions, which stabilizes the familiar regular secondary structures of helices and sheets, as well as β-turns and γ-turns which are formed by cyclic 10-membered ring (C10) or cyclic 7-membered ring (C7) H-bonds (Fig. 1).10–12 Recently, there has been an increased awareness of the importance of contributions to secondary structure stabilization from other types of non-covalent interactions, including less conventional H-bonds such as intra-residue C5 H-bonds (Fig. 1),13–15 or C–H⋯O interactions,16,17 as well as phenomena such as n → π* hyperconjugative interactions,18 C-bonds,19 cation–π interactions,20,21 and others.22
 | ||
| Fig. 1 Background for this study. | ||
Reports of non-covalent interactions in proteins which specifically implicate the side-chain heteroatom of sulfur-containing residues are rare and do not involve H-bonds; rather, they take the form of sulfur–aromatic interactions23,24 or chalcogen bonds.25 Studies on small peptide systems have revealed that backbone–backbone H-bonding interactions may exist in thioamides.26,27 Nonetheless, it was generally assumed for some time that a side-chain thioether was a poor H-bond acceptor and that N–H⋯S interactions between amide backbones and the sulfur atom of methionine were weak and of little significance.28,29
Recent observations on model systems have challenged this credo, however. Gas phase spectroscopic studies supported by quantum calculations suggested a significant role for short-range C7δ inter-residue and C6δ intra-residue N–H⋯S interactions, implicating the sulfur in the δ-position of the side chain, in derivatives of methionine (Fig. 1).30 An assessment of the strength of these H-bonds found them to be comparable with those of classical N–H⋯OC interactions.31 Related studies showed that C5γ intra-residue N–H⋯S H-bonds, implicating the sulfur in the γ-position, may contribute to the non-covalent interactions on the conformational landscape of cysteine derivatives in the gas phase (Fig. 1).32,33 Solution state evidence for such interactions has been scarce. C5 and C6 H-bond interactions implicating sulfur have been suggested in non-peptide arylthioether oligomers.34,35 In early work on peptides, it was suggested that intra-residue C5γ N–H⋯S interactions may exist in short oligomers of S-methylcysteine,36 although this had no apparent impact on secondary structure, since the appearance of helical conformations was indicated for oligomers longer than the pentamer.37 Likewise, short oligomers of methionine (smaller than the hexamer) do not show any conformational preferences,38 while short peptide derivatives of 4-aminotetrahydrothiopyran-4-carboxylic acid (Thp) adopt standard C7 (or C10) conformers (Fig. 1) with no apparent role for sulfur.39,40
In order to probe the potential of short-range N–H⋯S interactions with a view to controlling model peptide edifices we examined derivatives of 3-aminothietane-3-carboxylic acid (Attc) (Fig. 2). In principle, we felt that Attc might support C5 or C6γ N–H⋯S interactions, although the geometrical constraints imposed by the 4-membered ring might result in a bias for one or other of these features. In the event, we have discovered that short oligomers of this α-amino acid (up to a trimer) adopt a previously unknown combined C5–C6γ interaction, featuring two types of H-bond concurrently, enhanced by hyperconjugation effects and leading to well-defined low-energy conformer shapes in gas and solution phases (Fig. 1).
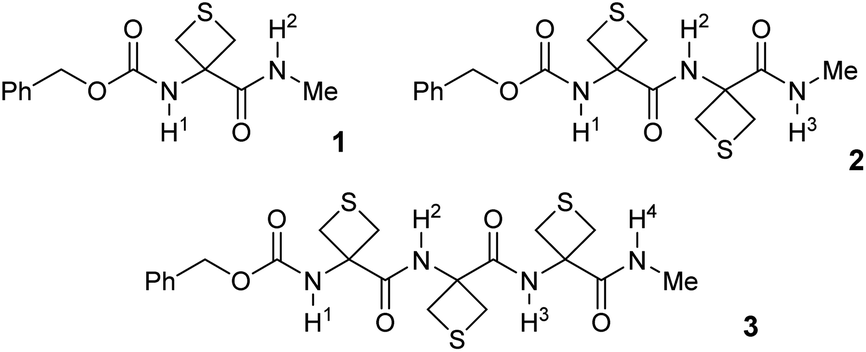 | ||
| Fig. 2 Structures of the compounds 1–3 used in this study. | ||
Methods
Attc was prepared by an adaptation of the literature method41 and its derivatives 1–3, each capped at the N-terminal with a benzylcarbamate group (Cbz-cap) and at the C-terminal as a methylamide, were prepared using standard solution phase procedures (see ESI Section 1 for syntheses†) (Fig. 2). Standard IR and NMR spectroscopic methods were used to perform solution state conformational analysis using dilute sample solutions in a low polarity solvent (chloroform) and solid state analysis using single crystal X-ray diffraction. Details are provided in the ESI document.†Experimental characterization focused on gas phase spectroscopy because in these conditions vibrationally resolved spectra can be obtained for systems composed of several residues, providing that care is taken to use a soft vaporisation technique along with an efficient rotational and vibrational cooling. This was achieved by combining a laser desorption set-up, bringing intact molecules into the gas phase, with the adiabatic expansion of a molecular jet.42 Furthermore using the sophisticated arsenal of gas phase laser techniques, which combines both UV and IR spectroscopy, namely the so-called IR/UV double resonance laser spectroscopy, conformational selectivity can be achieved.42 Using this strategy, conformer-specific IR spectra of each isolated compound 1–3 were obtained.
Comparison of the vibrationally resolved spectra with the theoretical IR spectra of low energy conformations allowed assignment of the observed conformers to specific conformations and thereby provided a detailed description of the intramolecular H-bond network of each conformation. The theoretical IR spectra were obtained by a three-step strategy combining several theoretical chemistry methods (see ESI Section 3†). First, exploration of the potential energy landscape of each compound was performed combining the OPLS-2005 force field with the Monte-Carlo multiple minima method.43 Second, geometry of the conformations selected during the exploration step was then optimized using the DFT-D quantum chemistry method (RI-B97-D3(BJ)-abc/def2-TZVPPD level).44–46 Finally, at the same level of theory, a frequency calculation was performed within the harmonic approximation for each optimized geometry. These calculations verified that the conformations corresponded to “true” minima and the harmonic frequencies derived were rescaled using scaling factors in order to generate theoretical IR spectra. In addition, detailed NBO analyses47–50 were performed for each conformation, leading to quantitative information on the strength of the intramolecular H-bonds as well as on hyperconjugation effects. These experimental and computational techniques have been described previously51–53 (details are given in ESI Sections 2 and 3†).
The Cbz-cap, which provides the UV chromophore required for the IR/UV double resonance spectroscopy, gives rise to rotamers (a phenomenon that we refer to as Cbz-rotamerism) which share essentially the same amide backbone and hence very similar, although distinct, IR spectra. Narrow UV spectral signatures were observed in the origin region of the first ππ* transition of the phenyl group (Fig. 3a), reflecting the rotational and vibrational cooling provided by the jet. Each was used in IR/UV double resonance spectroscopy to acquire conformer-selective IR spectra (see ESI Section 4.5†), whose analysis showed that the doublet bands, labelled A and A1, in compounds 1 and 2, were respectively consistent with origins and less intense vibronic bands (involving a low frequency torsional mode of the Cbz-cap) of the same Cbz-cap rotamer.
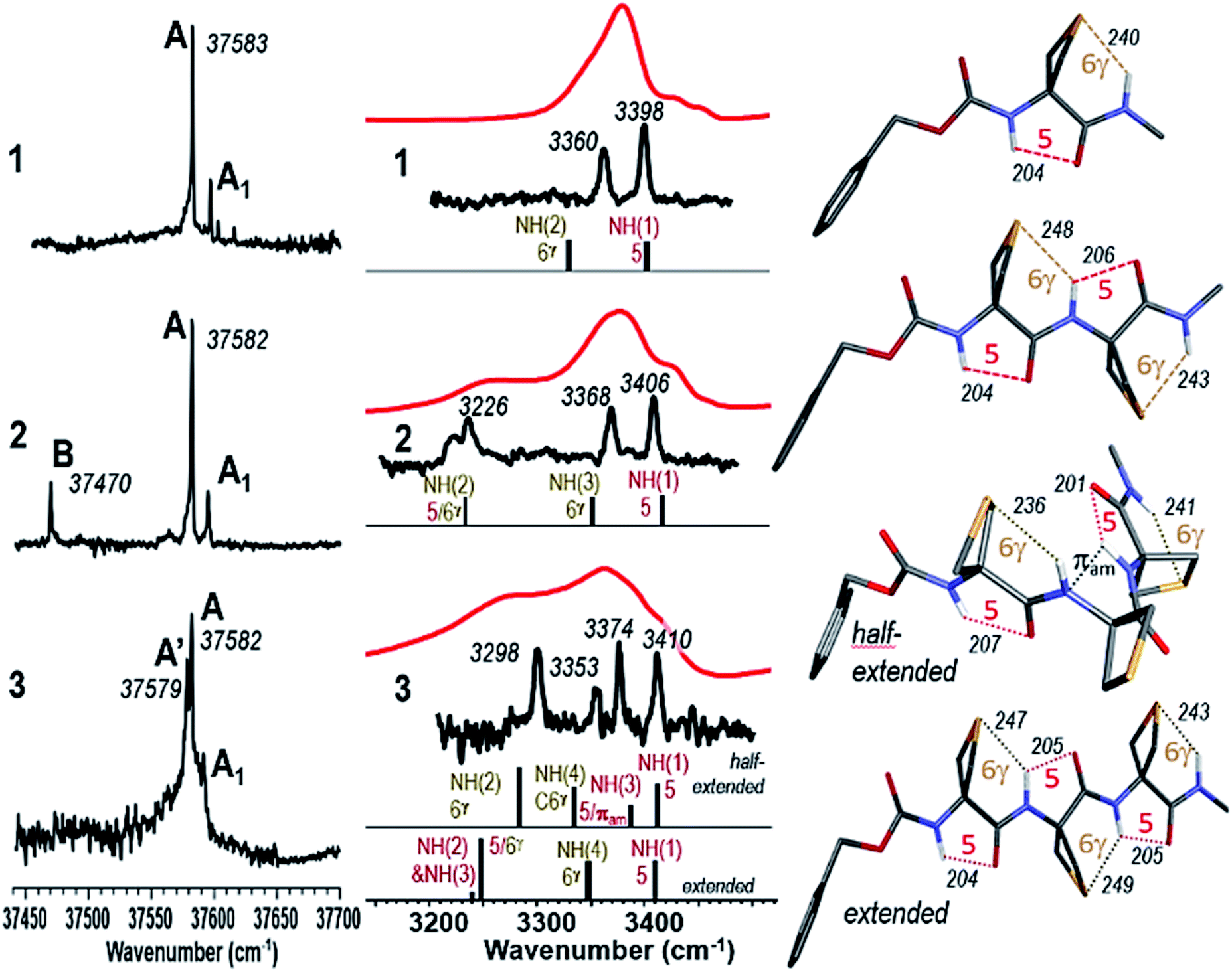 | ||
| Fig. 3 (left) Gas phase UV spectra of jet-cooled compounds 1–3. (centre) Gas phase IR spectra (black), obtained on UV bands A (1 and 2) or A′ (3), and solution phase spectra (red) of compounds 1–3, compared to the theoretical IR spectrum (stick) of the Cbz-cap trans rotamer of the lowest energy conformations. (right) Corresponding calculated lowest energy conformations of 1, 2, 3 (half-extended conformer) and 3 (extended conformer). | ||
This illustrated the efficient conformational cooling achieved in the supersonic expansion, which funnels the rotamer population into the most stable species. In the UV spectrum of compound 2, the red-shifted isolated band, labelled B, suggested a different interaction between the amide backbone and the side chain. Intriguingly, in 3, the complex feature observed nearly coincides with the doublets observed for compounds 1 and 2, with a splitting of the intense band into two components labelled A and A′.
The conformer-specific IR spectra collected from the several band systems (A, A′ and B) were assigned to the most stable conformations, obtained from the exploration of the potential energy landscape (see ESI Sections 4.1–4.3†) for which a best agreement was met between experimental and theoretical IR spectra, usually with discrepancies not larger than 20 cm−1 for each NH stretch band,52 and generally with an accuracy of 30 cm−1 in the case of N–H⋯S H-bonds.31 (see ESI Section 4 for complete sets of spectra.†)
Results and discussion
Conformer-specific NH stretch IR spectra of compound 1 revealed the presence of only one conformer family (ESI Section 4.1†), characterized by two strong vibrational bands at 3360 and 3398 cm−1, implicating two distinct H-bonds simultaneously (Fig. 3). Comparison with the most stable calculated conformations (see Table S2, Fig. S2 and S3†) enabled their respective assignments to a 6-membered ring N–H⋯S H-bond (designated C6γ, where the superscript refers to the γ-position occupied by the S atom in the side chain) implicating NH(2) and an intra-residue C5 N–H⋯OC interaction implicating NH(1), which together constitute a C5–C6γ structural feature characterized by an extended planar backbone (Fig. 3). The strengths of these H-bonds, as judged by the calculated H-bond interatomic distances (Table 1), were comparable with those observed in species where only one such interaction exists – the C5 form of Ac-Aib-NHMe53 and in the N–H⋯S intermolecular H-bond of the N-methylacetamide·SMe2 complex (see Fig. S4†) – suggesting the mutual compatibility of these interactions. NBO analysis of the stabilization energies induced by the H-bonding (Tables 1 and S5†) confirmed the unusual stabilization of the extended planar backbone (C5 H-bond) and emphasized the role of hyperconjugation effects between each of the Cα–Cβ bonds and vicinal covalent bonds or sites of the backbone, in particular that originating from the lone pair of N(1) which provides a stabilisation energy in the 40 kJ mol−1 range (see details in ESI section 4.4†).
| Compound | Residue | Distance NH(i)⋯O (pm) | Distance NH(i + 1)⋯S (pm) | Distance NH(i)⋯πam(i) (pm) | ϕ (°) | ψ (°) | E (2) C5 (kJ mol−1) | E (2) C6γ (kJ mol−1) | E (2) πam (kJ mol−1) |
|---|---|---|---|---|---|---|---|---|---|
| Ac-Aib-NHMe (C5)53 | 204 | 180 | 179 | 14.6 | |||||
| AcNHMe·SMe2 | 247 | 35.6 | |||||||
| 1 | 1 | 204 | 240 | 180 | 180 | 13.9 | 32.3 | ||
| 2 | 1 | 204 | 248 | 179 | 180 | 13.0 | 26.9 | ||
| 2 | 206 | 243 | 180 | 180 | 19.1 | 29.2 | |||
| 3 half-extended | 1 | 207 | 236 | — | 180 | 179 | 11.4 | 42.7 | — |
| 2 | — | — | 230 | 65 | 29 | — | — | 3.1 | |
| 3 | 201 | 241 | — | 174 | 177 | 18.6 | 31.6 | — | |
| 3 extended | 1 | 204 | 247 | 180 | 180 | 13.4 | 27.8 | ||
| 2 | 205 | 249 | 180 | 180 | 17.7 | 24.8 | |||
| 3 | 205 | 243 | 180 | 180 | 19.1 | 28.6 |
Conformer-specific NH stretch IR spectra of compound 2 revealed the presence of two conformer families (ESI Section 4.2†). The major family was assigned to the most stable calculated conformation (Fig. 3) which displayed an extended planar backbone composed of two successive C5–C6γ monomer motifs (designated C5–C6γ/C5–C6γ). This structure gives rise to two H-bonds in the same spectral range implicating NH(1) in a C5 interaction and NH(3) in a C6γ interaction, and a third which was considerably more red-shifted (at 3226 cm−1) implicating NH(2) which was engaged simultaneously in two contiguous C5–C6γ structures (Fig. 3). The bond distances (Table 1) showed significant elongation of the C6γ bonds, especially that involving NH(2), signalling some frustration. Nonetheless, in this conformation NH(2) was engaged in a three-centre H-bond of the type CO⋯H(N)⋯SR2, an H-bonding pattern which, as far as we are aware, is without precedent. NBO analysis of the significant stabilization energies (Tables 1 and S5†) confirmed the juxtaposition of two successive C5–C6γ monomer motifs, slightly weakened compared to 1 (see details in ESI Section 4.4†) and connected by a three-centre H-bond, with NH(2) as the double donor. The minor family, accounting for about 16% of the vaporized sample (B band in the UV spectrum of Fig. 3a), had a backbone which showed two successive γ-turns (Fig. S5 and S6†). It was found to be favoured by a dispersive contact between a Cbz-cap, in a gauche rotamer, and the C-terminus end of the backbone.
Conformer-specific NH stretch IR spectra of compound 3 revealed the presence of a single backbone family (ESI Section 4.3†), characterized by four H-bonds of various strengths spanning a spectral range of 120 cm−1 (Fig. 3). The theoretical conformational landscape (Fig. S7†) suggested that two nearly isoenergetic (ΔG = 2.2 kJ mol−1) low energy conformation families might exist, namely a half-extended (i.e. partly folded) conformer featuring two C5–C6γ motifs separated by a Ramachandran α-type backbone configuration (a local feature with a double donor role for NH(3), designated C5-πam, which has been observed only once before, in a stable conformation of a capped Aib dipeptide54), and an extended, planar C5–C6γ/C5–C6γ/C5–C6γ structure, perceived as a propagation of the structure adopted by 2. Comparison of experimental and theoretical IR spectra (Fig. 3) revealed that 3 adopts the half-extended structure, whose non-planar backbone furthermore accounts for the splitting of the UV features (A and A′ bands) due to a degeneracy lifting between gauche+ and gauche− Cbz-cap rotamers (see ESI Section 4.5†). The elongated N–H⋯S distances in the extended form of 3 compared to that in 1 (by 3–9 pm) suggested increased frustration in the former. In contrast, the NH(2)⋯S(1) distance in the half-extended form of 3 was shorter (by 4 pm) and the H-bond appeared commensurately stronger (by 11 kJ mol−1) than in 1 (Table 1). NBO analysis of the significant stabilization energies (Tables 1 and S5†) showed that the “missing” H-bond in the half-extended conformer was compensated by (i) an enhancement of the two remaining C6γ interactions, (ii) by specific hyperconjugation interactions from the thietane rings to the backbone, in particular those vicinal interactions not involving sulphur (see Table S5,† interactions HCα and HCβ) and (iii) more marginally by a weak interaction of NH(3) with the π system that implicates N(2) (see details in ESI Section 4.4†).
We looked for evidence for the C5–C6γ structural features in solution phase using IR and NMR spectroscopic techniques. The IR absorption spectra of compounds 1–3 were recorded in chloroform solution (5 mM) and showed no significant changes upon 10-fold dilution, pointing to the intramolecular nature of any non-covalent interactions contributing to the main spectral features. In all cases broad amide NH stretch bands were in evidence and, notwithstanding the lower spectral resolution, good overall correlation was observed with gas phase experimental and theoretical spectra for the lowest-energy conformations (Fig. 3). The IR spectrum of compound 1 showed one broad band centred at around 3375 cm−1 which we assigned to the overlap of NH(1) in a medium strength C5 interaction and NH(2) implicated in a C6γ feature. In compound 2 the NH(3) C6γ vibration remained at around 3370 cm−1 and the red-shifted NH(2) now appeared as a plateau at around 3250 cm−1. The appearance of a shoulder at 3410 cm−1 was attributed to an attenuated C5 interaction of NH(1). For compound 3 the IR spectrum was consistent with a weak C5 vibration for NH(1) at 3410 cm−1, C6γ features for NH(2) and NH(4), and some type of H-bonding for NH(3); these data were in good agreement with the half-extended conformation although they did not formally exclude the extended form. For comparison purposes, the derivative Cbz-Attc-OMe 4 was prepared and its IR spectrum recorded in chloroform (see ESI Sections 1.2 and 5.1†). With a now weaker H-bond acceptor than that in 1 (an ester instead of an amide), the main absorption band at 3440 cm−1 was assigned to a free NH vibration while a minor band at 3410 cm−1 indicated a contribution from a weak C5 H-bonded conformation. The absence of strong absorbance bands for free NH vibrations above 3420 cm−1 for compounds 1–3 suggested that conformations having no intramolecular H-bonds are not present. Likewise, conformations implicating larger-ring interactions, such as a C7 structure in 2 or C7 or C10 structures in 3, are of little or no significance, since these would be require at least one free NH group. Collectively, the IR data indicated that the C5–C6γ conformational feature was present for compounds 1–3 in solution, albeit with a weakened C5 interaction at the N-terminal in 2 and 3.
Further support was forthcoming from 1H NMR experiments on compounds 1–3 in solution in CDCl3 (5 mM) (ESI Section 5.2†). All signals were assigned using standard 1D and 2D pulse sequences and were well defined, suggesting the presence of one predominant conformation for each compound. In the spectrum of compound 1 both the carbamate NH(1) signal (δ = 6.45 ppm) and the amide NH(2) signal (δ = 7.98 ppm) were deshielded, suggesting their implication in H-bonds. Titration with DMSO-d6 induced a non-negligible downfield shift (Δδ = 0.60 ppm for 10% added DMSO) for NH(1), compatible with its involvement in a moderate strength C5 H-bond, while the upfield shift observed for NH(2) (Δδ = −0.68 ppm) suggested no solvent exposure, commensurate with it being H-bonded to sulfur. These data are in full agreement with the proposed C5–C6γ conformer for 1 (Fig. 1). In comparison, titration of a CDCl3 solution of ester 4 with DMSO-d6 induced a greater downfield shift of the NH signal (Δδ = 1.79 ppm for 10% added DMSO), pointing to extensive solvent exposure. In compound 2 the carbamate NH(1) was more shielded (δ = 6.23 ppm) than in 1. Amide NH(3) was deshielded (δ = 7.84 ppm) while amide NH(2) was strongly deshielded (δ = 8.95 ppm), this latter corresponding to a 3-centre H-bonded system. Titration with DMSO-d6 induced a considerable downfield shift (Δδ = 1.38 ppm) for NH(1), confirming the weakness of its C5 H-bond, while upfield shifts were observed for NH(2) and NH(3) (Δδ = −0.79 and −0.99 ppm respectively). Data for compound 3 were more difficult to interpret unequivocally but were fully compatible with the adoption of the combined C5–C6γ structural features suggested above. The carbamate NH(1) signal (δ = 6.26 ppm) was comparable to that of 2, while NH(4) was deshielded (δ = 7.52 ppm) and NH(2) and NH(3) were strongly deshielded (δ = 8.32 and 8.39 ppm). Titration with DMSO-d6 induced a downfield shift (Δδ = 1.89 ppm) for the weakly C5 H-bonded NH(1) and small downfield (or upfield) shifts for NH(2), NH(3) and NH(4) (Δδ = 0.30, −0.94 and −0.89 ppm, respectively).
A NOESY experiment carried out on compound 2 (ESI Section 5.3†) provided a correlation map that was in complete agreement with the C5–C6γ/C5–C6γ conformation. Medium cross peaks were observed between NH(i) and both CβH(a) protons for each Attc(i) and strong cross peaks were observed between NH(i + 1) and both CβH(b) protons for each Attc(i) (Fig. 4).
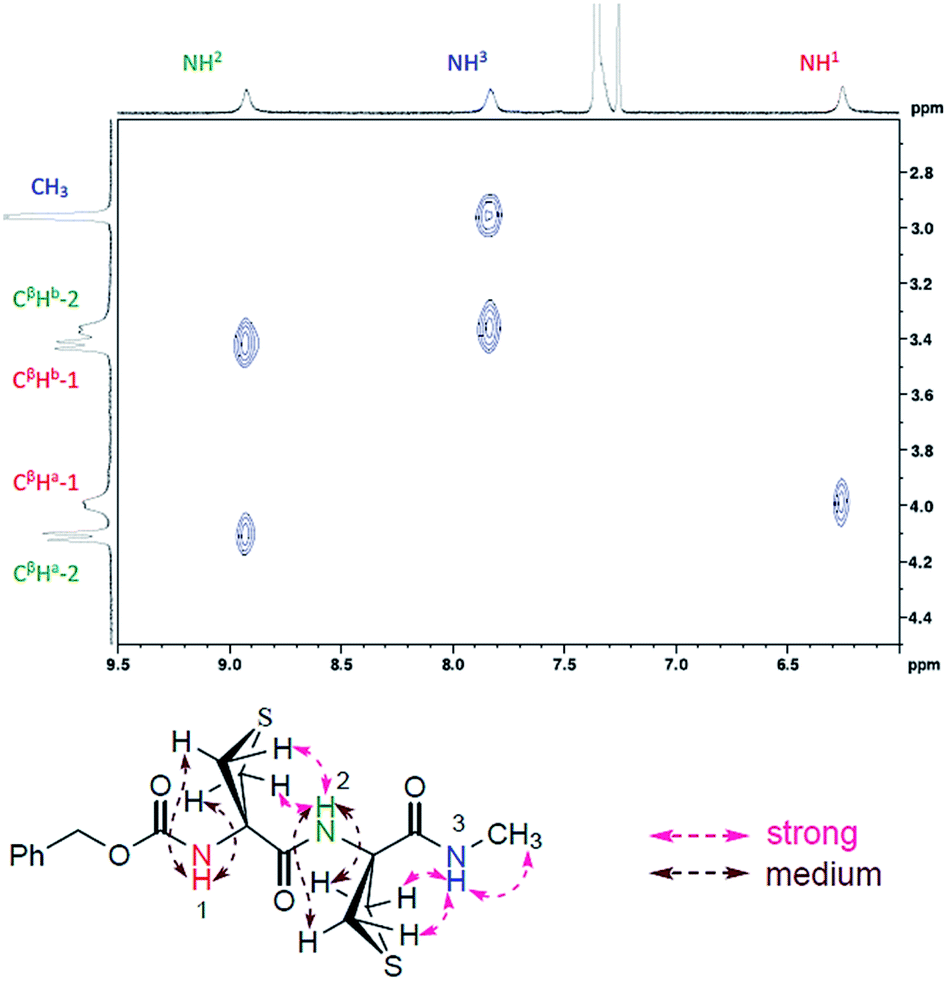 | ||
| Fig. 4 Significant NOESY correlations observed for compound 2 and their assignments. | ||
Compounds 1–3 were solids and for completeness we conducted X-ray diffraction analysis on single crystals. The C5–C6γ feature was not present in the crystal lattices; instead, strong intermolecular H-bonding networks were in evidence in all three cases (ESI Section 6†). The molecular structures of the three compounds are shown in Fig. 5. Compound 1 showed only intermolecular N–H⋯OC interactions in the lattice, implicating both NH and both carbonyl groups of each molecule. For compound 2 the asymmetric unit contained three molecules, each of which displayed an intramolecular C10 H-bonding interaction between NH(3) and the Cbz-cap CO, with N–H⋯OC distances of 219, 233 and 230 pm in the three distinct molecules. The crystal structure of compound 3 showed a conformation with two consecutive C10 interactions, which constituted a short 310-helix. NH(3) and NH(4) interacted with the Cbz-cap CO and the carbonyl of Attc(1), respectively, with N–H⋯OC distances of 208 and 218 pm, respectively. In all three crystal structures, the closest contacts for the sulfur atoms were with aromatic hydrogen atoms on the Cbz-cap phenyl rings of neighbouring molecules in the lattices, with H⋯S distances in the range 280–295 pm (ESI Section 6†). Such interactions, in which a thioether sulfur atom is located near the edge and slightly above the plane of the aromatic ring, have been observed before in crystal structures of proteins55 and of small molecules.56 The conformations adopted by 1–3 in the solid state are incompatible with the solution phase and gas phase data presented and discussed above.
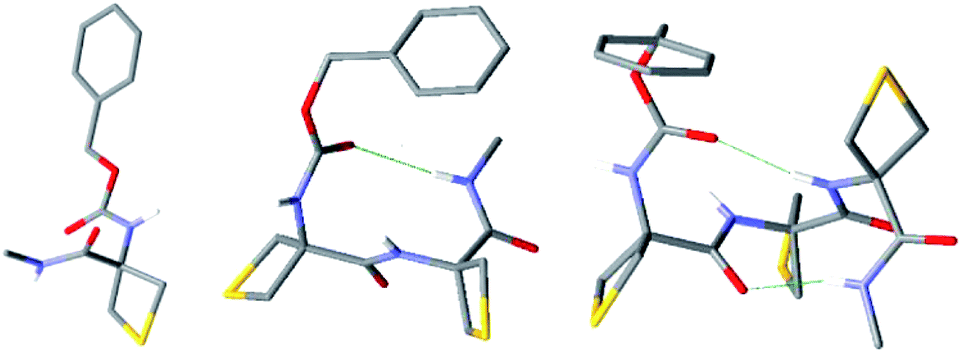 | ||
| Fig. 5 X-ray crystal structures of 1–3 (left to right). Only one of the three molecules in the unit cell is shown for 2. Intramolecular C10 H-bonding interactions are shown for 2 and 3. Only NH hydrogen atoms are shown for clarity. | ||
Conclusion
In summary, the unique properties of the thietane ring of Attc derivatives lead to the adoption of hyperconjugation-assisted C5–C6γ locally-planar structures in the gas phase and in solution phase, providing a new axiom for H-bonding. This discovery may facilitate the development of new tools for contributing to conformation control in small peptides, e.g. in the stabilization of extended backbone geometries57 or for the design of new peptidomimetic or foldamer architectures in a non-polar environment, and should provide a stimulus for further reflections on the significance of the less conventional N–H⋯S H-bond.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Ms Roxanne Berthin and Ms Anna Kriukova for their theoretical contribution to the conformational explorations. Support from the French National Research Agency (ANR; grant ANR-17-CE29-0008 “TUNIFOLD-S”) and from the “Investissements d'Avenir” Funding program (LabEx PALM; grant ANR-10-LABX-0039-PALM; DIRCOS) are acknowledged. This work was granted access to the HPC facility of [TGCC/CINES/IDRIS] under the grant 2019-A0050807540 awarded by GENCI (Grand Equipement National de Calcul Intensif) and to the CCRT High Performance Computing (HPC) facility at CEA under the grant CCRT2019-p606bren.Notes and references
- G. Gilli and P. Gilli, The Nature of the Hydrogen Bond. Outline of a Comprehensive Hydrogen Bond Theory, Oxford University Press, New-York, USA, 2009 Search PubMed.
- P. M. Pihko, Hydrogen Bonding in Organic Synthesis, Wiley-VCH, Weinhein, Germany, 2009 Search PubMed.
- A. D. Burrows, in Supramolecular Assembly Via Hydrogen Bonds I, ed. D. M. P. Mingos, 2004, vol. 108, pp. 55–95 Search PubMed.
- D. Braga, L. Maini, M. Polito and F. Grepioni, in Supramolecular Assembly Via Hydrogen Bonds II, ed. D. M. P. Mingos, 2004, vol. 111, pp. 1–32 Search PubMed.
- S. Hecht and I. Huc, Foldamers: Structure, Properties, and Applications, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed.
- Y. Lan, M. G. Corradini, R. G. Weiss, S. R. Raghavan and M. A. Rogers, Chem. Soc. Rev., 2015, 44, 6035–6058 RSC.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, New York, USA, 1999 Search PubMed.
- C. L. Perrin and J. B. Nielson, Annu. Rev. Phys. Chem., 1997, 48, 511–544 CrossRef CAS PubMed.
- G. A. Jeffrey and W. Sänger, Hydrogen Bonding in Biological Structures, Springer-Verlag, Berlin, Germany, 1991 Search PubMed.
- A. Karshikoff, Non-covalent Interactions in Proteins, Imperial College Press, London, UK, 2006 Search PubMed.
- E. N. Baker and R. E. Hubbard, Prog. Biophys. Mol. Biol., 1984, 44, 97–179 CrossRef CAS PubMed.
- G. D. Rose, L. M. Gierasch and J. A. Smith, Adv. Protein Chem., 1985, 37, 1–109 CrossRef CAS PubMed.
- C. Peggion, A. Moretto, F. Formaggio, M. Crisma and C. Toniolo, Biopolymers, 2013, 100, 621–636 CrossRef CAS PubMed.
- R. W. Newberry and R. T. Raines, Nat. Chem. Biol., 2016, 12, 1084 CrossRef CAS PubMed.
- M. Crisma, F. Formaggio, C. Aleman, J. Torras, C. Ramakrishnan, N. Kalmankar, P. Balaram and C. Toniolo, Pept. Sci., 2018, 110, e23100 CrossRef.
- Z. S. Derewenda, L. Lee and U. Derewenda, J. Mol. Biol., 1995, 252, 248–262 CrossRef CAS PubMed.
- S. Horowitz and R. C. Trievel, J. Biol. Chem., 2012, 287, 41576–41582 CrossRef CAS PubMed.
- G. J. Bartlett, A. Choudhary, R. T. Raines and D. N. Woolfson, Nat. Chem. Biol., 2010, 6, 615–620 CrossRef CAS PubMed.
- V. R. Mundlapati, D. K. Sahoo, S. Bhaumik, S. Jena, A. Chandrakar and H. S. Biswal, Angew. Chem., Int. Ed., 2018, 57, 16496–16500 CrossRef CAS PubMed.
- D. A. Dougherty, Science, 1996, 271, 163–168 CrossRef CAS PubMed.
- T. W. Craven, M. K. Cho, N. J. Traaseth, R. Bonneau and K. Kirshenbaum, J. Am. Chem. Soc., 2016, 138, 1543–1550 CrossRef CAS PubMed.
- R. W. Newberry and R. T. Raines, ACS Chem. Biol., 2019, 14, 1677–1686 CrossRef CAS PubMed.
- C. C. Valley, A. Cembran, J. D. Perlmutter, A. K. Lewis, N. P. Labello, J. Gao and J. N. Sachs, J. Biol. Chem., 2012, 287, 34979–34991 CrossRef CAS PubMed.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- M. Iwaoka, S. Takemoto, M. Okada and S. Tomoda, Bull. Chem. Soc. Jpn., 2002, 75, 1611–1625 CrossRef CAS.
- F. Formaggio, M. Crisma, C. Toniolo and C. Peggion, Eur. J. Org. Chem., 2013, 3455–3463 CrossRef CAS.
- V. R. Mundlapati, S. Gautam, D. K. Sahoo, A. Ghosh and H. S. Biswal, J. Phys. Chem. Lett., 2017, 8, 4573–4579 CrossRef CAS PubMed.
- L. M. Gregoret, S. D. Rader, R. J. Fletterick and F. E. Cohen, Proteins, 1991, 9, 99–107 CrossRef CAS PubMed.
- P. Zhou, F. F. Tian, F. L. Lv and Z. C. Shang, Proteins, 2009, 76, 151–163 CrossRef CAS PubMed.
- H. S. Biswal, E. Gloaguen, Y. Loquais, B. Tardivel and M. Mons, J. Phys. Chem. Lett., 2012, 3, 755–759 CrossRef CAS PubMed.
- V. R. Mundlapati, S. Ghosh, A. Bhattacherjee, P. Tiwari and H. S. Biswal, J. Phys. Chem. Lett., 2015, 6, 1385–1389 CrossRef CAS PubMed.
- B. Yan, S. Jaeqx, W. J. van der Zande and A. M. Rijs, Phys. Chem. Chem. Phys., 2014, 16, 10770–10778 RSC.
- M. Alauddin, H. S. Biswal, E. Gloaguen and M. Mons, Phys. Chem. Chem. Phys., 2015, 17, 2169–2178 RSC.
- H. Tang, R. J. Doerksen, T. V. Jones, M. L. Klein and G. N. Tew, Chem. Biol., 2006, 13, 427–435 CrossRef CAS PubMed.
- P. Du, X. K. Jiang and Z. T. Li, Tetrahedron Lett., 2009, 50, 320–324 CrossRef CAS.
- M. Palumbo, S. Darin, G. M. Bonora and C. Toniolo, Macromol. Chem. Phys., 1976, 177, 1477–1492 CrossRef CAS.
- C. Toniolo, G. M. Borona and A. Scatturin, Gazz. Chim. Ital., 1975, 105, 1063–1071 CAS.
- A. A. Ribeiro, M. Goodman and F. Naider, Int. J. Pept. Protein Res., 1979, 14, 414–436 CrossRef CAS PubMed.
- M. De Zotti and J. Clayden, Org. Lett., 2019, 21, 2209–2212 CrossRef CAS PubMed.
- I. Torrini, G. P. Zecchini, M. P. Paradisi, G. Lucente, G. Mastropietro, E. Gavuzzo, F. Mazza, G. Pochetti, S. Traniello and S. Spisani, Biopolymers, 1996, 39, 327–337 CrossRef CAS PubMed.
- A. P. Kozikowski and A. H. Fauq, Synlett, 1991, 783–784 CrossRef CAS.
- E. Gloaguen and M. Mons, Top. Curr. Chem., 2015, 364, 225–270 CrossRef CAS PubMed.
- Macromodel Schrödinger, LLC, New York, NY, USA, Schrödinger Release 2019-3, and references therein Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- F. Weinhold, J. Comput. Chem., 2012, 33, 2363–2379 CrossRef CAS PubMed.
- I. V. Alabugin, G. dos Passos Gomes and M. A. Abdo, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1389 Search PubMed.
- I. V. Alabugin, K. M. Gilmore and P. W. Peterson, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 109–141 CAS.
- E. Gloaguen, H. Valdes, F. Pagliarulo, R. Pollet, B. Tardivel, P. Hobza, F. Piuzzi and M. Mons, J. Phys. Chem. A, 2010, 114, 2973–2982 CrossRef CAS PubMed.
- E. Gloaguen and M. Mons, Top. Curr. Chem., 2015, 364, 225–270 CrossRef CAS PubMed.
- V. Brenner, E. Gloaguen and M. Mons, Phys. Chem. Chem. Phys., 2019, 21, 24601–24619 RSC.
- J. R. Gord, D. M. Hewett, A. O. Hernandez-Castillo, K. N. Blodgett, M. C. Rotondaro, A. Varuolo, M. A. Kubasik and T. S. Zwier, Phys. Chem. Chem. Phys., 2016, 18, 25512–25527 RSC.
- K. S. C. Reid, P. F. Lindley and J. M. Thornton, FEBS Lett., 1985, 190, 209–213 CrossRef CAS.
- R. J. Zauhar, C. L. Colbert, R. S. Morgan and W. J. Welsh, Biopolymers, 2000, 53, 233–248 CrossRef CAS PubMed.
- C. Toniolo, M. Crisma, F. Formaggio and C. Peggion, Biopolymers, 2001, 60, 396–419 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1985233–1985235. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03339a |
| ‡ These authors contributed equally to this work. |
| § Current address: ISMO, CNRS, Université Paris-Saclay, 91405 Orsay, France. |
| This journal is © The Royal Society of Chemistry 2020 |