
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multicomponent and multicatalytic asymmetric synthesis of furo[2,3-b]pyrrole derivatives: further insights into the mode of action of chiral phosphoric acid catalysts†‡
Lara
Cala§
 a,
Pedro
Villar§
b,
Ángel R.
de Lera
b,
Francisco J.
Fañanás
a,
Rosana
Álvarez
*b and
Félix
Rodríguez
*a
a,
Pedro
Villar§
b,
Ángel R.
de Lera
b,
Francisco J.
Fañanás
a,
Rosana
Álvarez
*b and
Félix
Rodríguez
*a
aInstituto Universitario de Química Organometálica “Enrique Moles”, Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universidad de Oviedo, Julián Clavería, 8, 33006-Oviedo, Spain. E-mail: frodriguez@uniovi.es
bDepartamento de Química Orgánica (CINBIO), Universidade de Vigo, As Lagoas-Marcosende, E-36310 Vigo, Spain. E-mail: rar@uvigo.es
First published on 12th August 2020
Abstract
Multicomponent and multicatalytic reactions are those processes that try to imitate the way the enzymatic machinery transforms simple building blocks into complex products. The development of asymmetric versions of these reactions is a step forward in our dream of mirroring the exquisite selectivity of biological processes. In this context, the present work describes a new reaction for the asymmetric synthesis of furo[2,3-b]pyrrole derivatives from simple 3-butynamines, glyoxylic acid and anilines in the presence of a dual catalytic system, formed from a gold complex and a chiral phosphoric acid. Computations, aimed to understand the exceptional performance of 9-anthracenyl-substituted BINOL-derived phosphoric acid catalyst, suggest a fundamental role of non-covalent interactions being established between the catalyst and the reagents for the outcome of the multicomponent process. The linear geometry of the anthracenyl substituent along with the presence of an electron-withdrawing group in the aniline and an aromatic substituent in the 3-butynamine derivative seem to be key structural factors to explain the experimental results and, particularly, the high stereoselectivity.
Introduction
Catalysis has become, without question, a major subject of modern chemistry. In traditional catalytic strategies, a single catalyst interacts with a reagent lowering the energetic barrier of the subsequent reaction with another reagent. Although this strategy has delivered vast numbers of new reactions over many decades, new directions in the field of catalysis are necessary in order to address the demands of sustainability and global well-being of our society. In this context, multicatalytic reactions (one-pot catalysis), defined as those processes where several reactions occur in a single flask as a result of the action of multiple catalyst, have become a powerful synthetic tool.1 On the other hand, multicomponent reactions, which are those processes where several starting materials react together to yield a product that retains the majority of atoms of the reactants, have also become of significant interest in the field of organic synthesis.2 As a result of merging both tools, multicomponent and multicatalytic reactions have recently emerged as one of those new concepts in the area of catalysis.3 In these processes, three or more starting materials react by means of two or more catalysts (all present from the onset of the reaction) to produce structurally and functionally complex products (Scheme 1a). These reactions try to mimic the way the enzymatic machinery (multicatalytic system) transforms in Nature a series of simple molecules (multicomponent system) into intricate structures. The exquisite stereoselectivity of biosynthetic reactions is particularly difficult to imitate and thus, the development of asymmetric multicomponent and multicatalytic reactions remains challenging.4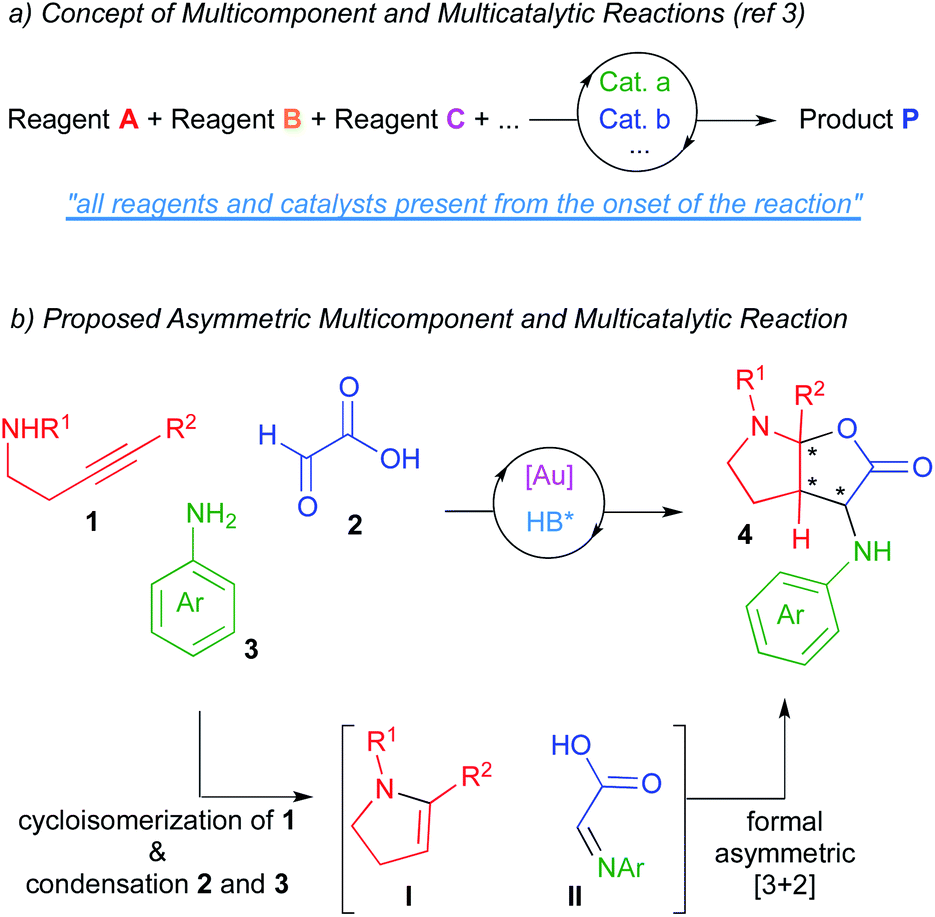 | ||
| Scheme 1 Multicomponent and multicatalytic reactions. Concept and our proposal. | ||
In this context and based on previous works form our laboratories,5 we envisioned that the multicomponent coupling of alkynamines 1, glyoxylic acid 2 and anilines 3 in the presence of a multicatalytic gold/chiral Brønsted acid system could deliver enantioenriched furo[2,3-b]pyrrole derivatives 4 comprising three contiguous stereocenters (one of them quaternary; Scheme 1b). We thought that the gold-catalysed cycloisomerization reaction of alkynamines 1 should deliver the enamine intermediates I.6 Moreover, the condensation reaction between glyoxylic acid and amines 3 should give imines II. Further asymmetric reaction between these in situ formed intermediates would lead to our desired furo[2,3-b]pyrrole derivatives 4, which are targets of interest owing to the prevalence of this structural motif in numerous biologically pharmacophores and natural products.7
In addition to reporting the development of this process, we are also presenting computational studies aimed to structurally justify the origin of the very high diastereo and enantioselectivities observed with just a single catalyst of the series of phosphoric acids surveyed, thus providing new vistas to rationalize the behaviour of chiral phosphoric acids in asymmetric processes.8
Results and discussion
At the outset of this study, we selected tert-butyl[4-(4-chlorophenyl)-3-butyn-1-yl]carbamate 1a, glyoxylic acid 2 and 3-nitroaniline 3a as model reagents in order to find optimal reaction conditions. As multicatalytic system, we chose a combination of AuMe(JohnPhos) and a Brønsted acid (HB; particularly, phosphoric acid derivatives). Initial experiments were performed at room temperature, in toluene as solvent and in the presence of 4 Å molecular sieves (Table 1).|
|
|||||
|---|---|---|---|---|---|
| Entry | HB | Yielda (%) | d.r. (4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diast-4a)b :diast-4a)b |
e.r. (4a)c | e.r. (diast-4a)c |
| a Based on 1a. b Determined by 1H NMR analysis of the crude reaction mixture. c Determined by HPLC using a chiral stationary phase. | |||||
| 1 | DPP | 91 | 1:1.5 |
— | — |
| 2 | CPAA | 88 | 1.3:1 |
86:14 |
36:64 |
| 3 | CPAB | 91 | 1.3:1 |
49:51 |
32:68 |
| 4 | CPAC | 95 | 1:1 |
74:26 |
50:50 |
| 5 | CPAD | 92 | 1.9:1 |
99:1 |
47:53 |
| 6 | CPAE | 87 | 1:1.4 |
19:81 |
4:96 |
| 7 | CPAF | 98 | >20:1 |
99:1 |
— |
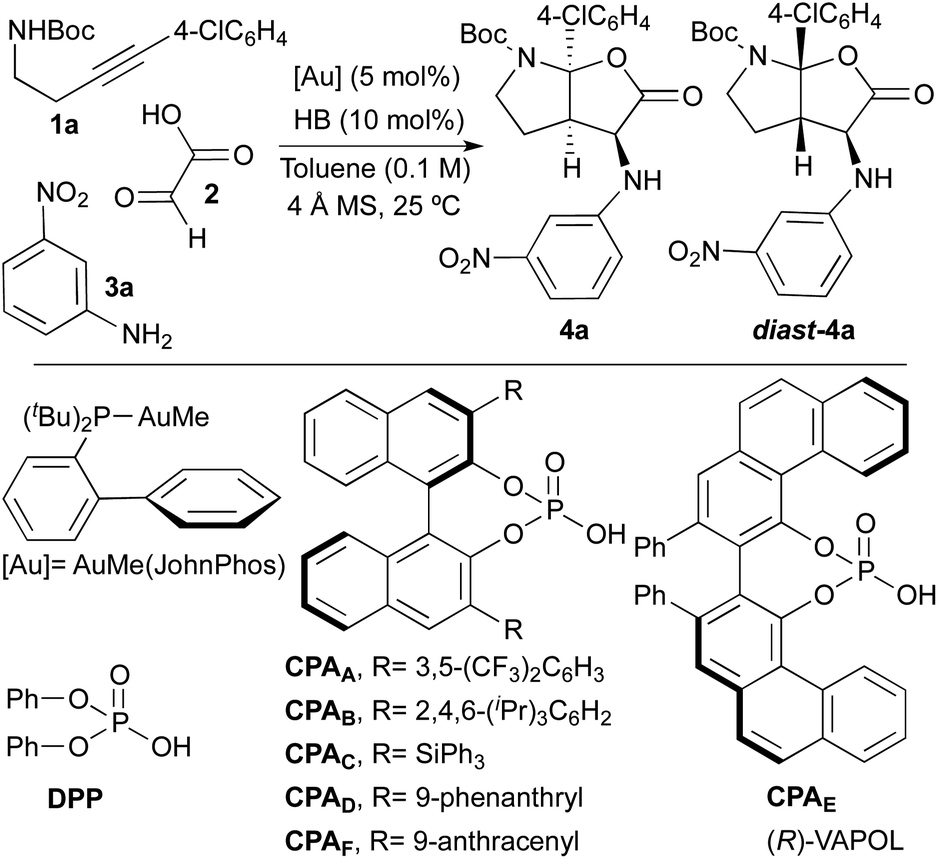
To check the viability of our proposal, we firstly tried the reaction in its racemic version by using diphenyl hydrogen phosphate [DPP, (PhO)2PO2H] as the acid catalyst. Although the reaction led to the desired product in high yield (91%), unfortunately, an almost equimolar mixture of the two diastereoisomers 4a and diast-4a was obtained (d.r. = 1:1.5; Table 1, entry 1). Considering that our objective was the development of a truly selective reaction (diastereo and enantioselective), this preliminary result was highly discouraging since we suspected that the use of chiral phosphoric acid-derived catalysts (CPAs) to perform the asymmetric version of the reaction, could in turn lead to the same diastereoselectivity problems. In fact, when we tried the reaction with BINOL-derived phosphoric acids substituted at C3- and C3′-positions with 3,5-bis(trifluoromethyl)phenyl (CPAA), 2,4,6-triisopropylphenyl (CPAB) or triphenylsilyl (CPAC) groups, we also observed the formation of roughly equimolar amounts of the two diastereoisomers 4a and diast-4a (Table 1, entries 2–4). Furthermore, the low levels of enantioselectivity found in both 4a and diast-4a were additional discouraging issues at this point.
Although the same problems of diastereoselectivity were observed when using the phenanthryl-substituted BINOL-derivative CPAD and the VAPOL-derived phosphoric acid CPAE (d.r. = 1.9:1 and 1:1.4 respectively; Table 1, entries 5 and 6), at least in these cases one of the diastereoisomers was obtained with high enantioselectivity [e.r.(4a) = 99:1 with CPAD; e.r.(diast-4a) = 96:4 with CPAD]. Nevertheless, none of the above mentioned phosphoric acid catalysts led to satisfactory results in terms of selectivity. Surprisingly, when the reaction was performed with the 9-anthracenyl-substituted BINOL-derived phosphoric acid CPAF, we only observed the formation of compound 4a in almost quantitative yield (98%) and, more importantly, with complete stereoselectivity (d.r. > 20:1; e.r. = 99:1).
The extraordinary performance of catalyst CPAF should be remarked. Thus, while all other acid catalysts (achiral or chiral) produced basically equimolecular mixtures of two diastereoisomers, CPAF proved unique in performing this transformation in a highly diastereoselective way. The very high enantioselectivity achieved with this catalyst (only one enantiomer was observed) should also be noted. Thus, from the eight possible stereoisomers, we only observed the formation of one of them.
Once we had verified that the 9-anthracenyl-substituted BINOL-derived phosphoric acid CPAF was an optimal acid catalyst to perform the desired asymmetric multicomponent and multicatalytic process, we addressed the scope of the transformation. Thus, a range of experiments were performed by mixing different 4-arylbut-3-yn-1-amines 1, glyoxylic acid 2 and aniline derivatives 3 in toluene (0.1 M) with 4 Å molecular sieves at room temperature in the presence of a mixture of AuMe(JohnPhos) (5 mol%) and chiral phosphoric acid CPAF (10 mol%; Scheme 2). We were delighted to find that, under these conditions, a series of furo[2,3-b]pyrrole derivatives 4 with diverse substitution patterns were obtained in excellent yields, as single diastereoisomers and with very high enantioselectivities in most cases. Alkynamines 1 substituted at the triple bond with different aromatic rings likewise performed efficiently and led to the corresponding products 4 with very high selectivity.9 Regarding the substitution on the nitrogen atom, tert-butoxycarbonyl (4a–h) and methoxycarbonyl groups (4i–k) were used. Anilines 3 bearing electron-withdrawing substituents were appropriate substrates. However, the reaction did not proceed when simple aniline or aniline derivatives containing electron-donating groups were used. The reaction with alkylamine derivatives instead of anilines was unsuccessful. The structure and absolute configuration of 4a was unambiguously determined by X-ray crystallographic analysis.10 The absolute configuration of the remaining furo[2,3-b]pyrrole derivatives 4 was assigned by analogy.
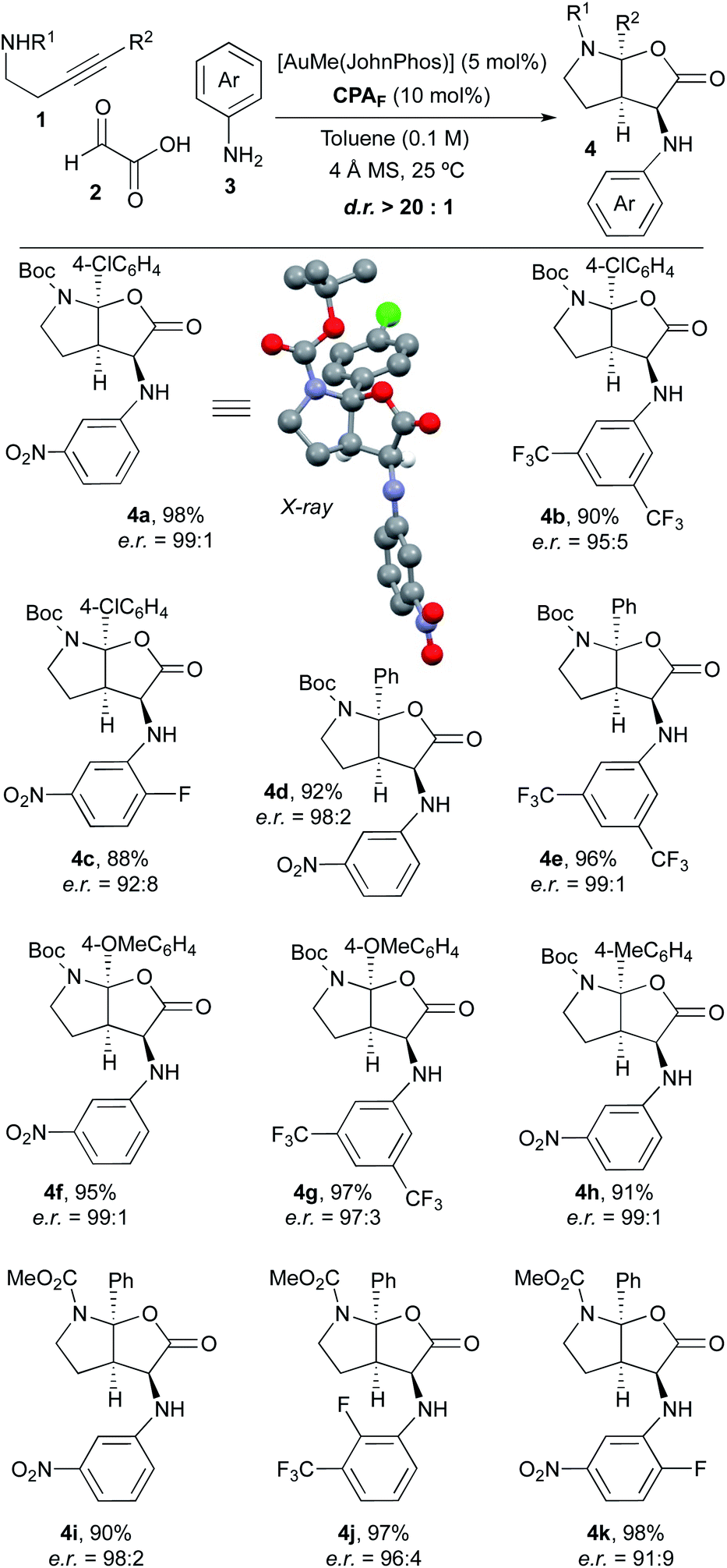 | ||
| Scheme 2 Scope of the reaction. | ||
A mechanistic proposal to explain this multicomponent and multicatalytic reaction is shown in Scheme 3. We considered the initial cycloisomerization of amine derivative 1 to give the enamine 7 in a process promoted by the cationic gold(I) complex generated from AuMe(JohnPhos) by reaction with the Brønsted acid (DPP or CPA). Thus, the initial coordination of the gold catalyst to the triple bond of 1 leads to intermediate 5. Intramolecular addition of the amino group to the distal carbon of the activated alkyne generates intermediate 6. Finally, a protodemetallation process, likely assisted by the anion (B−), affords the enamine derivative 7 regenerating the gold catalyst and closing the first catalytic cycle. Simultaneously, the Brønsted acid catalyst (HB) promotes the condensation of glyoxylic acid 2 and aniline derivative 3 to form imine 8.
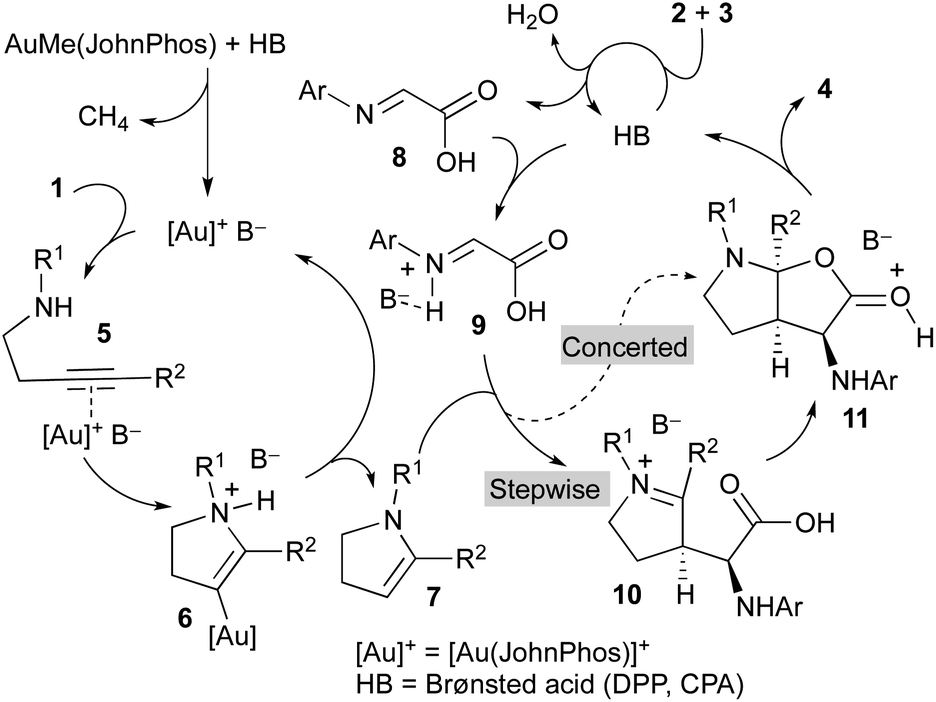 | ||
| Scheme 3 Mechanistic proposal. | ||
Both intermediates generated in the two previous independent processes, namely the N-protected 2,3-dihydro-1H-pyrrole 7 and the 2-(arylimino)acetic acid 8, enter a third catalytic cycle where the three stereogenic centres of the final product 4 are stereoselectively generated. Being both components achiral, the chiral Brønsted acid catalyst (HB) should be involved in this process. Thus, reaction of the imine 8 with the acid affords activated imine derivative 9. The subsequent reaction of this intermediate with enamine 7 could occur through a concerted mechanism to directly deliver intermediate 11, which evolves providing the final product 4 and regenerating the acid catalyst. Alternatively, intermediate 11 could be formed through a stepwise process initiated by a Mannich-type addition to produce iminium ion intermediate 10 followed by a cyclization reaction. Regardless of the mechanism, concerted or stepwise, the extraordinary efficiency of the chiral acid catalyst CPAF should be remarked because it favours the reaction through a single face of the activated imine 9 and a single face of the enamine 7 to form all three stereogenic centres of the final product 4 with exquisite selectivity.
The high specificity exhibited by catalyst CPAF is probably the most remarkable feature of the reaction here described. With this catalyst, we observed very high diastereo and enantioselectivities. On the contrary, basically equimolecular mixtures of diastereo and/or enantiomers were observed with the other catalysts used (Table 1). Although variations in stereoselectivity values are usually observed upon changing the substituents at the 3- and 3′-positions of BINOL-derived phosphoric acids (CPA), the contrasting differences we observed are unusual. Apparently, the specificity exhibited by the 9-anthracenyl-substituted BINOL-derived phosphoric acid CPAF cannot be just attributed to steric effects because other catalysts containing large groups at 3- and 3′-positions, i.e. the triphenylsilyl-substituted CPAC, did not perform satisfactorily. It is also surprising that phenanthryl-substituted chiral phosphoric acid CPAD, apparently very similar to CPAF, behaves so differently. Intrigued, we wondered why the 9-anthracenyl-substituted BINOL-derived phosphoric acid CPAF was so specific, and we considered performing a computational study to find the particular features of this catalyst in our reaction. In a more general sense, we also pursued with this investigation to better understand the behaviour of BINOL-derived phosphoric acids in asymmetric processes.
In this context, the reaction of methyl 5-phenyl-2,3-dihydro-1H-pyrrole-1-carboxylate 7a and imine 8a derived from the condensation of glyoxylic acid and 3,5-bis(trifluoromethyl)phenylaniline was selected as model to perform DFT calculations (Scheme 4).11 This model reaction, with both reagents in their more stable s-trans conformation, mimics the third catalytic cycle of the mechanism shown in Scheme 3 where all stereocenters of the final product 4 are generated.
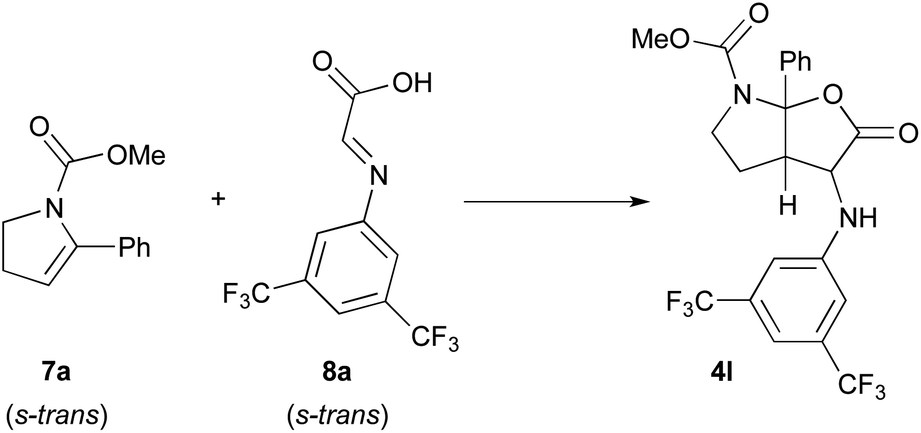 | ||
| Scheme 4 Model reaction selected for the computational studies. | ||
Initially, the reaction with the achiral acid catalyst dimethyl hydrogen phosphate (DMP; Scheme 5) was studied. Formation of a binary complex (aDC), in which the acid interacts with the imine 8a in the s-trans conformation through two hydrogen bonds in a bifunctional activation fashion, was considered to be the most likely. Two alternative mechanisms, namely concerted and stepwise, for the reaction of the binary complex aDC with enamine derivative 7a to give the final racemic products rac-4l or rac-diast-4l were evaluated. For the concerted [3 + 2]-cycloaddition mechanism, and prior to bond formation, we considered the formation of two possible three-component complexes (aTCendo and aTCexo), which would be created when aDC and 7a approach each other. These two three-component complexes differ by the orientation of the phenyl group of the enamine 7a when facing aDC [inwards (endo) or outwards (exo) the catalytic region]. Formation of rac-4l or rac-diast-4l from aTCexo or aTCendo proceeds through transition states aTSexo or aTSendo, respectively. Despite our efforts, only the concerted exo-approach could be characterized, and transition state aTSexo was found to be highly asynchronous with a computed activation energy of 15.9 kcal mol−1 (for details see the ESI‡). Detailed structural analysis of aTSexo allowed to identify a stabilizing CArH–π interaction (3.12 Å), the so-called T-shaped edge-to-face interaction (Scheme 5; blue strand in aTSexo).12 Taking into account that this arene–arene interaction is electrostatic, the presence of the trifluoromethyl substituents contributes to decrease the electron density of the CAr–H bonds favouring the mentioned interaction. We also noticed that the orientation of the 3,5-bis(trifluoromethyl)phenyl group appears to be controlled by hydrogen bonding interactions between one of the hydrogen atoms at the ortho-position and one of the oxygen atoms of the phosphoric acid (2.23 Å, Scheme 5, green strand in aTSexo).
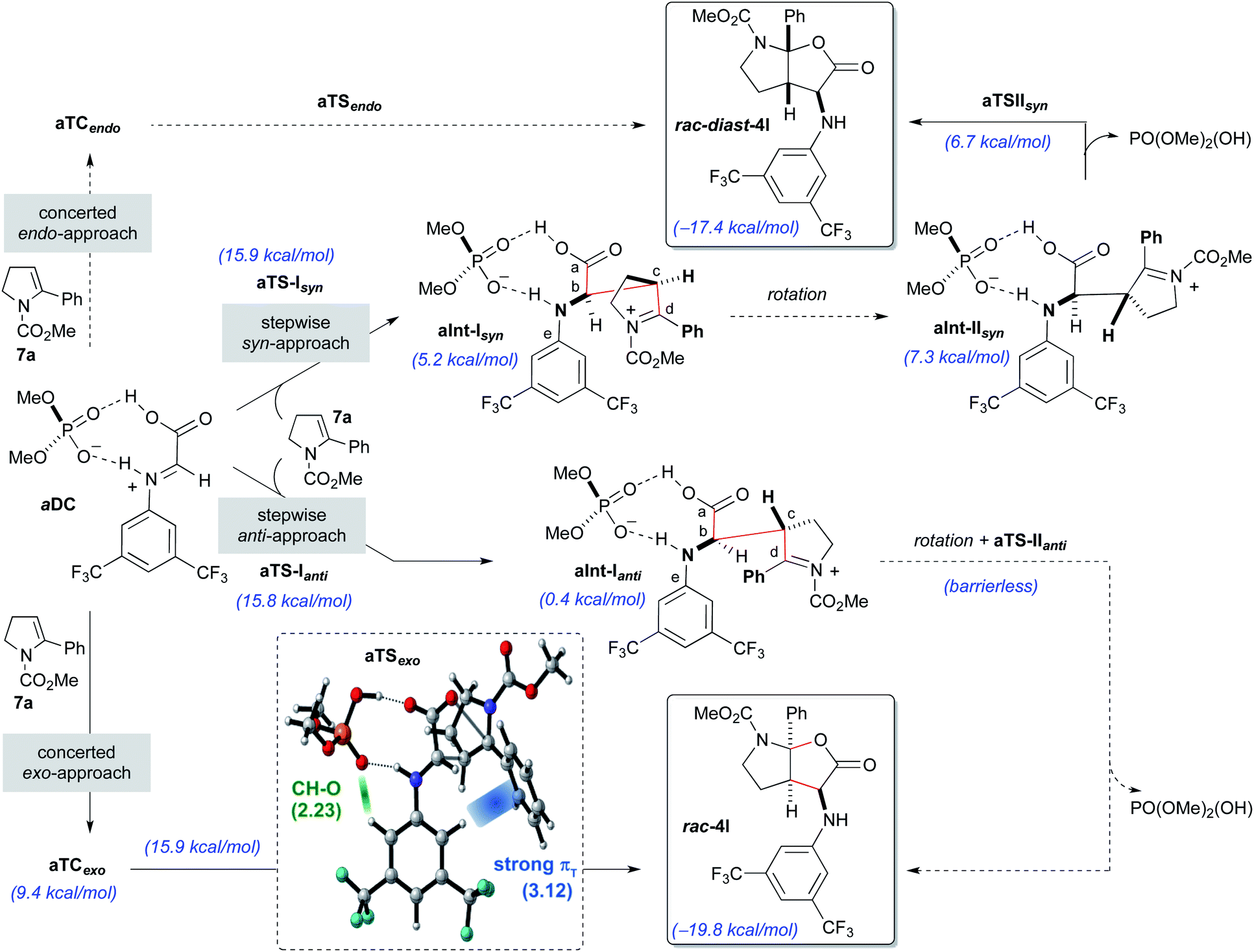 | ||
| Scheme 5 DMP-promoted reaction of 8a and 7a by concerted and stepwise mechanisms computed at the [wB97XD/DEF2TZVPP(SMD,toluene)//wB97XD/DEF2SVPP(SMD,toluene)] level of theory. Transition state aTSexo with distances indicated in Å, computed at the level shown above, is highlighted. | ||
In contrast, the endo-approach does not allow similar arene–arene stabilizing interactions and that might explain why we were unable to find the alternative concerted endo-approach pathway.
The stepwise mechanism for the [3 + 2]-cycloaddition could take place through two alternative orientations of the enamine 7a relative to the binary complex aDC (syn- or anti-approaches; Scheme 5). The reaction was considered to start with the stereodetermining nucleophilic addition of enamine 7a through its C3-position (Cc) to the electrophilic carbon (Cb) of the iminium ion of the aDC complex. Different orientations of the enamine 7a and complex aDC, as well as the s-cis and s-trans conformations of the imine 8a were taken into consideration. Several transition states, which showed different conformations around the dihedral angle CaCbCcCd and the s-cis and s-trans conformers of the carbamate, were optimized and turned out to be almost isoenergetic (ΔΔG = 2.0–0.1 kcal mol−1). The lowest energy transition state for each approach (aTS-Isyn and aTS-Ianti) were selected and found to have very similar activation energies (15.9 and 15.8 kcal mol−1 respectively). Once the first intermediates aInt-Isyn and aInt-Ianti are formed (note that these intermediates are similar to 10 in Scheme 3), a conformational change by bond rotation of dihedral angles CaCbCcCd and CaCbNCe should occur to allow the nucleophilic attack of the carboxylate onto the iminium ion (Scheme 5). Initially, we assumed that the energy required for the conformational change from aInt-I to aInt-II could not compete with the energy required for the C–C bond formation.
For the syn approach, we were able to characterize the intermediate aInt-IIsyn and the transition state aTS-IIsyn corresponding to the formation of the C–O bond. However, the energy of this transition state (6.7 kcal mol−1) is lower than that of intermediate aInt-IIsyn (7.5 kcal mol−1), suggesting that, under standard conditions, the second bond formation is a barrierless process. Interestingly, for the alternative anti-approach, the corresponding intermediate aInt-IIanti and the subsequent C–O bond formation transition state aTS-IIanti could not be characterized and we observed the direct formation of the final product from aInt-Ianti in a barrierless process. Most likely, the proximity of the electrophilic iminium carbon (Cd) and the carboxylate favours the collapse and fast formation of the C–O bond.
Considering the global study, the rate-determining steps of the processes going through aTSexo (15.9 kcal mol−1), aTS-Ianti (15.8 kcal mol−1) and aTS-Isyn (15.9 kcal mol−1) were isoenergetic and therefore, a low anti/syn diastereoisomeric ratio was predicted (d.r. = 2.1:1) for the formation of the furo[2,3-b]pyrrole derivatives rac-4l or rac-diast-4l. This is in complete agreement with our experimental results (see Table 1, entry 1).
This initial study performed with the achiral catalysts dimethyl hydrogen phosphate (DMP) served not only to verify the agreement between the computational results and the experimental data but also to show that our reaction could proceed through alternative concerted and/or stepwise mechanisms. With this information in hand, we faced the more challenging study on the origin of the diastereo and enantioselectivity of the formal [3 + 2]-cycloaddition when performed with the chiral 9-anthracenyl-substituted BINOL-derived phosphoric acid catalyst CPAF. More precisely, we evaluated the reaction between the enamine derivative 7a and a chiral binary complex (cDC), itself formed by the interaction of the imine 8a with CPAF. The two possible diastereomeric chiral binary complexes Re-cDC and Si-cDC, generated by the interaction of chiral acid CPAF and imine 8a (Fig. 1) were then analysed. These two complexes differ by the facial orientation of the imine towards the phosphoric acid. Whereas in Re-cDC, the Re-face of the imine is available for the subsequent reaction with enamine 7a, in Si-cDC is the Si-face. In both cases, the imine is stabilized by hydrogen bonding interactions through a bifunctional activation mode and by parallel-displacement (PD)–π-stacking interactions. Binary complex Si-cDC was found to be 2.7 kcal mol−1 more stable than Re-cDC. The greater relative stabilization of Si-cDC is likely due to the more effective overlap of the anthracenyl π-orbitals of the catalyst and the aryl fragment of the imine (PD-type; blue and green strands in Fig. 1). As shown, the distance between the 3,5-bis(trifluoromethyl)phenyl and the fused benzene rings of the anthracenyl group is shorter for Si-cDC relative to Re-cDC (3.68 and 3.81 vs. 3.95 Å, respectively). Additionally, the bis(trifluoromethyl)phenyl group of the imine appears to be conformationally restrained due to the hydrogen bonding interactions established between one of the hydrogen atoms at the ortho-position and one of the oxygen atoms of the phosphoric acid (with distances of 2.39 Å for Si-cDC and 2.44 Å for Re-cDC).
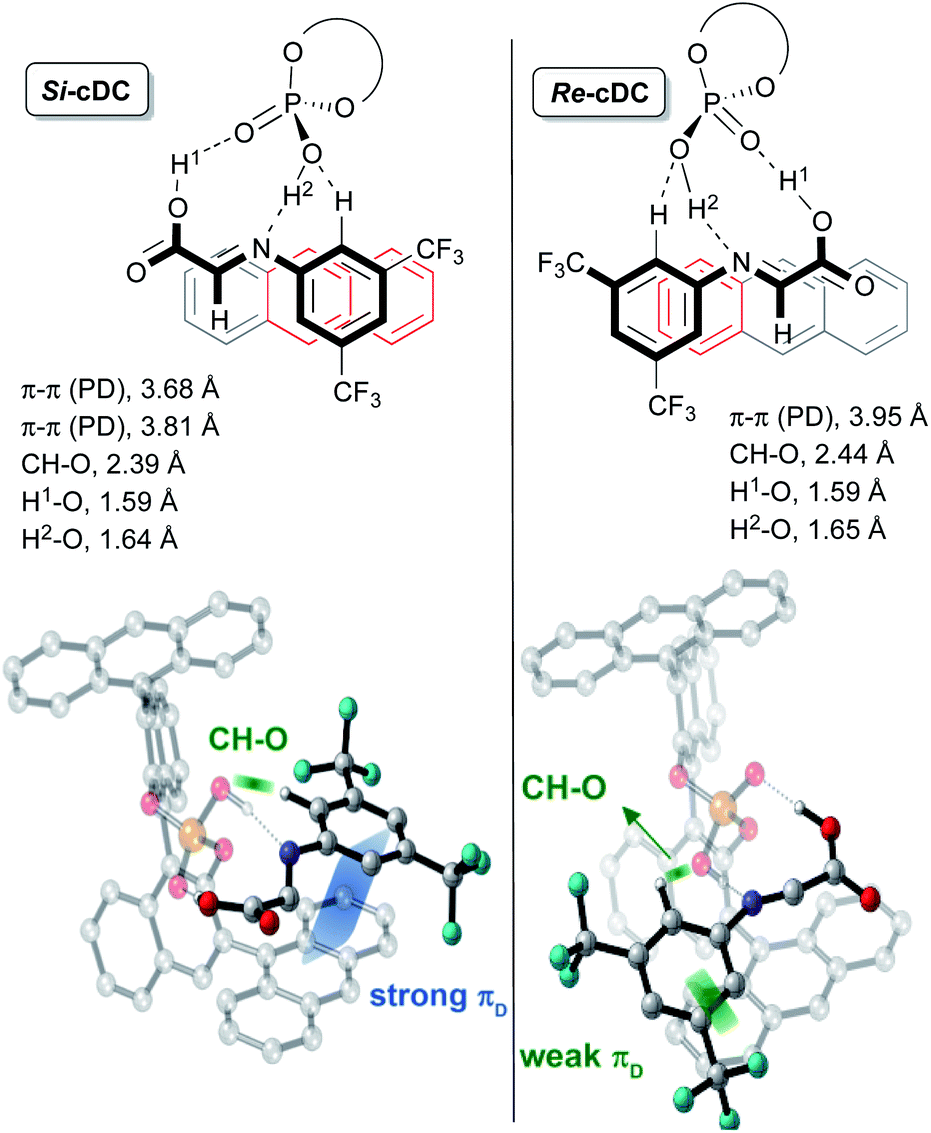 | ||
| Fig. 1 Representation of binary complexes Si-cDC and Re-cDC computed at the wB97XD/DEF2TZVPP(SMD,toluene)//wB97XD/DEF2SVPP (SMD,toluene) level of theory for (R)-3,3′-bis(9-anthracenyl)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate as CPAF. | ||
The coplanar orientation of the imine and the anthracenyl group, favoured by the PD–π interactions established between the latter and the 3,5-bis(trifluoromethyl)phenyl group, contributes to shield one of the enantiotopic faces of the imine, thus leaving the opposite one fully accessible for the reaction with enamine derivative 7a.13 These non-covalent interactions were evaluated using the NCI-index based on electron density described by Yang and co-workers,14 and we found that they were particularly relevant for the binary complex Si-cDC (for details, see the ESI‡).
The particular geometry of the anthracenyl group with three rings arranged in a linear sequence appears to be critical to the stabilization of the binary complexes, and particularly of Si-cDC, by the above-mentioned stabilizing π-interactions. The extraordinary results we obtained with anthracenyl-substituted BINOL-derived phosphoric acid CPAF could be associated to this unique structural feature of the anthracenyl group.15 For the sake of comparison, we performed a similar analysis of the binary complexes derived from other chiral phosphoric acids with different aryl substituents, in particular the 2,4,6-triisopropylphenyl-substituted and the 9-phenanthryl-substituted BINOL-derived phosphoric acids (CPAB and CPAD, respectively). For these skeletons, the non-covalent interactions appear to be less important and the computed energy values predicted a poor to modest stereoselectivity for the formation of the final product, in agreement with our experimental results (for details, see the ESI‡).
We had experimentally found that electron-withdrawing groups at the aryl ring of the imine (i.e. at the starting aniline 3) were required in order to obtain good results. This observation was computationally addressed by evaluation of the previously mentioned π-stacking interactions of the binary complexes as a function of the substituents at the imine aryl ring. In this regard, it has been previously suggested that the π-stacking interaction is enhanced when one of the aromatic rings contains electron-withdrawing substituents because the associated decrease of the electron density of the aromatic ring diminishes the electrostatic repulsion between the aryl groups.16 Our computations confirmed that the absence of the trifluoromethyl substituents led to a weakened π-interaction between the anthracenyl and the aryl group of the imine (for details, see ESI‡). Specifically, we evaluated the electron density distribution of Si-cDC (with a 3,5-bis(trifluoromethyl)phenyl group) in comparison with that of Si-cDC_Ph (with a phenyl group). This study allowed us to confirm the superior electronic complementarity when the 3,5-bis(trifluoromethyl)phenyl group was involved. This can be easily observed in the molecular electrostatic potential (MESP) representation shown in Fig. 2.17
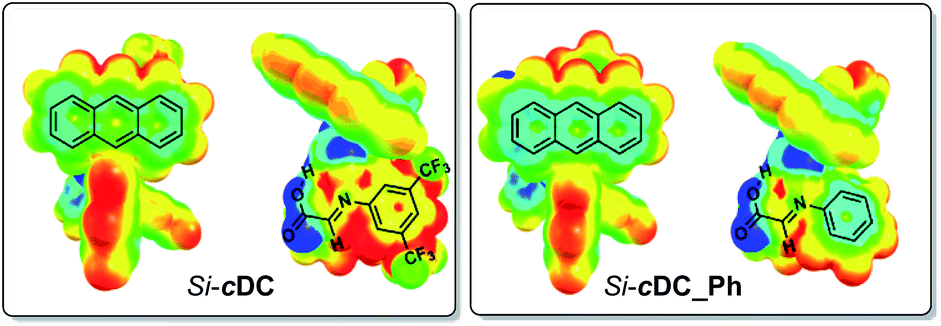 | ||
| Fig. 2 Representation of computed molecular electrostatic potential (MESP) of binary complex Si-cDC and Si-cDC_Ph. Colour codes: −0.04 a.u. (blue colour) to 0.04 a.u. (red colour) (computations at the wB97XD/DEF2SVPP(SMD,toluene) level). | ||
Once the binary complexes formed by coordination of the imine 8a to the 9-anthracenyl-substituted BINOL-derived phosphoric acid CPAF were analysed, we addressed their subsequent [3 + 2]-cycloaddition reactions with enamine derivative 7a (Fig. 3). Since 7a can react with binary complexes Re-cDC and Si-cDC through its Re- or Si-face, four possible ternary complexes (Re,Re-cTC, Si,Si-cTC, Si,Re-cTC and Re,Si-cTC) were characterized. Considering our previous studies with the achiral dimethyl hydrogen phosphate (DMP; see Scheme 5), we expected to be able to analyse the evolution of these intermediates through the two possible mechanistic options, namely the concerted and the stepwise. However, despite having characterized several transition states for the first bond formation of the stepwise mechanism, both the size and the structural rigidity of the chiral pocket of phosphoric acid CPAF hindered the conformational changes required for the formation of the C–O bond in the second step of the process. Thus, the stepwise pathway was discarded for this complex system, and we focused on the concerted mechanism.
 | ||
| Fig. 3 (A) Reaction free energy profiles (in kcal mol−1) and representation of transitions states for the concerted pathway of the [3 + 2]-cycloaddition (Ar = 3,5-(CF3)2–C6H3) catalysed by CPAF computed at the wB97XD/DEF2TZVPP(SMD,toluene)//wB97XD/DEF2SVPP(SMD,toluene) level. The ReSi (green), SiRe (red), ReRe (blue) and SiSi (purple) paths are shown. CPAF has been removed for clarity. (B) Representation of computed transition states for the four approaches (irrelevant hydrogen atoms have been removed for clarity). | ||
For this mechanism, two transition states for each of the four facial approaches (ReRe, ReSi, SiRe and SiSi), which differ by the conformation of the carbamate (s-cis or s-trans), were characterized. Although we were able to locate all the transition states, only the results with the more stable conformer will be shown (i.e. Re,Re-cTS, Si,Si-cTS, Si,Re-cTS and Re,Si-cTS; Fig. 3).
Interestingly, the approaches of 7a to the most stable binary complex Si-cDC leading to 4l or diast-4l were favoured over the alternative pathways involving Re-cDC (Fig. 3). Thus, the (ReSi)-cTS and (SiSi)-cTS (13.4 and 16.2 kcal mol−1, respectively) were found to be highly favoured when compared to the diastereoisomeric (SiRe)-cTS and (ReRe)-cTS transition states (19.1 and 22.5 kcal mol−1, respectively). The strongest π-interactions between one of the anthracenyl groups and the aryl group of imine 8 in (ReSi)-cTS and (SiSi)-cTS when compared to the same interaction in (SiRe)-cTS and (ReRe)-cTS seems to justify the preferential pathways leading to 4l and diast-4l. In other words, it seems that the highly stabilizing π-interactions we had observed in the binary complex Si-cDC, structurally associated to the particular geometry of the anthracenyl group, are maintained along the course of the reaction, contributing to further stabilize the corresponding transition states (ReSi)-cTS and (SiSi)-cTS.
As in the achiral model system, the global processes shown in Fig. 3A are highly exergonic, being the reaction energy values of −17.5, −19.6, −17.5 and −22.2 kcal mol−1 for ent-diast-4l, diast-4l, ent-4l and 4l, respectively. From the transition state energies computed for this multicomponent reaction, the Curtin–Hammett principle predicts that the expected diastereomeric ratio 4l/diast-4l would be ca. 99:1, and the enantiomeric ratio 4l/ent-4l would be >99:1, which is in excellent agreement with the experimental results.
In our reaction, the diastereoselectivity (formation of 4l or ent-4lversusdiast-4l or ent-diast-4l) is determined by the orientation of the enamine 7a upon reaction with the binary complexes Si-cDC or Re-cDC (endo or exo approaches). As shown, transition states (SiSi)-cTS and (ReRe)-cTS, in which the phenyl group of enamine 7a is oriented towards the catalytic pocket (a formal endo-cycloaddition), are destabilized relative to the (ReSi)-cTS and (SiRe)-cTS (a formal exo-cycloaddition) due to severe steric interactions between the phenyl substituent of the enamine 7a and one of the anthracenyl groups of the catalyst. Thus, it seems that the steric factors play an important role in determining the diastereoselectivity of the reaction.
At this point, the activation strain model was used to provide further semi-quantitative insight into the stereoinduction and the computed energies are shown in Table 2.18 These results provide additional support to the assumption that diastereoselectivity is determined by the distortion in the transition state, favoring those that show lower values. This is reflected in ΔΔEstrain for (ReRe)-cTS and (SiSi)-cTS, namely 2.92 and 1.76 kcal mol−1, respectively, which is related to the above-mentioned steric interactions between the phenyl group of 7a and the anthracenyl fragment.
| ΔΔE‡a | ΔΔEstrainb | ΔΔEintc | |
|---|---|---|---|
| a ΔΔE‡ is the activation energy relative to the lower energy transition state (Re,Si)-cTS. b ΔΔEstrain is the relative total distortion energy of the transition states. c ΔΔEint is the relative interaction energy of the transition states calculated with the corrected energies. | |||
| (Re,Re)-cTS | 9.51 | 2.92 | 2.34 |
| (Si,Re)-cTS | 6.33 | 0.64 | 1.45 |
| (Si,Si)-cTS | 2.62 | 1.76 | 0.87 |
| (Re,Si)-cTS | 0.00 | 0.00 | 0.00 |
The enantioselectivity of the process (formation of 4lversusent-4l, or of diast-4lversusent-diast-4l), appears to mainly be determined by electronic factors. Thus, (ReSi)-cTS (13.4 kcal mol−1) leading to 4l is favoured over (SiRe)-cTS (19.1 kcal mol−1) affording ent-4l) due to the stronger π-interactions noticed in the former and being established between one of the anthracenyl groups and the 3,5-bis(trifluoromethyl)phenyl group of the imine 8 (see Fig. 3). The same trend can be noticed when comparing (SiSi)-cTS (16.2 kcal mol−1) leading to diast-4l with (ReRe)-cTS (22.5 kcal mol−1) generating ent-diast-4l (see Fig. 3). As previously noted, those strong π-interactions can ultimately be associated to the particular linear geometry of the anthracenyl group.
Finally, some other particular features of transition state (ReSi)-cTS that leads to 4l should be remarked. In addition to the previously commented strong π-interaction between one of the anthracenyl groups and the 3,5-bis(trifluoromethyl)phenyl group of the imine 8, another strong T-shaped edge-to-face CArH–π interaction between the 3,5-bis(trifluoromethyl)phenyl group of imine 8 and the phenyl group of the enamine 7a was observed (see Fig. 3B). This is in agreement with our experimental observation that showed that an aryl substituent at the terminal position of the alkyne of amine 1 (eventually, an aryl substituent in enamine 7) was required in order to get good results.9
To further analyse all these non-covalent interactions, we studied the topological distribution of electron density in the optimized geometries of the four transition states using QTAIM (see the ESI‡).19 For the transition state of lower activation energy, namely (Re,Si)-cTS, two additional interactions between 7a and Si-cDC (c and g, Fig. 4) could be identified along with those already noted before (a, b and d–f, Fig. 4). In particular, the trifluoromethyl substituent favors the Hortho–π interactions. The ρBCP for π–π and H–X (X is N or O) interactions were found to be in the range of 0.014–0.005 au and 0.093–0.046 au respectively. For the lowest energy transition state, the π–π interaction between the imine and the anthracenyl fragment becomes 0.006 au. This is consistent with the lower ΔΔEint predicted for this transition state using the activation strain model (Table 2). In contrast, the lower number of non-covalent interactions predicted by QTAIM for the less favored transition state (Re,Re)-cTS (see the ESI‡), translates into higher ΔΔEint (2.34 kcal mol−1; Table 2).
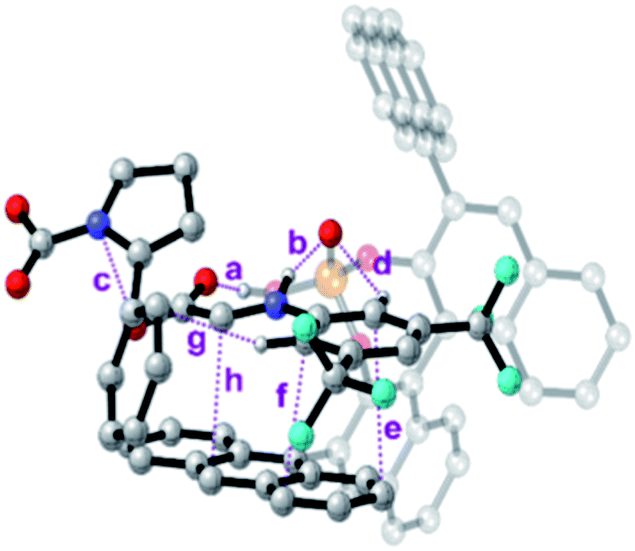 | ||
| Fig. 4 Bond critical points for the most important non-covalent interactions characterized (QTAIM) on the (Re,Si)-cTS. | ||
Overall, the computational study indicates that the preferential formation of compounds 4 over the alternative isomers (ent-4, diast-4 and ent-diast-4) appears to be a summation of key factors related to the specific structure of the anthracenyl-substituted BINOL-derived phosphoric acid catalyst CPAF and the reagents. Thus, the particular geometry of the anthracenyl group of the catalyst along with the presence of electron-withdrawing groups in the starting aniline allows the optimization of parallel-displacement π-interactions. This stabilization is less important with alternative enantiopure phosphoric acids having a shorter bicyclic aromatic ring (i.e., naphthyl) or ortho-fused (i.e. phenanthryl) or analogues with other substituents (i.e., 2,4,6-triisopropylphenyl or triphenylsilane). The rigidity and steric hindrance of the anthracenyl groups of the catalyst likely contributes to the diastereoselectivity of the process, since it determines the size of the catalytic pocket. In this regard, the steric interactions between the aryl group of enamine 7 when approaching the binary complex formed between the catalyst and the imine 8 seems to be determinant for the diastereoselectivity of the reaction. Finally, the excellent enantioselectivity of our process appears to be associated to the strong π-interactions established between the aryl group of imine 8 with one of the anthracenyl substituents of the CPAF catalyst and also with the aryl substituent of enamine 7.
Conclusions
We have developed a distereo and enantioselective synthesis of furo[2,3-b]pyrrole derivatives by means of a multicomponent and multicatalytic process. Thus, the reaction of a 3-butynamine derivative with glyoxylic acid and an aniline derivative in the presence of a dual catalytic system, formed by a gold complex and chiral BINOL-derived phosphoric acid, led to the formation of the corresponding furo[2,3-b]pyrrole derivative in very high yield and stereoselectivity. These products, containing three contiguous stereocenters, were selectively obtained only when the BINOL-derived phosphoric acid catalyst was substituted at 3- and 3′-positions with anthracenyl groups (CPAF). The extraordinary and unique performance of this catalyst was computationally studied in order to further understand the mode of action of chiral phosphoric acids in asymmetric processes. This study further supports the crucial influence of non-covalent interactions, established between the chiral phosphoric acid and the reagents, on the control of the stereoselectivity of asymmetric reactions. These interactions seem to be at least as important as the usually referred steric effects. In the reaction here described, features intimately related to those non-covalent interactions such as the linear geometry of the anthracenyl group of the catalyst, the electron density of the aniline, the electronic complementarity of aromatic rings involved in van der Waals interactions or the presence of an aryl substituent in the starting 3-butynamine derivative seem to be essential in order to justify the excellent results in terms of yield, diastereo and enantioselectivity experimentally observed. The work here presented not only demonstrates the power of asymmetric multicomponent and multicatalytic reactions to transform simple materials into complex products, but also provides new vistas to better understand how chiral phosphoric acids operate in asymmetric processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from Spanish Ministerio de Ciencia e Innovación (grant CTQ2016-76794-P and FPU-predoctoral grant to L. C.), Consejería de Ciencia, Innovación y Universidad del Principado de Asturias (grant IDI/2018/000231) and Xunta de Galicia (Consolidación GRC ED431C2017/61 from DXPCTSUG; ED-431G/02-FEDER “Unha maneira de facer Europa” to CINBIO, a Galician research center 2016–2019), We also thank Centro de Supercomputación de Galicia (CESGA) for generous allocation of computing resources.Notes and references
- For a review on one-pot catalysis, see: (a) N. T. Patil, V. S. Shinde and B. Gajula, Org. Biomol. Chem., 2012, 10, 211–224 RSC. For the use of multicatalytic systems comprising a metal complex and a chiral organocatalyst, see: (b) Z. Shao and H. Zhang, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC; (c) C. C. Loh and D. Enders, Chem.–Eur. J., 2012, 18, 10212–10225 CrossRef CAS PubMed; (d) H. Pellissier, Tetrahedron, 2013, 69, 7171–7210 CrossRef CAS; (e) Y. Deng, S. Kumar and H. Wang, Chem. Commun., 2014, 50, 4272–4284 RSC; (f) S. M. Inamdar and N. T. Patil, Chem. Commun., 2014, 50, 15124–15135 RSC; (g) D.-F. Chen, Z.-Y. Han, X.-L. Zhou and L.-Z. Gong, Acc. Chem. Res., 2014, 47, 2365–2377 CrossRef CAS PubMed.
- (a) Multicomponent Reactions, ed. J. Zhu and H. Bienaymé, Wiley-VCH, Weinheim, 2005 Search PubMed. For a review, see: (b) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390–2431 CrossRef CAS PubMed.
- A. Galván, F. J. Fañanás and F. Rodríguez, Eur. J. Inorg. Chem., 2016, 1306–1313 CrossRef.
- For some examples of Asymmetric Multicomponent and Multicatalytic Reactions, see: (a) S. Chercheja and P. Eilbracht, Adv. Synth. Catal., 2007, 349, 1897–1905 CrossRef CAS; (b) C. Wang, Z.-Y. Han, H.-W. Luo and L.-Z. Gong, Org. Lett., 2010, 12, 2266–2269 CrossRef CAS PubMed; (c) H. Wu, H.-P. He and L.-Z. Gong, Org. Lett., 2013, 15, 460–463 CrossRef CAS PubMed; (d) L. Cala, A. Mendoza, F. J. Fañanás and F. Rodríguez, Chem. Commun., 2013, 49, 2715–2717 RSC; (e) J. Calleja, A. B. González-Pérez, A. R. de Lera, R. Álvarez, F. J. Fañanás and F. Rodríguez, Chem. Sci., 2014, 5, 996–1007 RSC.
- For an account, see: (a) F. Rodríguez and F. J. Fañanás, Synlett, 2013, 24, 1757–1771 CrossRef. See also reference 4e and the following: (b) J. Barluenga, A. Mendoza, F. Rodríguez and F. J. Fañanás, Angew. Chem., Int. Ed., 2009, 48, 1644–1647 CrossRef CAS PubMed; (c) A. Galván, J. Calleja, F. J. Fañanás and F. Rodríguez, Angew. Chem., Int. Ed., 2013, 52, 6038–6042 CrossRef PubMed; (d) A. Galván, J. Calleja, A. B. González-Pérez, R. Álvarez, A. R. de Lera, F. J. Fañanás and F. Rodríguez, Chem.–Eur. J., 2015, 21, 16769–16774 CrossRef PubMed; (e) A. Galván, A. B. González-Pérez, R. Álvarez, A. R. de Lera, F. J. Fañanás and F. Rodríguez, Angew. Chem., Int. Ed., 2016, 55, 3428–3432 CrossRef PubMed.
- For some representative reviews on gold catalysis, see: (a) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed; (b) C. Obradors and A. M. Echavarren, Acc. Chem. Res., 2014, 47, 902–912 CrossRef CAS PubMed; (c) A. Fürstner, Acc. Chem. Res., 2014, 47, 925–938 CrossRef PubMed; (d) L. Fensterbank and M. Malacria, Acc. Chem. Res., 2014, 47, 953–965 CrossRef CAS PubMed; (e) A. S. K. Hashmi and F. D. Toste, Modern Gold Catalyzed Synthesis, Wiley-VCH, Weinheim, 2012 CrossRef; (f) M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448–2462 RSC; (g) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208–3221 RSC; (h) S. M. A. Sohel and R.-S. Liu, Chem. Soc. Rev., 2009, 38, 2269–2281 RSC; (i) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS PubMed; (j) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239–3265 CrossRef CAS PubMed; (k) A. Arcadi, Chem. Rev., 2008, 108, 3266–3325 CrossRef CAS PubMed; (l) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed; (m) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS PubMed; (n) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef PubMed; (o) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed.
- (a) A. Z. M. Saleh, K. L. Greenman, S. Billings, D. L. Van Vranken and J. J. Krolewski, Biochemistry, 2005, 44, 10822–10827 CrossRef CAS PubMed; (b) D. Yamamoto, T. Sunazuka, T. Hirose, N. Kojima, E. Kaji and S. Omura, Bioorg. Med. Chem. Lett., 2006, 16, 2807–2811 CrossRef CAS PubMed; (c) D. Barak, A. Ordentlich, D. Stein, Q.-S. Yu, N. H. Greig and A. Shafferman, Biochem. J., 2009, 417, 213–222 CrossRef CAS PubMed; (d) S. S. Soman and T. H. Thaker, Med. Chem. Res., 2013, 22, 4223–4227 CrossRef CAS; (e) Y. Li, S. Zhu, J. Li and A. Li, J. Am. Chem. Soc., 2016, 138, 3982 CrossRef CAS PubMed.
- For some reviews on the use of chiral phosphoric acids in organic synthesis, see: (a) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (b) D. Kampen, C. M. Reisinger and B. List, Top. Curr. Chem., 2010, 291, 395–456 CrossRef CAS PubMed; (c) M. Terada, Synthesis, 2010, 1929–1982 CrossRef CAS; (d) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- Reactions performed with alkynamines 1 substituted at the triple bond with alkyl groups were unsuccessful. Particularly, the reaction with R2= Me (Scheme 2) led to a mixture of several inseparable products from which the corresponding products 4 and diast-4 could be identified.
- CCDC 1550279 contains the supplementary crystallographic data for this paper.
- For a recent review on the application of computational studies to the field of chiral phosphoric acid catalysis, see: R. Maji, S. C. Mallojjala and S. E. Wheeler, Chem. Soc. Rev., 2018, 47, 1142–1158 RSC.
- The stabilization energy for typical T-shaped geometries has been estimated between 1.5 and 2.5 kcal mol−1 depending on the nature of the interacting arene fragments. See, L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- (a) J. S. Murray and P. Politzer, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 153–163 CAS; (b) S. S. Pingale, Phys. Chem. Chem. Phys., 2011, 13, 15158–15165 RSC.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed; (b) J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- Interestingly, other authors have found (R)-3,3′-bis(9-anthracenyl)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate (CPAF in the present work) as the optimal catalyst in reactions with substrates containing aryl groups that potentially could be involved in non-covalent interactions similar to those here proposed. See for example: (a) T. Akiyama, H. Morita and K. Fuchibe, J. Am. Chem. Soc., 2006, 128, 13070–13071 CrossRef CAS PubMed; (b) P. Maity, R. P. Pemberton, D. J. Tantillo and U. K. Tambar, J. Am. Chem. Soc., 2013, 135, 16380–16383 CrossRef CAS PubMed; (c) J. Y. See, H. Yang, Y. Zhao, M. W. Wong, Z. Ke and Y.-Y. Yeung, ACS Catal., 2018, 8, 850–858 CrossRef CAS.
- (a) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 Search PubMed; (b) S. L. Cockroft, C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Am. Chem. Soc., 2005, 127, 8594–8595 CrossRef CAS PubMed; (c) S. L. Cockroft and C. A. Hunter, Chem. Soc. Rev., 2007, 36, 172–188 RSC; (d) S. L. Cockroft, J. Perkins, C. Zonta, H. Adams, S. E. Spey, C. M. R. Low, J. G. Vinter, K. R. Lawson, C. J. Urch and C. A. Hunter, Org. Biomol. Chem., 2007, 5, 1062–1080 RSC; (e) C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC; (f) S. E. Wheeler, Acc. Chem. Res., 2013, 46, 1029–1038 CrossRef CAS PubMed.
- The molecular electrostatic potential (MESP) has been a widely effective tool for interpreting and predicting electron-rich and electron-poor regions of a molecular structure. See, (a) G. Merino, A. Vela and T. Heine, Chem. Rev., 2005, 105, 3812–3841 CrossRef CAS PubMed; (b) N. Mohan, C. H. Suresh, A. Kumar and S. R. Gadre, Phys. Chem. Chem. Phys., 2013, 15, 18401–18409 RSC; (c) F. Guégan, P. Mignon, V. Tognetti, L. Joubert and C. Morell, Phys. Chem. Chem. Phys., 2014, 16, 15558–15569 RSC.
- (a) W.-J. van Zeist and F. M. Bickelhaupt, Org. Biomol. Chem., 2010, 8, 3118–3127 RSC; (b) M. Pareek and R. B. Sunoj, ACS Catal., 2016, 6, 3118–3126 CrossRef CAS; (c) F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed; (d) R. Maji, P. A. Champagne, K. N. Houk and S. E. Wheeler, ACS Catal., 2017, 7, 7332–7339 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
Footnotes |
| † In memory of our friend Professor Kilian Muñiz (1970–2020). |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, full compound characterization and computational details. CCDC 1550279. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03342a |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |