
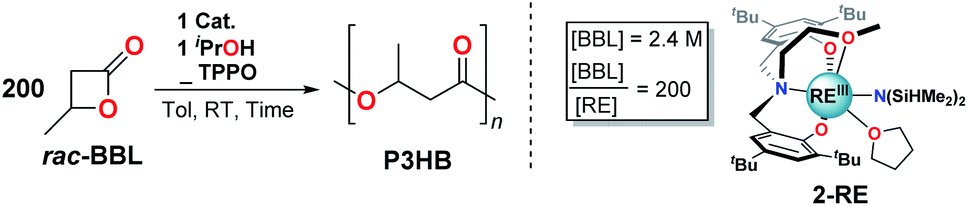
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of neutral donor ligands in the isoselective ring-opening polymerization of rac-β-butyrolactone†
Xiang
Dong
 and
Jerome R.
Robinson
*
and
Jerome R.
Robinson
*
Department of Chemistry, Brown University, 324 Brook St., Providence, RI 02912, USA. E-mail: jerome_robinson@brown.edu
First published on 16th July 2020
Abstract
Isoenriched poly-3-hydroxybutyrate (P3HB) is a biodegradable material with properties similar to isotactic polypropylene, yet efficient routes to this material are lacking after 50+ years of extensive efforts in catalyst design. In this contribution, a novel lanthanum aminobisphenolate catalyst (1-La) can access isoenriched P3HB through the stereospecific ring-opening polymerization (ROP) of rac-β-butyrolactone (rac-BBL). Replacing the tethered donor group of a privileged supporting ligand with a non-coordinating benzyl substituent generates a catalyst whose reactivity and selectivity can be tuned with inexpensive achiral neutral donor ligands (e.g. phosphine oxides, OPR3). The 1-La/OPR3 (R = n-octyl, Ph) systems display high activity and are the most isoselective homogeneous catalysts for the ROP of rac-BBL to date (0 °C: Pm = 0.8, TOF ∼190 h−1). Combined reactivity and spectroscopic studies provide insight into the active catalyst structure and ROP mechanism. Both 1-La(TPPO)2 and a structurally related catalyst with a tethered donor group (2-Y) operate under chain-end stereocontrol; however, 2-RE favors formation of P3HB with opposite tacticity (syndioenriched) and its ROP activity and selectivity are totally unaffected by added neutral donor ligands. Our studies uncover new roles for neutral donor ligands in stereospecific ROP, including suppression of chain-scission events, and point to new opportunities for catalyst design.
Introduction
Polyolefins enable numerous applications and benefits to society; however, there is growing concern over the immense amount of polymer waste entering landfills and waterways and their unfavorable environmental persistence.1 Poly-3-hydroxybutyrate (P3HB, Fig. 1), the most common member of naturally occurring polyhydroxyalkanoates, is a biodegradable aliphatic polyester which can have properties similar to isotactic polypropylene and applications ranging from packaging to bio-medical applications.2 Central to these applications is the polymer's relative stereochemistry (tacticity), which plays a critical role in its observed thermal,3 mechanical,3d–g,4 and degradation3d,5 properties. For example, atactic P3HB is an amorphous material with a glass transition temperature (Tg) of ∼5 °C with limited applications, while highly-enriched isotactic and syndiotactic P3HB are crystalline with melting temperatures (Tm) up to 183 °C. (R)-P3HB is synthesized naturally by many organisms through microbial fermentation; however, industrial production costs with this method remain high and only perfectly isotactic P3HB can be accessed.2a,6 The high degree of crystallinity and Tm of highly-enriched isotactic or syndiotactic P3HB leads to brittle materials with processing challenges due to the proximity of the material's Tm and decomposition temperature. Alternatively, isoenriched P3HB with Pm (percentage of meso diads) ranging from 0.65–0.8 can suitably balance mechanical and thermal properties, making the development of efficient and economically viable methods to access this material highly desirable.3c–f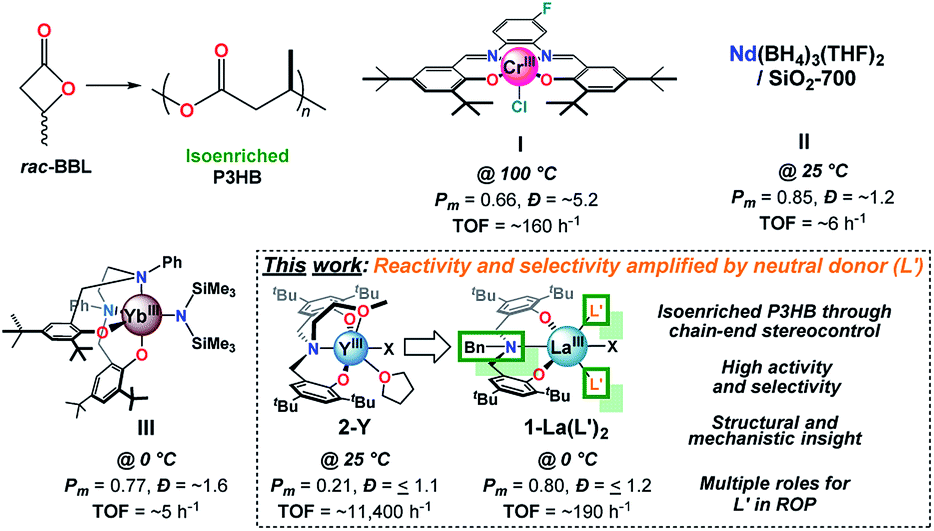 | ||
| Fig. 1 Key advances in catalyst development for the ROP of rac-BBL to access iso-enriched P3HB with I, II, III, and the current work. | ||
The stereospecific ring-opening polymerization (ROP) of rac-β-butyrolactone (rac-BBL) represents one such approach. This racemic monomer can be readily derived from abundant and inexpensive feedstocks (e.g. propylene oxide and carbon monoxide),7 displays favorable thermodynamics towards ROP thanks to its significant ring-strain (ΔGp = −59.2 kJ mol−1),8 and highly efficient catalysts have been developed for the stereospecific ROP of a variety of other lactone monomers.2a,9 Despite these desirable attributes and a diverse array of catalysts being explored over 50+ years,10 the development of efficient catalysts with high levels of stereocontrol has proven challenging.9,10n,11 Many catalysts which are stereoselective for other lactone monomers (e.g. rac-lactide) display much lower (or no) reactivity and/or stereoselectivity towards rac-BBL,10f,j,s,12 and both monomer and polymer are prone to side-reactions (e.g. transesterification, chain scission, deprotonation, elimination).
Extensive efforts in catalyst design have led to systems which can access syndioenriched P3HB;3l,10n,o,13 however, few systems have led to isoenriched P3HB.3i–k,9a,10q,11,14 Early work by Tani, Agostini, Lenz, and others identified that partial hydrolysis of alkyl aluminum species leads to catalysts which can produce a minor fraction of crystalline P3HB with properties similar to natural P3HB, albeit over prolonged reaction times (≥7 d) and with broad molecular weight distributions (Ð, Mw/Mn).3i,10c,14h–k Rieger and coworkers discovered that Cr salophen catalysts (Fig. 1, I) are capable of producing isoenriched P3HB with high Mn and broad Ð through a complex dual-site mechanism.3j,14e,f Thomas and coworkers developed the most isoselective heterogeneous catalyst to date (Pm = 0.85) by grafting Nd(BH4)3(THF)2 to nonporous SiO2 dehydroxylated at 700 °C (Fig. 1, II), but the system displays modest reactivity.10q Most recently, Yao and coworkers reported a series of REIII salan catalysts whose stereospecific ROP is highly sensitive to N-substitution.14g The Yb N-Ph derivative (Fig. 1, III) displays modest reactivity and limited control over Mn, but is the most isoselective homogeneous catalyst to date (Pm = 0.77 at 0 °C). Notably, Chen and coworkers reported the ROP of a designer 8-membered diolide as an elegant alternative to circumvent selectivity challenges associated with rac-BBL; however, optimized catalysts produce perfectly isotactic P3HB.3k
Of all the catalyst platforms, trivalent rare-earth (REIII) supported by tetradentate tripodal amino-bisphenolate ligands developed by Carpentier and coworkers (Fig. 1, 2-Y) stand out as “privileged” structures due to their exceptional activity and syndioselectivity.3l,10n,o Though numerous covalent modifications to the ligand scaffold have been explored (e.g. aryloxide substitution, donor identity, tether/linker, initiator, and REIII), none have provided access to isoenriched P3HB – even though such modifications have led to syndio- and iso-enriched polymers of other β-lactones.15 Amongst these modifications, the role of the tethered donor ligand remains obscure,16 and more broadly speaking, our understanding of how neutral achiral donor ligands influence the reactivity and stereoselectivity of ROP catalysts remains underdeveloped. In the field of asymmetric catalysis, introduction of achiral and meso neutral donor ligands can augment stereocontrol for a variety of metal-based catalysts,17 including rare-earths,18 in a facile and cost-effective manner. While donor-related effects in ROP have been noted,13a,19 connections between catalyst structure and function have been limited.
Herein, we report the synthesis, characterization, and catalytic activity of REIII benzyl-substituted amino-bisphenolate complexes for the isoselective ROP of rac-BBL. Replacing the tethered donor fragment of a tetradentate aminobisphenolate ligand with a non-coordinating benzyl substituent leads to a La catalyst whose reactivity and selectivity are amplified by the addition of inexpensive neutral achiral donor ligands (e.g. phosphine oxides, OPR3). The LaIII/OPR3/iPrOH (R: Ph, n-octyl) species display high activity and are the most isoselective homogeneous catalysts for the ROP of rac-BBL to date (Pm = 0.8 at 0 °C, TOF = ∼190 h−1). Despite the prevalence of such ligands in the coordination chemistry of RE's20 and other metal-ions,21 this is the first report of added phosphine oxides enhancing catalyst reactivity or selectivity in ROP. Evidence that strong neutral donors can suppress unwanted side-reactions such as chain-scission through base-promoted elimination are also presented for the first time. Statistical analysis of P3HB microstructure confirms that 1-La(TPPO)2 is the first catalyst to access isoenriched P3HB through chain-end stereocontrol. While a structurally related catalyst with a tethered donor (2-Y) also operates under chain-end stereocontrol, 2-RE favor the opposite polymer tacticity (syndioenriched P3HB) and their performance in ROP are unaffected by added neutral donor ligands. Our studies uncover the effects of neutral donors on catalyst structure and function, and provide new opportunities for the design of catalysts for stereospecific ROP.
Results and discussion
Catalyst synthesis
The white crystalline benzyl-amino-bisphenol ligand (H21L) was synthesized in one step by a Mannich condensation of benzyl amine, 2,4-ditertbutylphenol and paraformaldehyde in 47% yield (Scheme 1). H21L was then treated with one equivalent of REIII amide (REIII: La, Y), REIII[N(SiHMe2)2]3(THF)2, to afford the corresponding REIII complexes, LaIII(1L)[N(SiHMe2)2](THF)2 (1-La) and {YIII(1L)[N(SiHMe2)2]}2 (1-Y2), in nearly quantitative yields (Scheme 1). 1-La is a monomer in both the solid- and solution-state as determined by single-crystal X-ray diffraction (Fig. 2) and Diffusion Ordered NMR Spectroscopy (DOSY, Fig. S10†). While X-ray quality crystals could not be grown of the THF adduct, slow evaporation of Et2O solutions enabled the structural determination of the mixed THF/Et2O adduct (Fig. 2). The geometry of the six-coordinate LaIII center in the solid-state is best described as a distorted trigonal prism comprised of tridentate 1L, –N(SiHMe2)2, and two coordinated solvent molecules (THF and Et2O). 1L adopts a propeller-like conformation at nitrogen and enforces fac-coordination. Agostic β-H–Si interactions (La↼H–Si) were observed in the solid-state as supported by the smaller angle ∠La(1)–N(2)–Si(1) compared to ∠La(1)–N(2)–Si(2) (∼15°), close La(1)–Si(1)–H contact (3.3497(13) Å), and lower energy Si–H stretch in the IR spectrum (non-agostic: 2075 cm−1, agostic: 2011 cm−1, Fig. S3d†).22 Unlike 1-La, the yttrium derivative (1-Y2) exists as a dimer in solution as determined by 1H-DOSY NMR (Fig. S11†).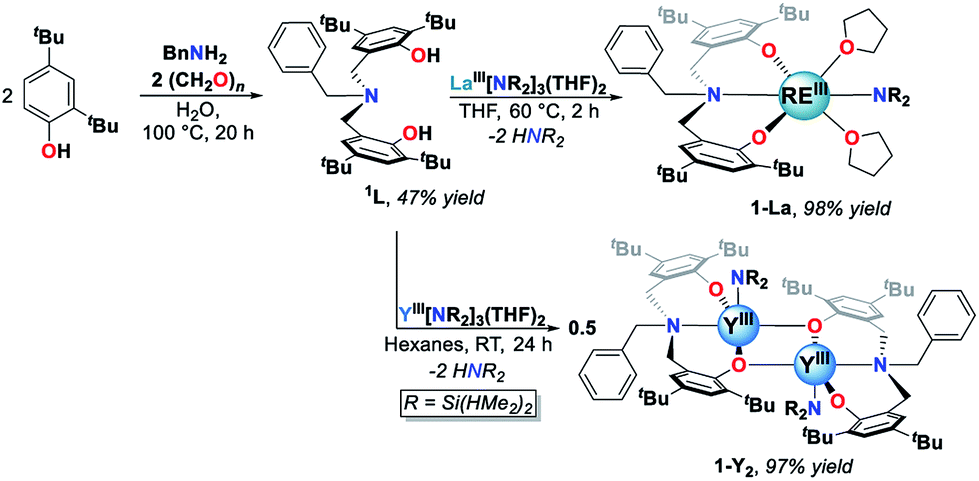 | ||
| Scheme 1 Synthesis of 1L and REIII complexes (1-La and 1-Y2). | ||
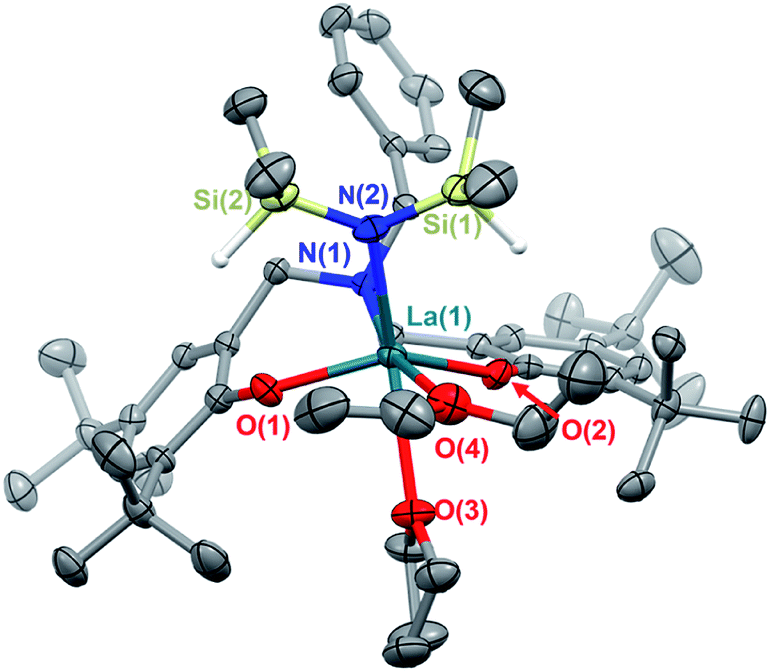 | ||
| Fig. 2 Thermal ellipsoid plot of 1-La (THF, Et2O adduct) displayed at 50% probability. | ||
Reaction optimization and neutral donor ligand effects
1-La and 1-Y2 were evaluated as catalysts for the ROP of rac-BBL (Tables 1 and S1†). Amide initiators displayed low efficiency for the ROP of rac-BBL, where 1-Y2 was completely inactive and 1-La formed 35% P3HB in 48 h at RT (Table S1,† entries 1 and 5). While 1-La was sluggish compared to Carpentier's 2-Y,3l,10o formation of P3HB was encouraging as the related SmIII borohydride complex supported by the propyl-amino-bisphenolate ligand reported by Mountford and coworkers was inactive for ROP of an arguably easier substrate, rac-lactide.16a Similar to other REIII catalysts,3l,10o,23in situ generation of alkoxide initiators by adding one equiv. iPrOH with respect to REIII (Table 1, entries 1 and 2 vs. Table S1,† entries 1 and 5) increased reactivity and furnished polymers with narrow Mw/Mn (Đ). Microstructural analysis of P3HB determined by integration of polymer C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O resonances by inverse-gated 13C-NMR revealed a slight isotactic preference (Pm = 0.57) using 1-La. While modest, the polymer tacticity was opposite that generated by 2-Y and other amino-bisphenolate catalysts with tethered donors.10n,o,24 Given the increased degree of coordinative unsaturation of 1-RE compared to 2-RE and the attributed importance of steric congestion to selectivity for other stereospecific ROP,3l,14g,25 we posited that added neutral donor ligands could improve catalyst reactivity and selectivity through enhanced steric pressure.
O resonances by inverse-gated 13C-NMR revealed a slight isotactic preference (Pm = 0.57) using 1-La. While modest, the polymer tacticity was opposite that generated by 2-Y and other amino-bisphenolate catalysts with tethered donors.10n,o,24 Given the increased degree of coordinative unsaturation of 1-RE compared to 2-RE and the attributed importance of steric congestion to selectivity for other stereospecific ROP,3l,14g,25 we posited that added neutral donor ligands could improve catalyst reactivity and selectivity through enhanced steric pressure.
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Cat. | [BBL]/[RE] | Ligand | Temp (°C) | Timeb (h) | Conv.c (%) | M n,calc (kg mol−1) | M n,exp (kg mol−1) | Đ , | P m |
a [BBL] = 2.4 M.
b Reaction times not optimized.
c Determined by 1H-NMR integration of BBL and PHB methine resonances in the crude reaction mixture.
d [BBL]/[RE]/[iPrOH] × Conv. × 0.08609 + 0.0601 kg mol−1.
e Determined by gel permeation chromatography (GPC) at 30 °C in THF using polystyrene standards and corrected by Mark–Houwink factor of 0.54.51
f
M
w/Mn.
g Probability of meso-linkages between repeat units. Determined by integration of P3HB ![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) O resonances using inverse gated (IG) 13C-NMR.
h 0.52 at 6 h (36% conversion). O resonances using inverse gated (IG) 13C-NMR.
h 0.52 at 6 h (36% conversion).
|
||||||||||
| 1 | 1-Y2 | 200 | — | 25 | 1 | 5 | 0.9 | n.d. | n.d. | n.d. |
| 2 | 1-La | 200 | — | 25 | 1 | 21 | 3.6 | 2.9 | 1.04 | 0.57 |
| 3 | 1-La | 200 | DMAP | 25 | 1 | 22 | 3.8 | 3.6 | 1.07 | 0.59 |
| 4 | 1-La | 200 | DABCO | 25 | 1 | 7 | 1.2 | n.d. | n.d. | n.d. |
| 5 | 1-La | 200 | PPh3 | 25 | 1 | 25 | 4.3 | 1.7 | 1.38 | n.d.h |
| 6 | 1-La | 200 | TPPO | 25 | 1 | 97 | 16.7 | 9.6 | 1.18 | 0.71 |
| 7 | 1-La | 200 | HMPA | 25 | 1 | 99 | 17.0 | 9.4 | 1.29 | 0.73 |
| 8 | 1-La | 200 | TOPO | 25 | 1 | 99 | 17.0 | 9.5 | 1.23 | 0.75 |
| 9 | 1-La | 200 | OP(OPh)3 | 25 | 1 | 25 | 4.3 | 1.6 | 1.35 | 0.63 |
| 10 | 1-La | 200 | TPPO | 0 | 1 | 96 | 16.5 | 11.2 | 1.15 | 0.76 |
| 11 | 1-La | 400 | TPPO | 0 | 4 | 77 | 26.5 | 15.3 | 1.20 | 0.75 |
| 12 | 1-La | 200 | TPPO | −30 | 24 | 99 | 17.0 | 11.5 | 1.12 | 0.80 |
| 13 | 1-La | 200 | TOPO | 0 | 1 | 99 | 17.0 | 12.9 | 1.20 | 0.80 |
| 14 | 1-La | 400 | TOPO | 0 | 4 | 96 | 33.1 | 19.2 | 1.09 | 0.80 |
| 15 | 1-La | 200 | TOPO | −30 | 6 | 99 | 17.0 | 13.0 | 1.09 | 0.80 |
We initially tested this hypothesis by screening 1-La with two equiv. of neutral monodentate ligands. Representative classes included ethers (tetrahydrofuran, THF), tertiary amines (1,4-diazabicyclo-[2.2.2]octane, DABCO), pyridines (4-dimethylaminopyridine, DMAP), phosphines, (PPh3), and phosphine oxides (OPPh3, TPPO). Unlike some literature reports for group 13, 14, and REIII-based systems,13a,19a–i weaker neutral ligands had a minor impact on ROP reactivity and stereoselectivity (Table 1, entries 2–5). In contrast, the harder phosphine oxide ligand, TPPO (entry 6), led to nearly quantitative conversion in 1 h (97%) and improved isoselectivity (Pm = 0.71). Notably [1-La]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[TPPO] ratios of at least 1:2 were needed to achieve maximum reactivity and selectivity, where a 1:1 ratio only led to 71% conversion and a Pm of 0.67 after 6 h (Table S1,† entries 6–9). While simple monodentate phosphine oxides have been reported as additives in asymmetric catalysis with hard metal-ions,18a–d,26 this is the first time they have been used to enhance reactivity and/or selectivity in ROP.
:[TPPO] ratios of at least 1:2 were needed to achieve maximum reactivity and selectivity, where a 1:1 ratio only led to 71% conversion and a Pm of 0.67 after 6 h (Table S1,† entries 6–9). While simple monodentate phosphine oxides have been reported as additives in asymmetric catalysis with hard metal-ions,18a–d,26 this is the first time they have been used to enhance reactivity and/or selectivity in ROP.
Given the unprecedented and dramatic enhancement in catalyst performance, we evaluated representative classes of phosphine oxides (aromatic, aliphatic, phosphoramide, and phosphate; Table 1, entries 6–9). Electron-rich donors, such as hexamethylphosphoramide (HMPA, entry 7) and trioctylphosphine oxide (TOPO, entry 8) increased reactivity and isoselectivity (Pm = 0.73 and 0.75 respectively). In contrast, triphenylphosphate (OP(OPh)3, entry 9), a weaker donor, didn't increase reactivity and showed small improvements in selectivity (Pm = 0.63). Our results suggest both electronic and steric contributions to catalyst reactivity and selectivity, and a more comprehensive evaluation is warranted in future studies. This is affirmed by reports of stereospecific ROP of (rac)-lactide by REIII and group 13 complexes supported by chelating alkoxides,27 amides,28 and pyrazolyl scorpionates29 with tethered phosphine-oxides, which display varied catalyst response depending on ligand substituents. Given the diverse array of phosphine oxides that can be derived from commercially available phosphines, this represents an exciting untapped opportunity to optimize catalyst performance in stereoselective ROP.
Key polymer attributes could be tuned by adjusting reaction temperature, catalyst loading, and chain-transfer agent. Lowering the reaction temperature from RT to 0 and −30 °C with TPPO and TOPO (entries 10–15) increased catalyst isoselectivity to a maximum (Pm = 0.80). This represents the highest values achieved for the ROP of rac-BBL by a homogeneous catalyst to date.14g Furthermore, increased [rac-BBL]/[RE] ratios (400) lead to higher molecular weight P3HB with reasonable rates, identical selectivities, and narrow Ð (Table 1, entries 11 and 14). Overall, 1-La/OPR3/iPrOH systems display excellent reactivity (TOF up to 200 h−1) and selectivity (Pm up to 0.80) with respect to the state-of-the-art for isoselective ROP of rac-BBL (Pm up to 0.77 (ref. 14g) and 0.85,10q TOF ∼5–6 h−1; Fig. 1).
Alcohols can serve as chain-transfer agents in living polymerizations to access “immortal” polymerization conditions,30 offering further opportunities to control polymer molecular weight.10g,23,31 A La catalyst was isolated from a toluene solution of 1-La and TPPO in a 1:2 molar ratio (vide infra), which displays similar reactivity in the ROP of rac-BBL as the in situ generated catalyst from adding 2 equivalents of TPPO to 1-La (Table S2,† entry 2 and Table 1, entry 6). The ROP of rac-BBL (200 equiv.) catalyzed by 1-La(TPPO)2 (1 equiv.) with iPrOH (1 equiv.) displayed characteristics of a living polymerization, such as narrow Ð throughout the reaction and reasonable agreement between experimental and calculated Mn (Table S4 and Fig. S24†). Adding iPrOH (0–4 equiv.) to 1-La(TPPO)2 maintained high catalyst activity and Pm, while producing P3HB with the expected changes in molecular weight (Table S2,† entries 1–4).
Mechanistic studies
![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) resonances, consistent with weakening and displacement of the β-H–Si interactions (La↼H–Si) and TPPO coordination (Fig. 3, left).22d The bis-TPPO adduct displays a single significantly broadened 31P signal, indicative of exchange on the NMR timescale. Isolation of crystalline bis-TPPO adducts, REIII(1L)(N(SiHMe2)2)(TPPO)2 (1-RE(TPPO)2; REIII: Y, La), was accomplished in high yields by adding two equiv. TPPO per REIII (1-La or 1-Y2) in toluene followed by layering with hexanes (Fig. 4a). Although under active investigation, attempts to crystallize the mono-TPPO adduct, 1-La(TPPO), have only led to isolation of crystalline 1-La(TPPO)2.
resonances, consistent with weakening and displacement of the β-H–Si interactions (La↼H–Si) and TPPO coordination (Fig. 3, left).22d The bis-TPPO adduct displays a single significantly broadened 31P signal, indicative of exchange on the NMR timescale. Isolation of crystalline bis-TPPO adducts, REIII(1L)(N(SiHMe2)2)(TPPO)2 (1-RE(TPPO)2; REIII: Y, La), was accomplished in high yields by adding two equiv. TPPO per REIII (1-La or 1-Y2) in toluene followed by layering with hexanes (Fig. 4a). Although under active investigation, attempts to crystallize the mono-TPPO adduct, 1-La(TPPO), have only led to isolation of crystalline 1-La(TPPO)2.
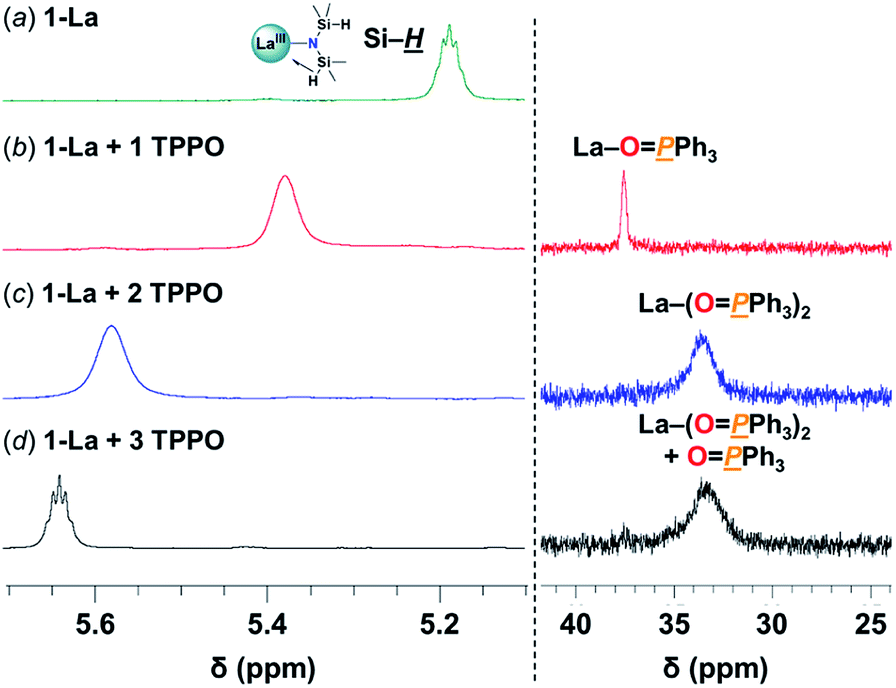 | ||
| Fig. 3 Selected spectral regions of (left) 1H- and (right) 31P{1H}-NMR studies in C6D6 at RT of: (a) 1-La(TPPO)2 (27 mM) (b) + 1 TPPO (c) + 2 TPPO (d) + 3 TPPO. Full spectra are displayed in Fig. S7.† | ||
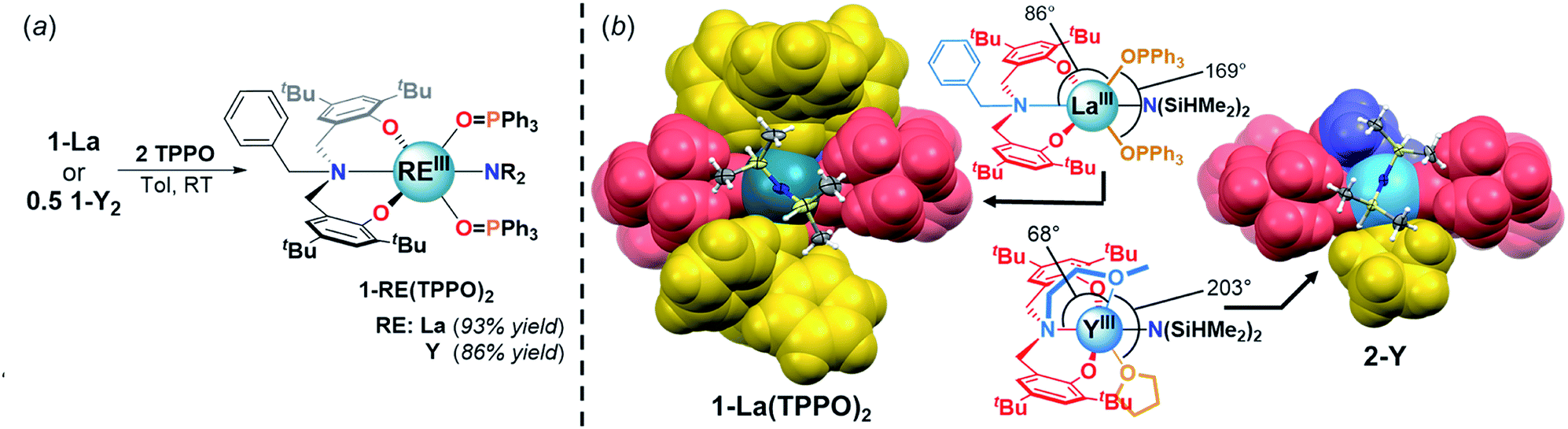 | ||
| Fig. 4 (a) Synthesis of 1-RE(TPPO)2. (b) Partial space-filling diagrams comparing 1-La(TPPO)2 and 2-Y. Fragment color coding: phenolate (red), amine (blue), labile neutral donors (gold). N(SiHMe2)2 shown as ellipsoids (50% probability). | ||
The solid-state structures of 1-RE(TPPO)2 were determined unambiguously by single crystal X-ray diffraction experiments (Fig. 4b, S25, and S26†). In the solid-state, 1L coordinates in a mer-arrangement for 1-RE(TPPO)2 rather than the fac-arrangement for 1-La. The isostructural compounds contain six-coordinate REIII centers in a distorted octahedron with equatorial sites occupied by 1L and –N(SiHMe2)2 and axial sites occupied by TPPO. Comparison of 1-RE(TPPO)2 and tethered donor system, 2-Y,32 revealed largely conserved equatorial sites and significantly perturbed axial sites (Fig. 4b). In 2-Y, the geometrically constrained tethered donor leads to a small NL2–Y–OMe angle (68°) and a large OOMe–Y–OTHF angle (203°) compared to 1-La(TPPO)2 (Fig. 4b, ∠NL1–La–OTPPO: 86°, ∠OTPPO–La–OTPPO: 169°). The differences in bond angles reflect increasing steric pressure from the axial donors, and suggest a plausible structural origin for the selectivity in 1-La/OPR3.
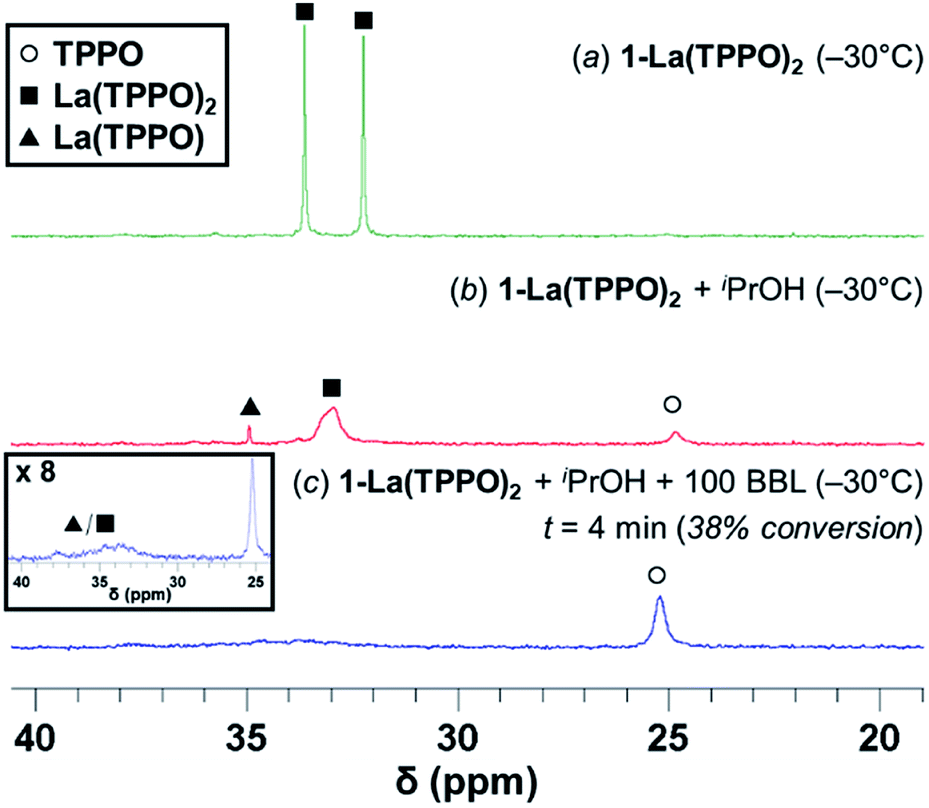 | ||
| Fig. 5 31P{1H}-NMR studies in toluene-d8 at −30 °C of: (a) 1-La(TPPO)2 (25 mM) (b) 1-La(TPPO)2 + iPrOH (c) 1-La(TPPO)2 + iPrOH + 100 equiv. (rac)-BBL (38% conversion). All spectra are internally referenced and intensity-normalized to PPh3 (4.0 μmol; Fig. S16b†). | ||
Addition of rac-BBL (100 equiv.) increases the signal for free TPPO significantly, while resonances associated with La(TPPO)n (n = 1, 2) were dramatically broadened (Fig. 5c and S16†). Warming the reaction mixture from −30 °C to −15 °C and 0 °C (Fig. S17†) increased exchange of free and bound TPPO as evidenced by the increasing line-width of free TPPO (half-width at half-maximum, HWHM; 25, 80, and 150 Hz respectively), and RT experiments produced similar species (Fig. S15b†). Reactions performed at RT with one equiv. TPPO formed similar species without generation of free TPPO (Fig. S15a†); however, optimal catalyst reactivity and selectivity required at least two equiv. of TPPO (vide supra, Table S1,† entries 6–9). Taken together, our reaction optimization and in situ spectroscopic studies support dissociation of one equiv. TPPO from the pre-catalyst, 1-La(TPPO)2, and dynamic phosphine oxide exchange during catalysis. While La(TPPO) was identified as a catalyst resting state, the observed TPPO-dependent reactivity and observed speciation implicates both La(TPPO)n (n = 1, 2) as catalytically relevant species.
Stereocontrol. Statistical analysis of P3HB microstructure can distinguish enantiomorphic site or chain-end stereocontrol mechanisms based on the distribution of stereoerrors.35 Bernoullian analysis of the meso (m) and racemic (r) content in triad distributions of P3HB, B = 4(mm)(rr)/[(rm) + (mr)]2, predicts a value of 1 for perfect chain-end control.35b A B value of 1.05 was obtained from quantitative 13C-NMR of the methylene region of P3HB generated by 1-La/TPPO at RT (Table 1, entry 6; Fig. S1†), which is close to the theoretical prediction and confirms chain-end stereocontrol.3l,10e,36 While chain-end stereocontrol is operative for several syndioselective catalysts,3l,37 this is the first example with an isoselective catalyst. Other catalysts which produce isoenriched P3HB proceed through enantiomorphic site-control3k,38 or their mechanism of stereocontrol have not yet been determined.
End-group analysis. Additional insights into the polymerization mechanism were facilitated by end-group analyses using 1H-NMR and MALDI-TOF techniques. ROP of rac-BBL and other β-lactones catalyzed by neutral metal alkoxides commonly proceed through coordination–insertion or anionic pathways.39 The coordination–insertion mechanism proceeds through acyl cleavage (ester and alcohol end-groups), while the anionic mechanism proceeds through alkyl cleavage (ether and carboxylate end-groups). 1H-NMR spectra of isolated P3HB samples revealed the presence of an isopropyl ester end-group (Fig. S19 and S23†), while signals for an isopropyl ether were notably absent. These observations are consistent with a coordination–insertion mechanism for ROP with initiation occurring from a metal-isopropoxide.
Following a coordination–insertion mechanism, the other end-group should be a terminal secondary alcohol, which would be obtained upon hydrolysis of the propagating metal-alkoxide. As expected, the secondary alcohol end-group (–CHOHCH3) was observed in a ∼1:1 molar ratio with respect to the ester end-group (COOiPr). However, additional C–H resonances which correspond to a crotyl end-group were observed by 1H-NMR spectroscopy (crotyl:CHOHCH3:COOiPr; ∼1:1:1). Further evidence of the crotyl end-group was established by MALDI-TOF measurements of P3HB obtained from ROP of 40 equiv. rac-BBL using the 1-La(TPPO)2/iPrOH catalyst system. MALDI-TOF spectra corroborated 1H-NMR end-group assignments, and clearly supported crotyl end-group formation during the reaction (Fig. S22 and S23†).
Elimination studies. Generation of crotyl end-groups after polymerization could proceed through several possible pathways: (i) elimination of water, hydroxide, or oxide from the alcohol end-group under acidic or basic conditions respectively,10g,40 (ii) thermal scission,41 or (iii) base-induced elimination of internal ester units,10f,42 (iv) terminal elimination from a metal alkoxide. While pathway (i) has been proposed to explain generation of crotyl end-groups after quenching polymerizations with weak acids,3k,10g this stands in contrast to the stability of such 3-hydroxybutanoate monomer and oligomers under strong-acid conditions.43 Furthermore, elimination from a metal alkoxide during the reaction would convert the secondary alcohol end-group to an inactive crotyl end-group and broaden Ð. The relative amounts of crotyl, secondary alcohol, and isopropyl ester end groups observed (∼1
:1:1, vide supra) and narrow Ð are inconsistent with expectations for this pathway. Pathway (ii) can be excluded due to the reaction temperatures evaluated in our studies (ambient or below).
Side-reactions such as deprotonation, transesterification, and elimination were proposed in early reports for the ROP of rac-BBL;40b,44 however, detailed examination of the elimination pathway (iii) under mild temperatures (<100 °C) has been limited to the independent studies of Kricheldorf (K catalysts) and Coates (Zn beta-diketiminate catalysts).10f,42a Reactivity of internal P3HB linkages were established by the use of small molecule models, which enabled detailed identification of the resulting organic products. Despite the superior performance of many RE-based catalysts and the observation of crotyl formation in several reports,3k,14g,24,45 investigations into elimination pathways for RE-based catalysts are notably absent. Therefore, we examined the reactivity of 1-La and 1-La(TPPO)2 with a new small molecule model, (R)-3-acetoxybutyric acid methylester [(R)-3-OAcBMe], to establish the viability of such side-reactions [e.g. pathway (iii)] during ROP.
Addition of one equiv. iPrOH at RT to 1-La and 1-La(TPPO)2 cleanly generated the La isopropoxide species, 1′-La and 1′-La(TPPO)2, and one equiv. HN(SiHMe2)2 (▲). 15 equiv. of (R)-3-OAcBMe was added, and reactivity was monitored by 1H-NMR after 0.5 and 7 h (Fig. 6 and S25†). Our initial expectations were that (R)-3-OAcBMe would react with 1′-La and 1′-La(TPPO)2 through base-promoted elimination to form crotonate (trans-CrotMe), iPrOH, and a La acetate species. 1′-La readily produced crotonate (0.5 h: 0.6 equiv.; 7 h: 1.2 equiv.); however, free iPrOH was not observed. Instead, the transesterification products, isopropyl butyrate/crotonate [(R)-3-OAcBiPr/trans-CrotiPr] and methyl acetate (MeOAc), were readily identified (Fig. S25† for detailed assignments). Transesterification between the La isopropoxide and (R)-3-OAcBMe/trans-CrotMe would lead to a La methoxide and (R)-3-OAcBiPr/trans-CrotiPr, while transesterification between the La methoxide and the 3-acetoxy group of (R)-3-OAcBiPr would generate MeOAc and a La 3-alkoxybutyrate species. The observed reactivity is consistent with reports of neutral La alkoxides as extremely efficient transesterification catalysts under mild conditions.46 In addition to the aforementioned products, quantifiable amounts of free ligand (H21L; 0.5 h: 0.1 equiv., 7 h: 0.6 equiv.) were also detected. The formation of crotonate and the direct (conjugate acids) or indirect (transesterification) products of base-promoted elimination provide clear evidence for pathway (iii) occurring readily at RT with 1′-La.
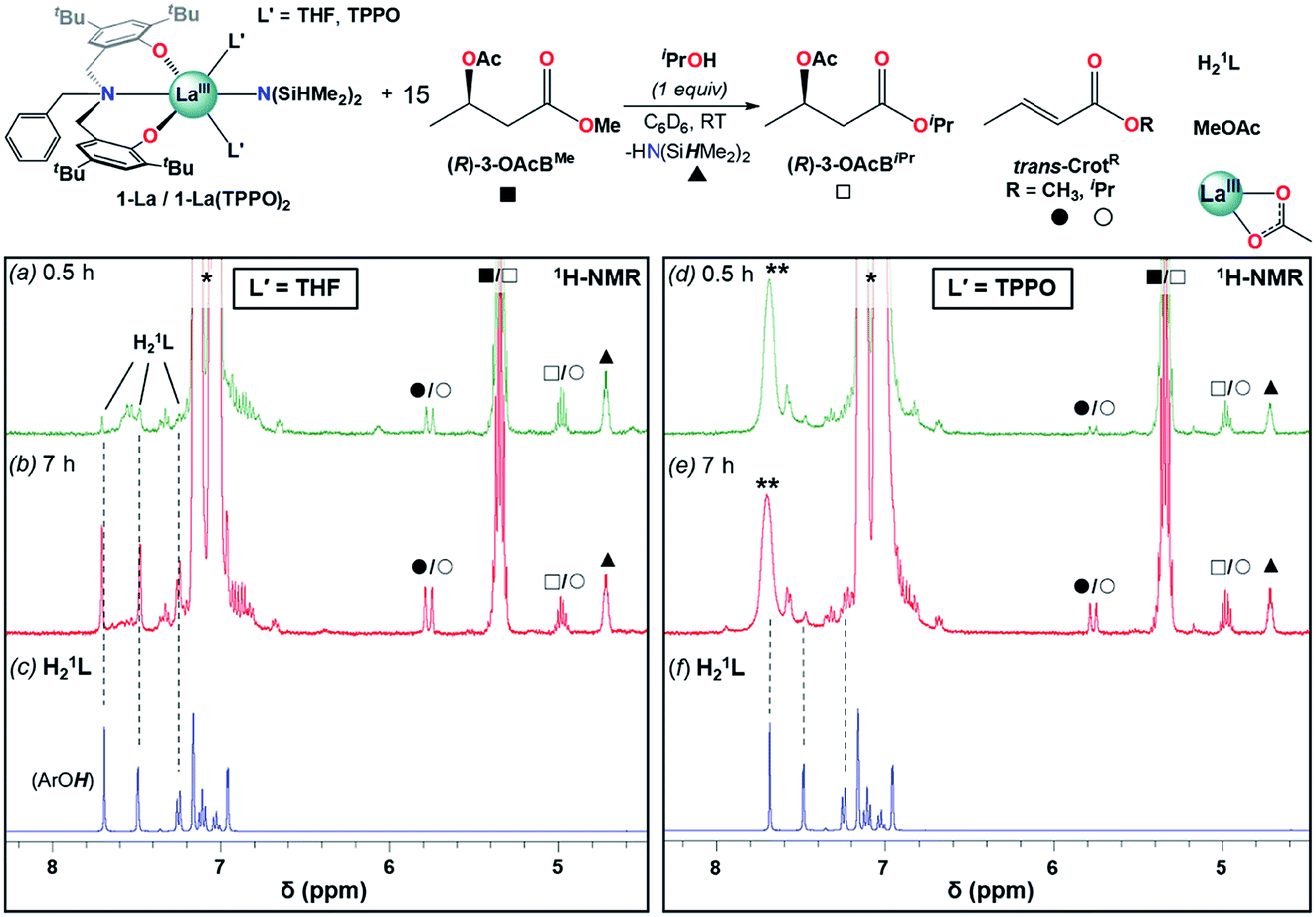 | ||
| Fig. 6 Reactivity studies of 1-La and 1-La(TPPO)2 in the presence of one equiv. iPrOH and 15 equiv. (R)-3-OAcBMe followed by 1H-NMR after 0.5 h (a and d), 7 h (b and e). Dashed lines provided to help track the formation of H21L (c and f) during the reaction time course. * = toluene (from iPrOH stock solution), ** = TPPO. Detailed assignments of full spectra provided as Fig. S25.† | ||
In contrast to 1′-La, 1′-La(TPPO)2 generated less crotonate (0.5 h: 0.14 equiv., 7 h: 0.73 equiv.) and only trace amounts of H21L after 7 h. These results highlight two additional and beneficial roles that strong neutral donor ligands (e.g. TPPO) can play in the ROP of rac-BBL. First, strong neutral donors can suppress elimination, as evidenced by the significantly decreased amount of crotonate formed with 1′-La(TPPO)2 compared to 1′-La. Supressing this side-reaction is critical, as the resulting La carboxylates would be inactive towards coordination–insertion ROP at RT (i.e. dormant chains), while chain-scission would also broaden Ð and lower Mn. Second, strong neutral donors can effectively suppress the kinetic basicity of the supporting ligand, as evidenced by significant amounts of H21L generated with 1′-La. While RE aryloxides have been leveraged as efficient multi-functional catalysts through cooperative Lewis-acid/Lewis-base reactions (e.g. Michael, aldol, hydrophosphination),47 RE aryloxides have been considered as innocent supporting ligands for the ROP of rac-BBL. Although rapid transesterification was observed for 1-La and 1-La(TPPO)2, the low Ð and high Pm support that this side-reaction is less significant under catalytic conditions. The pronounced tendency towards transesterification should correspond to the lower steric-bulk of the methyl ester found in (R)-3-OAcBMe compared to the more hindered ester linkages in P3HB.
The effect of neutral donors on ROP catalyzed by 1-RE and 2-RE. Given the observed benefits of adding strong neutral donor ligands to 1-RE (vide supra), we set out to evaluate whether similar enhancements would occur in structurally related catalysts with a tethered donor group (2-Y and 2-La). This was motivated by reports of solvent-dependent10o and tethered-donor dependent24,48 ROP reactivity for 2-RE and its derivatives. 1H- and 31P{1H}-NMR studies indicate that TPPO readily binds to 2-RE in solution (Fig. S18†); however, unlike 1-RE, rates and selectivity for the ROP of rac-BBL were totally unaffected by added TPPO (Table 2, entries 3–5 and 8–10). While initially unanticipated, we suspect this is due to the much high concentrations of the weaker donor ligands (solvent) compared to our studies (two equiv.).3l,10o Under our experimental conditions, the tethered donor of 2-RE dominates the observed reactivity and stereoselectivity, indicating that propagation from the corresponding TPPO adducts of 2-RE is a higher energy pathway.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | [Cat.] | [TPPO]/[RE] | Timea (h) | Conv.b (%) | M n,exp (kg mol−1) | Đ , | P m |
|
a Reaction times not optimized.
b Determined by 1H-NMR integration of BBL and PHB methine resonances in the crude reaction mixture.
c Determined by gel permeation chromatography (GPC) at 30 °C in THF using polystyrene standards and corrected by Mark–Houwink factor of 0.54.51
d
M
w/Mn.
e Probability of meso-linkages between repeat units. Determined by integration of P3HB O resonances using inverse gated (IG) 13C-NMR.
|
|||||||
| 1 | 1-La | 0 | 24 | 40 | 2.2 | 1.23 | 0.57 |
| 2 | 1-La | 2 | 1 | 97 | 9.6 | 1.18 | 0.71 |
| 3 | 2-La | 0 | 24 | 22 | 1.4 | 1.17 | 0.45 |
| 4 | 2-La | 1 | 24 | 21 | 1.6 | 1.14 | 0.49 |
| 5 | 2-La | 2 | 24 | 21 | 1.7 | 1.16 | 0.48 |
| 6 | 1-Y2 | 0 | 24 | 33 | 5.9 | 1.15 | 0.55 |
| 7 | 1-Y2 | 2 | 3 | 95 | 14.0 | 1.18 | 0.51 |
| 8 | 2-Y | 0 | 1 | 91 | 14.2 | 1.16 | 0.22 |
| 9 | 2-Y | 1 | 1 | 99 | 17.6 | 1.12 | 0.22 |
| 10 | 2-Y | 2 | 1 | 99 | 15.9 | 1.14 | 0.22 |
The presence or absence of a tethered donor also manifests opposite size-dependent reactivity and selectivity trends for 1-RE and 2-RE. Smaller ions are more reactive and selective for 2-RE,3l,25,37b while larger ions are more reactive for 1-RE/OPR3. While both catalysts display chain-end stereocontrol and feature labile coordination sites cis to an initiator (two for 1-RE, one for 2-RE), amplified selectivity is only observed with the largest and most coordinatively unsaturated catalyst, 1-La. Furthermore, the presence (2-RE) or absence (1-RE/OPR3) of a tethered donor group favors opposite polymer tacticities (2-RE: syndio, 1-RE: iso).
Proposed mechanism. Given the results of our catalytic and mechanistic studies, we propose the following mechanism for the ROP of rac-BBL catalyzed by 1-La & 1-La(TPPO)2 (Fig. 7; L′ = THF or TPPO). Addition of iPrOH to 1-La or 1-La(TPPO)2 leads to a highly reactive initiator, 1′-La or 1′-La(TPPO)2. Ligand exchange of L′ for rac-BBL generates A, which can then undergo insertion of the La alkoxide to generate B. Ring-opening would lead to C, which is involved in two competing ligand-exchange equilibria that gates productive (propagation) and unproductive (elimination) pathways. Upon binding of one equiv. L′ to C, the catalytic cycle is successfully completed with the regeneration of 1′-La. Our low-temperature NMR studies of 1-La(TPPO)2 support a mono-TPPO resting state during ROP, while our catalytic studies indicate that more than one equiv. of TPPO is required to achieve maximum rate and selectivity enhancements (Table S1,† entries 6–9). While we have depicted A, B, and C as mono-L′ adducts, we cannot exclude the possibility that one or more of these intermediates may be bis-L′ adducts.
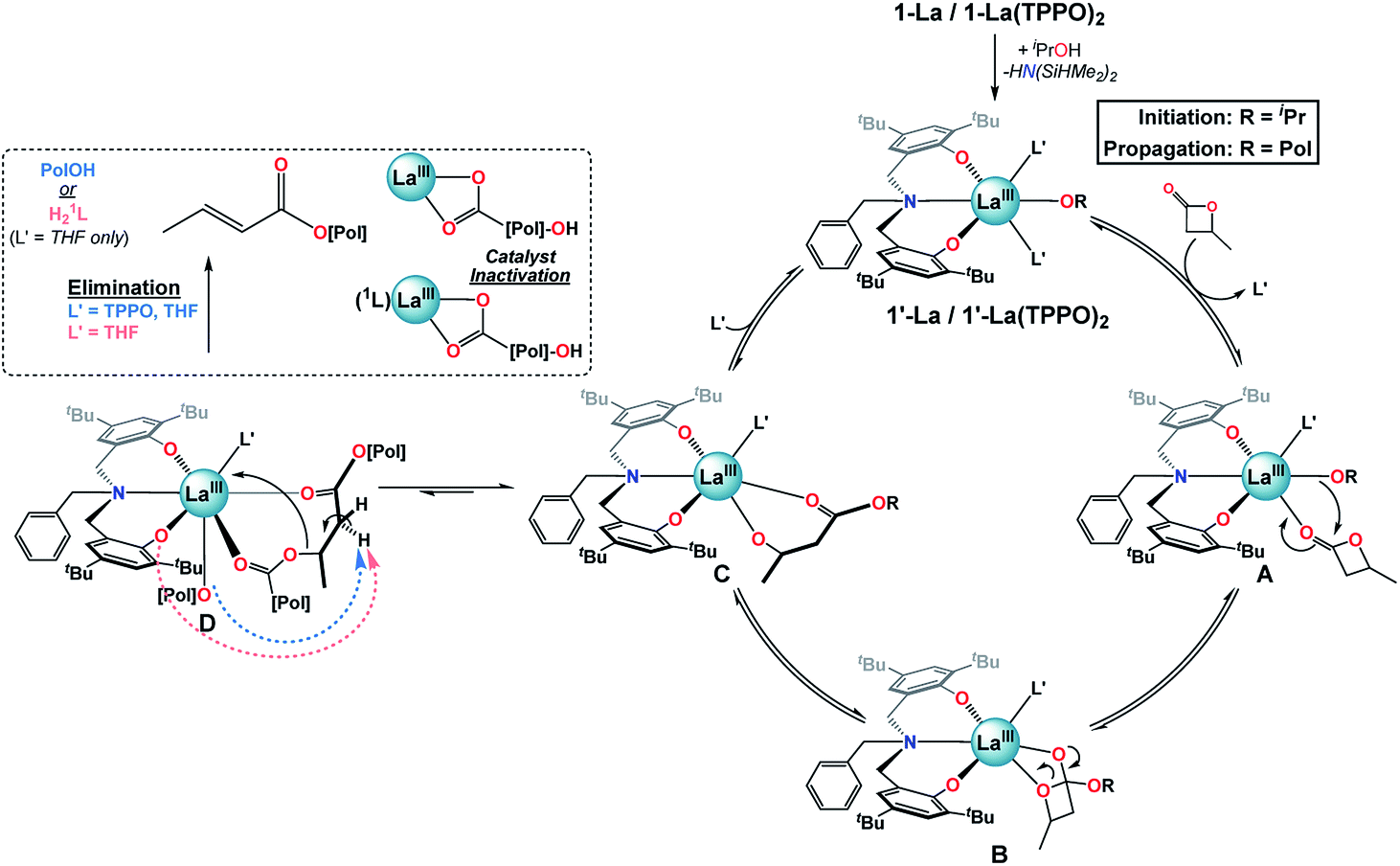 | ||
| Fig. 7 Proposed mechanism for the ROP of rac-BBL catalyzed by 1-La and 1-La(TPPO)2. | ||
Alternatively, at high reaction conversions or with weaker donor ligands, binding of ester linkages to C may become competitive with L′ to form the key intermediate for base-promoted elimination, D. Chain cleavage through elimination would generate two polymer fragments that are inactive for further ROP at RT: (i) a terminated polymer with ester and crotyl end-groups, and (ii) a dormant polyester chain terminated by rare-earth carboxylate and secondary alcohol end-groups. With weak and sterically unencumbered donors (e.g. L′ = THF), both the propagating alkoxide chain and 1L could act as competent bases, while the kinetic basicity of 1L is suppressed with strong and bulky neutral donors (e.g. L′ = TPPO). While intermediate D is depicted with coordination of a neighboring polyester chain, we expect that both intra- and inter-molecular pathways are viable.
While the exact origin of the unique isoselectivity remains unresolved, we hypothesize that strong neutral donors such as TPPO lead to a sterically crowded axial environment in 1-La(TPPO)2 compared to 1-La and 2-RE (Fig. 7). Non-covalent C–H⋯π (arene) interactions between ligand and substrate have been proposed to explain the high syndioselectivity for the ROP of rac-BBL with a yttrium catalyst supported by a cumyl-substituted tetradentate amino-bisphenolate ligand.37b In contrast, we observed similar reactivity and selectivity with phosphine oxides containing aromatic (TPPO) or aliphatic (TOPO) substituents, which suggests other origins for the unique isoselective chain-end stereocontrol. Rieger and coworkers recently carried out an extensive computational study investigating the ROP of rac-BBL catalyzed by 2-Y.25 The syndioselective pathway is favored kinetically and thermodynamically by the propagating P3HB chain adopting a κ3 binding mode. The authors suggest that alternative P3HB binding modes (e.g. κ1 or κ2) may lead to iso-enriched P3HB. Such intermediates (Fig. 7: A or B) could be favored by the stronger binding and enhanced steric bulk of phosphine oxides, opening up new pathways that are disfavored for 2-Y.
Finally, our mechanistic studies uncover new roles for neutral donor ligands in ROP. Previous studies have provided evidence for decreased transesterification19a,b,e,49 and control of catalyst aggregation state19h,j,49a,50 with added neutral donor ligands; however, their role in suppressing base-promoted elimination (i.e. crotyl end-group) was previously unknown. Our results suggest that neutral donor groups play a critical role in suppressing or shifting ligand-exchange equilibria for both productive and non-productive pathways in the ROP of rac-BBL, and addition of these simple ligands provide a facile and inexpensive way to further modulate catalyst performance.
Conclusions
In summary, we have synthesized, characterized, and evaluated the reactivity of novel benzyl-substituted amino-bisphenolate rare-earth complexes, 1-RE, as catalysts for the isoselective ROP of rac-BBL. 1-RE display ROP rates and selectivities that are tuned by the identity of exogenous neutral donor ligands (e.g. OPR3). 1-La/OPR3/iPrOH display excellent reactivity and selectivity (Pm = 0.8 at 0 °C, TOF = ∼190 h−1), and are the most isoselective homogeneous catalysts for ROP of rac-BBL (R: n-octyl, Ph). The use of simple monodentate OPR3 to enhance catalyst performance in stereoselective ROP is unprecedented, and the relative ease and accessibility to a diverse array of phosphine oxides makes this an attractive and operationally simple strategy to further optimize catalyst performance.Our preliminary mechanistic studies indicate that (i) 1-La(TPPO)2 is a precatalyst for the isoselective ROP of rac-BBL, (ii) La(TPPO)n (n = 1, 2) are implicated as catalytically relevant species, (iii) isoselective ROP proceeds with chain-end stereocontrol through a coordination–insertion mechanism, and (iv) addition of neutral donor ligands can suppress elimination side-reactions. This is the first investigation into elimination pathways of RE-based catalysts in the ROP of rac-BBL, and 1-La(TPPO)2 is the first catalyst to access isoenriched P3HB with chain-end stereocontrol. While structurally related catalysts with a tethered donor group (2-RE) also operate under chain-end stereocontrol, ROP activity and selectivity of 2-RE (i) are unaffected by added neutral donor ligands and (ii) display opposite stereoselectivity (syndioselective) compared to 1-La/OPR3. Our study uncovers new roles for neutral donor ligands in stereospecific ROP, and begins to connect their effect on catalyst structure and function. Removing the tethered donor fragment and increasing axial steric bulk with strong neutral donor ligands favors isoenriched P3HB. Similar donor-related enhancements may require catalysts with enhanced metal accessibility (i.e. several labile coordination sites), and highlight new opportunities in catalyst design and optimization. Extension of this approach to other catalysts for stereoselective ROP, the stereoselective synthesis of other oxygenated (co)polymers, and further mechanistic studies are currently underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Brown University for support of this research. We thank Savannah Snyder and Chrys Wesdemiotis, University of Akron, for performing high-resolution MALDI-TOF measurements used for chain-end analysis. We thank Patrick J. Carroll, Eric J. Schelter, and Chris R. Graves for helpful discussions. X. D. and J. R. R. are inventors on U.S. patent application 62/950,702 submitted by Brown University, which covers the catalysts and process described herein.Notes and references
- (a) R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed; (b) B. Worm, H. K. Lotze, I. Jubinville, C. Wilcox and J. Jambeck, Annu. Rev. Environ. Resour., 2017, 42, 1–26 CrossRef; (c) A. A. de Souza Machado, W. Kloas, C. Zarfl, S. Hempel and M. C. Rillig, Global Change Biol., 2018, 24, 1405–1416 CrossRef PubMed; (d) S. Chiba, H. Saito, R. Fletcher, T. Yogi, M. Kayo, S. Miyagi, M. Ogido and K. Fujikura, Marine Policy, 2018, 96, 204–212 CrossRef.
- (a) B. Rieger, A. Künkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, Synthetic Biodegradable Polymers, Springer Berlin, Berlin, 2014, pp. 49–90 Search PubMed; (b) R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS PubMed; (c) E. Bugnicourt, P. Cinelli, A. Lazzeri and V. Alvarez, eXPRESS Polym. Lett., 2014, 8, 791–808 CrossRef; (d) Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354 CrossRef CAS PubMed; (e) Z. A. Raza, S. Abid and I. M. Banat, Int. Biodeterior. Biodegrad., 2018, 126, 45–56 CrossRef CAS; (f) A. Sangroniz, J.-B. Zhu, X. Tang, A. Etxeberria, E. Y. X. Chen and H. Sardon, Nat. Commun., 2019, 10, 3559 CrossRef PubMed.
- (a) Z. Jedliński, M. Kowalczuk, P. Kurcok, L. Brzoskowska and J. Franek, Macromol. Chem. Phys., 1987, 188, 1575–1582 CrossRef; (b) S. Bloembergen, D. A. Holden, T. L. Bluhm, G. K. Hamer and R. H. Marchessault, Macromolecules, 1989, 22, 1656–1663 CrossRef CAS; (c) N. Tanahashi and Y. Doi, Macromolecules, 1991, 24, 5732–5733 CrossRef CAS; (d) Y. Kumagai and Y. Doi, Macromol. Rapid Commun., 1992, 13, 179–183 CrossRef CAS; (e) H. Abe, I. Matsubara, Y. Doi, Y. Hori and A. Yamaguchi, Macromolecules, 1994, 27, 6018–6025 CrossRef CAS; (f) M. Haslböck, M. Klotz, J. Sperl, V. Sieber, C. Zollfrank and D. Van Opdenbosch, Macromolecules, 2019, 52, 5407–5418 CrossRef; (g) H. R. Kricheldorf and S. Eggerstedt, Macromolecules, 1997, 30, 5693–5697 CrossRef CAS; (h) C. Jaimes, M. Arcana, A. Brethon, A. Mathieu, F. Schue and J. M. Desimone, Eur. Polym. J., 1998, 34, 175–185 CrossRef CAS; (i) B. Wu and R. W. Lenz, Macromolecules, 1998, 31, 3473–3477 CrossRef CAS; (j) M. Zintl, F. Molnar, T. Urban, V. Bernhart, P. Preishuber-Pflügl and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 3458–3460 CrossRef CAS PubMed; (k) X. Tang and E. Y. X. Chen, Nat. Commun., 2018, 9, 1–11 CrossRef CAS PubMed; (l) N. Ajellal, M. Bouyahyi, A. Amgoune, C. M. Thomas, A. Bondon, I. Pillin, Y. Grohens and J.-F. Carpentier, Macromolecules, 2009, 42, 987–993 CrossRef CAS.
- S. Kusaka, T. Iwata and Y. Doi, J. Macromol. Sci., Part A: Pure Appl. Chem., 1998, 35, 319–335 CrossRef.
- (a) J. E. Kemnitzer, S. P. McCarthy and R. A. Gross, Macromolecules, 1992, 25, 5927–5934 CrossRef CAS; (b) H. Abe and Y. Doi, Macromolecules, 1996, 29, 8683–8688 CrossRef CAS; (c) M. R. Timmins, R. W. Lenz, P. J. Hocking, R. H. Marchessault and R. C. Fuller, Macromol. Chem. Phys., 1996, 197, 1193–1215 CrossRef CAS; (d) Y. He, X. Shuai, K.-i. Kasuya, Y. Doi and Y. Inoue, Biomacromolecules, 2001, 2, 1045–1051 CrossRef CAS PubMed; (e) S. I. Vagin, A. Kronast, P. T. Altenbuchner, F. Adams, C. Sinkel, P. Deglmann, R. Loos, T. Schuffenhauer, B. Sommer, T. Brück and B. Rieger, Polym. Degrad. Stab., 2017, 143, 176–185 CrossRef CAS.
- (a) Y. K. Leong, P. L. Show, C. W. Ooi, T. C. Ling and J. C.-W. Lan, J. Biotechnol., 2014, 180, 52–65 CrossRef CAS PubMed; (b) J. Możejko-Ciesielska and R. Kiewisz, Microbiol. Res., 2016, 192, 271–282 CrossRef PubMed.
- (a) Y. D. Y. L. Getzler, V. Mahadevan, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1174–1175 CrossRef CAS PubMed; (b) F. Molnar, G. A. Luinstra, M. Allmendinger and B. Rieger, Chem.–Eur. J., 2003, 9, 1273–1280 CrossRef CAS PubMed; (c) J. W. Kramer, E. B. Lobkovsky and G. W. Coates, Org. Lett., 2006, 8, 3709–3712 CrossRef CAS PubMed.
- A. Duda and S. Penczek, Mechanisms of Aliphatic Polyester Formation, in Biopolymers Online, Wiley VCH, 2002, vol. 3b, pp. 371–429 Search PubMed.
- (a) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC; (b) S. M. Guillaume, E. Kirillov, Y. Sarazin and J.-F. Carpentier, Chem.–Eur. J., 2015, 21, 7988–8003 CrossRef CAS PubMed; (c) H. Li, R. M. Shakaroun, S. M. Guillaume and J.-F. Carpentier, Chem.–Eur. J., 2020, 26, 128–138 CrossRef CAS PubMed.
- Select representative examples. Historical examples: (a) H. K. Hall and A. K. Schneider, J. Am. Chem. Soc., 1958, 80, 6409–6412 CrossRef CAS; (b) S. Inoue, Y. Tomoi, T. Tsuruta and J. Furukawa, Macromol. Chem. Phys., 1961, 48, 229–233 CrossRef CAS; (c) D. E. Agostini, J. B. Lando and J. R. Shelton, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9, 2775–2787 CrossRef CAS; (d) T. Yasuda, T. Aida and S. Inoue, Macromol. Rapid Commun., 1982, 3, 585–588 CrossRef CAS; (e) J. E. Kemnitzer, S. P. McCarthy and R. A. Gross, Macromolecules, 1993, 26, 6143–6150 CrossRef CAS; Zn: (f) L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239–15248 CrossRef CAS PubMed; (g) M. Shaik, J. Peterson and G. Du, Macromolecules, 2019, 52, 157–166 CrossRef CAS; group I, II, 13: (h) J. Gao, D. Zhu, W. Zhang, G. A. Solan, Y. Ma and W.-H. Sun, Inorg. Chem. Front., 2019, 6, 2619–2652 RSC; (i) S. M. Quan and P. L. Diaconescu, Chem. Commun., 2015, 51, 9643–9646 RSC; (j) T. Ebrahimi, S. G. Hatzikiriakos and P. Mehrkhodavandi, Macromolecules, 2015, 48, 6672–6681 CrossRef CAS; group IV: (k) B. J. Jeffery, E. L. Whitelaw, D. Garcia-Vivo, J. A. Stewart, M. F. Mahon, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 12328–12330 RSC; Au: (l) E. Brulé, S. Gaillard, M.-N. Rager, T. Roisnel, V. Guérineau, S. P. Nolan and C. M. Thomas, Organometallics, 2011, 30, 2650–2653 CrossRef; REIII (m) D. M. Lyubov, A. O. Tolpygin and A. A. Trifonov, Coord. Chem. Rev., 2019, 392, 83–145 CrossRef CAS; (n) J.-F. Carpentier, Organometallics, 2015, 34, 4175–4189 CrossRef CAS; (o) A. Amgoune, C. M. Thomas, S. Ilinca, T. Roisnel and J.-F. Carpentier, Angew. Chem., Int. Ed., 2006, 45, 2782–2784 CrossRef CAS PubMed; (p) A. Alaaeddine, A. Amgoune, C. M. Thomas, S. Dagorne, S. Bellemin-Laponnaz and J.-F. Carpentier, Eur. J. Inorg. Chem., 2006, 2006, 3652–3658 CrossRef; (q) N. Ajellal, G. Durieux, L. Delevoye, G. Tricot, C. Dujardin, C. M. Thomas and R. M. Gauvin, Chem. Commun., 2010, 46, 1032–1034 RSC; organocatalysts: (r) M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS; (s) W. Jeong, J. L. Hedrick and R. M. Waymouth, J. Am. Chem. Soc., 2007, 129, 8414–8415 CrossRef CAS PubMed; (t) C. G. Jaffredo, J. F. Carpentier and S. M. Guillaume, Macromol. Rapid Commun., 2012, 33, 1938–1944 CrossRef CAS PubMed.
- J.-F. Carpentier, Macromol. Rapid Commun., 2010, 31, 1696–1705 CrossRef CAS PubMed.
- (a) M. Cheng, A. B. Attygalle, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11583–11584 CrossRef CAS; (b) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed; (c) A. F. Douglas, B. O. Patrick and P. Mehrkhodavandi, Angew. Chem., Int. Ed., 2008, 47, 2290–2293 CrossRef CAS PubMed; (d) T. R. Jensen, L. E. Breyfogle, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2004, 2504–2505 RSC; (e) A. P. Dove, H. Li, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2006, 2881–2883 RSC; (f) P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS PubMed; (g) P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pugh and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343–15348 CrossRef CAS PubMed; (h) E. D. Cross, L. E. N. Allan, A. Decken and M. P. Shaver, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1137–1146 CrossRef CAS; (i) C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 9226–9230 CrossRef CAS PubMed; (j) C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Inorg. Chem., 2015, 54, 2204–2212 CrossRef CAS PubMed; (k) K. Nie, W. Gu, Y. Yao, Y. Zhang and Q. Shen, Organometallics, 2013, 32, 2608–2617 CrossRef CAS.
- (a) J. S. Klitzke, T. Roisnel, E. Kirillov, O. d. L. Casagrande and J.-F. Carpentier, Organometallics, 2014, 33, 309–321 CrossRef CAS; (b) J. Fang, M. J. L. Tschan, T. Roisnel, X. Trivelli, R. M. Gauvin, C. M. Thomas and L. Maron, Polym. Chem., 2013, 4, 360–367 RSC; (c) H. Wang, J. Guo, Y. Yang and H. Ma, Dalton Trans., 2016, 45, 10942–10953 RSC.
- (a) T. Takeichi, Y. Hieda and Y. Takayama, Polym. J., 1988, 20, 159–162 CrossRef CAS; (b) A. Le Borgne and N. Spassky, Polymer, 1989, 30, 2312–2319 CrossRef CAS; (c) N. Spassky, C. Pluta, V. Simic, M. Thiam and M. Wisniewski, Macromol. Symp., 1998, 128, 39–51 CrossRef CAS; (d) M. Terrier, E. Brule, M. J. Vitorino, N. Ajellal, C. Robert, R. M. Gauvin and C. M. Thomas, Macromol. Rapid Commun., 2011, 32, 215–219 CrossRef CAS PubMed; (e) R. Reichardt, S. Vagin, R. Reithmeier, A. K. Ott and B. Rieger, Macromolecules, 2010, 43, 9311–9317 CrossRef CAS; (f) S. Vagin, M. Winnacker, A. Kronast, P. T. Altenbuchner, P. Deglmann, C. Sinkel, R. Loos and B. Rieger, ChemCatChem, 2015, 7, 3963–3971 CrossRef CAS; (g) Z. Zhuo, C. Zhang, Y. Luo, Y. Wang, Y. Yao, D. Yuan and D. Cui, Chem. Commun., 2018, 54, 11998–12001 RSC; (h) H. Tani, S. Yamashita and K. Teranishi, Polym. J., 1972, 3, 417–418 CrossRef CAS; (i) M. Iida, T. Araki, K. Teranishi and H. Tani, Macromolecules, 1977, 10, 275–284 CrossRef CAS; (j) K. Teranishi, M. Iida, T. Araki, S. Yamashita and H. Tani, Macromolecules, 1974, 7, 421–427 CrossRef CAS; (k) R. A. Gross, Y. Zhang, G. Konrad and R. W. Lenz, Macromolecules, 1988, 21, 2657–2668 CrossRef CAS.
- R. Ligny, M. M. Hänninen, S. M. Guillaume and J.-F. Carpentier, Angew. Chem., Int. Ed., 2017, 56, 10388–10393 CrossRef CAS PubMed.
- (a) H. E. Dyer, S. Huijser, N. Susperregui, F. Bonnet, A. D. Schwarz, R. Duchateau, L. Maron and P. Mountford, Organometallics, 2010, 29, 3602–3621 CrossRef CAS; (b) L.-Q. Deng, Y.-X. Zhou, X. Tao, Y.-L. Wang, Q.-S. Hu, P. Jin and Y.-Z. Shen, J. Organomet. Chem., 2014, 749, 356–363 CrossRef CAS.
- P. J. Walsh, A. E. Lurain and J. Balsells, Chem. Rev., 2003, 103, 3297–3344 CrossRef CAS PubMed.
- (a) T. Nemoto, T. Ohshima, K. Yamaguchi and M. Shibasaki, J. Am. Chem. Soc., 2001, 123, 2725–2732 CrossRef CAS PubMed; (b) H. Kakei, R. Tsuji, T. Ohshima and M. Shibasaki, J. Am. Chem. Soc., 2005, 127, 8962–8963 CrossRef CAS PubMed; (c) H. Kakei, R. Tsuji, T. Ohshima, H. Morimoto, S. Matsunaga and M. Shibasaki, Chem.–Asian J., 2007, 2, 257–264 CrossRef CAS PubMed; (d) K. Hara, S.-Y. Park, N. Yamagiwa, S. Matsunaga and M. Shibasaki, Chem.–Asian J., 2008, 3, 1500–1504 CrossRef CAS PubMed; (e) J. R. Robinson, J. Yadav, X. Fan, G. R. Stanton, E. J. Schelter, M. A. Pericàs and P. J. Walsh, Adv. Synth. Catal., 2014, 356, 1243–1254 CrossRef CAS.
- Reactivity: (a) P. Dubois, I. Barakat, R. Jerome and P. Teyssie, Macromolecules, 1993, 26, 4407–4412 CrossRef CAS; (b) P. Dubois, R. Jérôme and P. Teyssié, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1994, 35, 536–537 CAS; (c) P. Dubois, N. Ropson, R. Jérôme and P. Teyssié, Macromolecules, 1996, 29, 1965–1975 CrossRef CAS; (d) L. S. Boffa and B. M. Novak, Macromolecules, 1997, 30, 3494–3506 CrossRef CAS; (e) P. Degée, P. Dubois, S. Jacobsen, H.-G. Fritz and R. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2413–2420 CrossRef; (f) P. Ravi, T. Gröb, K. Dehnicke and A. Greiner, Macromolecules, 2001, 34, 8649–8653 CrossRef CAS; selectivity: (g) M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717–6725 CrossRef CAS PubMed; (h) P. Horeglad, P. Kruk and J. Pécaut, Organometallics, 2010, 29, 3729–3734 CrossRef CAS; (i) M. H. Chisholm, K. Choojun, A. S. Chow and G. Fraenkel, Angew. Chem., Int. Ed., 2013, 52, 3264–3266 CrossRef CAS PubMed; (j) P. Horeglad, G. Szczepaniak, M. Dranka and J. Zachara, Chem. Commun., 2012, 48, 1171–1173 RSC.
- A. W. G. Platt, Coord. Chem. Rev., 2017, 340, 62–78 CrossRef CAS.
- M. B. Smith, Phosphorus Ligands, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2013 Search PubMed.
- (a) R. Anwander, O. Runte, J. Eppinger, G. Gerstberger, E. Herdtweck and M. Spiegler, J. Chem. Soc., Dalton Trans., 1998, 847–858 RSC; (b) H. M. Dietrich, C. Meermann, K. W. Törnroos and R. Anwander, Organometallics, 2006, 25, 4316–4321 CrossRef CAS; (c) H. F. Yuen and T. J. Marks, Organometallics, 2008, 27, 155–158 CrossRef CAS; (d) C. Meermann, G. Gerstberger, M. Spiegler, K. W. Törnroos and R. Anwander, Eur. J. Inorg. Chem., 2008, 2008, 2014–2023 CrossRef; (e) Y. Chapurina, J. Klitzke, O. d. L. Casagrande Jr, M. Awada, V. Dorcet, E. Kirillov and J.-F. Carpentier, Dalton Trans., 2014, 43, 14322–14333 RSC.
- H. Ma and J. Okuda, Macromolecules, 2005, 38, 2665–2673 CrossRef CAS.
- K. Nie, L. Fang, Y. Yao, Y. Zhang, Q. Shen and Y. Wang, Inorg. Chem., 2012, 51, 11133–11143 CrossRef CAS PubMed.
- P. T. Altenbuchner, A. Kronast, S. Kissling, S. I. Vagin, E. Herdtweck, A. Pöthig, P. Deglmann, R. Loos and B. Rieger, Chem.–Eur. J., 2015, 21, 13609–13617 CrossRef CAS PubMed.
- L. Wang, D. Yang, D. Li, X. Liu, P. Wang, K. Wang, H. Zhu, L. Bai and R. Wang, Angew. Chem., Int. Ed., 2018, 57, 9088–9092 CrossRef CAS PubMed.
- (a) P. L. Arnold, J.-C. Buffet, R. P. Blaudeck, S. Sujecki, A. J. Blake and C. Wilson, Angew. Chem., Int. Ed., 2008, 47, 6033–6036 CrossRef CAS PubMed; (b) P. L. Arnold, J. C. Buffet, R. Blaudeck, S. Sujecki and C. Wilson, Chem.–Eur. J., 2009, 15, 8241–8250 CrossRef CAS PubMed; (c) J.-C. Buffet, J. Okuda and P. L. Arnold, Inorg. Chem., 2010, 49, 419–426 CrossRef CAS PubMed.
- (a) R. H. Platel, A. J. P. White and C. K. Williams, Chem. Commun., 2009, 4115–4117 RSC; (b) R. H. Platel, A. J. P. White and C. K. Williams, Inorg. Chem., 2011, 50, 7718–7728 CrossRef CAS PubMed.
- (a) Z. Zhang and D. Cui, Chem.–Eur. J., 2011, 17, 11520–11526 CrossRef CAS PubMed; (b) Z. Mou, B. Liu, X. Liu, H. Xie, W. Rong, L. Li, S. Li and D. Cui, Macromolecules, 2014, 47, 2233–2241 CrossRef CAS.
- S. Asano, T. Aida and S. Inoue, J. Chem. Soc., Chem. Commun., 1985, 1148–1149 RSC.
- N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC.
- C.-X. Cai, L. Toupet, C. W. Lehmann and J.-F. Carpentier, J. Organomet. Chem., 2003, 683, 131–136 CrossRef CAS.
- H. Friebolin, Basic One- and Two-Dimensional NMR Spectroscopy, WILEY-VCH, Weinheim, 4th edn, 2005 Search PubMed.
- (a) A. V. Pawlikowski, A. Ellern and A. D. Sadow, Inorg. Chem., 2009, 48, 8020–8029 CrossRef CAS PubMed; (b) J. R. Robinson, Z. Gordon, C. H. Booth, P. J. Carroll, P. J. Walsh and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 19016–19024 CrossRef CAS PubMed.
- (a) T. Fueno, R. A. Shelden and J. Furukawa, J. Polym. Sci., Part A: Polym. Chem., 1965, 3, 1279–1288 CAS; (b) F. A. Bovey and P. A. Mirau, NMR of Polymers, Elsevier, 1996 Search PubMed.
- M. Arcana, O. Giani-Beaune, F. Schué, W. Amass and A. Amass, Polym. Int., 2000, 49, 1348–1355 CrossRef CAS.
- (a) N. Ajellal, D. M. Lyubov, M. A. Sinenkov, G. K. Fukin, A. V. Cherkasov, C. M. Thomas, J.-F. Carpentier and A. A. Trifonov, Chem.–Eur. J., 2008, 14, 5440–5448 CrossRef CAS PubMed; (b) M. Bouyahyi, N. Ajellal, E. Kirillov, C. M. Thomas and J.-F. Carpentier, Chem.–Eur. J., 2011, 17, 1872–1883 CrossRef CAS PubMed; (c) D. Pappalardo, M. Bruno, M. Lamberti and C. Pellecchia, Macromol. Chem. Phys., 2013, 214, 1965–1972 CrossRef CAS; (d) Y. Hori and T. Hagiwara, Int. J. Biol. Macromol., 1999, 25, 237–245 CrossRef CAS PubMed.
- (a) X. Tang, A. H. Westlie, E. M. Watson and E. Y.-X. Chen, Science, 2019, 366, 754–758 CrossRef CAS PubMed; (b) X. Tang, A. H. Westlie, L. Caporaso, L. Cavallo, L. Falivene and E. Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 7881–7890 CrossRef CAS PubMed.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed.
- (a) P. Kurcok, Z. Jedlinski and M. Kowalczuk, J. Org. Chem., 1993, 58, 4219–4220 CrossRef CAS; (b) P. Kurcok, P. Dubois and R. Jèrôme, Polym. Int., 1996, 41, 479–485 CrossRef CAS; (c) A. Dominski, T. Konieczny, M. Zieba, M. Klim and P. Kurcok, Polymers, 2019, 11, 1121 CrossRef PubMed.
- (a) H. Morikawa and R. H. Marchessault, Can. J. Chem., 1981, 59, 2306–2313 CrossRef CAS; (b) A. Ballistreri, D. Garozzo, M. Giuffrida, G. Impallomeni and G. Montaudo, J. Anal. Appl. Pyrolysis, 1989, 16, 239–253 CrossRef CAS; (c) M. Kunioka and Y. Doi, Macromolecules, 1990, 23, 1933–1936 CrossRef CAS; (d) H. Ariffin, H. Nishida, Y. Shirai and M. A. Hassan, Polym. Degrad. Stab., 2008, 93, 1433–1439 CrossRef CAS.
- (a) H. R. Kricheldorf, N. Scharnagl and Z. Jedlinski, Polymer, 1996, 37, 1405–1411 CrossRef CAS; (b) M. Kawalec, G. Adamus, P. Kurcok, M. Kowalczuk, I. Foltran, M. L. Focarete and M. Scandola, Biomacromolecules, 2007, 8, 1053–1058 CrossRef CAS PubMed.
- B. M. Bachmann and D. Seebach, Helv. Chim. Acta, 1998, 81, 2430–2461 CrossRef CAS.
- (a) H. R. Kricheldorf and N. Scharnagl, J. Macromol. Sci., Part A: Pure Appl. Chem., 1989, 26, 951–968 CrossRef; (b) P. Kurcok, M. Kowalczuk, K. Hennek and Z. Jedlinski, Macromolecules, 1992, 25, 2017–2020 CrossRef CAS; (c) Z. Jedliński, P. Kurcok and R. W. Lenz, J. Macromol. Sci., Part A: Pure Appl. Chem., 1995, 32, 797–810 CrossRef.
- (a) T. Zeng, Q. Qian, B. Zhao, D. Yuan, Y. Yao and Q. Shen, RSC Adv., 2015, 5, 53161–53171 RSC; (b) K. Nie, T. Feng, F. Song, Y. Zhang, H. Sun, D. Yuan, Y. Yao and Q. Shen, Sci. China: Chem., 2014, 57, 1106–1116 CrossRef; (c) E. Grunova, E. Kirillov, T. Roisnel and J.-F. Carpentier, Dalton Trans., 2010, 39, 6739–6752 RSC; (d) M. A. Sinenkov, G. K. Fukin, A. V. Cherkasov, N. Ajellal, T. Roisnel, F. M. Kerton, J.-F. Carpentier and A. A. Trifonov, New J. Chem., 2011, 35, 204–212 RSC; (e) I. D'auria, M. Mazzeo, D. Pappalardo, M. Lamberti and C. Pellecchia, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 403–413 CrossRef.
- (a) A. A. Neverov, T. McDonald, G. Gibson and R. S. Brown, Can. J. Chem., 2001, 79, 1704–1710 CAS; (b) M. Hatano and K. Ishihara, Chem. Commun., 2013, 49, 1983–1997 RSC; (c) R. Zeng, H. Sheng, Y. Zhang, Y. Feng, Z. Chen, J. Wang, M. Chen, M. Zhu and Q. Guo, J. Org. Chem., 2014, 79, 9246–9252 CrossRef CAS PubMed.
- (a) M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Acc. Chem. Res., 2009, 42, 1117–1127 CrossRef CAS PubMed; (b) J. R. Robinson, J. Gu, P. J. Carroll, E. J. Schelter and P. J. Walsh, J. Am. Chem. Soc., 2015, 137, 7135–7144 CrossRef CAS PubMed; (c) N. Kumagai, M. Kanai and H. Sasai, ACS Catal., 2016, 6, 4699–4709 CrossRef CAS.
- J.-B. Zhu and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2019, 58, 1178–1182 CrossRef CAS PubMed.
- (a) N. Ropson, P. Dubois, R. Jerome and P. Teyssie, Macromolecules, 1995, 28, 7589–7598 CrossRef CAS; (b) G. Montaudo, M. S. Montaudo, C. Puglisi, F. Samperi, N. Spassky, A. LeBorgne and M. Wisniewski, Macromolecules, 1996, 29, 6461–6465 CrossRef CAS.
- (a) P. Horeglad, M. Cybularczyk, A. Litwińska, A. M. Dąbrowska, M. Dranka, G. Z. Żukowska, M. Urbańczyk and M. Michalak, Polym. Chem., 2016, 7, 2022–2036 RSC; (b) P. Horeglad, M. Cybularczyk, B. Trzaskowski, G. Z. Żukowska, M. Dranka and J. Zachara, Organometallics, 2015, 34, 3480–3496 CrossRef CAS.
- M. Save, M. Schappacher and A. Soum, Macromol. Chem. Phys., 2002, 203, 889–899 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1980000–1980002. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03507f |
| This journal is © The Royal Society of Chemistry 2020 |