
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible OH-bond activation and amphoterism by metal–ligand cooperativity of calix[4]pyrrolato aluminate†
Lukas M.
Sigmund
 and
Lutz
Greb
*
and
Lutz
Greb
*
Ruprecht-Karls-Universität Heidelberg, Anorganisch-Chemisches Institut, Im Neuenheimer Feld 275, 69126 Heidelberg, Germany. E-mail: greb@uni-heidelberg.de
First published on 20th August 2020
Abstract
Most p-block metal amides irreversibly react with metal alkoxides when subjected to alcohols, making reversible transformations with OH-substrates a challenging task. Herein, we describe how the combination of a Lewis acidic square-planar-coordinated aluminum(III) center with metal–ligand cooperativity leverages unconventional reactivity toward protic substrates. Calix[4]pyrrolato aluminate performs OH-bond activation of primary, secondary, and tertiary aliphatic and aromatic alcohols, which can be fully reversed under reduced pressure. The products exhibit a new form of metal–ligand cooperative amphoterism and undergo counterintuitive substitution reactions of a polar covalent Al–O bond by a dative Al–N bond. A comprehensive mechanistic picture of all processes is buttressed by isolation of intermediates, spectroscopy, and computation. This study delineates how structural constraints can invert thermodynamics for seemingly simple addition reactions and invert common trends in bond energies.
Introduction
In the field of transition metal–ligand cooperativity (MLC), ligand aromatization/dearomatization is a paradigmatic modus operandi for substrate activation.1,2 The Milstein group exploited this concept for remarkable catalytic transformations such as the direct synthesis of amides from primary alcohols and amines,3 or the light-supported splitting of water into its elements.4 A critical step during these transformations is the reversible interaction of the catalyst with the protic OH group of the substrate (Fig. 1a).5–7 Upon coordination to the Lewis-acidic metal center, the alcohol proton gets acidified and shifts to the ligand backbone, resulting in the aromatization of the ligand framework. The obtained alkoxo complexes are then the starting points for further reactions. In contrast to the well-developed field of d-block MLC, p-block element–ligand cooperativity (ELC)8 and the conceptual transfer of ligand aromatization/dearomatization into the p-block is happening only very slowly.9–12 Irreversible ELC reactivity of main-group element-based systems with OH-substrates was reported for a few examples: Milstein and coworkers found a 1,2-dihydrodiazaborolopyridine capable of adding benzoic acid through boron–ligand cooperation.10 The Berben group observed aluminum–ligand cooperative addition of phenol, benzyl alcohols,13 water,14 and formic acid15 to their bis(iminopyridine)-ligated aluminum hydride.16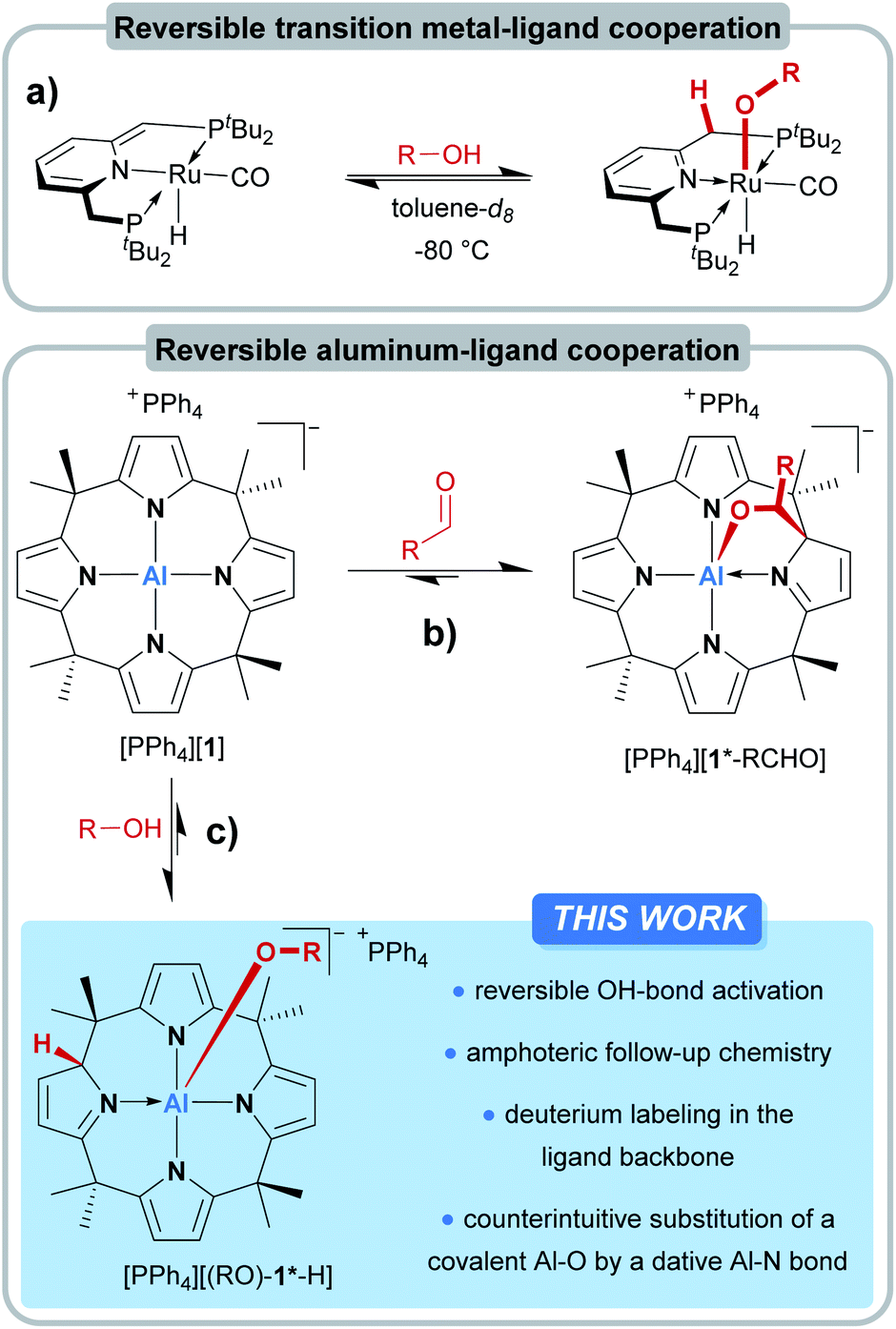 | ||
| Fig. 1 (a) Reversible alcohol activation by transition metal complexes with a metal–ligand cooperative aromatization/dearomatization strategy. (b) Reversible aluminum–ligand cooperativity for the binding of carbonyl compounds through dearomatization/aromatization at meso-octamethylcalix[4]pyrrolato aluminate ([1]−) and (c) against protic substrates, described in this work. | ||
Other examples of aluminum-based cooperative OH-addition reactions were provided by Fedushkin17 and recently by Zhu.18 The formal oxidative addition of ROH to phosphorus(III), likely assisted by ELC, was reported for structurally constrained systems by Goicoechea19 and Dobrovetsky.20 However, the reversible activation of OH-bonds by p-block elements poses a more challenging task. Given the much stronger aluminum–oxygen bond in comparison to the aluminum–nitrogen bond, the reversible interaction of an OH-substrate with an aluminum amide appears even counterintuitive and implausible. Indeed, reports of reversible OH-bond activation by ELC are currently restricted to a xanthene-derived B/P frustrated Lewis pair21 and a structurally constrained trisamido phosphorous(III) system.22
We introduced meso-octamethylcalix[4]pyrrolato aluminate ([1]−), an anionic Lewis acid with a square-planar “anti-van't-Hoff–Le-Bel” coordination environment around the Al(III) center.23 More recently, [1]− was shown to undergo dearomatization during the interaction with carbonyls – the inverse process in comparison to the Milstein complexes (Fig. 1b).24 In the present work, we tackle the questions on how [1]− interacts with protic substances and whether the concept of dearomatization/aromatization can lead to p-block ELC-based reversible OH-bond activation (Fig. 1c).
Results and discussion
The treatment of [PPh4][1] with one equivalent of an alcohol in dichloromethane-d2 at room temperature resulted in an instant color change from colorless to yellow. 1H NMR spectroscopy displayed the formation of a C1-symmetric species with eight chemically inequivalent methyl groups, one dearomatized and three aromatic pyrrole moieties, and one bound alkoxy substituent (Fig. 2a). The observed splitting pattern in the 1H NMR spectrum and the cross-peaks in the 1H,1H COSY NMR spectrum strongly suggested the transfer of the alcohol proton to the 2-position of one of the pyrrole rings. An unambiguous piece of evidence for this aluminum–ligand cooperative motive was obtained from SCXRD measurements of the para-bromophenol addition product, [PPh4][(p-BrPhO)-1*-H] (Fig. 2c, * denotes the dearomatized state of one pyrrole ring). The aluminum atom resides in a distorted square-pyramidal coordination environment having the para-bromophenolato ligand in the apical position with an Al–O bond length of 179.1(2) pm. This distance closely resembles those found for the addition products of aldehydes to [1]− (176–180 pm).24 The Al–N bonds originating from the aromatic pyrrole rings in [(p-BrPhO)-1*-H]− have an average distance of 194.1(2) pm. In contrast, the nitrogen atom of the pyrrole ring bearing the former alcohol proton is separated from the aluminum center by 202.1(3) pm. Combined with the C–N and C–C distances in this ring, the structural picture of dearomatization, as well as the change in Al–N binding mode from polar covalent to dative upon alcohol addition, is supported.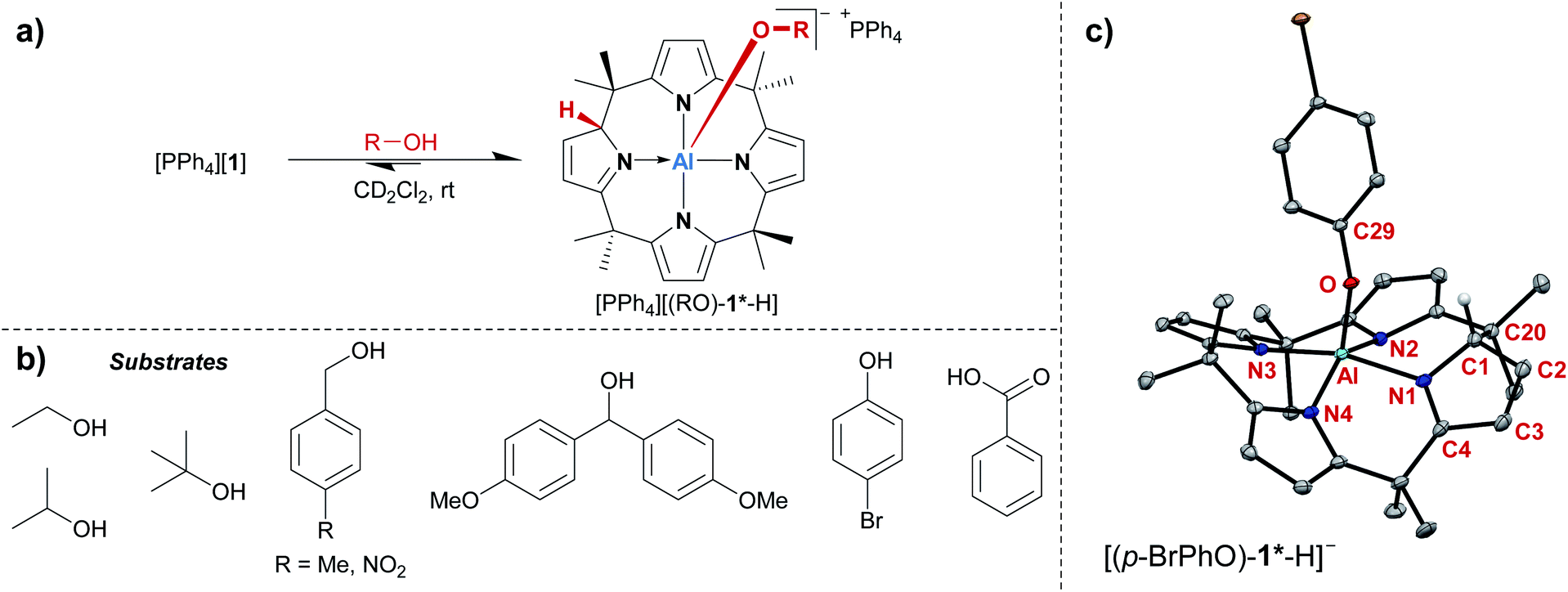 | ||
Fig. 2 (a) Aluminum–ligand cooperative binding of protic substrates (b) by [1]− and (c) molecular structure of the addition product of para-bromophenol obtained from SCXRD measurements. The PPh4+ counter cation, two cocrystallized CH2Cl2 molecules, and all hydrogen atoms except for the proton transferred to the ligand backbone are omitted. Thermal displacement ellipsoids are shown at the 50% probability level. Selected bond lengths [pm]: Al–N1: 202.1(3), Al–N2: 194.3(3), Al–N3: 193.0(2), Al–N4: 194.9(2), Al–O: 179.1(2), N1–C1: 145.7(4), C1–C2: 149.4(4), C2–C3: 132.6(5), C3–C4: 146.4(4), C4–N1: 130.0(4). Selected bond angles [°]: N3–Al–N1: 165.33(10), N2–Al–N4: 147.90(10), O–Al–N3: 104.23(10), O–Al–N2: 102.80(10), O–Al–N4: 107.97(10), O–Al–N1: 90.05(9), N1–C1–C20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :108.9(2), N1–C1–C2: 104.1(2), C2–C1–C20:114.1(2), C29–O–Al: 134.23(17). :108.9(2), N1–C1–C2: 104.1(2), C2–C1–C20:114.1(2), C29–O–Al: 134.23(17). | ||
A broad assortment of protic substrates was suitable for the addition reaction including primary, secondary, and tertiary alkyl and benzyl alcohols, para-bromophenol, and the more acidic benzoic acid (Fig. 2b). Additional functional groups, such as nitro or methoxy groups, were tolerated. Remarkably, [1]− withstood a large excess (>50 eq. relative to [1]−) of the alcohol substrates and did not succumb to alcoholysis.
In contrast, the reaction of the unbridged tetrapyrrolato aluminate with isopropanol resulted in the immediate appearance of the characteristic NH-triplet signal of free pyrrole at around 10 ppm in the 1H NMR spectrum, but there was no sign of cooperative proton transfer (see ESI Fig. S-1†). This contrasting reactivity in comparison to the observations for [1]− is nicely mirrored by COSMO-RS-corrected DFT calculations (Fig. 3a). The cooperative addition of iPrOH to the tetrapyrrolato aluminate was calculated to be energetically unfavorable (ΔRGDFT = 85 kJ mol−1) while the reaction with [(iPrO)-1*-H] is exergonic by 14 kJ mol−1. Consequently, the tetrapyrrolato aluminate cannot accommodate iPrOH through aluminum–ligand cooperativity and instead undergoes pyrrole elimination (ΔRGDFT = −33 kJ mol−1). In contrast, for [1]− the usually observed OH-bond splitting across the aluminum–nitrogen bond brings a thermodynamic disadvantage of more than 35 kJ mol−1 and is hence not found. Ultimately, these inverted thermodynamics can be traced back to the difference in the electronic structure and frontier molecular orbital energies of the tetrapyrrolato aluminate compared to [1]−. In both compounds, the HOMO is located on the ligand framework (see the ESI† for visualization). The structural constrained planar coordination environment of the aluminum center in [1]− results in an increase of the energy of the highest occupied molecular orbital (HOMO) compared to the tetrapyrrolato aluminate. On the other hand, the mainly Al-centered lowest unoccupied molecular orbital (LUMO) in [1]− is significantly lowered in its energy in comparison to the tetrapyrrolato aluminate. These findings underscore the vital impact of planarization on the reactivity of a pyrrolato-ligated Al(III) atom and highlight the robustness of [1]− imparted by the macrocyclic ligand.
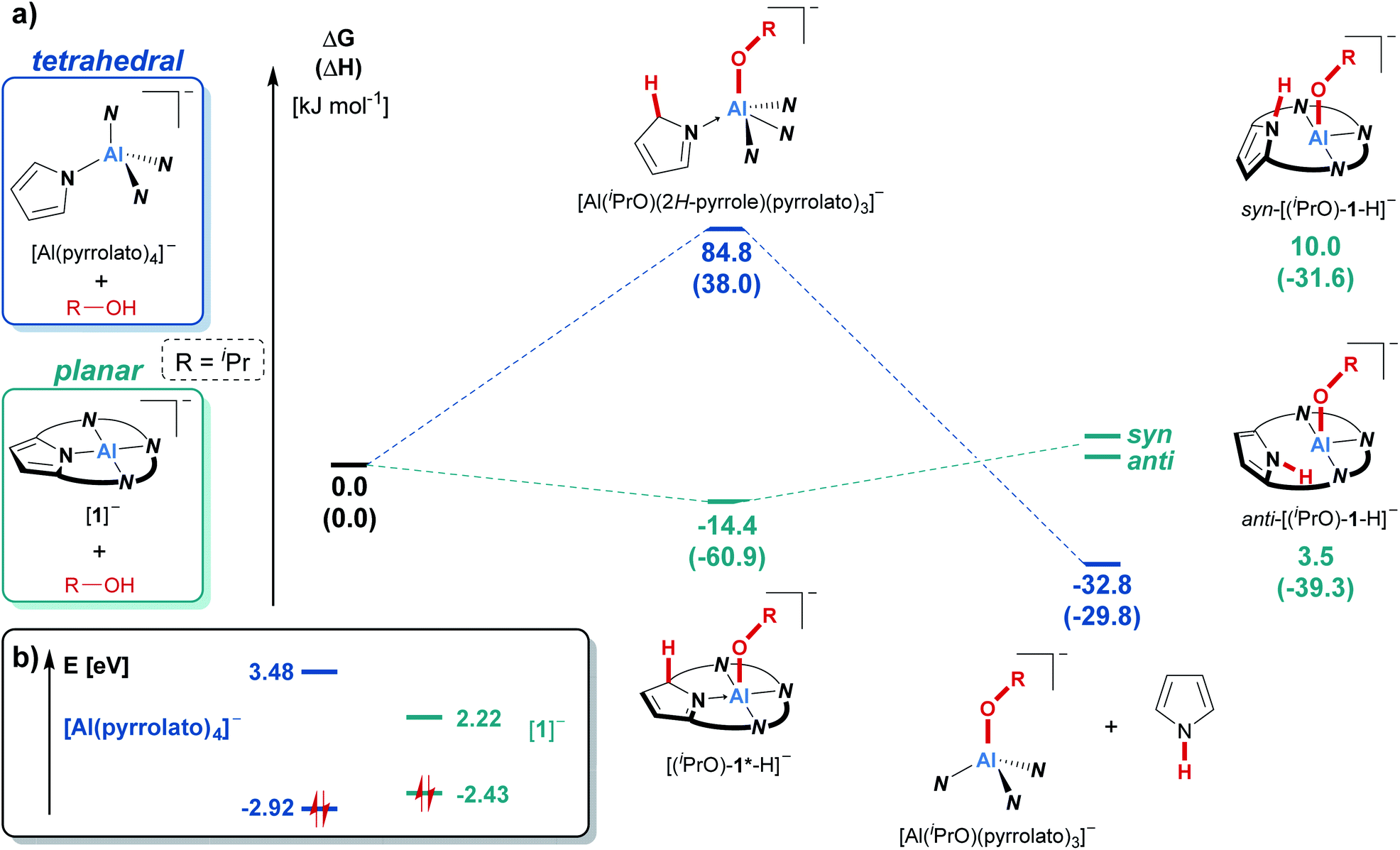 | ||
| Fig. 3 (a) Comparison of the reactivity of [1]− and tetrapyrrolato aluminate ([Al(pyrrolato)4]−) with isopropanol as the model substrate using density functional theory. Calculations were carried out at the PW6B95-D3(BJ)/def-QZVPP//PBEh-3c level of theory. The given values were obtained after considering the solvent environment (CH2Cl2) with the COSMO-RS scheme. For a visualization of the individual molecular structures see the ESI (Chapter S-18).† (b) Kohn–Sham frontier molecular orbital energies (highest occupied and lowest unoccupied molecular orbitals) of the tetrapyrrolato aluminate and of [1]−, obtained at the PW6B95-D3(BJ)/def-QZVPP//PBEh-3c level of theory. | ||
All tested alcohol substrates underwent quantitative addition to [1]− at room temperature, except for tert-butyl alcohol, which gave a mixture of free and bound substrates, with the bound form being clearly favored. It was possible to influence this equilibrium by temperature variation. 1H VT-NMR spectroscopy classified the tBuOH-addition reaction as exothermic by ΔRHobs = −37 kJ mol−1 and exergonic by ΔRGobs = −16 kJ mol−1, which is in reasonable agreement with the thermodynamic parameters calculated at the COSMO-RS-corrected DFT level (ΔRHDFT = −48 kJ mol−1, ΔRGDFT = 2 kJ mol−1, see the ESI† for computational details). The binding Gibbs free energies for all other substrates were computed to be more exergonic, in line with the experimental observations. The obtained ΔRGDFT values span from −14 and −24 kJ mol−1 for isopropanol and ethanol, respectively, down to −48 and −50 kJ mol−1 for benzoic acid and para-bromophenol, respectively. These values are in the same energetic regime as the Gibbs free reaction energies calculated for the addition of aldehydes to [1]− (−14 to −37 kJ mol−1).24
Besides influencing the equilibrium of the tBuOH-addition reaction by temperature variation, it was also possible to completely undo the alcohol binding to [1]− by the application of vacuum. Exposing a mixture of [1]− and tBuOH in tetrachloroethane-d2 to reduced pressure at room temperature for 90 min resulted in the disappearance of the 1H NMR signals of free tBuOH as well as of [(tBuO)-1*-H]−. Instead, the characteristic resonances of [1]− were found in the spectrum. This finding evidences the fact that the OH-bond activation process at [1]− is fully reversible.
To further study the observed reversibility, we examined [1]− in the presence of an excess of alcohol substrate (isopropanol) through 1H,1H exchange NMR spectroscopy (1H,1H EXSY). Chemical exchange cross-peaks for trapped and free isopropanol were found in the spectrum, thus proving the aluminum–ligand cooperative addition to be a highly dynamic low-barrier process. To investigate the nature of the self-exchange, samples with varying excesses of isopropanol were analyzed. As for the aldehyde exchange,24 the obtained EXSY data strongly suggested a dissociative mechanism for the self-exchange of alcohols. The overall rate constant for the release of iPrOH from [1]− was found to be constant irrespective of the amount of excess isopropanol present and was determined to be kobs = 0.122 s−1. It allowed us to obtain the Gibbs free activation energy for the isopropanol elimination (ΔRG‡obs = 78 kJ mol−1). In line with the experiment, the DFT-computed Gibbs free activation energy for the back transfer of the proton from the ligand backbone to iPrO amounts to 63 kJ mol−1, and that of the dissociation of iPrOH from [1]− to 40 kJ mol−1. Moreover, the initial step for an associative exchange mechanism, that is the coordination of an isopropanol molecule to [(iPrO)-1*-H]−trans to the bound isopropanolato ligand, was computed to be energetically disfavored, corroborating the associative character.
Subsequently, the alcohol addition process was elucidated by deuterium labeling. [PPh4][1] was reacted with a slight excess of EtOD (1.7 eq. relative to [1]−) in dichloromethane-d2 at room temperature, and the reaction mixture was analyzed by 1H NMR spectroscopy immediately after alcohol addition. As expected, the spectrum showed the characteristic signals of the addition product; however, there was also a singlet 1H resonance at 5.60 ppm, which is the chemical shift of the transferred proton. This signal grew over time, along with the simultaneous decrease of all pyrrolic β-proton signals (Scheme 1). Hence, we suspected the β-positions as the source of protons in the 2-position. Indeed, a 2H NMR spectrum of a sample of [PPh4][1] and EtOD in dichloromethane-h2 matched the 1H NMR signals of the β-protons as well as that of the transferred proton in [(EtO)-1*-H]−. A series of [1,5]H/D sigmatropic rearrangements was suggested as the mechanism (see the ESI† for a detailed discussion).
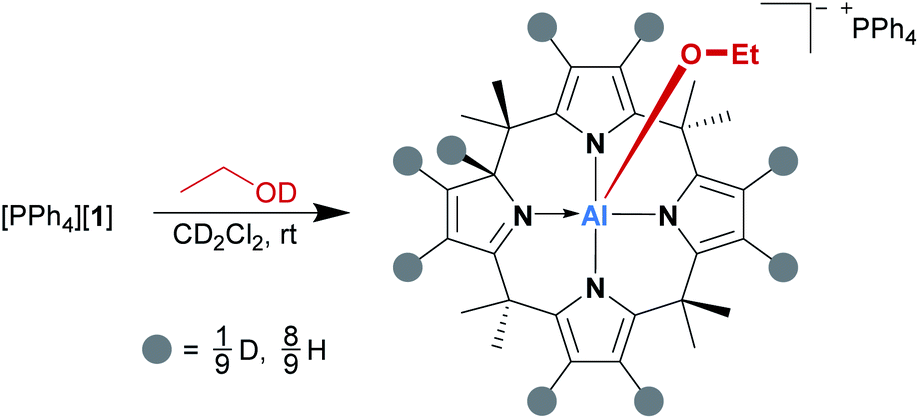 | ||
| Scheme 1 β-Pyrrole deuteration by the reaction of [PPh4][1] with ethanol-d1. | ||
Indeed, the computed Gibbs free activation energies for the involved elementary steps are in the region of 100 kJ mol−1, in line with the experimentally observed reaction at room temperature. The addition of an excess amount of EtOD to [1]− enabled deuteration up to 78%, offering an attractive and mild method for isotopic labeling of calix[4]pyrroles. It has been described that the free calix[4]pyrrole can be deuterated under strongly acidic conditions, such as H2SO4 in D2O.25 Strikingly, [1]− mimics this acidic scenario by synergy with the planar Lewis acidic aluminum center.
During the syntheses of the alcohol addition products, we consistently observed the partial formation of additional species. Although the corresponding 1H NMR signals occurred only with minute intensities (2 to 6% relative to [(RO)-1*-H]−), their formation could not be suppressed, but rather indicated another equilibrium inherent to the system. Serendipitously, we made an observation that turned out to explain this additional reaction pathway. When the alcohol addition products were prepared in 1 mL of dichloromethane, and 5 mL of pentane were added, the expected products [PPh4][(RO)-1*-H] precipitated as yellow solids. However, when these suspensions were allowed to stand at room temperature for several days, the precipitates almost entirely redissolved, and orange crystals developed at the bottom of the reaction vessels. With ethanol as the substrate, these crystals allowed us to determine the atom connectivity by SCXRD analysis, identifying the crystalline material as the dianionic ethanolato complex [PPh4]2[(EtO)-1] (Fig. 4c). The proton which had been transferred to the ligand backbone in the first step is now absent, and the Al(III) center is in a square-pyramidal N4O coordination environment. Of note, it represents the first dianionic AlN4O2− structural motif reported. The fate of the missing proton became clear from the SCXRD analysis of single crystals grown from the supernatant of the same sample. This second species represented the neutral (EtO)-1**-HH (Fig. 4b). It is C2-symmetric for the calix[4]pyrrolato aluminate system, with two protonated, dearomatized pyrrole rings vis-à-vis each other. When the 1H NMR characteristics of (EtO)-1**-HH were compared with the additional set of signals mentioned at the beginning of this section, perfect accordance was found. Hence, the additional signals in the 1H NMR spectra that were steadily observed throughout the OH-bond activations were stemming from an autoprotolysis process, which was found to be dependent on the solvent polarity (Fig. 4a, see the ESI† for a detailed discussion). It discloses another facet of the calix[4]pyrrolato aluminate alcohol interplay, and endows the addition products [(RO)-1*-H]− with an amphoteric character – they can behave either as a Brønsted acid or base.
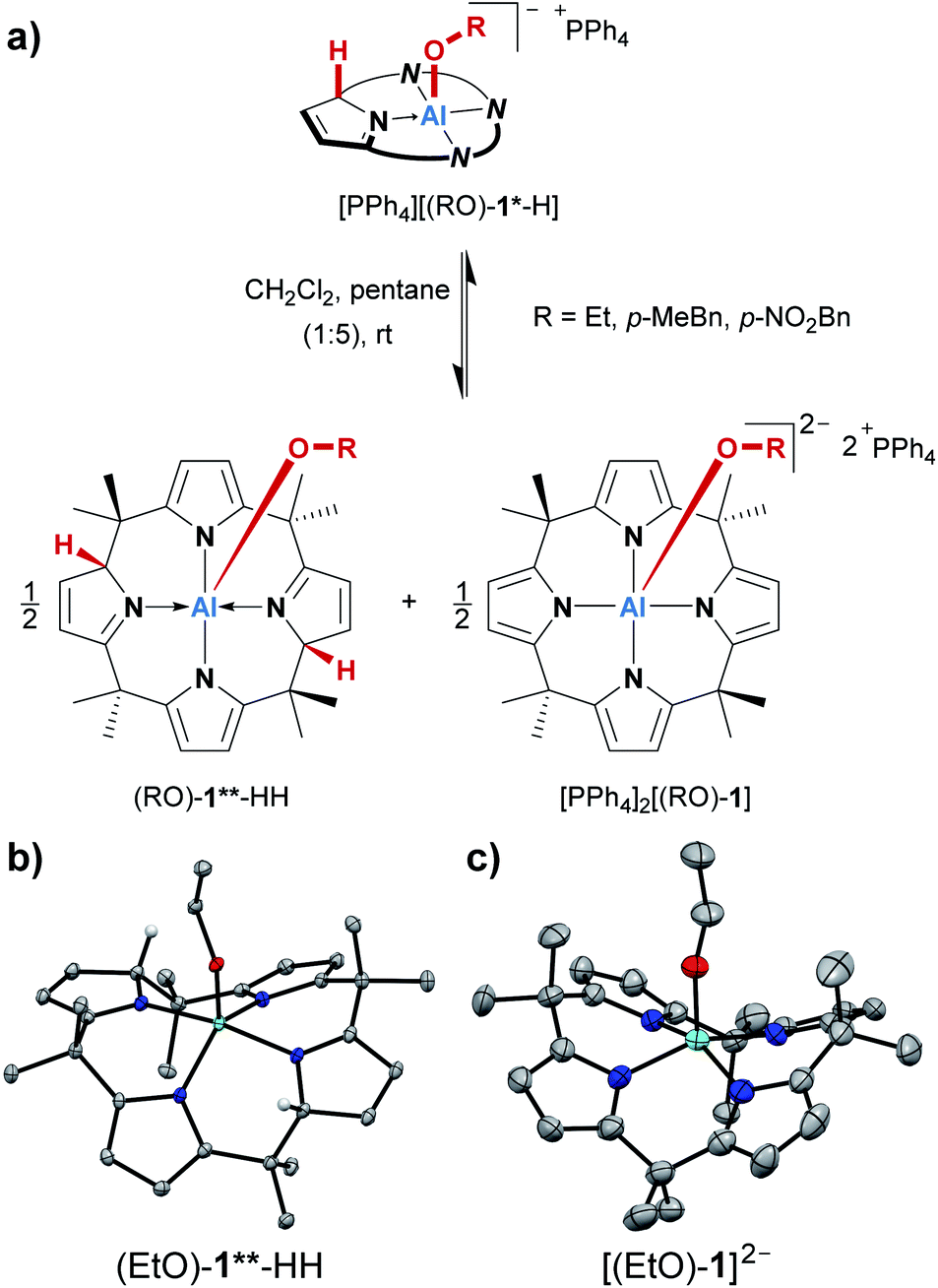 | ||
| Fig. 4 (a) Autoprotolysis equilibrium of the alcohol addition products, which can be enforced by a nonpolar solvent. (b and c) Molecular structures of the charge-separated products obtained from the autoprotolysis with ethanol as a substrate. The PPh4+ counter cations, cocrystallized solvent molecules, and all hydrogen atoms except for the two protons transferred to the ligand backbone in (b) are omitted. Thermal displacement ellipsoids are shown at the 50% probability level. For [PPh4]2[(EtO)-1], only the atom connectivity was determined. | ||
Consequently, the acid/base chemistry of the alcohol adducts was considered. Successive addition of portions of para-bromophenol to its addition product [(p-BrPhO)-1*-H]− in dichloromethane-d2 led to the increased formation of the neutral compound (p-BrPhO)-1**-HH at room temperature (Scheme 2a). Remarkably, even a weak Brønsted acid such as para-bromophenol is sufficiently acidic to protonate [(p-BrPhO)-1*-H]−. In turn, it was expected that the addition of a Brønsted base to the activation products [PPh4][(RO)-1*-H] would induce deprotonation and the formation of the dianionic [(RO)-1]2−. However, another unexpected observation was made. The treatment of the alcohol addition products [PPh4][(RO)-1*-H] (R = Et, iPr, p-MeBn) in dichloromethane-d2 with an excess of pyridine at room temperature resulted in the quantitative substitution of the alcohols and the formation of the classical Lewis adduct [(pyridine)-1]−, as observed by 1H NMR spectroscopy (Scheme 2b). This finding is remarkable: a polar covalent Al–O bond present in [(RO)-1*-H]−, which is commonly perceived as highly stable, is spontaneously cleaved at the expense of a dative Al–N bond, which is formed instead!
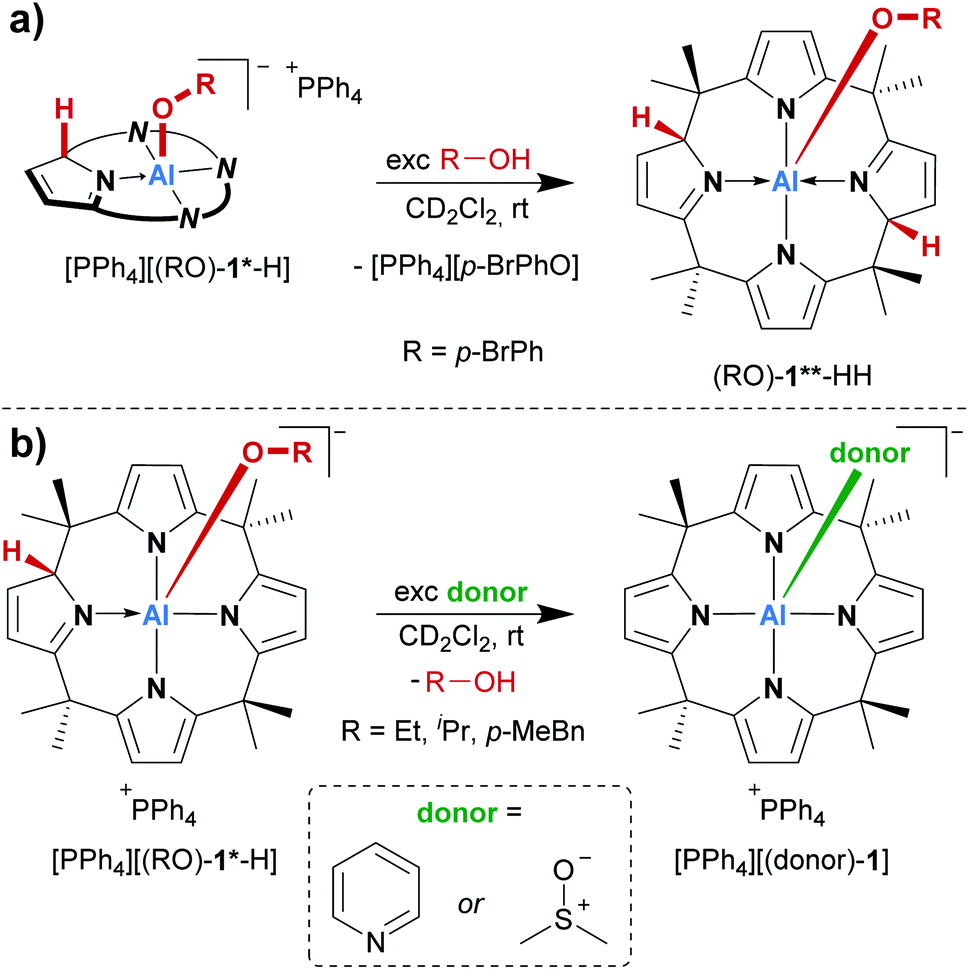 | ||
| Scheme 2 (a) Reactivity of the para-bromophenol addition product ([p-BrPhO]-1*-H) with an excess of p-BrPhOH, and (b) substitution of alcohols added to [1]− with pyridine and DMSO, respectively. | ||
A similar replacement was possible with DMSO as a substitution agent, leading to the Lewis adduct [(dmso)-1]− (Scheme 2b).
Conclusions
In conclusion, we present a clear-cut picture of the reactivity of calix[4]pyrrolato aluminate toward protic substrates. The synergy of the Lewis acidic square-planar aluminum(III) center with the electron-rich ligand backbone enables the rapid OH-bond activation of primary, secondary, and tertiary aliphatic and aromatic alcohols. In contrast to the previously observed activations that consistently occur by OH-bond addition across the Al–N bond, a suitable distal ligand–element cooperative mode is disclosed herein. The addition products undergo rapid low-barrier self-exchange, and the alcohol binding can be fully reversed upon application of vacuum or elevated temperature. Furthermore, the alcohol activation products exhibit a new form of element–ligand cooperative amphoterism and autoprotolysis. The addition of sterically unhindered Lewis/Brønsted bases to the addition products induces the counterintuitive cleavage of a polar covalent Al–O bond. Such processes are, to the best of our knowledge, unprecedented, and likely originate from the rearomatization occurring in the ligand backbone upon alcohol release. The dearomatization upon alcohol binding prevents the system from collapsing into deep energetic sinks and upholds a balanced energy landscape for reversibility. Hence, these inverted thermodynamics are the combined result of structural constraint and element–ligand cooperativity, and illustrate how ligand engineering allows annulling common bond energy preferences. It might enable unconventional sequences of substrate activation and product release during catalytic cycles. Noteworthily, we have observed alcohol oxidation upon heating of the activation products, though not yet in a catalytic fashion. Once more, these findings exemplify the evolution of an “anti-van't Hoff/Le Bel” species from a structural curiosity to a reagent with unique but well-defined reactivity and a multi-purpose platform for rapid dynamic covalent chemistry.Conflicts of interest
There are no conflicts to declare by the authors.Acknowledgements
We thank Prof. H.-J. Himmel for his constant support and Prof. M. Enders for helpful discussions. Financial support was provided by the FCI (Liebig fellowship, L. G.) and the DFG (Emmy-Noether program, GR5007/2-1, L. G.). The BWFor- and the BWUniCluster are acknowledged for computational resources, funded by the DFG.References
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed.
- S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, Y. Ben-David, M. A. Iron and D. Milstein, Science, 2009, 324, 74–77 CrossRef CAS PubMed.
- J. Li, Y. Shiota and K. Yoshizawa, J. Am. Chem. Soc., 2009, 131, 13584–13585 CrossRef CAS PubMed.
- X. Yang and M. B. Hall, J. Am. Chem. Soc., 2010, 132, 120–130 CrossRef CAS PubMed.
- M. Montag, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2012, 134, 10325–10328 CrossRef CAS PubMed.
- L. Greb, F. Ebner, Y. Ginzburg and L. M. Sigmund, Eur. J. Inorg. Chem., 2020, 3030–3047 Search PubMed.
- U. Gellrich, F. Wech and M. Hasenbeck, Chem.–Eur. J., 2020 DOI:10.1002/chem.202001276.
- U. Gellrich, Y. Diskin-Posner, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2016, 138, 13307–13313 CrossRef CAS PubMed.
- U. Gellrich, Angew. Chem., Int. Ed., 2018, 57, 4779–4782 CrossRef CAS PubMed.
- M. Hasenbeck, T. Müller and U. Gellrich, Catal. Sci. Technol., 2019, 9, 2438–2444 RSC.
- T. J. Sherbow, C. R. Carr, T. Saisu, J. C. Fettinger and L. A. Berben, Organometallics, 2016, 35, 9–14 CrossRef CAS.
- T. W. Myers and L. A. Berben, Organometallics, 2013, 32, 6647–6649 CrossRef CAS.
- T. W. Myers and L. A. Berben, Chem. Sci., 2014, 5, 2771–2777 RSC.
- E. J. Thompson, T. W. Myers and L. A. Berben, Angew. Chem., Int. Ed., 2014, 53, 14132–14134 CrossRef CAS PubMed.
- M. V. Moskalev, A. N. Lukoyanov, E. V. Baranov and I. L. Fedushkin, Dalton Trans., 2016, 45, 15872–15878 RSC.
- P. Wang, M. Zhang and C. Zhu, Organometallics, 2020, 39, 2732–2738 CrossRef CAS.
- T. P. Robinson, D. M. De Rosa, S. Aldridge and J. M. Goicoechea, Angew. Chem., Int. Ed., 2015, 54, 13758–13763 CrossRef CAS PubMed.
- S. Volodarsky and R. Dobrovetsky, Chem. Commun., 2018, 54, 6931–6934 RSC.
- P. Vasko, M. Á. Fuentes, J. Hicks and S. Aldridge, Dalton Trans., 2019, 48, 2896–2899 RSC.
- W. Zhao, S. M. McCarthy, T. Y. Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 17634–17644 CrossRef CAS PubMed.
- F. Ebner, H. Wadepohl and L. Greb, J. Am. Chem. Soc., 2019, 141, 18009–18012 CrossRef CAS PubMed.
- F. Ebner, L. M. Sigmund and L. Greb, Angew. Chem., Int. Ed. DOI:10.1002/anie.202007717.
- Y.-C. He, J.-G. Pan and D.-S. Liu, Tetrahedron Lett., 2016, 57, 3133–3136 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and experimental procedures, spectroscopic and crystallographic characterisation data, computational details, and further discussions. CCDC 2013196 and 2013197. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03602a |
| This journal is © The Royal Society of Chemistry 2020 |