
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShaping non-noble metal nanocrystals via colloidal chemistry
Valeria
Mantella
,
Laia
Castilla-Amorós
and
Raffaella
Buonsanti
 *
*
Laboratory of Nanochemistry for Energy (LNCE), Department of Chemical Sciences and Engineering, École Polytechnique Fedérale de Lausanne, CH-1950 Sion, Switzerland. E-mail: raffaella.buonsanti@epfl.ch
First published on 5th October 2020
Abstract
Non-noble metal nanocrystals with well-defined shapes have been attracting increasingly more attention in the last decade as potential alternatives to noble metals, by virtue of their earth abundance combined with intriguing physical and chemical properties relevant for both fundamental studies and technological applications. Nevertheless, their synthesis is still primitive when compared to noble metals. In this contribution, we focus on third row transition metals Mn, Fe, Co, Ni and Cu that are recently gaining interest because of their catalytic properties. Along with providing an overview on the state-of-the-art, we discuss current synthetic strategies and challenges. Finally, we propose future directions to advance the synthetic development of shape-controlled non-noble metal nanocrystals in the upcoming years.
1. Introduction
Metal nanocrystals (NCs) continue to be at the forefront of modern science and technology with their applications spanning from catalysis and energy conversion to photonics, biology and medicine.1–5 Their physico-chemical properties are strongly influenced by size, shape and composition, and thus, tailoring such features provides an effective strategy to boost their performances and broaden their applications.1–5 To this aim, colloidal chemistry has been largely exploited as it enables the design of NCs with atomic-level precision, while minimizing particle polydispersity under relatively mild conditions.6–10 A huge library of noble metal NCs with a large variety of structures and a continuously increasing compositional complexity has been assembled up to now.1,7,11–13 In parallel to synthetic progress, the development of advanced methods to study the impact of different shapes on the physicochemical properties of noble metal NCs is proceeding at a considerable rate. For example, single-particle microscopy and spectroscopy studies to investigate individual particle effects are emerging in the literature.14,15 On the other side, non-noble metal NCs are far less investigated and the level of shape control attainable for this class of NCs is still considerably scarce. Consequently, a deep understanding of their physical and chemical properties has yet to be achieved. Furthermore, the range of applications is still quite limited compared to that where these NCs can potentially make an impact.In this contribution, we focus on third row transition metal NCs, specifically Mn, Fe, Co, Ni and Cu. These metals are of particular interest for catalytic applications, in the quest for the use of more earth-abundant elements, as well as for their magnetic properties (i.e. Fe, Co, and Ni).16–20 First, we present a general overview on the state-of-the-art in the synthesis of these non-noble metal NCs. Then, because of the more extensive body of literature, we use Cu NCs to showcase the importance of shape control for applications. Finally, we conclude with an outlook on future directions towards advancing the synthetic development of this class of materials.
2. General principles of colloidal synthesis
A typical colloidal synthesis of metal NCs involves the reduction or decomposition of a metal precursor in the presence of ligands in either aqueous or organic media.6–9,21,22 Whether they serve only as surface passivating agents or take part into the reaction by complexing metal precursors, ligands play a crucial role in regulating the NC size and shape.6–9,21,22 Many reaction parameters need to be carefully tuned during the synthesis, including temperature, concentration, atmosphere, injection rate, and reactor volume, in order to tailor the shape and size of metal NCs at will. The topic has been extensively discussed in dedicated reviews and we refer to those for details.1,7,12,23 Below, we summarize some of the general principles to provide a basic background for readers who are not familiar with the field.2.1 Nanocrystal nucleation and growth
The bases of our understanding on the synthesis of uniform NCs are covered by the classical theories dating back to the 1870s and 1920s.24 According to these, the NC formation in a homogeneous solution occurs in three main steps: (1) conversion of the molecular precursor into monomers, such as atoms, ions or molecules, which are the active species accounting for nucleation; (2) increase of the monomer concentration until nucleation occurs at supersaturation, (3) growth of stable nuclei that have surpassed the critical size, via monomer diffusion and reaction at the surface to form the final NCs. Within the classical theories, the concept of ‘burst nucleation’ has been established as crucial for the synthesis of monodisperse NCs as it refers to the formation of nuclei within a short period of time, followed by their growth, without additional nucleation events.24While they provide fundamental knowledge, the classical theories do not always describe the actual reaction pathways underlying the formation of NCs.21,25,26 Recently, in situ methods, based on X-rays and on transmission electron microscopy (TEM), evidence the existence of multistep nucleation mechanisms during the synthesis of single and multi-component NCs that can be described within the framework of a nonclassical nucleation theories.22,27–31 Here, the free-energy landscape shows multiple local minima corresponding to reaction intermediates, pre-nucleation structures, (i.e. nanoclusters, amorphous phases, and lamellar polymers), which form as expedients to lower the homogeneous nucleation energy barrier.22,32–35 To cite one example, Au and Ag NCs have been shown to form through a spinodal structure evolving into amorphous clusters by in situ TEM measurements.36 In a different study, crystalline domains were observed to gradually evolve within an amorphous phase and finally transform into the crystalline Ni NCs.37 A recent in situ X-ray absorption and scattering investigation has revealed that spherical Cu NCs nucleate from coordination polymer lamellae.22 Overall, these and many other findings highlight the diversity in crystallization processes and, therefore, the importance of developing more accurate and broadly applicable nucleation models.
2.2 Parameters influencing the shape of colloidal nanocrystals
In general, the NC shape is dictated by the growth regime, which can occur under thermodynamic or kinetic control.1 Thermodynamics drive the reaction at high reaction temperatures and/or when the solution is supplied with a gradual monomer flux, while, kinetics dominate at low reaction temperatures and/or for a high monomer flux. The monomer flux can be adjusted by the delivery method of the metal precursor (i.e. fast or dropwise injection) or by controlling its decomposition or reduction rate.1,12,21,23 The conditions adopted in a thermodynamic regime ensure that the monomers have enough mobility and/or time to attain the global minimum for the Gibbs free energy. This implies that the NCs will ideally assume the equilibrium shape as defined by the Wulff construction, where the total interfacial free energy (γ) is minimized, at a fixed volume.1,12,23 In contrast, metastable shapes occupying local energy minima will be obtained under a kinetic growth regime.1,12,23Ligands acting as surface passivating agents will also greatly contribute to determining the final NC shape. Indeed, their preferential binding to certain crystalline planes modulates the interfacial energy consequently hindering the growth of particular faces and favouring others.1,12 Thus, shape control can be attained by exploiting capping ligand effects.
Particularly common for metal NCs, one strategy to achieve shape control is to introduce pre-formed seeds in the reaction vessels. Indeed, their structure will play an important role in directing the final shape of the NCs. For example, single crystal seeds will form octahedral or cubic NCs while plates with stacking faults will generate NCs as hexagonal or triangular plates.7,38
3. Shape control in non-noble metal nanocrystals
One of the reasons behind the delayed development in the synthesis of non-noble metals is their tendency to easily oxidize. Most of the synthetic development for noble metals has been carried out in water where varying the power of the reducing agent (i.e. ethylene glycol, ascorbic acid, citric acid, and poly(vinyl pyrrolidone)) provides a way to tune the monomer flux and therefore the shape of the final NCs.1,7,11–13,39–43 As for non-noble metals the choice is restrained to strong reducing agents when the synthesis is carried out in an aqueous environment, thus limiting the shape tunability. In the case of more oxophilic elements, such as Fe or Mn, obtaining NCs as pure metals is extremely challenging. Indeed, an oxide shell was found to form around Fe NCs when synthesized in an aqueous environment even when using a strong reducing agent such as NaBH4.44,45 With the exception of Cu, which is discussed below in detail, non-noble metal NCs have been mostly synthesized via high-temperature decomposition of organometallic precursors in organic solvents.A few early examples of Co NCs were published by Puntes et al.46–49 By hot-injection and consequent pyrolysis of cobalt carbonyl in the presence of oleic acid (OLAC) and trioctylphosphine oxide (TOPO), the authors synthesized thermodynamically stable spherical Co NCs (Fig. 1a); they could then access metastable shapes, including nanodisks (Fig. 1b), when quenching the reaction at the very early stages, and nanorods, when lowering the reaction temperature.46–50 The role of the surfactant mixture was explained as a way to modulate the relative growth rate of different facets. Later on, substituting the TOPO with linear amine was found to produce a higher yield of nanodisks which were stable for longer time periods compare to those obtained with TOP, before converting into the more stable spheres.66 Interestingly, Bao et al. pointed at the importance of Co-surfactant complexes forming during these reactions rather than only considering the surfactants as surface passivating agents.50 While not supported by experiments, the authors conducted calculations aiming at predicting the formation pathways based on the stability of Co-surfactant complexes. These complexes are expected to directly influence the monomer concentration and therefore the nucleation rate.70 As a proof that the organometallic complexes forming in solution are important, Co nanorods (Fig. 1c) and nanowires were successfully obtained by decomposition of a pre-synthesized olefinic complex [Co(η3-C8H13)(η4-C8H12)] or by reduction of [Co{N(SiMe3)2}2(thf)] in anisole under a pressure of 3 bar H2 in the presence of alkylamines and carboxylic acids as ligands.51,52 Cormary et al. have conducted a beautiful time dependent in situ X-ray absorption (XAS) study to follow the evolution of both the oxidation state and coordination environment of these precursors over the course of the reaction.52 These data suggested that the starting precursor rapidly exchanges ligands to the mixture of alkylamines before getting reduced by the hydrogen. Unfortunately, the dramatic differences in timescale between the laboratory and in situ XAS synthesis did not allow the correlation of the reduction with the nanorod growth. However, the authors proposed that the anisotropic growth is favoured by the inherent anisotropic hcp structure of the Co together with efficient passivation of the longitudinal facets by the laurate ligand.52
 | ||
| Fig. 1 Third row transition metal NCs. TEM images of (a) Co nanospheres prepared by the pyrolysis of Co2(CO)8 in dichlorobenzene with OLAC and TOPO at 182 °C for 30 minutes. Reprinted with permission from ref. 47. Copyright 2001 AAAS; (b) Co nano-disk product of the quenching of the previous reaction after 5 seconds. Reprinted from ref. 46. 2002 American Chemical Society; (c) Co nanorods obtained from the decomposition of [Co(η3-C8H13)(η4-C8H12)] in the presence of OLAC and octadecylamine in anisole under a pressure of 3 bar H2 at 150 °C for 48 hours. Reprinted with permission from ref. 50. Copyright 2010 John Wiley and Sons; (d) Ni nanorods synthesized from Ni(COD)2 (COD = cycloocta-1,5-diene) as the precursor in THF with hexadecylamine at 70 °C for 12 h, under dihydrogen pressure (3 bar). Reprinted from ref. 54. 2017 American Chemical Society; (e) Fe nanorods prepared by consecutive additions of Fe(CO)5 and TOP to 2 nm nanosphere seeds at 320 °C followed by aging for 30 minutes. Reprinted from ref. 45. 2005 American Chemical Society; (f) OLAC-capped Mn NCs synthesized by reducing MnCl2 with n-BuLi at 200 °C with a reaction time of 20 minutes. Reprinted from ref. 56. 2000 American Chemical Society. | ||
In one of the few relevant examples, size-tunable spherical Ni NCs were synthesized by reacting nickel acetylacetonate with oleylamine (OLAM) and trioctylphosphine (TOP).53 Varying the relative amount of OLAM and TOP allowed the control of size. The authors pointed at the role of TOP as a ligand forming Ni–TOP complexes, then getting reduced by OLAM, and also as the main surface passivating agent. However, the use of TOP in the synthesis of Ni can be problematic as nickel phosphides might form. An alternative synthetic route involves the thermal decomposition of bis(1,5-cyclooctadiene)-nickel(0) in OLAC to obtain nanospheres or in hexadecylamine (HDA) to obtain nanorods (Fig. 1d).54,55 In the latter, the shape control was attributed to specific surface coordination of the HDA.
As for the Fe NCs, there is only one example illustrating that Fe nanorods (Fig. 1e) could be obtained by the pyrolysis of Fe(CO)5 in the presence of TOP, respectively.56 The nanorods were formed by controlled coalescence of the spheres via consecutive injections of the iron precursor. Nanospheres were instead obtained with TOPO.
There is also only one relevant report for Mn nanospheres (Fig. 1f), where they were synthesized by reducing MnCl2 with n-BuLi in diphenyl ether with oleic acid as the ligand.57
4. The case of copper nanocrystals
Compared to Mn, Fe, Co and Ni, the synthesis of Cu NCs is the most advanced. A comprehensive survey on all the copper and copper oxide nanostructures produced by different synthetic approaches has been reported elsewhere.58,59 Here, we focus on recent examples where Cu NCs are synthesized by means of colloidal chemistry. Because of its physical and chemical properties being not far from those of noble metals, Cu is often referred to as a semi-noble metal. As a matter of fact, its synthesis has been developed both in the aqueous environment and in organic solvents.4.1 Synthesis in an aqueous environment
One of the challenges with Cu is to find a suitable reducing agent to convert Cu(II) to Cu(0).Jin et al. found glucose to be suitable to drive this reduction and HDA to work well as a capping agent for the Cu {100} facets when used in conjunction with it.60 Cu nanowires and nanocubes (Fig. 2a and b) were produced by adjusting the concentrations of these two reagents to control the surface passivation or crystalline habit of the stable nuclei forming during the synthesis.
 | ||
| Fig. 2 Cu NCs synthesized in aqueous solutions via direct nucleation. Electron microscopy images of (a) Cu nanocubes prepared after reduction of CuCl2·2H2O by glucose, in the presence of HDA in water for 6 hours at 100 °C, under magnetic stirring; (b) Cu nanowires synthesized as in (a) but using a higher concentration of HDA (18 mg ml−1vs. 9 mg ml−1). (a) and (b) are reprinted with permission from ref. 60. Copyright 2011 John Wiley and Sons; (c) Cu nanoplates obtained by reducing the Cu[N2H4O2]2+·2OAc− complex in DMF at 60 °C, in the presence of PVP. Reprinted with permission from ref. 61. Copyright 2009 John Wiley and Sons. | ||
Most of the other syntheses based on direct homogeneous nucleation reported for Cu NCs use hydrazine as the reductant for copper salts. For example, Cu nanoplates with a diameter of around 50 nm and an average thickness of around 20 nm (Fig. 2c) were obtained by reducing copper(II) acetate with hydrazine in the presence of N,N-dimethylformamide (DMF) and polyvinylpyrrolidone (PVP).61 While DMF was observed to be crucial for shape control, its exact role remained unclear.
Cu nanowires were instead obtained through the reduction of copper nitrate with hydrazine in a NaOH solution in the presence of ethylenediamine (EDA).62–65 In this mixture, the initially formed Cu(OH)4− complex turns into Cu(OH)2− with a small amount of Cu2O nanoparticles upon the addition of hydrazine. The latter are then further reduced to metallic Cu aggregates which serve as seeds for the growth of the nanowires. The EDA acts as the surface passivating ligand inducing preferential growth along the [100] direction.
Turning to the seeded-growth approach, penta-twinned Cu nanorods were obtained when using single-crystal Au and Pd twinned decahedra as seeds, with the latter leading to better size monodispersity.66,67 By changing the Cu precursor/Pd seed ratio, as well as the growth time, the nanorod length was tuned, while the diameter remained unchanged. Using Pd cubic seeds, Wang et al. synthesized Cu nanocubes. After that, controlled etching using a TOP–Se complex was utilized to eventually obtain rhombic dodecahedrons with enriched high-energy {110} facets (Fig. 3a).68
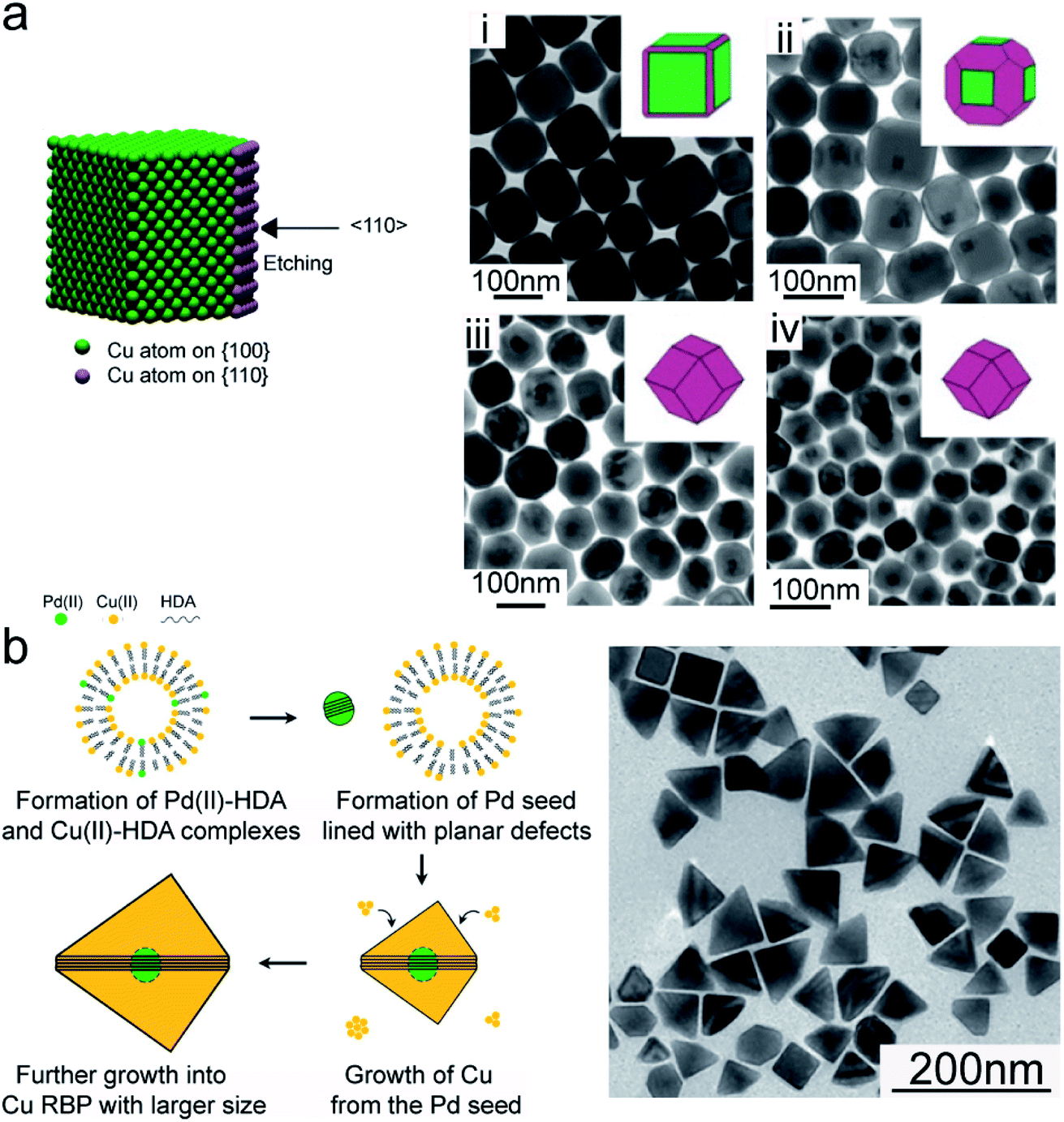 | ||
| Fig. 3 Cu NCs synthesized in aqueous solutions by seeded-growth. (a) Schematic illustration representing the preferred etching by TOP–Se and TEM images illustrating the gradual evolution of Cu nanocubes into Cu rhombic dodecahedrons as the etching time increases: (i) 4 h, (ii) 8 h, (iii) 12 h, and (iv) 24 h. Reprinted from ref. 68. 2016 American Chemical Society. (b) Schematic illustration of the formation mechanism and TEM image of Cu right bipyramids obtained by first reacting trace amounts of Na2PdCl4, CuCl2·2H2O, HDA and glucose at room temperature overnight and then heating the resulting mixture at 100 °C for 6 h, under magnetic stirring. Reprinted from ref. 69. 2018 American Chemical Society. | ||
In situ forming Pd seeds have instead been shown to lead to Cu right bipyramids (Fig. 3b).69 The reaction mixture included trace amounts of Na2PdCl4, copper(II) chloride, hexadecylamine and glucose. Herein, the coordination of the amine slows down the reduction rate of Pd(II) and leads to the formation of Pd seeds lined with multiple parallel planar defects, followed by the growth of Cu on them.69
4.2 Synthesis in organic solvents
The synthesis of Cu NCs in organic solvents has resulted in the formation of Cu spheres, cubes, octahedra, tetrahedra and wires.21,22,70–75To start with the simplest shape, Hung et al. have reported a reproducible and facile synthesis where copper(I) acetate (CuOAc) decomposes in trioctylamine (TOA) in the presence of tetradecylphosphonic acid (TDPA) to form highly monodisperse 8 nm Cu spheres.72 Mantella et al. have recently studied this reaction using in situ X-ray measurements and identified a Cu–TDPA coordination lamellar polymer as the reaction intermediate accounting for the achieved size monodispersity (Fig. 4a).22 Having identified this polymer to be crucial for the NC growth within a size-focusing regime, monodisperse Cu nanospheres with various sizes, 3, 12, 14 and 26 nm, were obtained by changing its concentration in solution.22
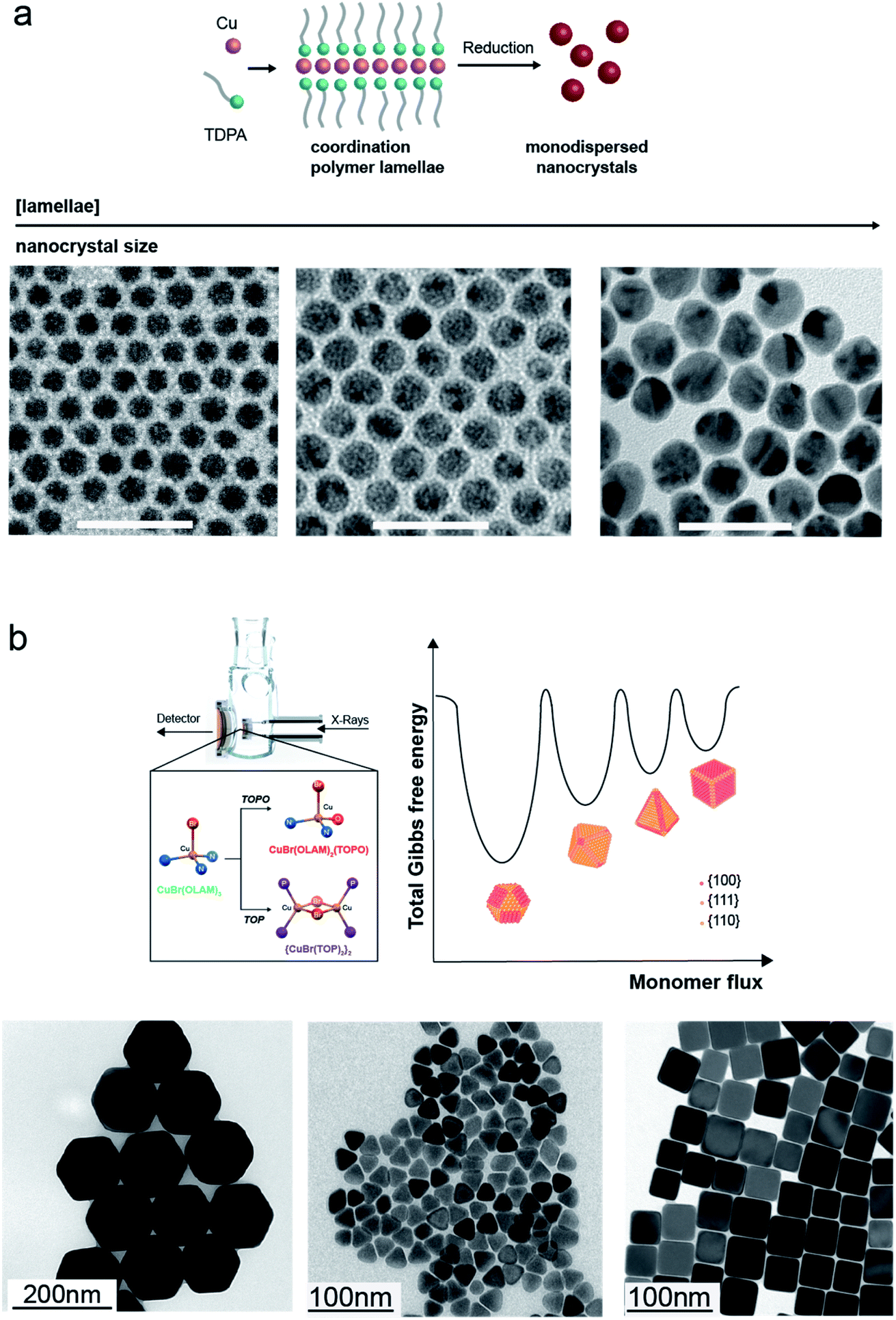 | ||
| Fig. 4 Cu NCs synthesized in organic solvents. (a) Top: Sketch of the conversion of the TDPA–Cu polymer lamellae into monodisperse Cu nanospheres. Bottom: TEM images of Cu nanospheres with different sizes (scale bar: 50 nm) obtained by changing the concentration of the polymer in the reaction flask; reprinted with permission from ref. 22. Copyright 2020 John Wiley and Sons. (b) Top: Rendering of the custom-made flask for in situ X-ray measurements and complexes formed during the reactions of CuBr(OLAM)3 with TOPO and TOP, the disproportionation of which regulates the monomer flux and thus controls the NC shape. Bottom: TEM images of Cu spheres, octahedra, tetrahedra and cubes obtained by controlling the injection of CuBr(OLAM)2(TOPO) and {CuBr(TOP)2}2 complexes in OLAM at different temperatures. Reprinted from ref. 21. Copyright 2019 American Chemical Society. | ||
Different studies have been reported on the synthesis of Cu nanocubes. For instance, Yang et al. addressed the preparation of monodisperse Cu nanocubes with an average edge length of 75.7 nm by rapidly injecting a mixture of CuCl and octadecylamine (ODA) in squalene into a hot OLAM/TOP solution at 330 °C.73 A small percentage of the sample, such as 5%, was composed of Cu rods and spheres. All ligands used during the synthesis (i.e. TOP, ODA, and OLAM) were found to act as surface passivating agents. Another strategy to synthesize Cu cubes is provided by Huang et al.75 In this case, a mixture of Cu(acac)2, ascorbic acid (AA), OLAM, and NH4Cl is heated up to 180 °C for 3 hours to yield monodisperse Cu cubes, with an average edge length of 45 nm. In the same work, the authors were able to shift the synthesis of Cu NCs from cubes to 5-fold twinned nanowires, by simply replacing NH4Cl with RuCl3. The shape control was explained considering the differences of these chloride agents in consuming O2 in the reaction mixture and thus in protecting the twinned nuclei from being etched. Such a protection ensured the continuous growth of the multiply twinned seeds into Cu nanowires.75 Cu nanowires, with a higher degree of monodispersity and a controllable aspect ratio, were also obtained by reacting CuCl2 and tris(trimethylsilyl)silane in OLAM at 160 °C.76
20 nm Cu spheres and 24 nm Cu cubes were obtained by Guo et al. via the reaction of CuBr in OLAM in the presence of TOP and TOPO at 260 °C, respectively.71 Here, the authors explained the shape control as resulting from the Br- and TOP/TOPO binding to the different crystallographic facets.
Cu octahedra of around 150 nm were obtained by Lu et al. via hot-injection of CuCl with TOP (pre-reacted 200 °C) in OLAM at 335 °C.74 The formation of these NCs is proposed to proceed through the disproportionation of a Cu(I)–OLAM complex, simply based on color changes in solution.
From these reports, it is clear that different parameters play a role in the shape control of Cu NCs. However, the provided explanations are often speculative in nature. Recent in situ investigation helped to gain more insights. Strach et al. have identified the formation of Cu–TOP and Cu–TOPO complexes and then correlated their conversion kinetics into Cu monomers with the subsequent nucleation and growth mechanism (Fig. 4b). The authors evidence that the disproportionation rate of such complexes governs the monomer flux and thus the final shapes, instead of the latter being dictated by the selective binding of capping agents to specific crystallographic facets. Furthermore, by tuning, in a predictive manner, the conditions that give octahedra, such as slowly injecting CuBr(OLAM)2(TOPO) at 300 °C, the synthesis of Cu tetrahedra as a novel shape was promoted.21
4.3 Properties and applications of shape-controlled copper nanocrystals
Cu NCs are highly appealing both for their optical and catalytic properties.58 As for their optical properties, very small clusters (2–3 nm) have shown interesting tunable photoluminescence in the IR range.77 As the size increases, they display an intense and narrow LSPR peak, which can be tuned in the region between 600 nm and 1000 nm by changing the particle diameter.78 However, the LSPR response of Cu is weaker than that of Ag or Au, because of the damping effects by the interband transitions of Cu.61,74,79 Here, the synthesis of Cu nanoplates, rhombic dodecahedrons and octahedrons is a successful strategy to avoid such overlap.61,74,79 Indeed, their UV-vis absorption spectra are dominated by well-defined plasmon resonances shifted away from interband transitions (Fig. 5a).61,74,79 As a result, a widespread use of these materials in various applications, such as photocatalysis and SERS is expected.79–82 Another interesting application involves the use of Cu nanowires to build transparent conductive electrodes as a lower cost alternative to Ag nanowires or indium tin oxide (Fig. 5b).63–65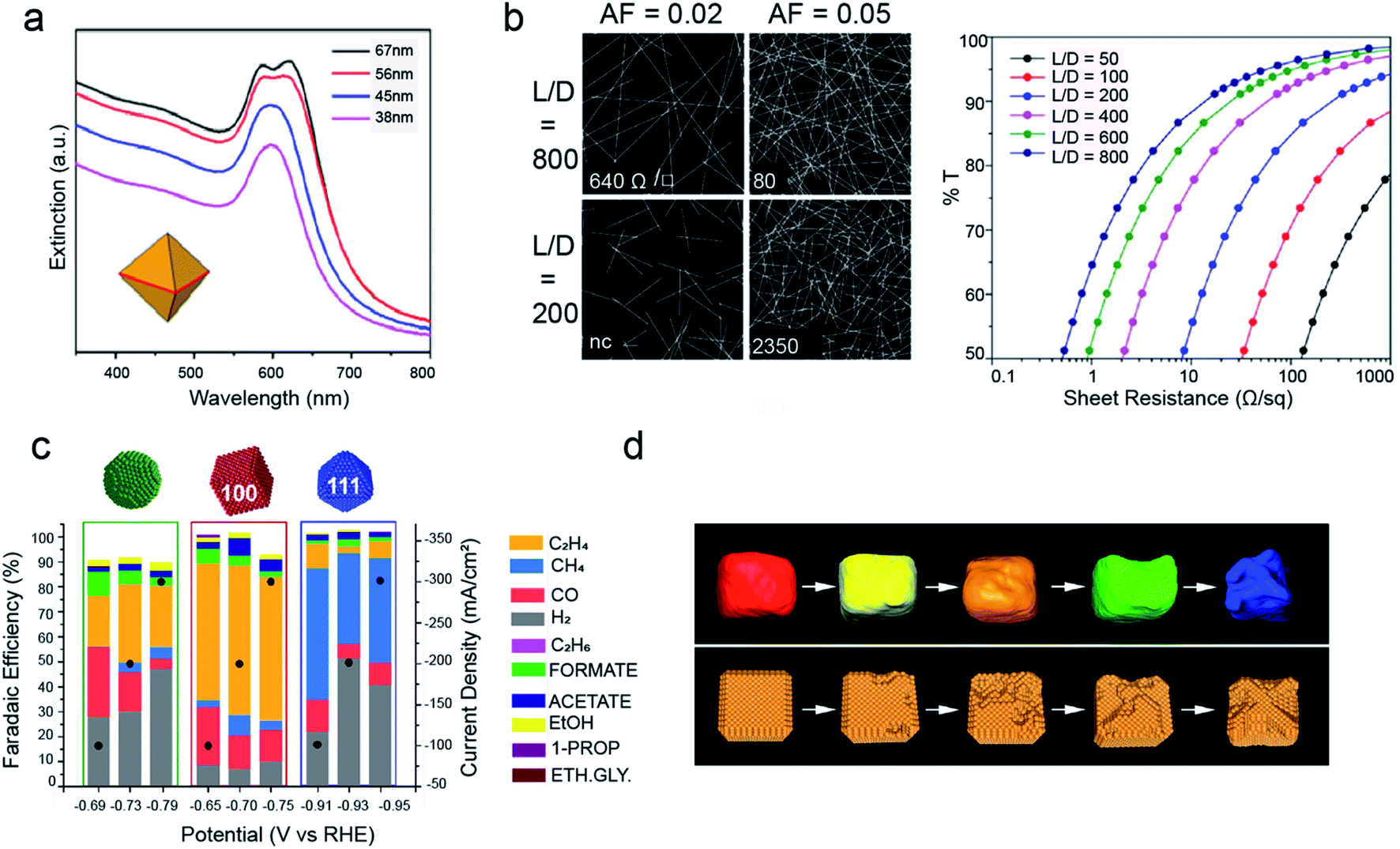 | ||
| Fig. 5 Applications of Cu NCs. (a) UV-vis extinction spectra of Cu right bipyramids with different sizes; reprinted from ref. 69. 2018 American Chemical Society; (b) on the left: images of the nanowire network for two different length/diameter ratios (L/D) and two different area fractions (AFs); on the right: plot of transmittance vs. sheet resistance for networks of nanowires with different L/D. Reprinted from ref. 65. 2016 American Chemical Society; (c) faradaic efficiencies versus applied potential for Cu spheres, cubes and octahedrons assembled as gas diffusion electrodes and measured in the gas-fed flow cell in 1 M KOH. Reprinted from ref. 86. 2020 American Chemical Society; (d) tomographic reconstruction of Cu nanocubes and corresponding schematic morphological models at different stages during 12 hours of CO2RR. Reprinted from ref. 87. 2018 Nature Publishing Group. | ||
Regarding the catalytic properties, Cu NCs are utilized in both thermal catalysis and electrocatalysis. In thermal catalysis, they are employed to drive CO2 hydrogenation, CO oxidation reaction; in these reactions, the NCs are normally deposited on metal oxide powders, which favour synergistic effects while avoiding sintering.83 As electrocatalysts, Cu NCs have received tremendous attention to drive the electrochemical CO2 reduction reaction (CO2RR) over the past five years. Cu is the only single metal able to convert CO2 into hydrocarbons and alcohols, yet copper foil produces 16 different products.84,90 Studies on single crystals had evidenced improved selectivity towards certain products on different Cu surfaces.85,90 Recently, shape-controlled Cu NCs have been demonstrated to translate these properties into actual electrolysers (Fig. 5c).70,86–88 Size, in addition to shape, has been found to play an unpredicted yet crucial role. Indeed, while the shape governs the facets exposed on the surface, the size regulates the area ratio between these facets.89 In addition, the exquisite size and shape monodispersity attainable by colloidal chemistry is also ideal to study degradation pathways through morphological evolution studies. Indeed, the investigation of the nanocubes has revealed a unique degradation mechanism during the CO2RR where nanoclustering instead of coalescence or dissolution/reprecipitation takes place (Fig. 5d).87 Grand-potential density functional theory calculations confirm the role of the negative potential applied to reduce CO2 as the main driving force for the clustering, which suggests that more active catalysts operating at more positive potentials are expected to be more stable.87
5. Conclusions and outlook
Shape-controlled non-noble metal NCs have been attracting increasingly more attention in the last decade as potential alternatives to noble metals, by virtue of their earth abundance combined with intriguing physical and chemical properties relevant for both fundamental studies and technological applications. However, the state of the art of the shape control synthesis of non-noble metal NCs is far from being mature when compared to noble metals.Overall, the examples reported so far for Mn, Fe, Co and Ni NCs are very few and most syntheses are based on high temperature decomposition of organometallic precursors. Being often referred to as a semi-noble metal, Cu is unique. Indeed, successful syntheses have been carried out in both the aqueous environment and organic solvents. Nevertheless, the variety of shapes is not even close yet to that achieved for noble metals. In aqueous environments, the choice of a suitable reducing agent is still a challenge. In organic solvents, a clear understanding of reaction pathways is still under development.
Generally for non-noble metal NCs, the synthesis in organic media seems to be more promising as it allows the mitigation of the impact of oxophilicity, which is the big challenge for this class of materials. As a future direction, it would be interesting to utilize some of the seeded-growth approaches, a strategy that has not been much explored so far for metal NCs in organic solvents. One additional consideration is that native oxide shells might still form during the synthesis of highly oxophilic metals. As their crystal habits are much different than the pure metals, forces such as strain and atom migration might play a role in the shape tuning. Therefore, one might consider to use capping agents suitable for the oxide rather than the metal itself or also take the oxide crystalline structures into account in the case of seeded-growth.
Various studies discussed above point at the crucial role of the precursor's chemical nature, speciation and reduction kinetics to control the NC shape in such environments. More investigations to identify such relationships will be crucial.
On a closely related topic, despite the recognized importance of capping agents in tuning the NC shape, their role is still unpredictable due to the limited knowledge of their molecular structures and binding modes on the NC surface. To move towards a more rational synthesis design, this knowledge gap must be filled. Solid-state nuclear magnetic resonance complemented by theory represents a promising tool to address this need.91
The in situ studies performed for Co NCs and, more recently, for Cu NCs highlight the importance of understanding the chemistry behind nucleation and growth to predict synthetic pathways for size- and shape-controlled non-noble metal NCs.21,22,52 More of these kinds of studies are needed to accelerate the progress via a rational and predictive design approach instead of the traditional trial and error avenues.
Finally, developing a comprehensive understanding of the chemistry of non-noble metal NCs will facilitate the the synthesis of multicomponent NCs (i.e. heterostructures, multimetallic alloys, multi-cation oxides and chalcogenides), including the same elements, which are becoming increasingly popular because of the additional property tunability offered by combining domains with a different chemical nature within the same nanodomain.92–101
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Swiss National Science Foundation (AP Energy Grant, Project Number PYAPP2_166897/1).References
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS.
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Nat. Catal., 2018, 1, 656–665 CrossRef.
- H. Tabassum, A. Mahmood, B. Zhu, Z. Liang, R. Zhong, S. Guo and R. Zou, Energy Environ. Sci., 2019, 12, 2924–2956 RSC.
- X. Wang, S.-C. Huang, S. Hu, S. Yan and B. Ren, Nat. Rev. Phys., 2020, 2, 253–271 CrossRef.
- M. Yadid, R. Feiner and T. Dvir, Nano Lett., 2019, 19, 2198–2206 CrossRef CAS.
- M. R. Buck, J. F. Bondi and R. E. Schaak, Nat. Chem., 2012, 4, 37–44 CrossRef CAS.
- Y. Xia, K. D. Gilroy, H. C. Peng and X. Xia, Angew. Chem., Int. Ed., 2017, 56, 60–95 CrossRef CAS.
- M. Cargnello, Chem. Mater., 2019, 31, 576–596 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS.
- P. C. Chen, M. Liu, J. S. Du, B. Meckes, S. Wang, H. Lin, V. P. Dravid, C. Wolverton and C. A. Mirkin, Science, 2019, 363, 959–964 CrossRef CAS.
- Y. N. Xia and Y. G. Sun, Science, 2002, 298, 2176–2179 CrossRef.
- Y. Xia, X. Xia and H.-C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS.
- M. N. O'Brien, M. R. Jones, K. A. Brown and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 7603–7606 CrossRef.
- A. Baldi, T. C. Narayan, A. L. Koh and J. A. Dionne, Nat. Mater., 2014, 13, 1143–1148 CrossRef CAS.
- S. Syrenova, C. Wadell, F. A. A. Nugroho, T. A. Gschneidtner, Y. A. D. Fernandez, G. Nalin, D. Świtlik, F. Westerlund, T. J. Antosiewicz, V. P. Zhdanov, K. Moth-Poulsen and C. Langhammer, Nat. Mater., 2015, 14, 1236–1244 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Soc. Rev., 2017, 46, 816–854 RSC.
- D. A. Torelli, S. A. Francis, J. C. Crompton, A. Javier, J. R. Thompson, B. S. Brunschwig, M. P. Soriaga and N. S. Lewis, ACS Catal., 2016, 6, 2100–2104 CrossRef CAS.
- M. Casavola, J. Hermannsdörfer, N. De Jonge, A. I. Dugulan and K. P. De Jong, Adv. Funct. Mater., 2015, 25, 5309–5319 CrossRef CAS.
- Y. Zhang, W. Huang, S. E. Habas, J. N. Kuhn, M. E. Grass, Y. Yamada, P. Yang and G. A. Somorjai, J. Phys. Chem. C, 2008, 112, 12092–12095 CrossRef CAS.
- C. S. Chen, C. S. Budi, H. C. Wu, D. Saikia and H. M. Kao, ACS Catal., 2017, 7, 8367–8381 CrossRef CAS.
- M. Strach, V. Mantella, J. R. Pankhurst, P. Iyengar, A. Loiudice, S. Das, C. Corminboeuf, W. van Beek and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 16312–16322 CrossRef CAS.
- V. Mantella, M. Strach, K. Frank, J. R. Pankhurst, D. Stoian, C. Gadyiar, B. Nickel and R. Buonsanti, Angew. Chem., Int. Ed., 2020, 59, 2–9 CrossRef.
- Y. Wang, J. He, C. Liu, W. H. Chong and H. Chen, Angew. Chem., Int. Ed., 2015, 54, 2022–2051 CrossRef CAS.
- S. G. Kwon and T. Hyeon, Small, 2011, 7, 2685–2702 CrossRef CAS.
- D. Gebauer and S. E. Wolf, J. Am. Chem. Soc., 2019, 141, 4490–4504 CrossRef CAS.
- Y. H. Liu, F. Wang, Y. Wang, P. C. Gibbons and W. E. Buhro, J. Am. Chem. Soc., 2011, 133, 17005–17013 CrossRef CAS.
- M. Gromova, A. Lefrançois, L. Vaure, F. Agnese, D. Aldakov, A. Maurice, D. Djurado, C. Lebrun, A. De Geyer, T. U. Schülli, S. Pouget and P. Reiss, J. Am. Chem. Soc., 2017, 139, 15748–15759 CrossRef CAS.
- L. Wu, J. J. Willis, I. S. McKay, B. T. Diroll, J. Qin, M. Cargnello and C. J. Tassone, Nature, 2017, 548, 197–201 CrossRef CAS.
- B. Ingham, Crystallogr. Rev., 2015, 21, 229–303 CrossRef.
- J. H. Yu, X. Liu, K. E. Kweon, J. Joo, J. Park, K. T. Ko, D. W. Lee, S. Shen, K. Tivakornsasithorn, J. S. Son, J. H. Park, Y. W. Kim, G. S. Hwang, M. Dobrowolska, J. K. Furdyna and T. Hyeon, Nat. Mater., 2010, 9, 47–53 CrossRef CAS.
- J. Yang, J. S. Son, J. H. Yu, J. Joo and T. Hyeon, Chem. Mater., 2013, 25, 1190–1198 CrossRef CAS.
- F. Wang, V. N. Richards, S. P. Shields and W. E. Buhro, Chem. Mater., 2014, 26, 5–21 CrossRef CAS.
- J. Lee, J. Yang, S. G. Kwon and T. Hyeon, Nat. Rev. Mater., 2016, 8, 16034 CrossRef.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS.
- D. Erdemir, A. Y. Lee and A. S. Myerson, Acc. Chem. Res., 2009, 42, 621–629 CrossRef CAS.
- N. D. Loh, S. Sen, M. Bosman, S. F. Tan, J. Zhong, C. A. Nijhuis, P. Král, P. Matsudaira and U. Mirsaidov, Nat. Chem., 2017, 9, 77–82 CrossRef CAS.
- J. Yang, J. Koo, S. Kim, S. Jeon, B. K. Choi, S. Kwon, J. Kim, B. H. Kim, W. C. Lee, W. B. Lee, H. Lee, T. Hyeon, P. Ercius and J. Park, J. Am. Chem. Soc., 2019, 141, 763–768 CrossRef CAS.
- A. N. Chen, M. M. Scanlan and S. E. Skrabalak, ACS Nano, 2017, 11, 12624–12631 CrossRef CAS.
- C. Li, R. Sato, M. Kanehara, H. Zeng, Y. Bando and T. Teranishi, Angew. Chem., Int. Ed., 2009, 48, 6883–6887 CrossRef CAS.
- H. Huang, Y. Wang, A. Ruditskiy, H. C. Peng, X. Zhao, L. Zhang, J. Liu, Z. Ye and Y. Xia, ACS Nano, 2014, 8, 7041–7050 CrossRef CAS.
- C. Li, K. L. Shuford, Q. H. Park, W. Cai, Y. Li, E. J. Lee and S. O. Cho, Angew. Chem., Int. Ed., 2007, 46, 3264–3268 CrossRef CAS.
- B. Wiley, T. Herricks, Y. Sun and Y. Xia, Nano Lett., 2004, 4, 1733–1739 CrossRef CAS.
- H. Zhang, M. Jin, Y. Xiong, B. Lim and Y. Xia, Acc. Chem. Res., 2013, 46, 1783–1794 CrossRef CAS.
- B. Carpinteiro-Carreto, B. M. Marín-Santibáñez, J. Pérez-González, F. Rodríguez-González, H. J. Dorantes-Rosales and R. Sánchez, Colloid Polym. Sci., 2017, 295, 37–43 CrossRef CAS.
- J. T. Nurmi, P. G. Tratnyek, V. Sarathy, D. R. Baer, J. E. Amonette, K. Pecher, C. Wang, J. C. Linehan, D. W. Matson, R. L. Penn and M. D. Driessen, Environ. Sci. Technol., 2005, 39, 1221–1230 CrossRef CAS.
- V. F. Puntes, D. Zanchet, C. K. Erdonmez and A. P. Alivisatos, J. Am. Chem. Soc., 2002, 124, 12874–12880 CrossRef CAS.
- V. F. Puntes, K. M. Krishnan and P. Alivisatos, Appl. Phys. Lett., 2001, 78, 2187–2189 CrossRef CAS.
- V. F. Puntes, K. M. Krishnan and A. P. Alivisatos, Science, 2001, 291, 2115–2117 CrossRef CAS.
- V. Puntes, K. Krishnan and A. P. Alivisatos, Top. Catal., 2002, 19, 145–148 CrossRef CAS.
- Y. Bao, W. An, C. Heath Turner and K. M. Krishnan, Langmuir, 2010, 26, 478–483 CrossRef CAS.
- F. Dumestre, B. Chaudret, C. Amiens, M. C. Fromen, M. J. Casanove, P. Renaud and P. Zurcher, Angew. Chem., Int. Ed., 2002, 41, 4286–4289 CrossRef CAS.
- B. Cormary, T. Li, N. Liakakos, L. Peres, P. F. Fazzini, T. Blon, M. Respaud, A. J. Kropf, B. Chaudret, J. T. Miller, E. A. Mader and K. Soulantica, J. Am. Chem. Soc., 2016, 138, 8422–8431 CrossRef CAS.
- S. Carenco, C. Boissière, L. Nicole, C. Sanchez, P. Le Floch and N. Mézailles, Chem. Mater., 2010, 22, 1340–1349 CrossRef CAS.
- E. Zacharaki, P. Beato, R. R. Tiruvalam, K. J. Andersson, H. Fjellvåg and A. O. Sjåstad, Langmuir, 2017, 33, 9836–9843 CrossRef CAS.
- N. Cordente, M. Respaud, F. Senocq, M. J. Casanove, C. Amiens and B. Chaudret, Nano Lett., 2001, 1, 565–568 CrossRef CAS.
- S. J. Park, S. Kim, S. Lee, Z. G. Khim, K. Char and T. Hyeon, J. Am. Chem. Soc., 2000, 122, 8581–8582 CrossRef CAS.
- J. F. Bondi, K. D. Oyler, X. Ke, P. Schiffer and R. E. Schaak, J. Am. Chem. Soc., 2009, 131, 9144–9145 CrossRef CAS.
- M. B. Gawande, A. Goswami, F.-X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS.
- P. Lignier, R. Bellabarba and R. P. Tooze, Chem. Soc. Rev., 2012, 41, 1708–1720 RSC.
- M. Jin, G. He, H. Zhang, J. Zeng, Z. Xie and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 10560–10564 CrossRef CAS.
- I. Pastoriza-Santos, A. Sánchez-Iglesias, B. Rodríguez-González and L. M. Liz-Marzán, Small, 2009, 5, 440–443 CrossRef CAS.
- Y. Chang, M. L. Lye and H. C. Zeng, Langmuir, 2005, 21, 3746–3748 CrossRef CAS.
- A. R. Rathmell, S. M. Bergin, Y. L. Hua, Z. Y. Li and B. J. Wiley, Adv. Mater., 2010, 22, 3558–3563 CrossRef CAS.
- S. Ye, A. R. Rathmell, I. E. Stewart, Y. C. Ha, A. R. Wilson, Z. Chen and B. J. Wiley, Chem. Commun., 2014, 50, 2562–2564 RSC.
- S. Ye, I. E. Stewart, Z. Chen, B. Li, A. R. Rathmell and B. J. Wiley, Acc. Chem. Res., 2016, 49, 442–451 CrossRef CAS.
- Z. Wang, Z. Chen, H. Zhang, Z. Zhang, H. Wu, M. Jin, C. Wu, D. Yang and Y. Yin, ACS Nano, 2015, 9, 3307–3313 CrossRef CAS.
- M. Luo, A. Ruditskiy, H. C. Peng, J. Tao, L. Figueroa-Cosme, Z. He and Y. Xia, Adv. Funct. Mater., 2016, 26, 1209–1216 CrossRef CAS.
- Z. Wang, G. Yang, Z. Zhang, M. Jin and Y. Yin, ACS Nano, 2016, 10, 4559–4564 CrossRef CAS.
- Z. Lyu, M. Xie, K. D. Gilroy, Z. D. Hood, M. Zhao, S. Zhou, J. Liu and Y. Xia, Chem. Mater., 2018, 30, 6469–6477 CrossRef CAS.
- P. Iyengar, J. Huang, G. L. De Gregorio, C. Gadiyar and R. Buonsanti, Chem. Commun., 2019, 55, 8796–8799 RSC.
- H. Guo, Y. Chen, M. B. Cortie, X. Liu, Q. Xie, X. Wang and D. L. Peng, J. Phys. Chem. C, 2014, 118, 9801–9808 CrossRef CAS.
- L. L. Hung, C. K. Tsung, W. Huang and P. Yang, Adv. Mater., 2010, 22, 1910–1914 CrossRef CAS.
- H. J. Yang, S. Y. He, H. L. Chen and H. Y. Tuan, Chem. Mater., 2014, 26, 1785–1793 CrossRef CAS.
- S.-C. Lu, M.-C. Hsiao, M. Yorulmaz, L.-Y. Wang, P.-Y. Yang, S. Link, W.-S. Chang and H.-Y. Tuan, Chem. Mater., 2015, 27, 8185–8188 CrossRef CAS.
- X. Huang, Y. Chen, C. Y. Chiu, H. Zhang, Y. Xu, X. Duan and Y. Huang, Nanoscale, 2013, 5, 6284–6290 RSC.
- Y. Li, F. Cui, M. B. Ross, D. Kim, Y. Sun and P. Yang, Nano Lett., 2017, 17, 1312–1317 CrossRef CAS.
- C. M. Andolina, S. E. Crawford, A. M. Smith, K. A. Johnston, P. J. Straney, L. E. Marbella, N. L. Tolman, T. J. Hochuli and J. E. Millstone, ChemNanoMat, 2018, 4, 265–268 CrossRef CAS.
- G. H. Chan, J. Zhao, E. M. Hicks, G. C. Schatz and R. P. Van Duyne, Nano Lett., 2007, 7, 1947–1952 CrossRef CAS.
- Z. Lyu, M. Xie, K. D. Gilroy, Z. D. Hood, M. Zhao, S. Zhou, J. Liu and Y. Xia, Chem. Mater., 2018, 30, 6469–6477 CrossRef CAS.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2009, 7, 308–319 Search PubMed.
- J. Li, S. K. Cushing, F. Meng, T. R. Senty, A. D. Bristow and N. Wu, Nat. Photonics, 2015, 9, 601–607 CrossRef CAS.
- A. Marimuthu, J. Zhang and S. Linic, Science, 2013, 340, 1590–1593 CrossRef.
- P. Losch, W. Huang, E. D. Goodman, C. J. Wrasman, A. Holm, A. R. Riscoe, J. A. Schwalbe and M. Cargnello, Nano Today, 2019, 24, 15–47 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS.
- J. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. L. De Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2018, 9, 1–9 CrossRef.
- Y. T. Guntern, J. R. Pankhurst, J. Vávra, M. Mensi, V. Mantella, P. Schouwink and R. Buonsanti, Angew. Chem., Int. Ed., 2019, 58, 12632–12639 CrossRef CAS.
- G. Mangione, J. Huang, R. Buonsanti and C. Corminboeuf, J. Phys. Chem. Lett., 2019, 10, 4259–4265 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- H. Al-Johani, E. Abou-Hamad, A. Jedidi, C. M. Widdifield, J. Viger-Gravel, S. S. Sangaru, D. Gajan, D. H. Anjum, S. Ould-Chikh, M. N. Hedhili, A. Gurinov, M. J. Kelly, M. El Eter, L. Cavallo, L. Emsley and J. M. Basset, Nat. Chem., 2017, 9, 890–895 CrossRef CAS.
- Z. Li, C. Yu, Y. Wen, Y. Gao, X. Xing, Z. Wei, H. Sun, Y. W. Zhang and W. Song, ACS Catal., 2019, 9, 5084–5095 CrossRef CAS.
- R. E. Schaak, A. K. Sra, B. M. Leonard, R. E. Cable, J. C. Bauer, Y. F. Han, J. Means, W. Teizer, Y. Vasquez and E. S. Funck, J. Am. Chem. Soc., 2005, 127, 3506–3515 CrossRef CAS.
- H. S. Jeon, J. Timosnenko, F. Scholten, I. Sinev, A. Herzog, F. T. Haase and B. R. Cuenya, J. Am. Chem. Soc., 2020, 141, 19879–19887 CrossRef.
- X. Sun, K. Jiang, N. Zhang, S. Guo and X. Huang, ACS Nano, 2015, 9, 7634–7640 CrossRef CAS.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS.
- C. Coughlan, M. Ibáñez, O. Dobrozhan, A. Singh, A. Cabot and K. M. Ryan, Chem. Rev., 2017, 117, 5865–6109 CrossRef CAS.
- C. Gadiyar, M. Strach, P. Schouwink, A. Loiudice and R. Buonsanti, Chem. Sci., 2018, 9, 5658–5665 RSC.
- X. Yang, E. A. Fugate, Y. Mueanngern and L. R. Baker, ACS Catal., 2017, 7, 177–180 CrossRef CAS.
- V. Mantella, S. Ninova, S. Saris, A. Loiudice, U. Aschauer and R. Buonsanti, Chem. Mater., 2019, 31, 532–540 CrossRef CAS.
- J. M. Lee, L. A. Kraynak and A. L. Prieto, Angew. Chem., Int. Ed., 2020, 59, 3038–3042 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |