DOI:
10.1039/D0SC04012F
(Perspective)
Chem. Sci., 2020,
11, 11380-11393
Copper-catalyzed functionalization of enynes
Received
22nd July 2020
, Accepted 7th October 2020
First published on 7th October 2020
Abstract
The copper-catalyzed functionalization of enyne derivatives has recently emerged as a powerful approach in contemporary synthesis. Enynes are versatile and readily accessible substrates that can undergo a variety of reactions to yield densely functionalized, enantioenriched products. In this perspective, we review copper-catalyzed transformations of enynes, such as boro- and hydrofunctionalizations, copper-mediated radical difunctionalizations, and cyclizations. Particular attention is given to the regiodivergent functionalization of 1,3-enynes, and the current mechanistic understanding of such processes.
1. Introduction
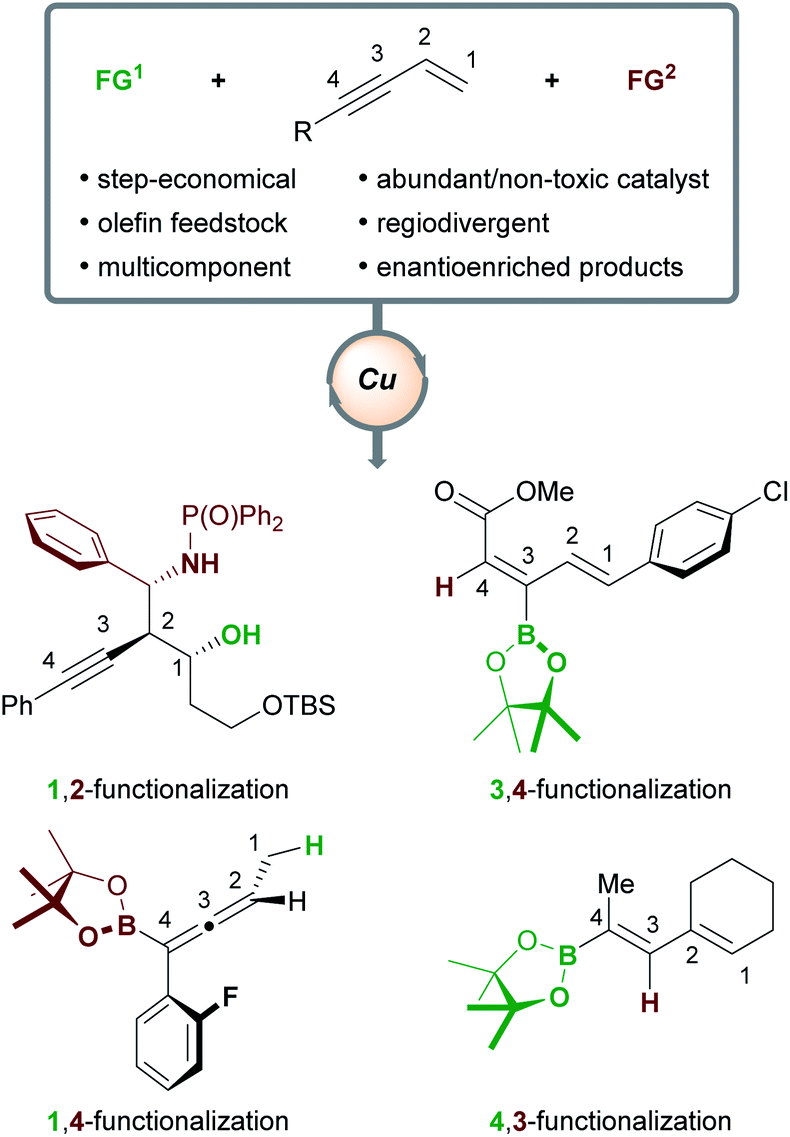
The desire to build complex, functionalized molecules rapidly and efficiently from simple precursors is a recurring theme in modern organic chemistry. Many envision a sustainable future in which the processing of feedstock substrates in catalytic and step-/atom-economical transformations will allow new regions of chemical space to be explored.1 The transition-metal catalyzed functionalization of olefins is seen as an approach with the potential to contribute to such a future,2 as it has proved capable of delivering high-value, multifunctionalized, stereodefined products. Copper catalysts hold particular promise for fulfilling goals pertaining to sustainability, as copper is more abundant3 and less toxic4 than other transition metals. Indeed, various olefins5 have been transformed into densely functionalized products6 using copper catalysis. In particular, enynes are highly versatile substrates. They can be obtained using numerous, efficient catalytic methods7 and their ambident reactivity has been exploited to deliver a multitude of products.5f,8
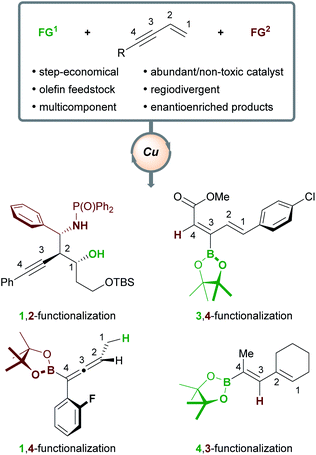
Herein, we will review the copper-catalyzed functionalization of enynes. This fast-growing field has seen the implementation of various strategies in copper catalysis, such as hydro- and borofunctionalizations, multicomponent reactions, radical difunctionalizations, and cyclizations. The copper-catalyzed functionalization of enynes will be discussed in four sections: (1) borofunctionalization, where a copper-boryl complex is the catalytically active species; (2) hydrofunctionalization, where a copper-hydride complex is the catalytically active species; (3) copper-mediated radical difunctionalization, and; (4) copper-catalyzed functionalization of non-conjugated 1,n-enynes. Generally, 1,3-enynes are the most common substrates in these processes and a variety of highly useful products are accessible (Scheme 1); for example, enantioenriched, (homo)propargylic compounds (1,2-functionalization), allenes (1,4-functionalization), and dienes (4,3-/3,4-functionalization), are important in synthetic9 and/or medicinal chemistry.10
 |
| | Scheme 1 Copper-catalyzed functionalization of enynes. | |
2. Mechanistic principles
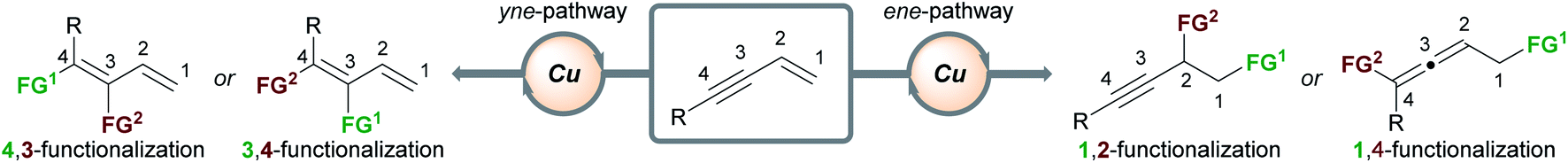
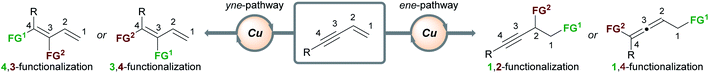
The outcome of the copper-catalyzed functionalization of 1,3-enynes is determined by the interplay of several factors at different stages of the catalytic cycle: the chemo- (alkene vs. alkyne) and regioselectivity (e.g. 1,2- vs. 2,1-addition) of the first functional group (FG1) addition, the possible isomerization of organocopper intermediates, and the mechanism of the second functional group (FG2) addition. Details of these mechanisms will be presented when appropriate and when experimental evidence or DFT calculations are available. In general terms, the possible products can be grouped into two families (Scheme 2) – it is the chemoselectivity of the reaction with the first functional group (FG1) that determines which family of product forms. Other products are theoretically possible, however, Scheme 2 summarises the types of products that have been accessed to date. If the initial functionalization occurs at the olefinic component of the enyne (the ene-pathway), the (homo)propargylic/allenic family of products is formed. Alternatively, reaction of FG1 with the alkyne component (yne-pathway) leads to the diene family of products. Within each family are sub-groups that are accessed through different modes of addition to the enyne. For example, in the ene-pathway, 1,2-functionalization leads to (homo)propargylic products, whereas 1,4-functionalization leads to allenic products. The notation used to describe the type of functionalization refers to the position of the attached functional groups in the product, i.e. the (homo)propargylic product is said to form via a 1,2-functionalization as the first functional group (FG1) is attached to C1 and the second functional group (FG2) is attached to C2. Using the same concepts, the yne-pathway leads to diene products of either 4,3-or 3,4-functionalization. This is a simplified model for describing the outcomes of these reactions and a similar nomenclature will be used throughout this perspective. There are numerous pathways by which enynes can react and a detailed description of each mechanistic pathway is beyond the scope of this review, however, key aspects will be highlighted.
 |
| | Scheme 2 Copper-catalyzed regiodivergent functionalization of enynes. | |
3. Borofunctionalization
Boron-containing compounds are involved in 11% of the C–C bond forming reactions used in process chemistry.1d Since the seminal reports of Miyaura11 and Hosomi12 on the borylation of enones, the copper-catalyzed borylation of olefins has become a versatile starting point for the design of important multicomponent reactions.5c,5d,13 Thus, it is not surprising to find these processes at the heart of several seminal reports on the copper-catalyzed functionalization of 1,3-enynes.
3.1. Regiodivergent processes
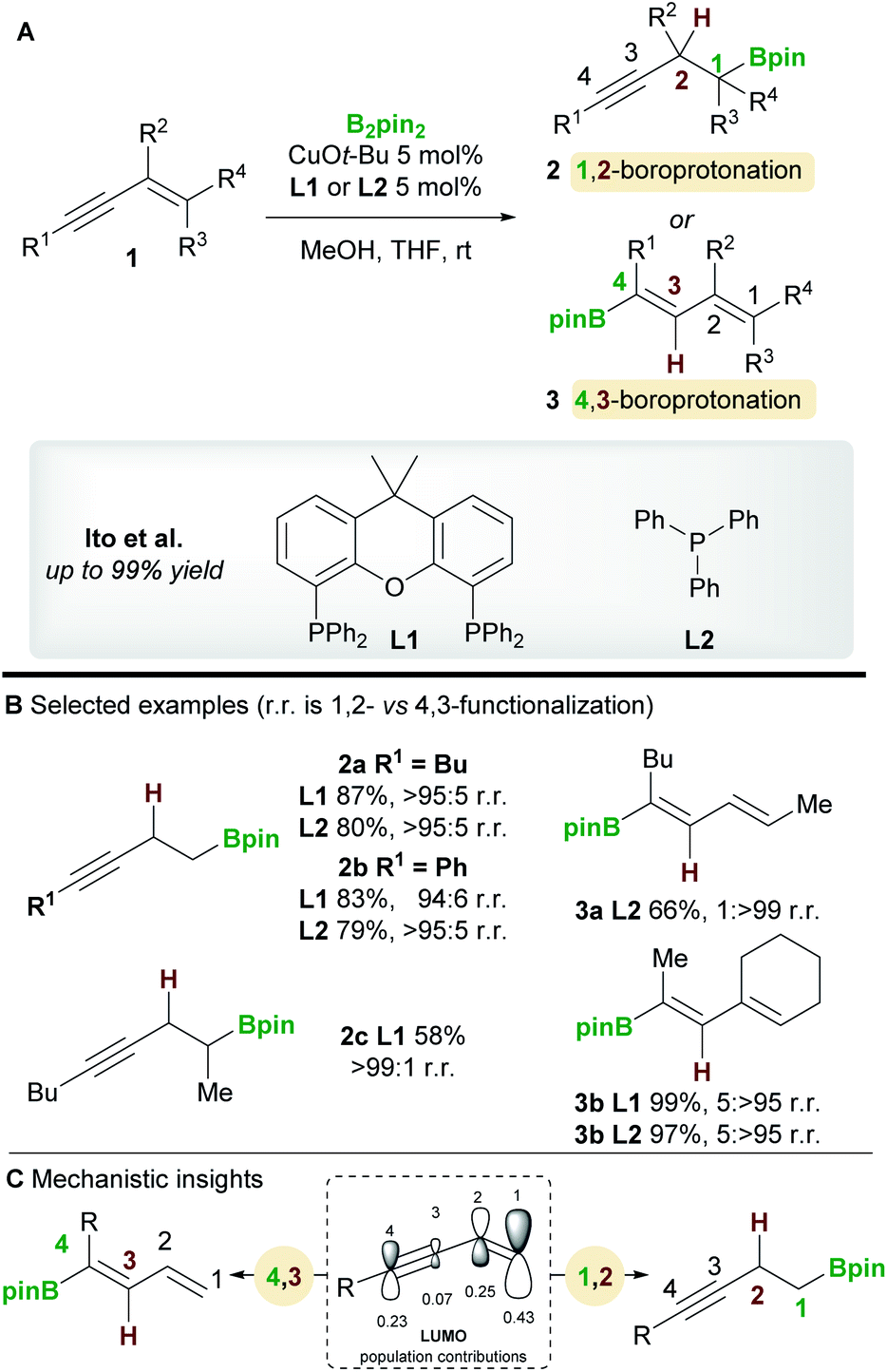
The copper-catalyzed boroprotonation of 1,3-enynes 1 reported by Ito and co-workers14 in 2011 introduced some useful concepts concerning the regioselectivity of enyne functionalization (Scheme 3A). The system used a Cu-Bpin catalyst, formed in situ from a Cu-alkoxide and B2pin2, and MeOH as a proton source. The reaction of relatively simple enynes, for example butyl-1,3-enyne 1a and phenyl-1,3-enyne 1b, provided the 1,2 products 2a/2b with high regioselectivity with ligands L1 and L2 (Scheme 3B). The authors then explored the effect of substitution on the alkene moiety of the enyne and observed a ligand-controlled regioselectivity switch. Thus, 1-substituted enynes gave the 1,2-product 2c with the bidentate XantPhos ligand L1, whereas the 4,3-product 3a was obtained when using monodentate PPh3L2. Conversely, 1,2-disubstituted enynes always gave the 4,3-product 3b no matter which ligand was used, possibly due to the steric hindrance around the olefin. The authors also reported a moderately enantioselective 1,2-boroprotonation. Frontier orbital population analysis from DFT calculations was used to explain the regioselectvity of these results (Scheme 3C). The most important interaction to consider is between the HOMO of the copper-boryl complex and the LUMO of the enyne. In this regard, the larger coefficients at C1 and C4 explain the preference for 1,2-/4,3-functionalization. However, a detailed discussion of the observed regiodivergency was not provided.
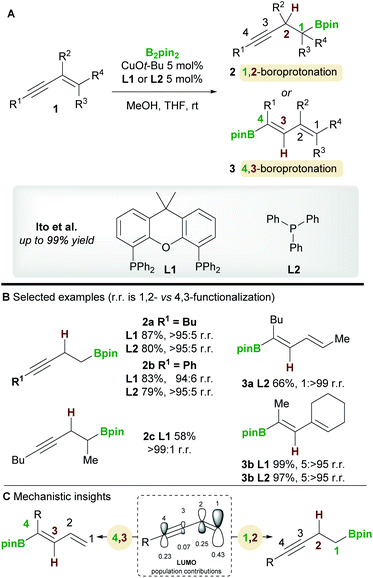 |
| | Scheme 3 Ito's regiodivergent boroprotonation of 1,3-enynes. | |
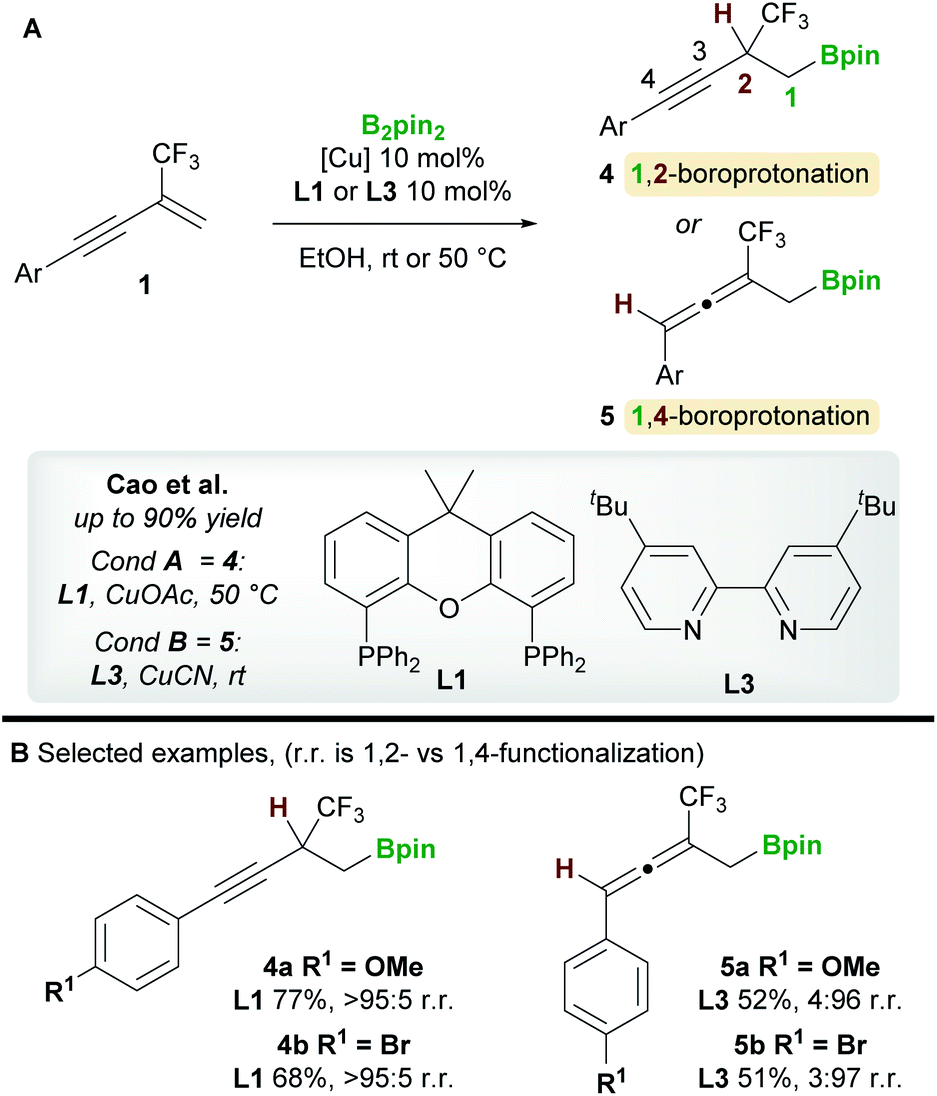
In 2020, a regiodivergent boroprotonation of 2-trifluoromethyl-1,3-enynes 1 was reported by Cao and co-workers (Scheme 4).15 In accordance with the previous work of Ito et al. (Scheme 3), they found that XantPhos L1 promoted the 1,2-boroprotonation of 2-substituted 1,3-enynes in good yields and with complete regiocontrol (see 4vs.2). Conversely, 1,4-products 5 were accessed with high selectivity and in moderate yield upon switching to the bidentate, nitrogen-based ligand 4,4′-di-tert-butyl-2,2′-bipyridine (dtbpy) L3.
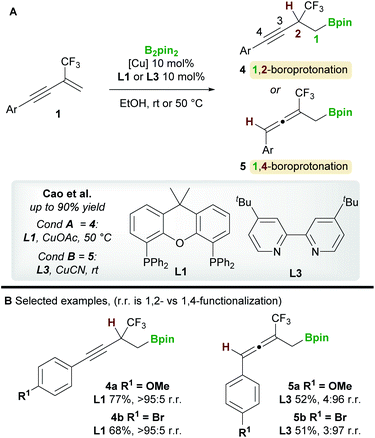 |
| | Scheme 4 Cao's regiodivergent boroprotonation of trifluoromethyl-substituted 1,3-enynes. | |
3.2. 1,2-Borofunctionalization processes
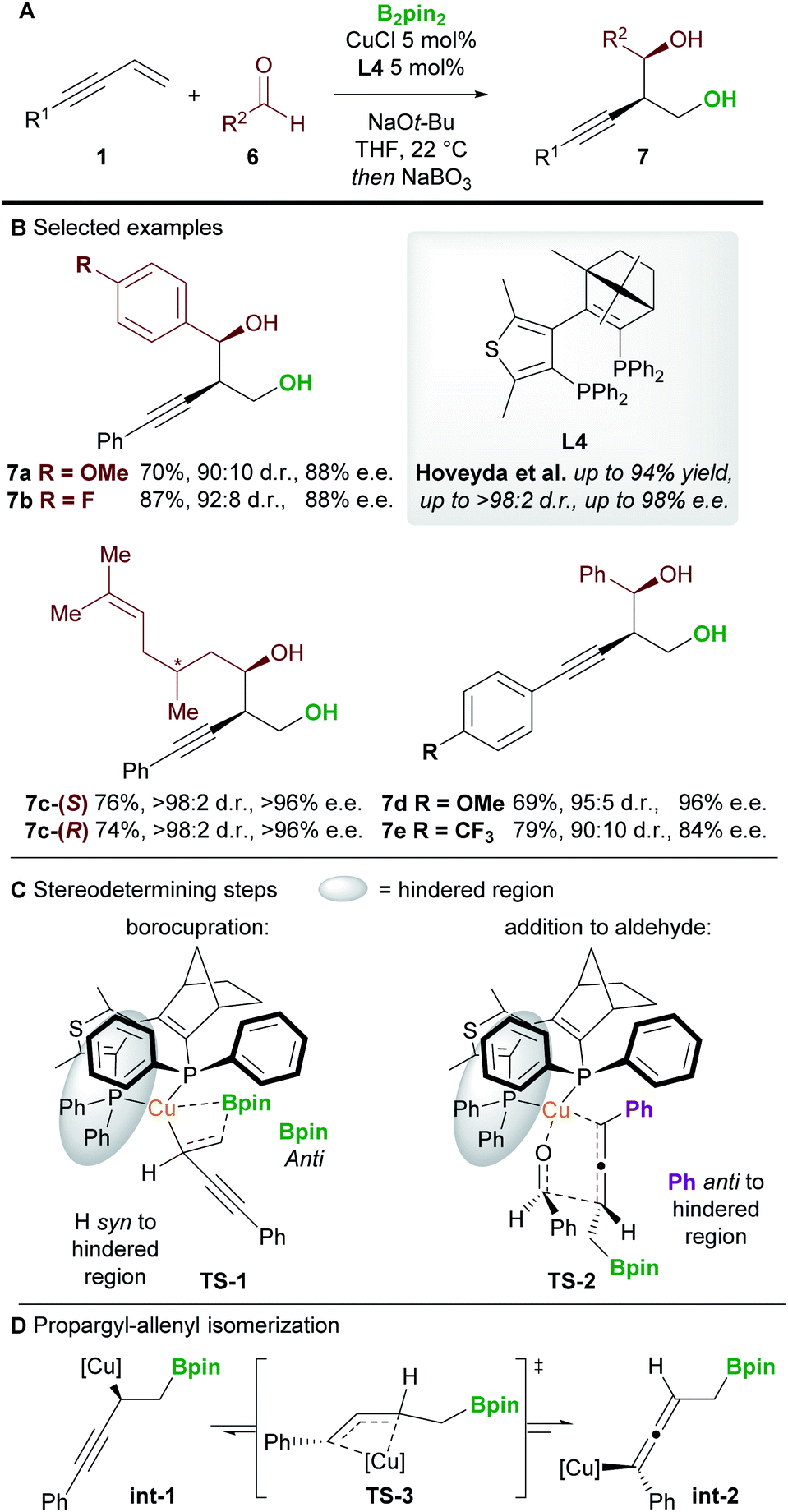
An important milestone in this field was achieved by Hoveyda and co-workers16 in 2014 when they realized the first enantioselective borylative 1,2-functionalization of 1,3-enynes 1 with aldehydes 6 (Scheme 5). The borylated products were oxidized in situ using NaBO3·4H2O to provide homopropargylic 1,3-diols 7 in good to excellent yield, and with high diastereo- and enantiocontrol. Aromatic and aliphatic aldehydes, and aryl-substituted enynes were all suitable coupling partners, but alkyl-substituted enynes proved more challenging substrates.
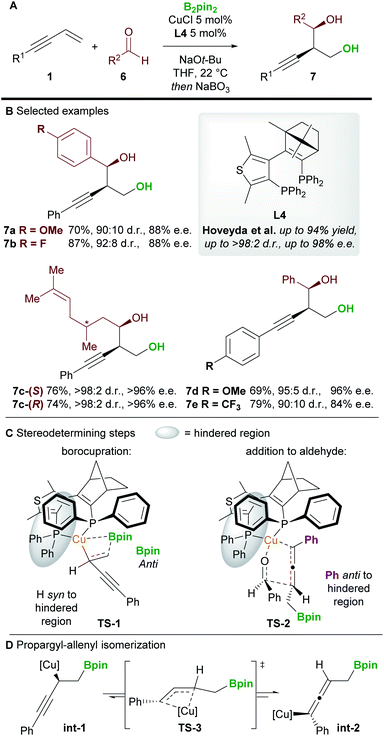 |
| | Scheme 5 Hoveyda's 1,2-borofunctionalization of 1,3-enynes with aldehydes. | |
DFT studies were carried out on the two stereodetermining steps of this reaction (Scheme 5C). Firstly, the 1,2-borocupration was proposed as the enantiodetermining step of the process. This step proceeds through attack of a Cu-Bpin species at the C–C double bond of the enyne through a 4-membered transition state TS-1. Attack on the Si face of the enyne was favoured by 2.1 kcal mol−1 over attack on the Re-face. The resulting propargyl-copper species int-1 can then isomerise viaTS-3 to give the corresponding allenyl-copper species int-2 (Scheme 5D). Although this step was not investigated in this report, the propargyl-allenyl isomerisation of transition metal complexes is a well-known phenomenon that has been studied using DFT calculations and X-ray crystallography.17 Finally, the desired product was proposed to form through the coupling of the allenyl copper species int-2 and the aldehyde 6 in a diastereo determining, closed, 6-membered transition-state (TS-2, Scheme 5C). Other transition states (e.g. involving attack on the other face of the aldehyde) were modelled but were disfavoured by at least 1.9 kcal mol−1.
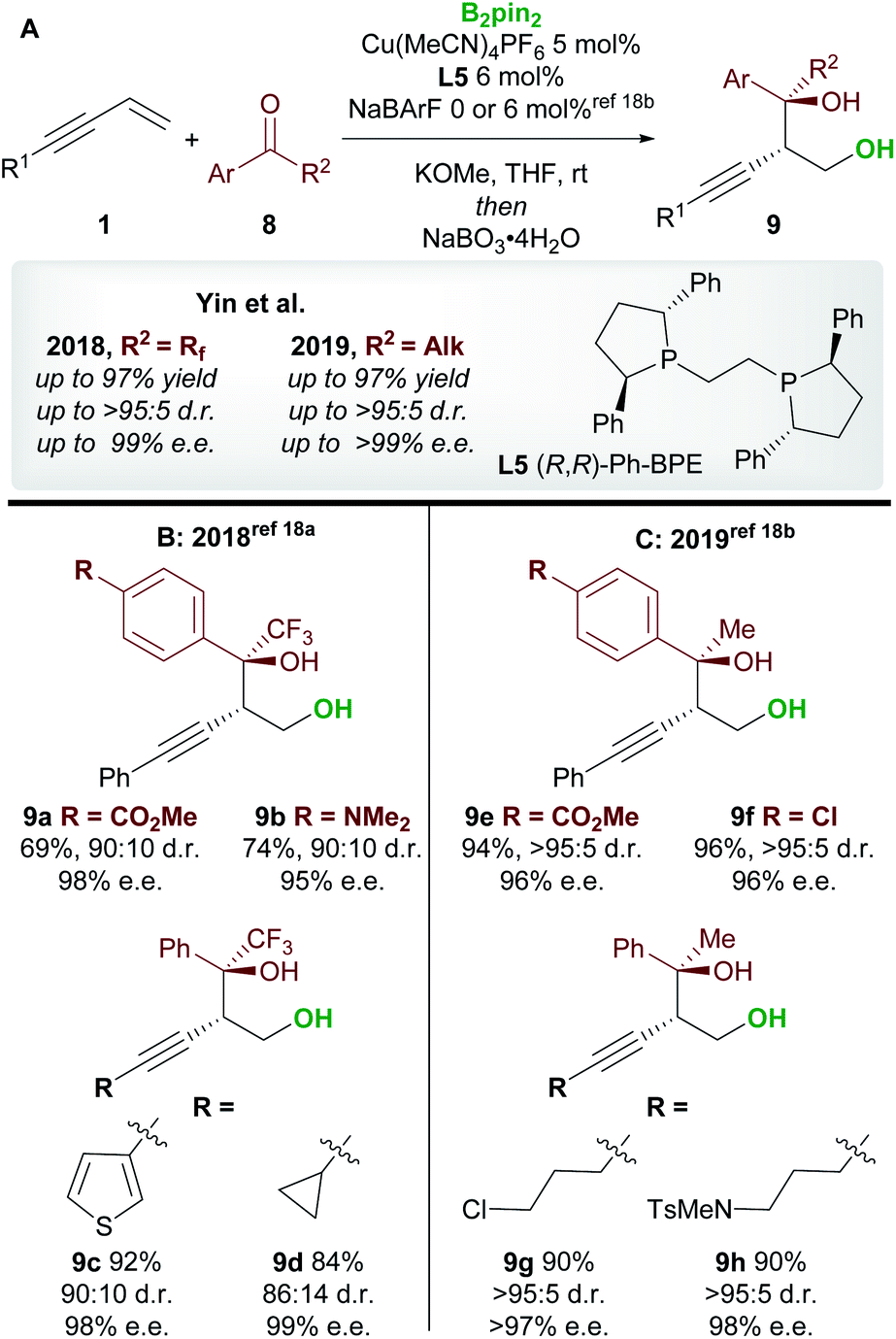
Following on from this report, Yin and co-workers18 published consecutive papers on the copper-catalyzed borylative 1,2-functionalization of 1,3-enynes 1 with ketones 8 (Scheme 6A). Firstly, Jia, Yin et al.18a described the coupling of aryl and alkyl-substituted enynes with perfluoroalkyl ketones (Scheme 6B). Using Ph-BPE L5, a ligand that has found much use in the field of copper-catalyzed functionalization of olefins, very good to excellent yields and excellent enantioselectivities were obtained across a wide range of aryl and alkenyl trifluoromethyl ketones. Similarly, longer perfluoroalkyl chains were also well tolerated. Yin and co-workers18b then extended their work towards the use of aryl, alkyl ketones (Scheme 6C). The conditions were similar to those used in their previous report, however, the addition of a non-coordinating counter-anion NaBArF was required to obtain high yields. Excellent enantioselectivities were observed across all ketone inputs, and both aryl and alkyl-substituted 1,3-enynes were good substrates. In both reports, the authors chose to oxidize the products upon workup to afford the anti-homopropargylic alcohols 9.
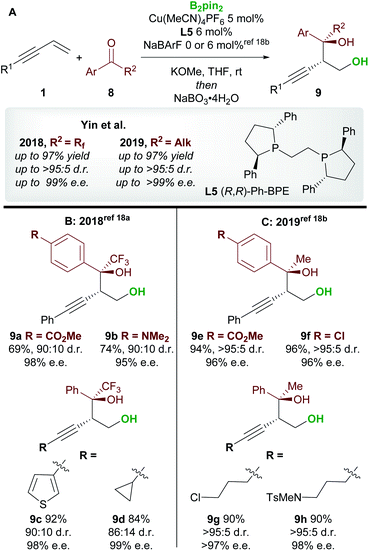 |
| | Scheme 6 Yin's 1,2-borofunctionalization of 1,3-enynes with fluorinated ketones and aryl, alkyl-ketones. | |
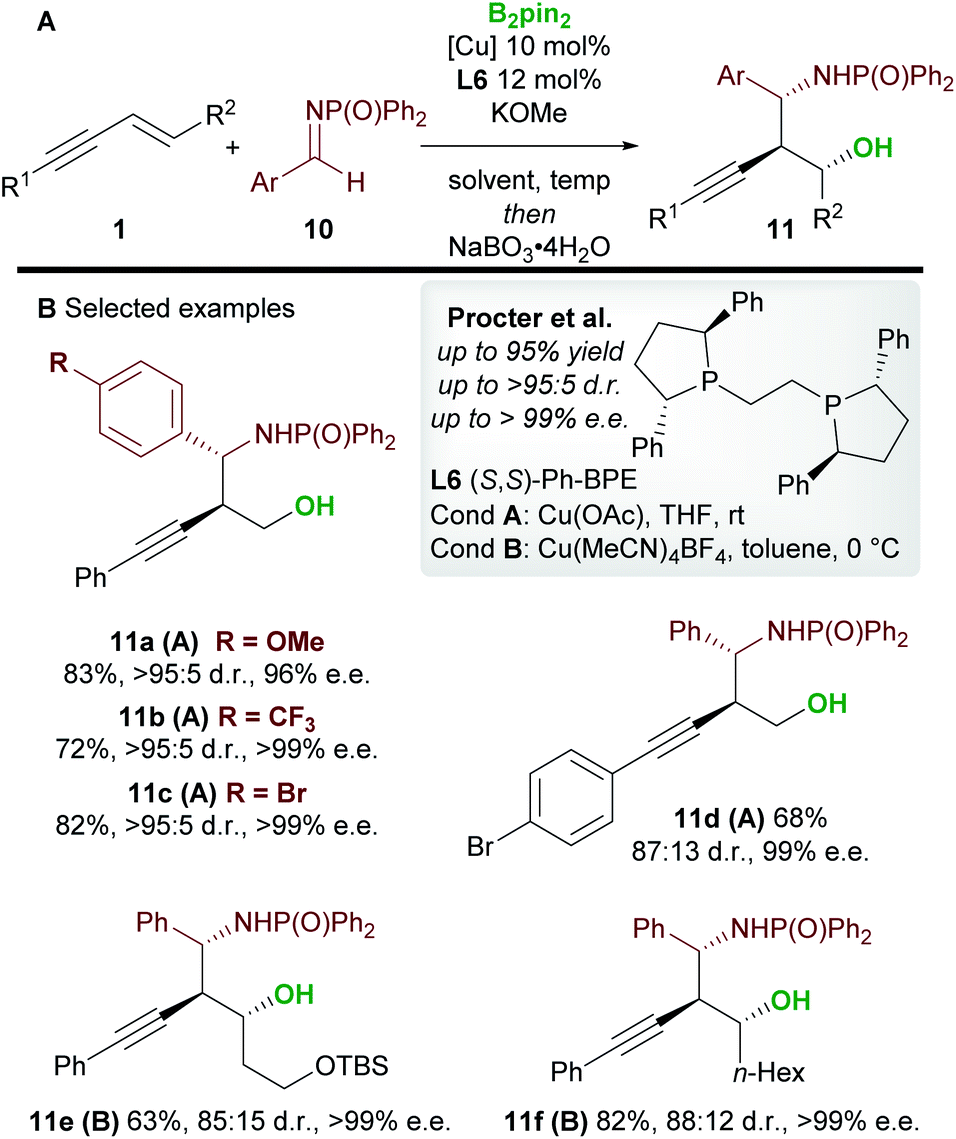
In 2020, Procter and co-workers19 developed the copper-catalyzed borylative 1,2-functionalization of 1,3-enynes 1 with aldimines 10 to give anti-homopropargylic amines 11 (Scheme 7). Interestingly, Ph-BPE L6 was again the ligand of choice. A range of electron-rich and electron-deficient N-phosphinoyl imines coupled to enynes in very good yield and with excellent diastereo- and enantiocontrol. The authors found that a switch in solvent, temperature and copper catalyst ensured favourable reactivity with a wide range of enynes. The products were oxidized during work-up to afford the 1,3-aminoalcohols 11. Importantly, the reaction was extended to 1,2-disubstituted (E)-enynes (11e and 11f) to give products containing 3 contiguous stereocenters with high enantiocontrol.
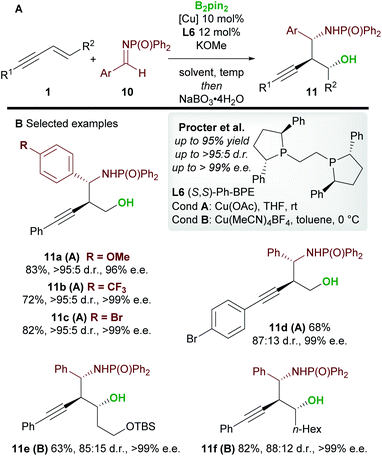 |
| | Scheme 7 Procter's 1,2-borofunctionalization of 1,3-enynes with imines. | |
3.3. 1,4-Borofunctionalization processes
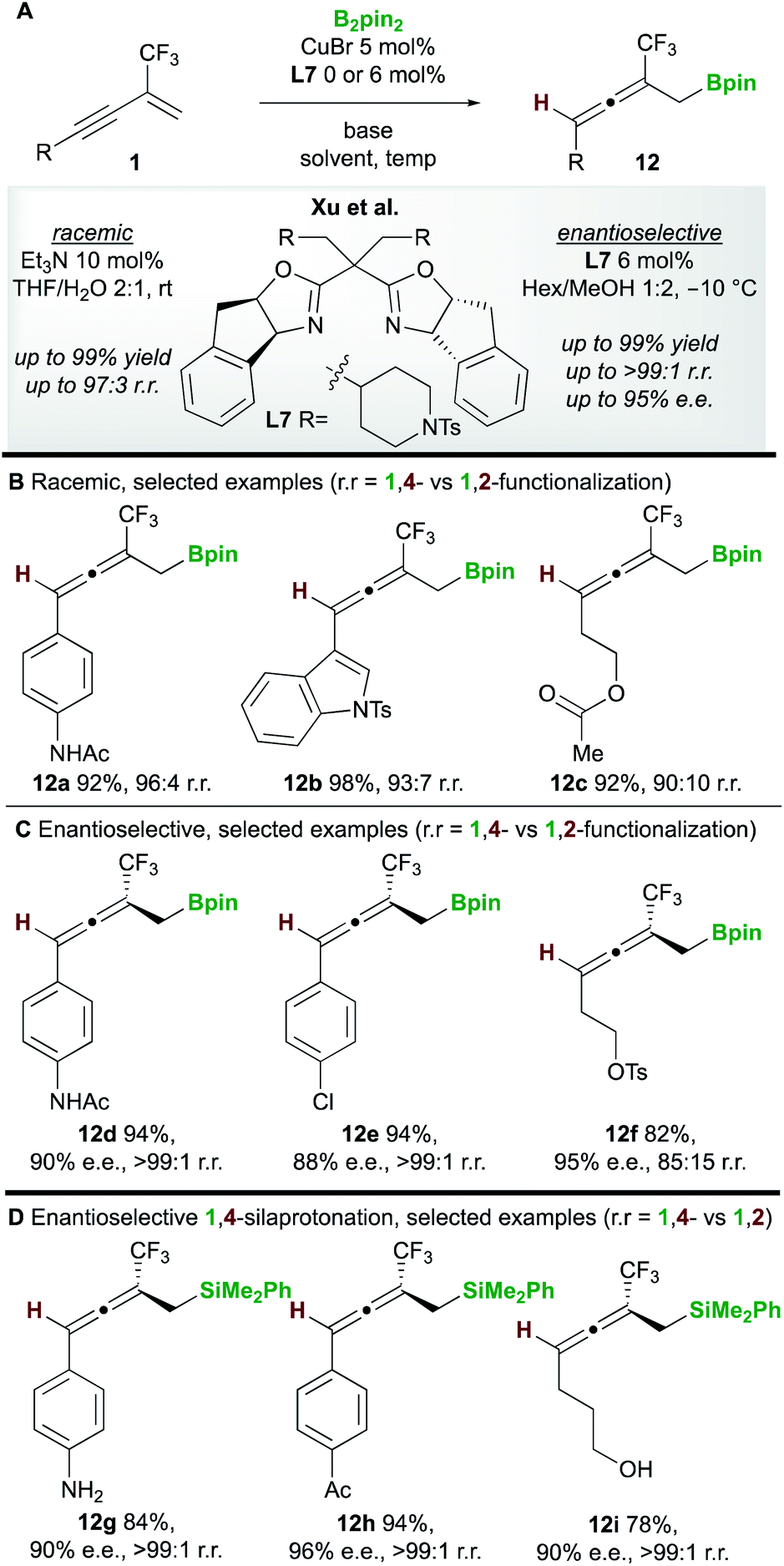
In 2020, Xu and co-workers20 developed the copper-catalyzed 1,4-boroprotonation of 2-trifluoromethyl-1,3-enynes 1. A ligand-free CuBr catalyst promoted the formation of aryl and alkyl-substituted allenes 12 in very good to excellent yields (Scheme 8A and B). An efficient enantioselective process that, unusually, did not require a base was also developed using the bisoxazoline ligand L7 (Scheme 8C). Interestingly, in some cases, the authors observed minor amounts of the 1,2-boroprotonated product, although no clear explanation for variation in regioselectivity was put forward. Furthermore, a related 1,4-silaprotonation operating under similar conditions exhibited high functional group tolerance and allowed access to the 1,4-silaprotonated products 12g–12i with high enantiocontrol (Scheme 8D).
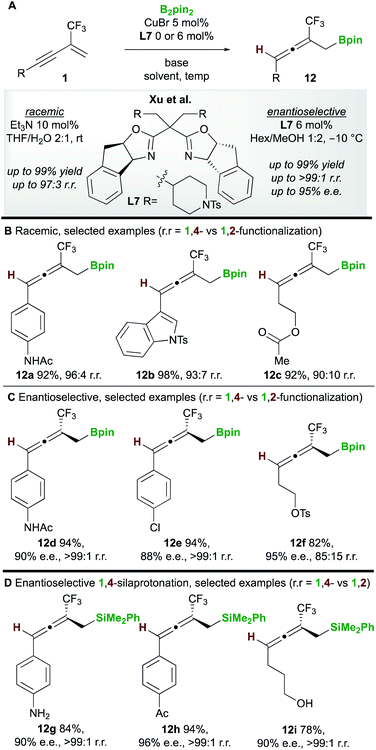 |
| | Scheme 8 Xu's boroprotonation of 2-trifluoromethyl-1,3-enynes. | |
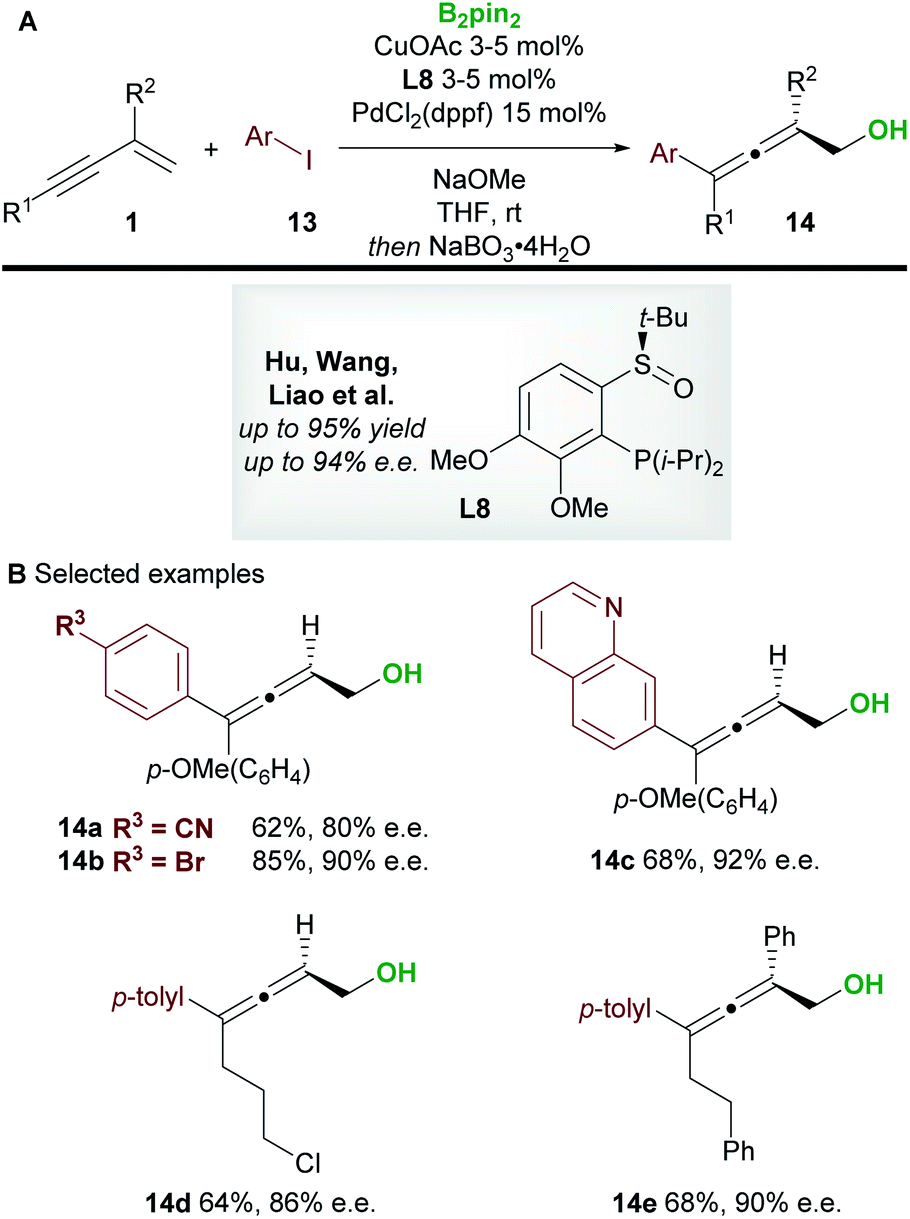
In 2020, Hu, Wang, Liao, and co-workers21 disclosed a borylative copper palladium co-catalyzed 1,4-boroarylation of 1,3-enynes 1 (Scheme 9). Using their previously developed sulfoxide–phosphine (SOP) ligand L8, in conjunction with CuOAc (3 to 5 mol%), PdCl2(dppf) (15 mol%) and aryl iodides 13, they obtained tri- and tetra-substituted enantioenriched allenes 14. The coupling of electron-rich/neutral, aryl substituted 1,3-enynes (R1 = aromatic, R2 = H) with a wide range of aryl iodides gave tri-substituted allenes (14a–14c) in high yield and with high enantiocontrol. When using 1,3-alkylenynes (R1 = Alk), both tri-substituted (R2 = H, 14d), and tetra-substituted (R2 = Ar, 14e) products were accessible with very good enantiocontrol. It was proposed that the copper catalyst and chiral SOP ligand promoted an enantioselective 1,2-borocupration and subsequent isomerisation gave an allenyl copper species (c.f.int-2, Scheme 5D). Then, after transmetallation, palladium catalyzed arylation gave the desired allene products 14.
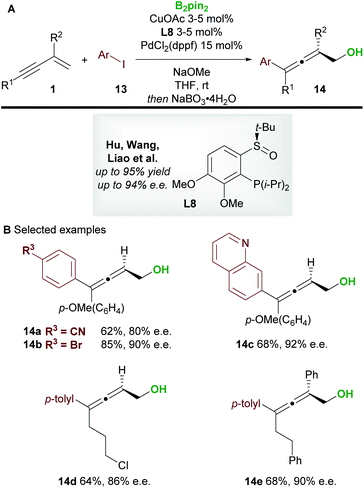 |
| | Scheme 9 Hu, Wang, Liao's boroarylation of 1,3-enynes with aryl iodides. | |
3.4. 3,4-Borofunctionalization processes
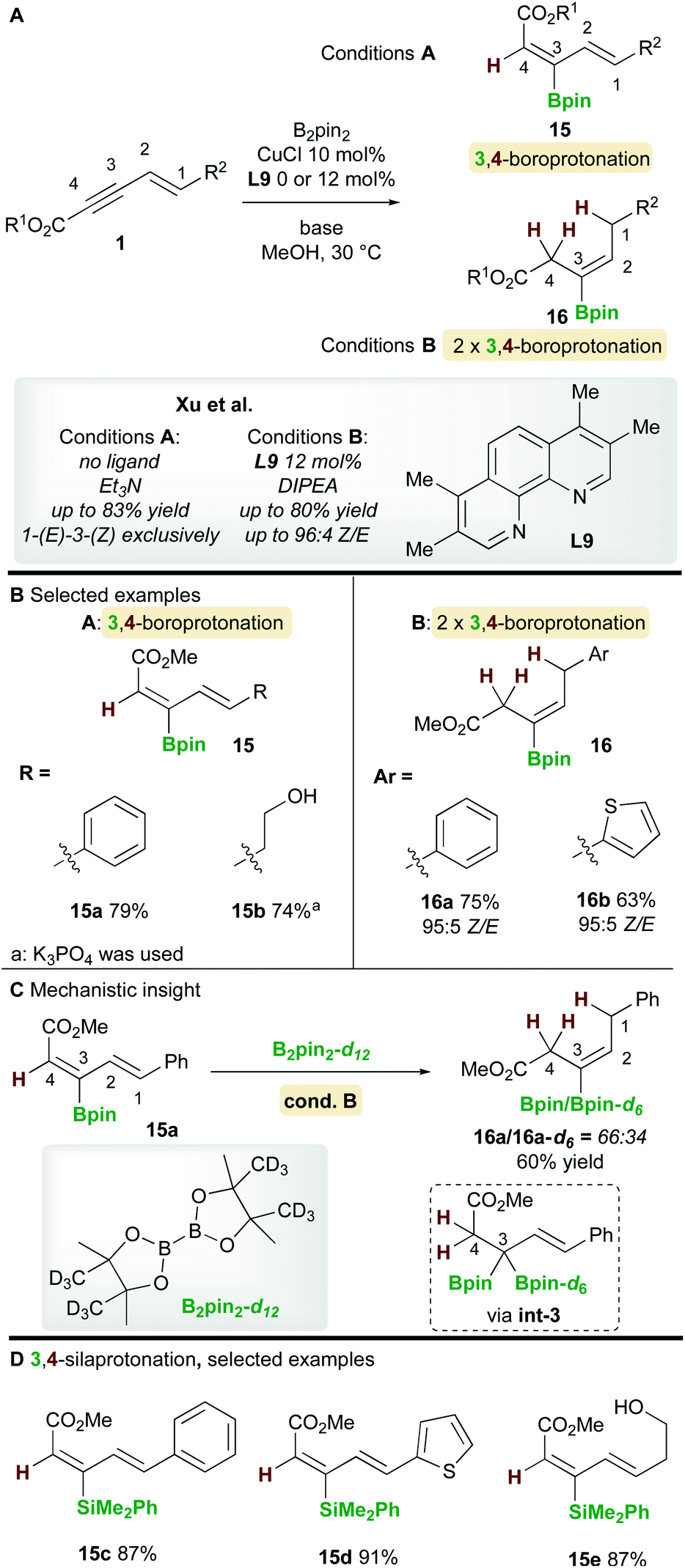
In 2019, Xu and co-workers22 described a copper-catalyzed boroprotonation of 1,3-enynoates 1 (Scheme 10). When a ligand-free CuCl catalyst and a substoichiometric amount of base were used in MeOH, the diene 15 from 3,4-boroprotonation was isolated with essentially perfect E,Z-selectivity (Scheme 10, cond. A). Moderate to very good yields were obtained with aryl- and alkyl-substituted 1,3-enynoates (Scheme 10B). Conversely, when using CuCl and 3,4,7,8-tetramethyl-1,10-phenanthroline ligand L9 in the presence of 2 equivalents of di-isopropylethylamine (DIPEA), alkene products 16 were formed. Very good yields and Z-selectivities were obtained in this process when using (hetero)aryl-substituted 1,3-enynoates. It was proposed that product 16 forms through two rounds of boroprotonation across the C3/C4 bond of the enyne, followed by protodeborylation i.e. the 3,4-boroprotonated product 15 is formed initially, but undergoes another boroprotonation followed by protodeborylation. A control experiment lent support to this hypothesis: when 3,4-boroprotonated product 15a was subjected to conditions B in the presence of B2pin2-d12, product 16a was obtained in 60% yield with significant Bpin-d6 incorporation at the 3 position. This indicated that boroprotonation occurs across the C3/C4 bond of 15a to give a 3,3-diborylated intermediate int-3, followed by an unselective protodeborylation to give a mixture of Bpin/Bpin-d6 products. A 3,4-silaprotonation using the ligand-free system was also developed (Scheme 10D). Products 15c–e, were isolated with complete E,E-selectivity.
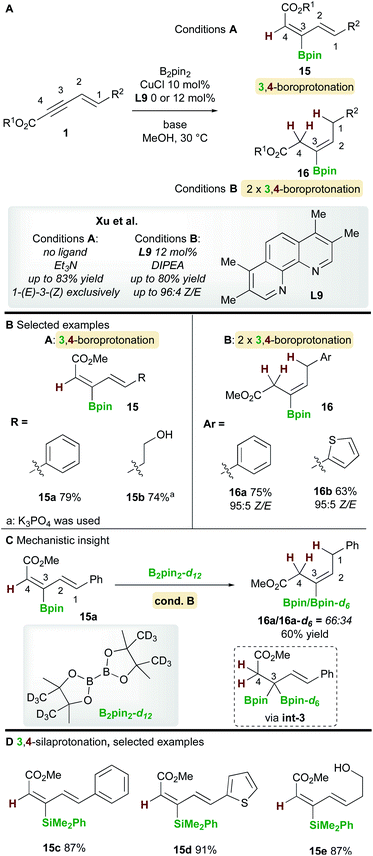 |
| | Scheme 10 Xu's boroprotonation of 1,3-enynoates. | |
4. Hydrofunctionalization
With origins in the 1850s,23 copper hydride chemistry gained popularity with the introduction of Stryker's reagent24 [(PPh3)CuH]6 and its use for the selective reduction of carbonyl derivatives. Significant advancements by Buchwald,25 Lipshutz26 and others23 led to catalytic asymmetric reactions using copper hydride species. The application of copper hydride chemistry in processes involving enynes is particularly appealing as it allows facile access to propargyl and allenyl metal species that react selectively under mild conditions.
4.1. 1,2-Hydrofunctionalization processes
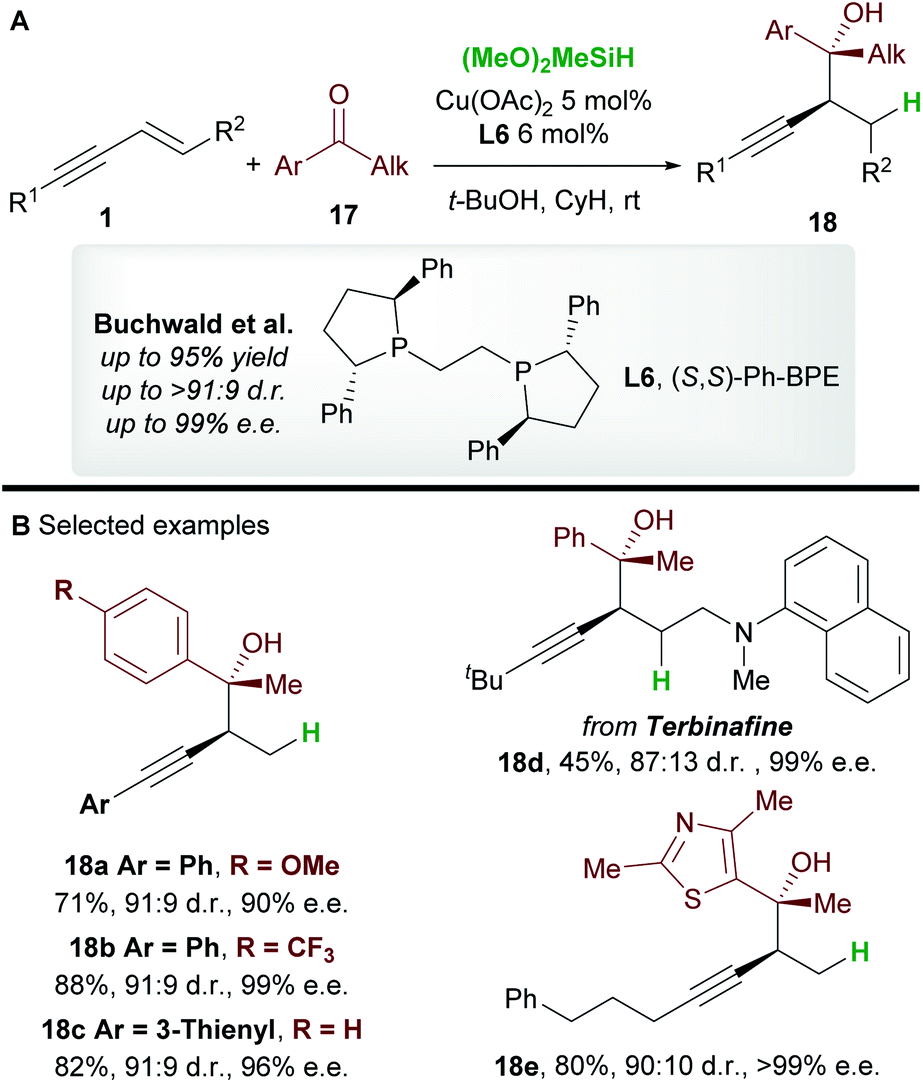
In 2016, Buchwald and co-workers27 reported an efficient copper-catalyzed 1,2-hydrofunctionalization of enynes 1 with ketones 17 to give highly substituted and enantioenriched homopropargyl alcohols 18 (Scheme 11A). The suitability of various enyne inputs was demonstrated and the process showed excellent diastereo- and enantioselectivity. The reaction tolerated both electron-donating and electron-withdrawing substituents on the aryl ring of the ketone. Notably, the antifungal medication terbinafine, which contains an enyne unit, responded well to hydrofunctionalization to give 18d (Scheme 11B).
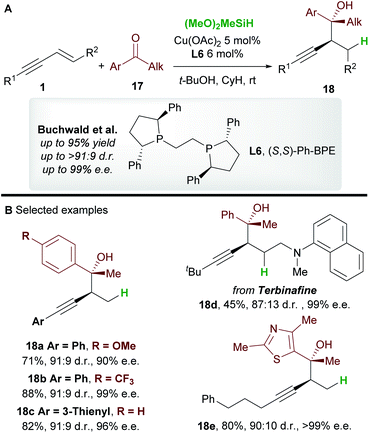 |
| | Scheme 11 Buchwald's hydrofunctionalization of 1,3-enynes with ketones. | |
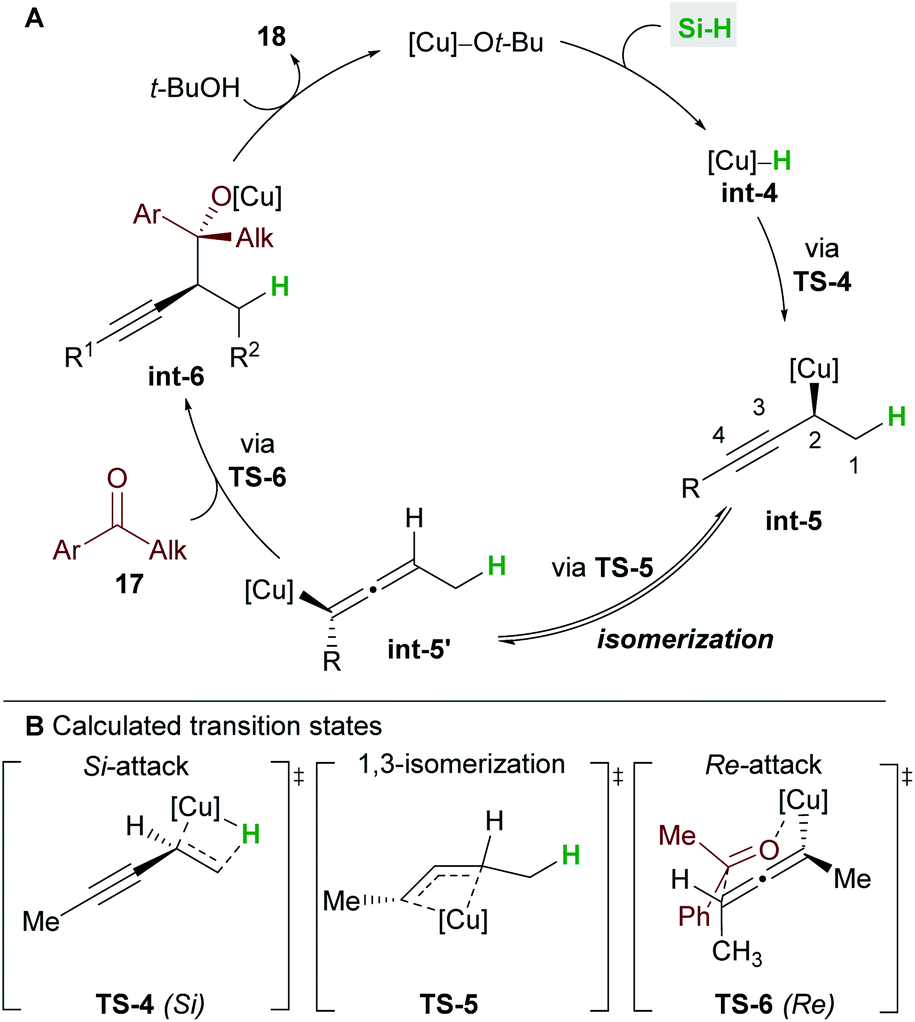
A full mechanistic profile for the reaction was provided by DFT calculations (Scheme 12). The catalytic cycle begins with an enantioselective 1,2-hydrocupration of the enyne by the in situ generated copper hydride int-4, to give propargyl-copper species int-5via a 4-membered transition state TS-4. It was found that attack on the Si face of the olefin was favoured by 5.5 kcal mol−1 and that the selectivity arises from minimisation of steric interactions between the alkynyl group and the phenyl groups of ligand L6. A facile, exergonic, and stereospecific 1,3-isomerization of int-5 through TS-5 led to the thermodynamically more stable allenyl-copper intermediate int-5′. The intermediate int-5′ then undergoes coupling with the Re face of the ketone 17 through a 6-membered cyclic transition state TS-6 to afford int-6. This step was rendered highly diastereoselective through the minimisation of unfavourable gauche interactions between the ketone and int-5′, and between the ligand and the allenyl substituents. Protonation of int-6 delivers the homopropargyl alcohol products 18. Clear parallels can be drawn between this mechanism and that proposed by the Hoveyda group (Scheme 5). These mechanisms also likely underpin the transformations in Schemes 6 and 7.
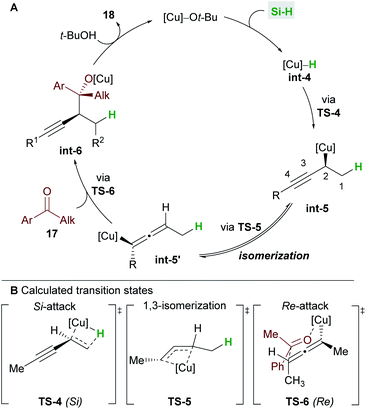 |
| | Scheme 12 Buchwald's catalytic cycle (A) and DFT calculations (B). | |
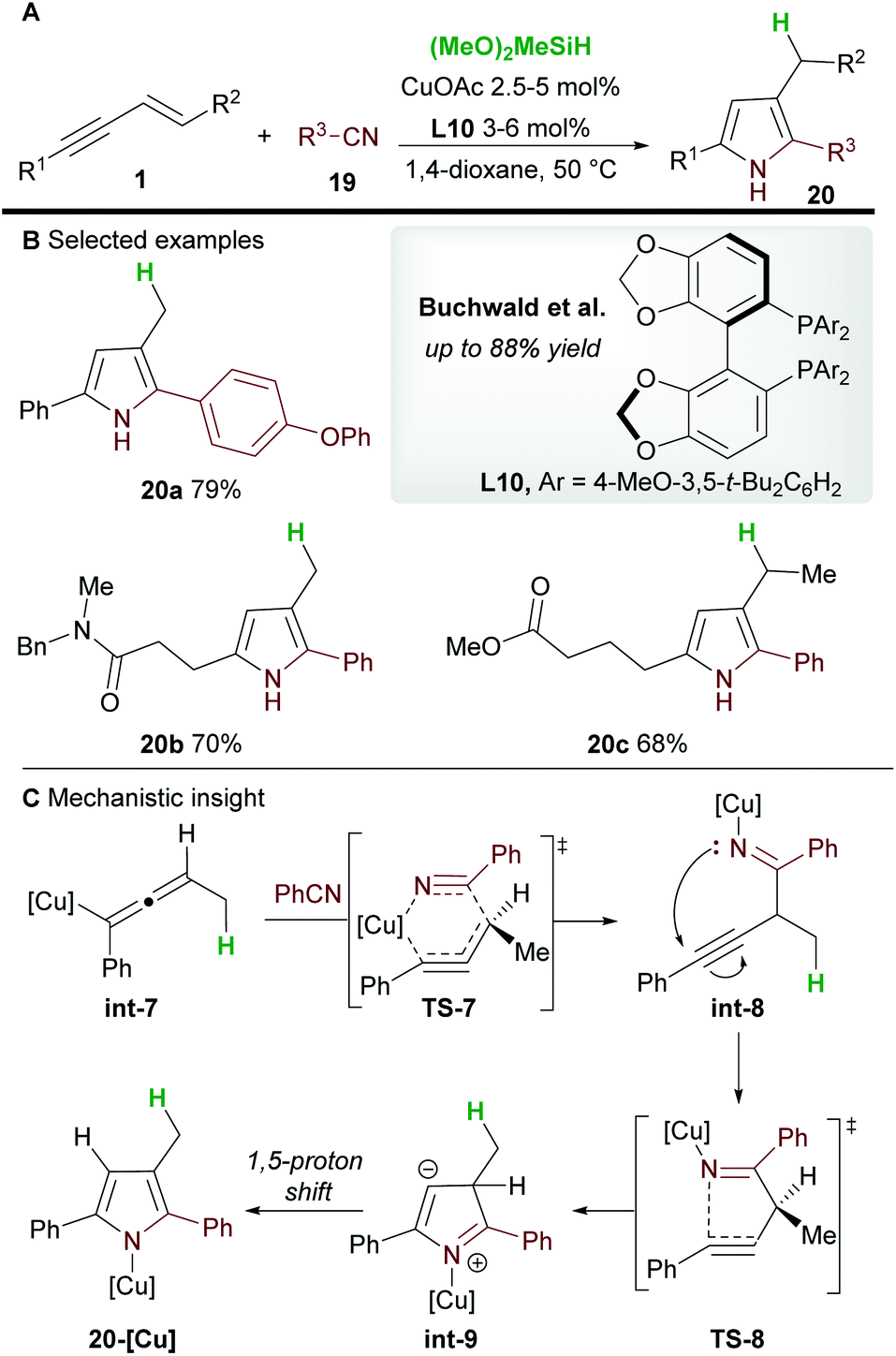
In 2020, Buchwald and co-workers28 used the copper-catalyzed 1,2-hydrofunctionalization of 1,3-enynes 1 with nitriles 19 to afford polysubstituted pyrroles 20 (Scheme 13). Both alkyl and aryl-substituted enynes, and aromatic and aliphatic nitriles afforded 2,3,5-trisubstituted pyrroles in moderate to good yield.
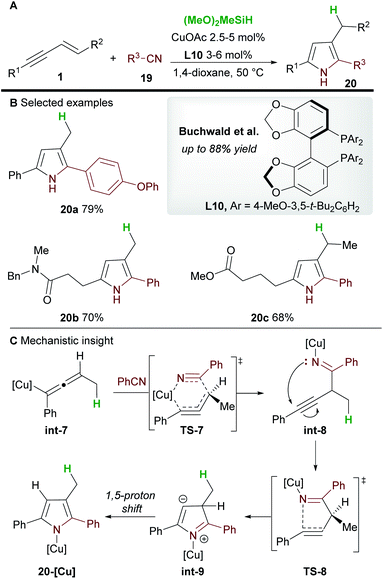 |
| | Scheme 13 Buchwald's hydrofunctionalization of enynes with nitriles for the synthesis of pyrroles. | |
The mechanism of the reaction was investigated by DFT (Scheme 13C). As previously described (Scheme 12), hydrocupration and 1,3-isomerization to the allenyl copper int-7 was found to be facile and exergonic. Subsequent coupling with nitrile 19 proceeds via the 6-membered transition state TS-7 to deliver the 1,2-functionalized species int-8 and is reminiscent of the addition to imines (Scheme 7). In related procedures, the catalytic cycle would usually close at this point to give functionalized (homo)propargylic products, however, in this case, a cyclization through transition state TS-8, followed by a 1,5-proton shift gave substituted pyrroles 20. Small quantities of a side product resulting from 1,4-functionalization were also observed during the reaction.
4.2. 1,4-Hydrofunctionalization processes
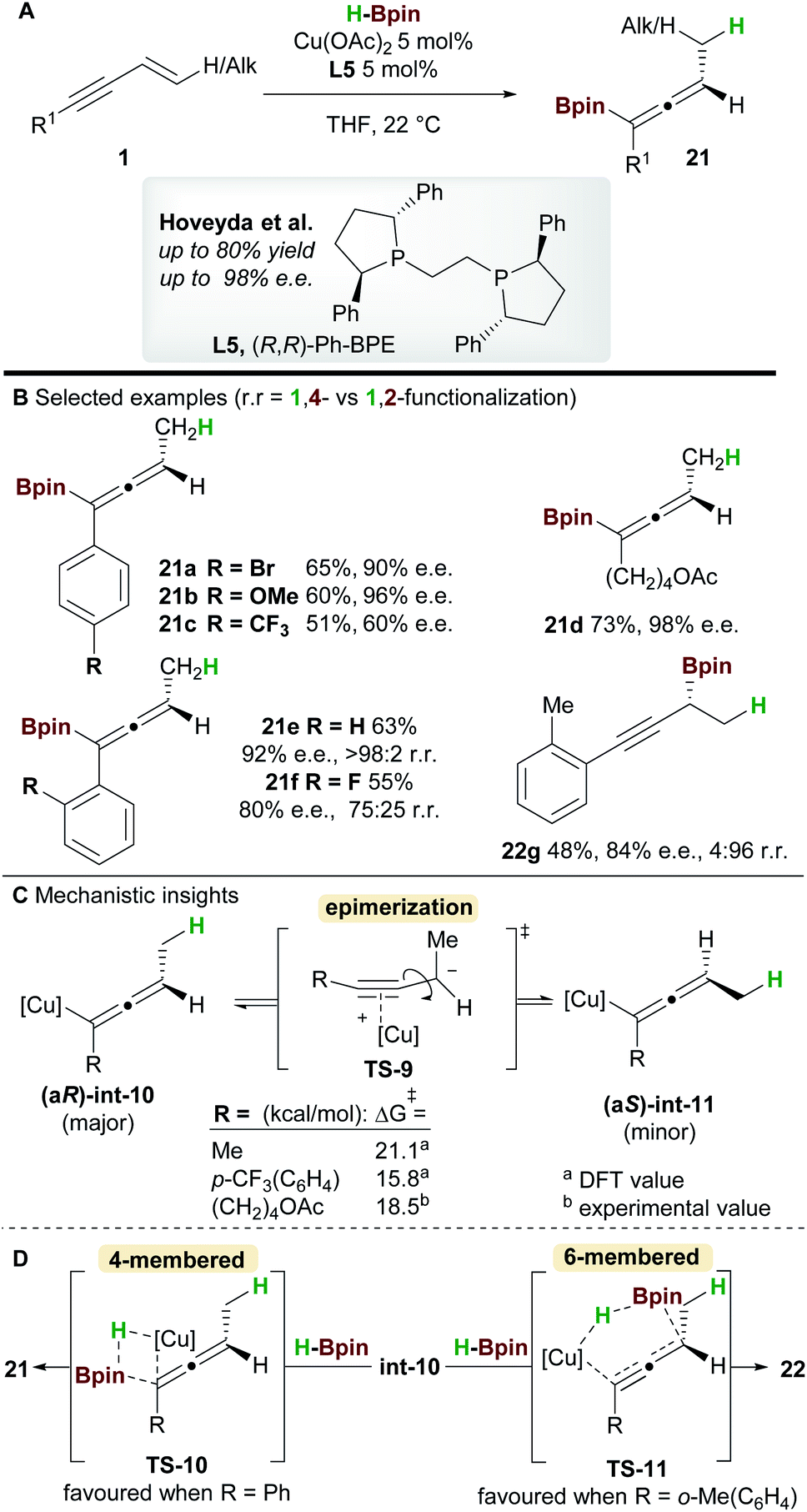
In 2018, an enantioselective copper-catalyzed hydroboration of 1,3-enynes 1 was developed by Hoveyda and co-workers29 using pinacolborane (HBpin) to synthesise substituted allenyl-Bpin products 21 (Scheme 14). Once again Ph-BPE L5 was the ligand of choice for this reaction. Electron-rich and electron-neutral aryl/alkyl-substituted 1,3-enynes delivered allene products with excellent enantiocontrol, however, the process was less efficient for electron-poor substrates such as 21c. Furthermore, 1,3-enynes bearing a bulky ortho-substituent on the aromatic ring afforded propargylic products, suggesting that steric hindrance about the alkynyl bond plays an important role in selectivity (products 21e, 21f, and 22g).
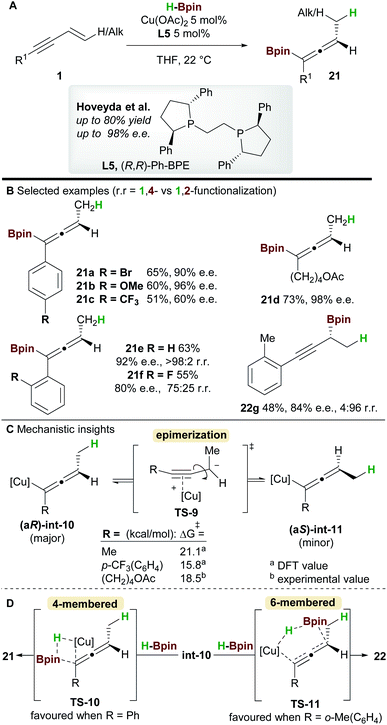 |
| | Scheme 14 Hoveyda's hydrofunctionalization of 1,3-enynes with pinacolborane. | |
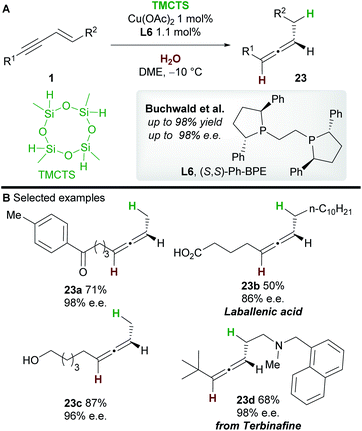 |
| | Scheme 15 Buchwald's hydrofunctionalization of 1,3-enynes with TMCTS and water. TMCTS = 2,4,6,8-tetramethylcyclotetrasiloxane. | |
Intrigued by the lower enantioselectivities observed for electron poor substrates, mechanistic experiments and DFT calculations were conducted and the results provided important insight into the stereochemical integrity of the allenyl-copper intermediate (Scheme 14C). In agreement with the work of Buchwald et al. (Scheme 12), hydrocupration of the (Re)-face of the enyne was found to be enantiodetermining. A facile, stereospecific 1,3-isomerization then led to the allenyl-copper species int-10. The lower enantioselectivities observed for some substrates was proposed to arise from epimerization of intermediate int-10via the propargyl anion-type transition state TS-9. Furthermore, this epimerization was shown to be more facile for electron-poor enynes e.g. that give 21c (Scheme 14C). Low-temperature NMR kinetic studies on the epimerization of the related allenyl-copper species derived from the substrate that gives 21d, found the epimerization barrier to be similar to those calculated by DFT (Scheme 14C). Finally, formation of the allenyl-Bpin product 21 and regeneration of the copper-hydride catalyst occurs via the 4-membered transition state TS-10. Conversely, when sterically demanding substituents are present, the corresponding 6-membered transition state TS-11 was favoured and delivered the propargylic product e.g.22g (Scheme 14D).
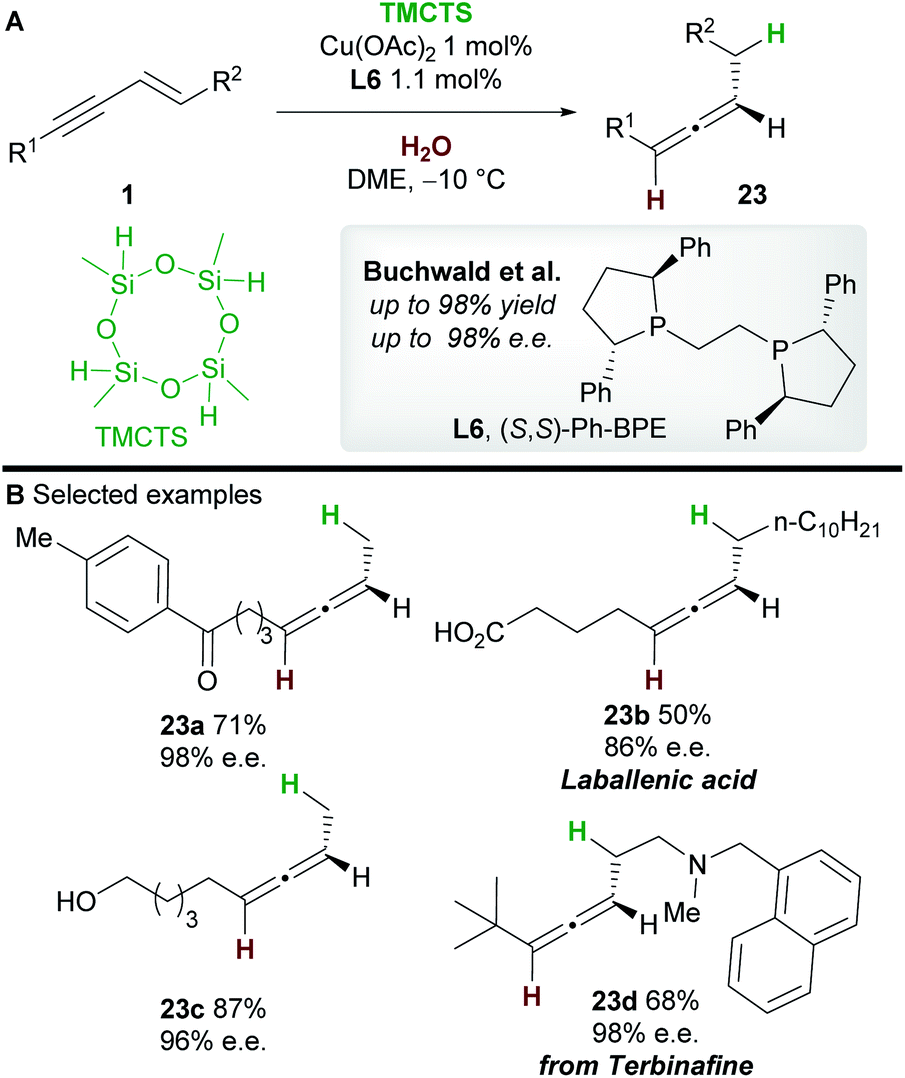
In 2019, an asymmetric semireduction of 1,3-enynes 1 was described by Buchwald and co-workers30 using 2,4,6,8-tetramethylcyclotetrasiloxane (TMCTS) as a hydride source. As is now established in these processes, the Ph-BPE ligand L6 provided the best results. A range of aryl and alkyl-substituted 1,3-enynes, including those bearing sensitive functional groups, gave the allene products 23 with high enantioselectivity. Furthermore, the methodology was applied in an enantioselective synthesis of the natural product laballenic acid 23b, without the need for protection of the acid group.
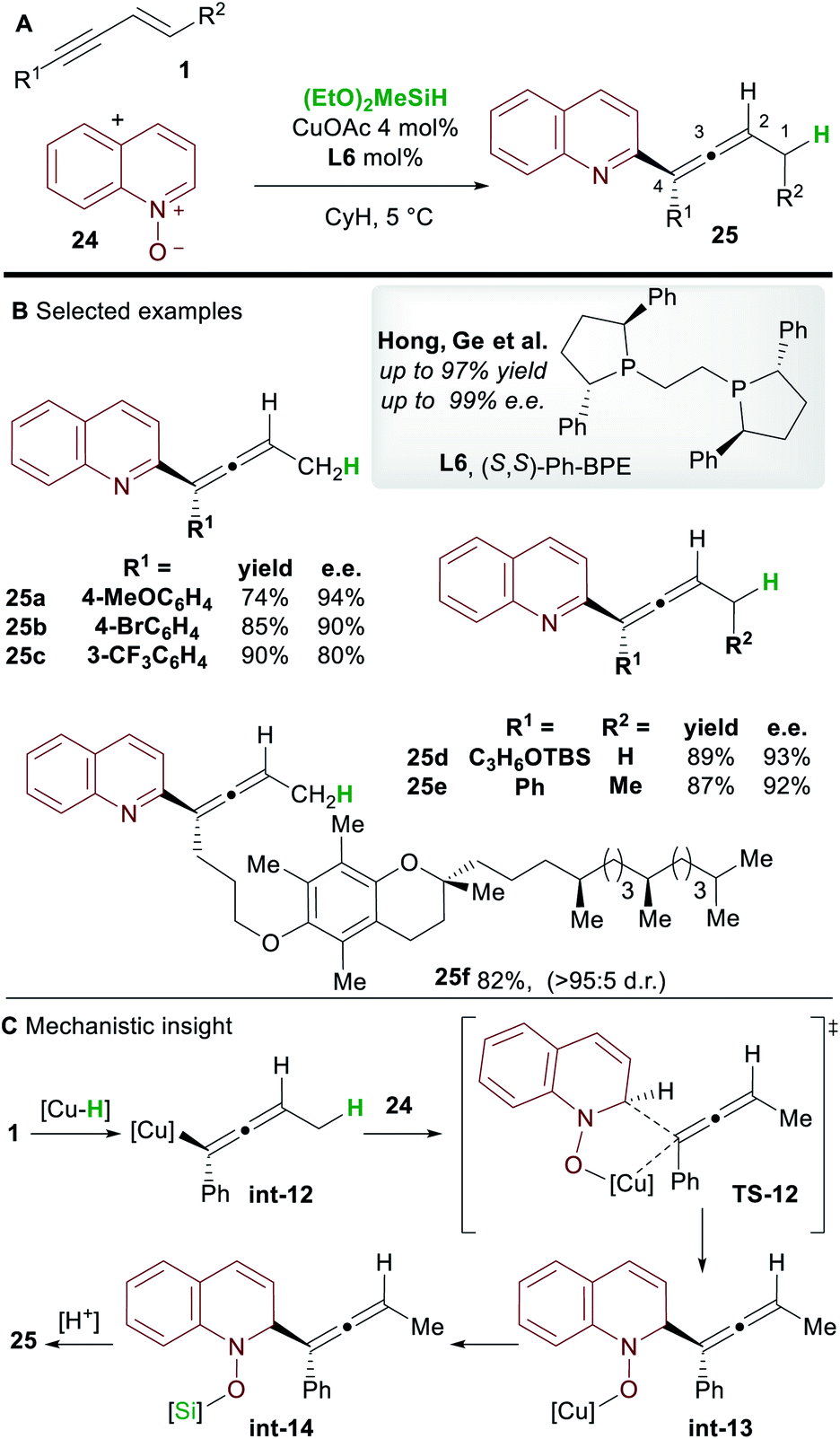
In 2018, Hong, Ge and co-workers31 reported the copper-hydride catalyzed allenylation of quinoline N-oxide 24 using enynes 1 and affording enantioenriched N-heteroaryl-substituted allenes 25 (Scheme 16). In this case also, the Ph-BPE ligand L6 provided the best results. Aryl and alkyl-substituted 1,3-enyne inputs, as well as various heterocyclic N-oxides, gave heteroaryl substituted allene derivatives with high enantioselectivity. Aryl-substituted 1,3-enynes bearing electron-donating (25a) and electron-withdrawing groups (25c) were tolerated, although, as seen by Hoveyda and co-workers (Scheme 5), electron-poor and ortho-substituted aryl-1,3-enynes gave lower enantioselectivities (25c). Compatibility with some natural products, such as a vitamin E (to give 25f) and a cholesterol derivative, was also shown. DFT calculations showed that, in accordance with previous reports by Buchwald and Hoveyda, stereospecific isomerization follows enantioselective 1,2-hydrocupration to give the allenyl copper complex int-12. Coupling of allenyl-copper int-12 with the N-oxide was proposed to go through a 5 membered-transition state TS-12 to give int-13. A related 7-membered transition state leading to the propargylic product was found to be disfavoured by 3.5 kcal mol−1. Finally, The N–O silylated adducts int-14 re-aromatized spontaneously during work-up.
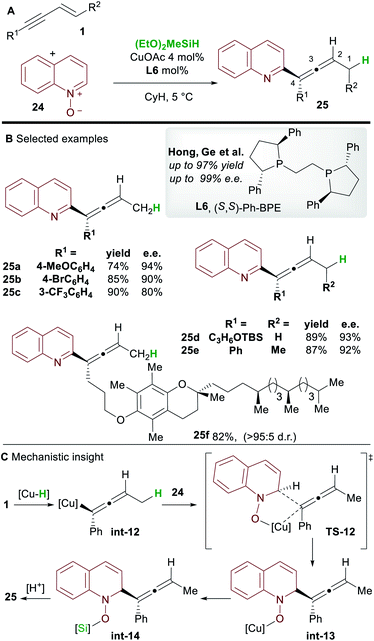 |
| | Scheme 16 Hong and Ge's hydrofunctionalization of 1,3-enynes with quinoline N-oxide. | |
5. Copper-catalyzed radical functionalization of enynes
The addition of radicals to olefins is an essential tool for C–C bond formation in organic synthesis.32 Copper, with several readily accessible oxidation states, can mediate single- or two-electron processes and thus facilitate an array of redox processes that convert enynes to functionalized products.33
5.1. Regiodivergent processes
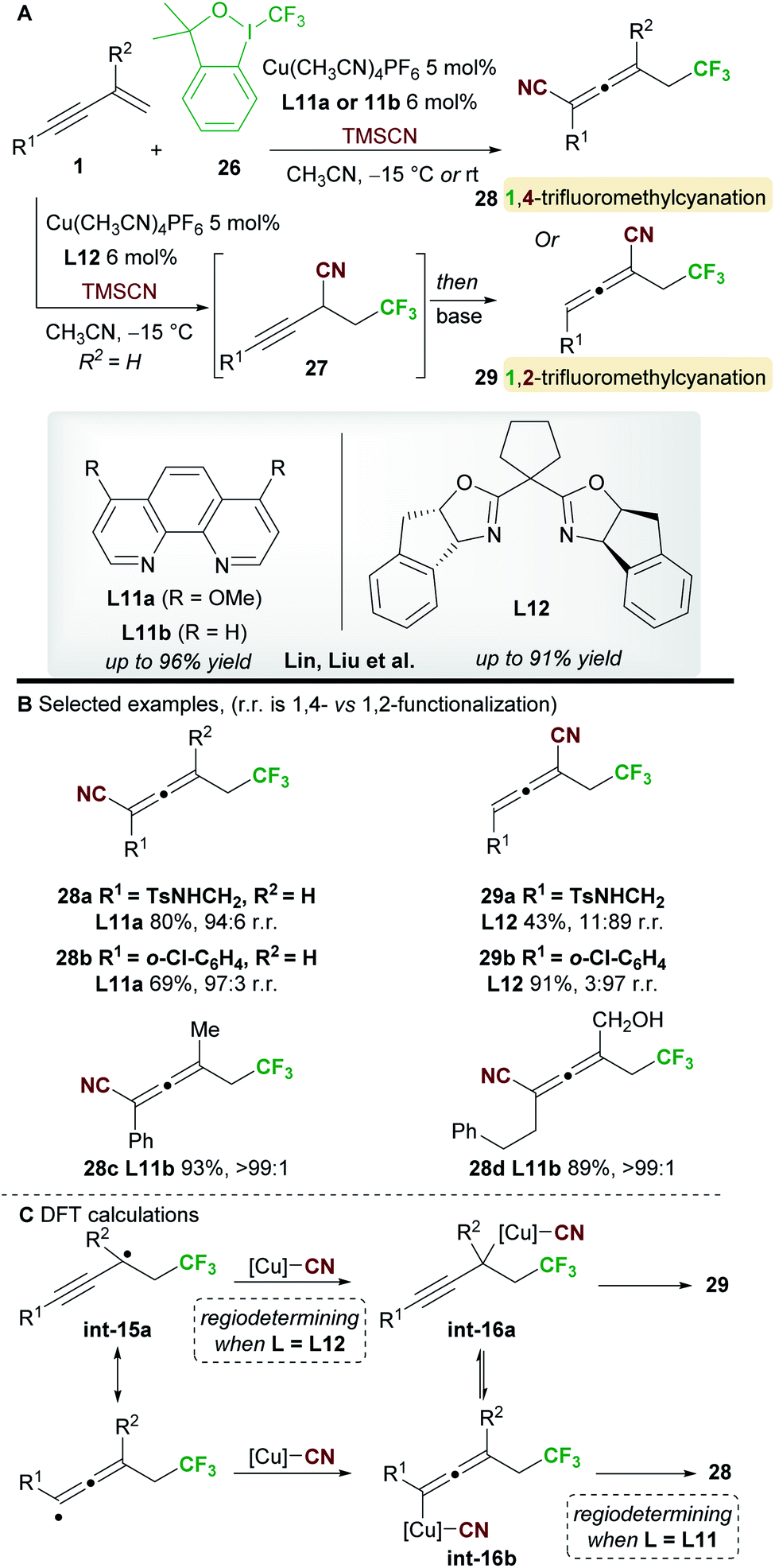
In 2018, Lin, Liu and co-workers34 reported a ligand-controlled, regiodivergent trifluoromethylcyanation of 1,3-enynes 1 using Togni's reagent 26 and TMSCN (Scheme 17). Simple 1,3-enynes (R2 = H) were converted to either 1,4-trifluorocyanated allenes 28, using a phenanthroline ligand L11a, or 1,2-trifluorocyanated allenes 29 using the bulkier bisoxazoline ligand L12. On the other hand, 2-substituted 1,3-enynes underwent 1,4-trifluoromethylcyanation only using ligand L11b, providing tetrasubstituted allenes 28. The selectivity and yields remained high with substrates bearing various aromatic, heteroaromatic, and aliphatic alkynyl substituents (R1). The enantioselectivity of processes using the chiral ligand L12 was not reported.
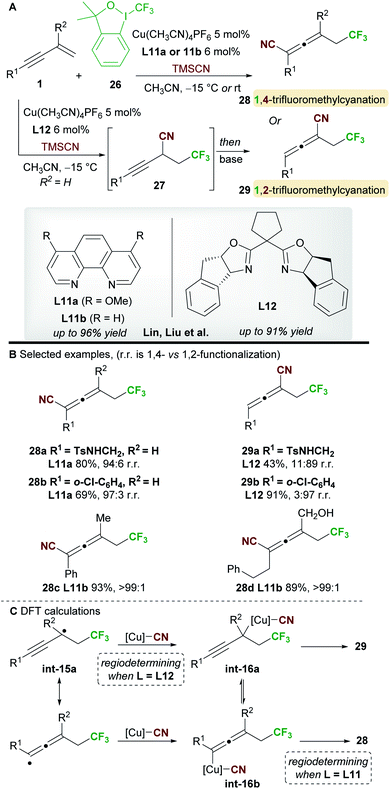 |
| | Scheme 17 Lin and Liu's trifluorocyanation of 1,3-enynes. | |
The authors studied the origin of regiodivergency using DFT (Scheme 17C). When using L11, delocalized radical int-15a can be trapped by the copper(II)cyanide complex at both the propargyl and allenic positions. Thus, the regiodetermining step was found to be the reductive elimination, which proved easier for the allenyl-copper species int-16b leading to the observed 1,4-disubstituted allenes 28. On the other hand, trapping of radical int-15a at the less congested, propargylic site with the bulky copper(II) complex derived from L12 was favoured. Thus, the authors suggest that regioselectivity is controlled by the reductive elimination step when using non-bulky ligands, while trapping of the radical int-15a determines the outcome of the reaction when using sterically demanding ligands.
5.2. 1,2-Functionalization
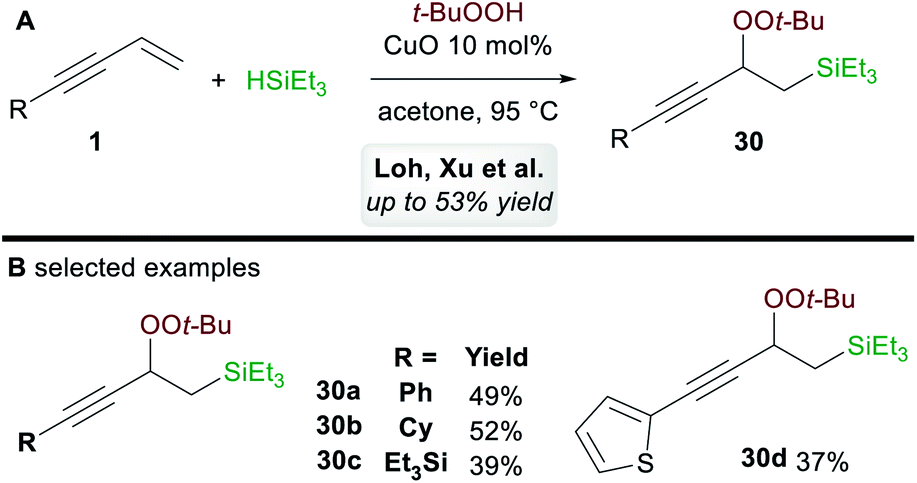
In 2017, Loh, Xu and co-workers35 disclosed a ligand-free copper-catalyzed regioselective 1,2-silylperoxidation of 1,3-enynes 1 using tert-butyl hydroperoxide (TBHP) and triethylsilane (Scheme 18A). Aryl, alkyl, and silyl-substituted 1,3-enynes were both compatible with the process and gave moderate yields of 1,2-functionalized products 30 (Scheme 18B). The process was also applied to other olefins such as α,β-unsaturated ketones, acrylates, and enamides.
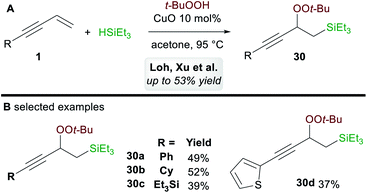 |
| | Scheme 18 Loh and Xu's silylperoxidation of 1,3-enynes. | |
5.3. 1,4-Functionalization
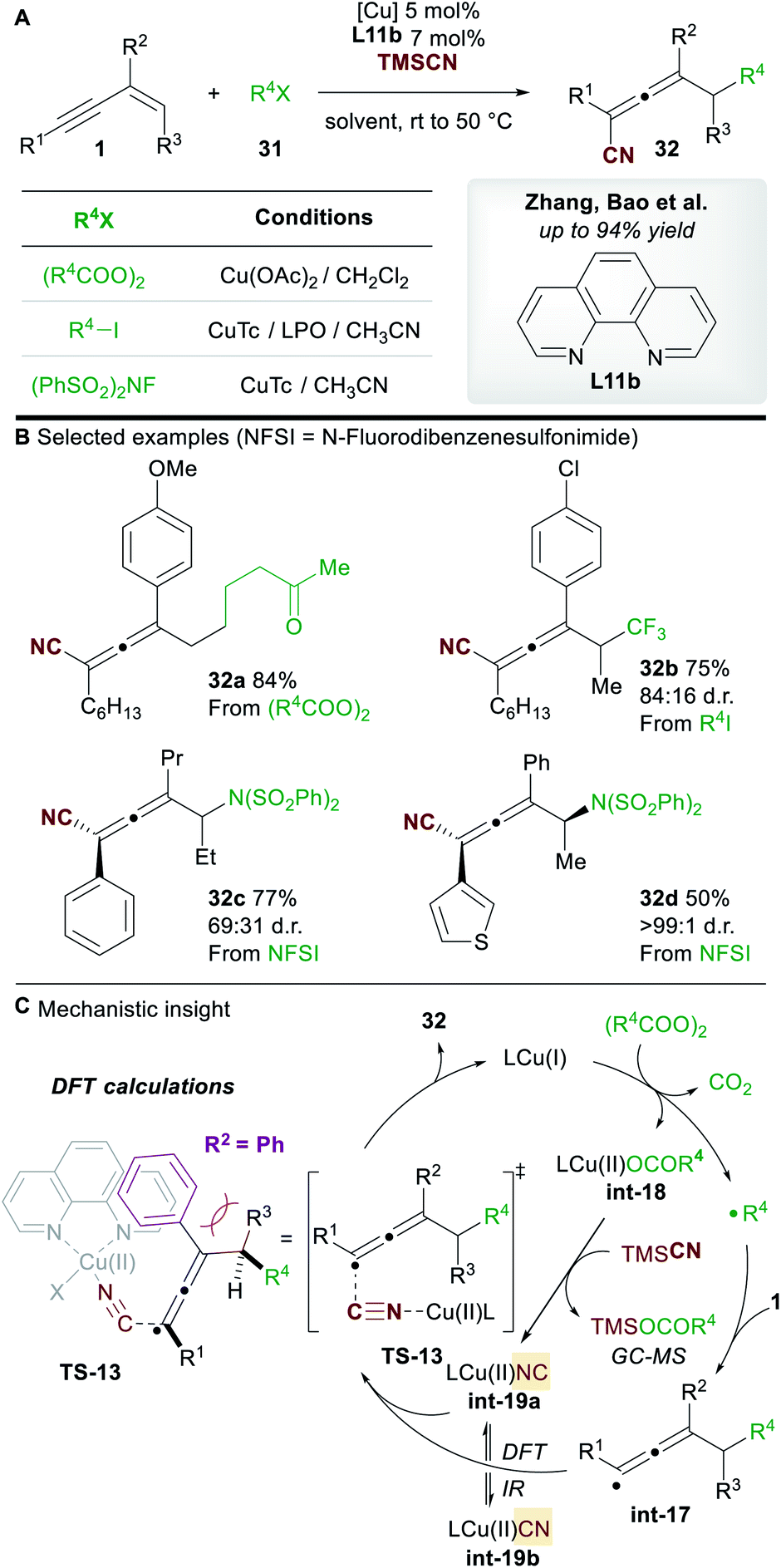
In 2019, Zhang, Bao and co-workers36 reported a copper-catalyzed regioselective 1,4-carbo-/aminocyanation of 1,3-enynes 1 using trimethylsilyl cyanide (TMSCN) (Scheme 19A). A wide range of 1,4-carbocyanated allenes 32 was obtained using alkyl diacyl peroxide (to give 32a) or alkyl iodide (to give 32b) radical precursors 31 (Scheme 19B). 1,4-Aminocyanated allenes 32c and 32d were also obtained in good yields using N-fluorobenzenesulfonimide (NFSI) as the precursor of a N-centered radical. 2-Substituted enynes were used in all cases (R2 = aryl/alkyl). Substitution on the terminus of the alkene in the 1,3-enynes was also tolerated (R3 ≠ H). In these cases, diastereocontrol was found to be dependent on the nature of R2. The authors proposed a mechanism in which copper promotes the formation of a C-centered (or N-centered) radical (Scheme 19C), followed by radical addition to the alkene moiety of the 1,3-enyne to give allenyl radical int-17, whose intermediacy was ascertained by radical probe experiments. Interestingly, the cyanation was proposed to occur from a copper(II)isocyano complex int-19a (detected by IR spectroscopy) rather than copper(II)cyanide complex int-19b. This raises the possibility that an alternative mechanism for cyanation might be occurring in the report by Lin, Liu et al. (Scheme 17). DFT studies suggested that the energy barrier to cyanation through TS-13, from int-19a, was 13.5 kcal mol−1 lower in energy than the analogous step with int-19b. The higher diastereoselectivites observed when R2 = aryl were proposed to arise from a favourable π–π stacking interaction with the ligand 1,10-phenanthroline L11b.
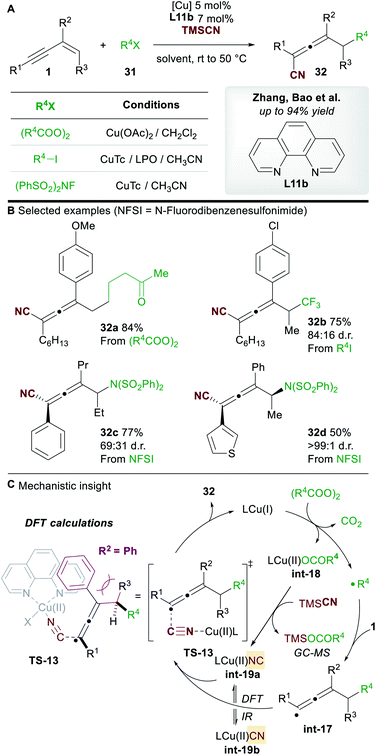 |
| | Scheme 19 Zhang and Bao's carbocyanation of 1,3-enynes. NFSI = N-fluorobenzenesulfonimide. | |
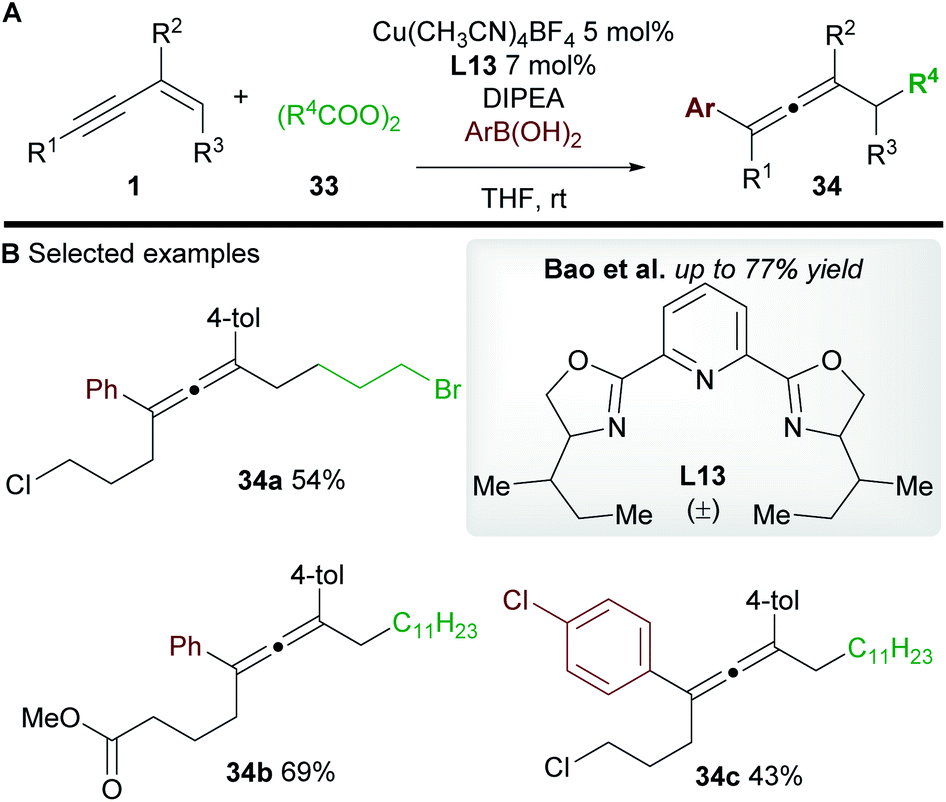
Bao and co-workers37 later reported a related transformation using aryl boronic acids in place of TMSCN to access 1,4-difunctionalized allenes 34 (Scheme 20A). A wide range of tetrasubstituted allenes bearing various aromatic and aliphatic groups was obtained in good yields (Scheme 20B). Mechanistic studies provided support for a radical mechanism and the authors suggested a catalytic cycle related to that shown in Scheme 19. Attempts to develop an asymmetric variant were unsuccessful.
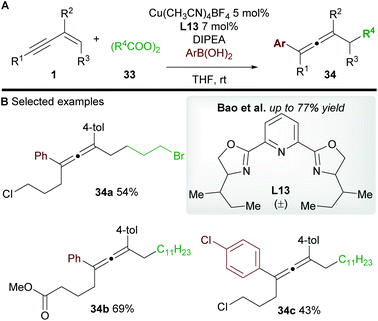 |
| | Scheme 20 Bao's carboarylation of 1,3-enynes. | |
6. Copper-catalyzed functionalization of 1,n-non-conjugated enynes
1,n-Non-conjugated enynes are important and versatile building blocks. For example, skipped enynes (1,4-enynes) have been used in the synthesis of important natural products,38 while other 1,n-enynes, following the work of Trost,39 have proved valuable in transition-metal catalyzed cycloisomerization reactions.8b,8c Herein, we will survey the copper-catalyzed functionalization of 1,n-enynes.
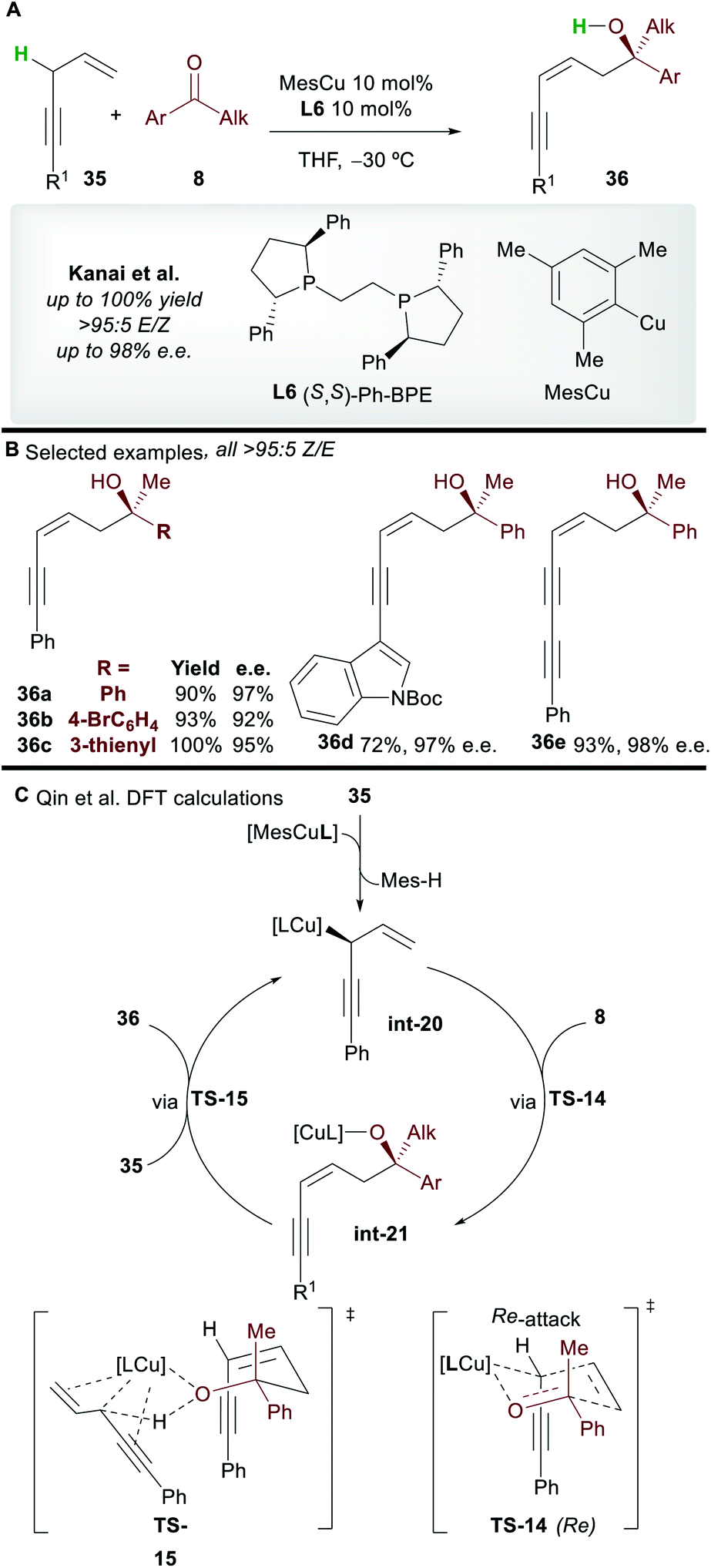
The copper-catalyzed asymmetric addition of 1,4-enynes to ketones was reported by Kanai and co-workers40 in 2017 (Scheme 21A). Alkyl and aryl-substituted 1,4-enynes gave functionalized 1,3-enyne products 36 with complete Z-selectivity, good to excellent enantioselectivity, and in very good yield with the commonly used ligand L6. A wide range of functional groups was tolerated and no base was required; the 1,4-enynes are initially deprotonated by the L6·CuMes complex, and then by the copper-alkoxide species int-21 formed during the catalytic cycle.
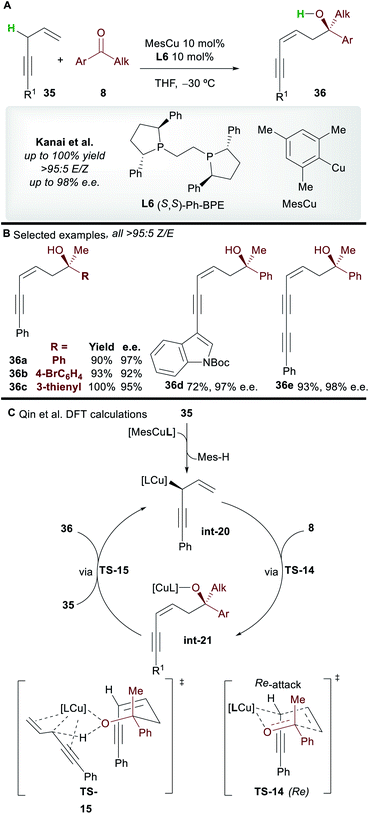 |
| | Scheme 21 Kanai's functionalization of 1,4-enynes with ketones and Qin's subsequent DFT study. | |
The mechanism was studied by Qin and co-workers41 using DFT (Scheme 21C). Calculations showed that pre-catalyst L6·CuMes deprotonates 35, producing the allylcopper int-20. A 6-membered chair-like transition state TS-14, in which the larger phenyl group of the ketone occupies a pseudo-equatorial position and the Re-face of the ketone is attacked is invoked to account for the selectivity of the process. The cycle is closed by deprotonation of 35 by the copper-alkoxide complex int-21viaTS-15. The deprotonation of the enyne viaTS-15 was proposed to be the rate determining step of the process.
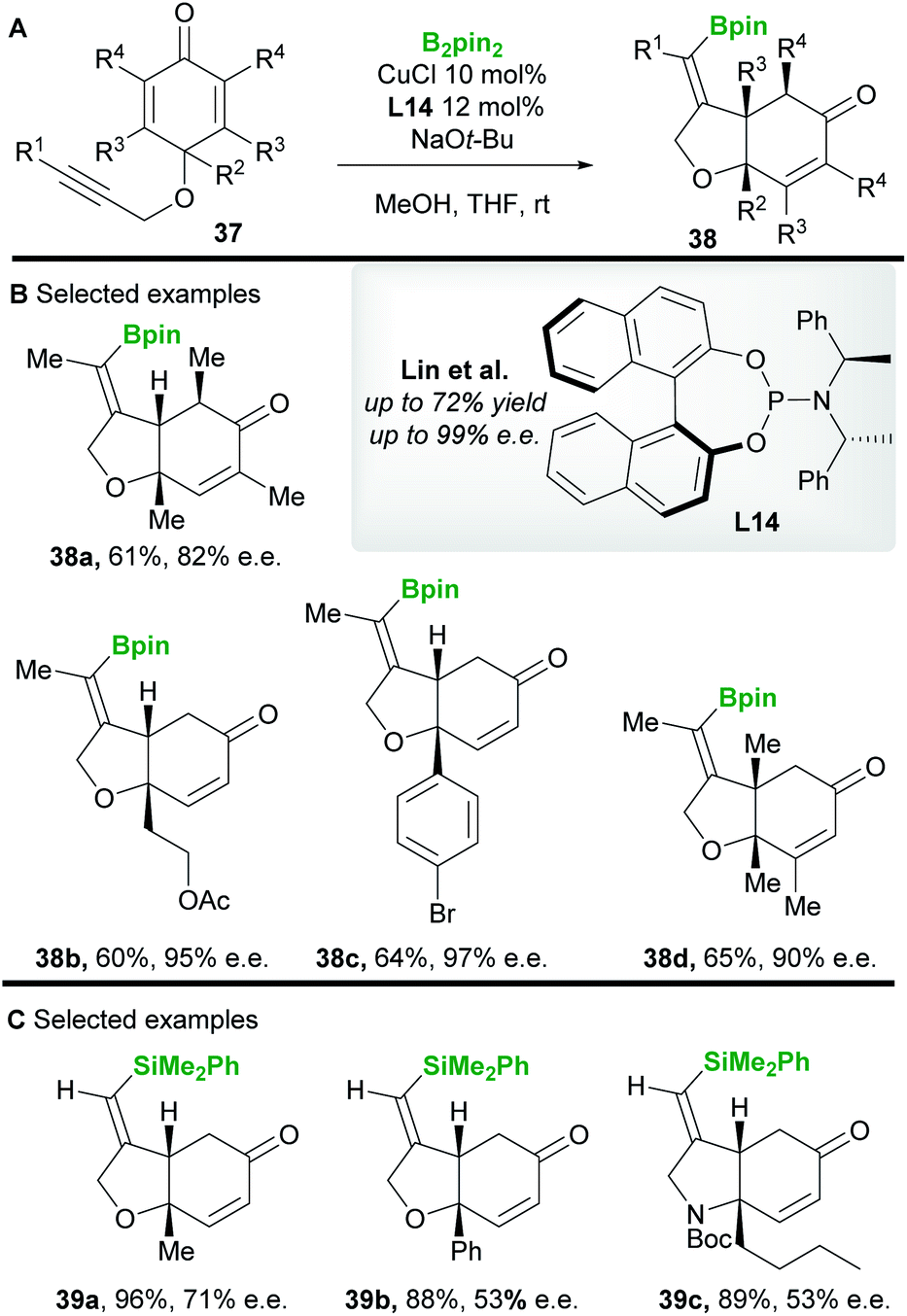
In 2013 Lin and co-workers42 reported a copper-catalyzed asymmetric borylative desymmetrization of cyclohexadienones 37 bearing a 1,6-enyne functional group. The cyclization reactions proceeded with high regio- and enantiocontrol to yield cis-dihydrobenzofuran derivatives 38 (Scheme 22A and B). cis-Products were exclusively obtained in moderate to good yield using the phosphoramidite ligand L14. Competing conjugate borylation is thought to be suppressed by the steric hindrance imposed by the neighbouring (R2) substituent. Furthermore, the regioselectivity of alkyne borylation is thought to arise from the oxygen in the tether, coordinating and directing copper to the β-position of the alkyne. In 2019, the same group43 (Scheme 22C) reported the silylative variant using Suginome's reagent (PhMe2Si-Bpin) and (R,R)-Ph-BPE L5.
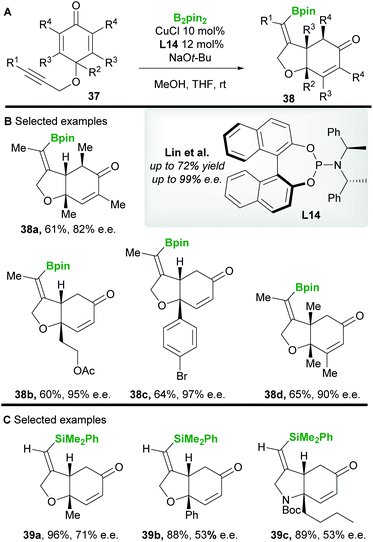 |
| | Scheme 22 Lin's borylative/silylative cyclization of 1,6-enynes. | |
Liu and co-workers44 have developed a copper-catalyzed tandem annulation of 1,6-enynes to give benzo[b]fluorenone derivatives (Scheme 23A). 5-Fluorinated benzo[b]fluorenones 41 were obtained when using tert-butyl-substituted alkynes 40 through a tandem annulation, C–(t-Bu) bond-cleavage and fluorination. On the other hand, aryl and other alkyl bearing 1,6-enynes 40′ exclusively provided 5-aryl-substituted benzo[b]fluorenones 41′ in moderate to excellent yield.
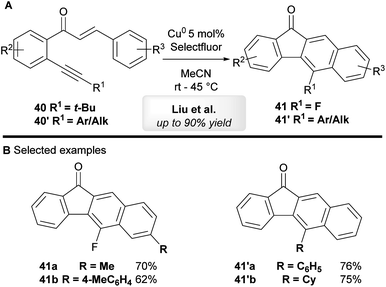 |
| | Scheme 23 Liu's copper-catalyzed cyclization of chalcone derivatives. | |
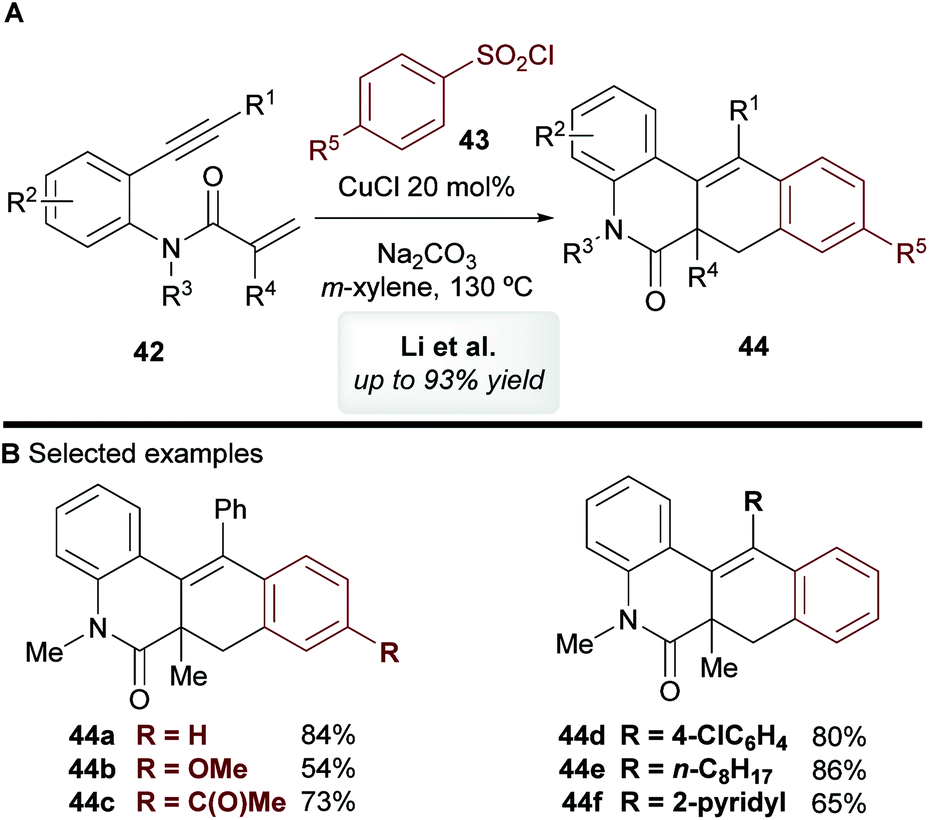
In 2014, Li and co-workers45 disclosed the copper-catalyzed cascade cyclization of 1,7-enynes 42 using sulfonyl chlorides 43 as coupling partners, affording substituted benzo[j]phenanthridin-6(5H)-one scaffolds 44 (Scheme 24). The mechanism of this reaction was not explored by the authors, but they proposed that an aryl radical or aryl-copper species, formed from the aromatic sulfonyl chloride, first reacts at the alkynyl moiety, which triggers a cascade process to deliver products 44
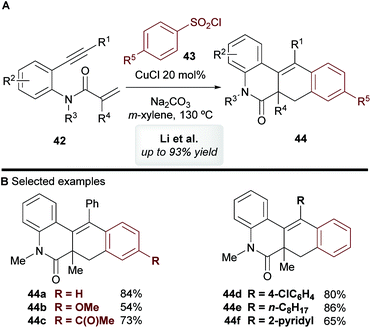 |
| | Scheme 24 Li's cyclization of 1,7-enynes with sulfonyl chlorides. | |
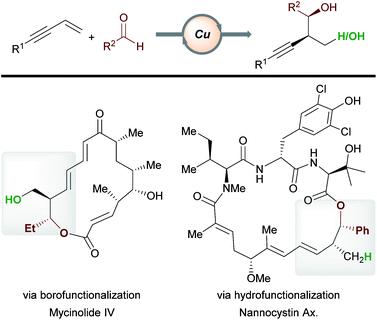 |
| | Scheme 25 Applications in target molecule synthesis. | |
7. Conclusions
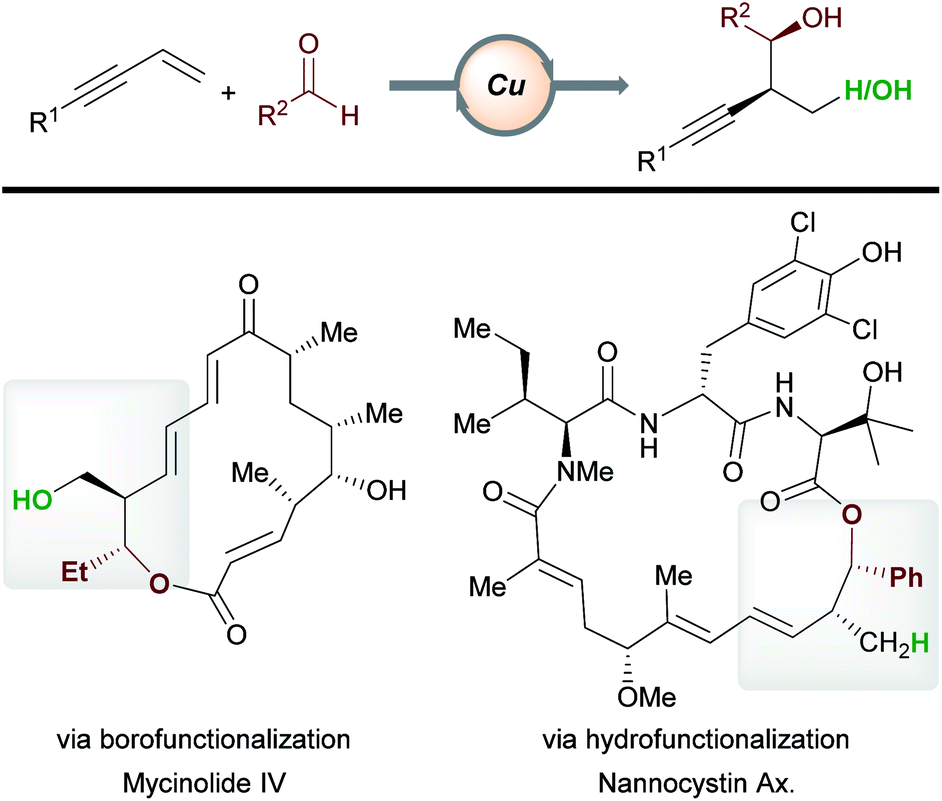
In recent years, copper-catalyzed transformations of readily available enynes have allowed the construction of densely functionalized scaffolds, often in an enantioselective fashion. Due to the ambident nature of enynes and the versatile reactivity of copper, a range of products are accessible by the careful marriage of substrate and catalyst class. We have highlighted examples of processes being used in the functionalization or preparation of bioactive molecules (e.g.Schemes 11, 15 and 16). In addition, copper-catalyzed functionalizations of enynes have begun to find application in total synthesis (Scheme 25). For example, Hoveyda et al.16 have constructed known fragments of tylonolide and mycinolide IV using the enantioselective borofunctionalization of enynes with aldehydes. Similarly, Fürstner, Müller and co-workers utilized a related hydrofunctionalization in an approach to the cytotoxic natural product nannocystin Ax.46 We expect to see further applications of copper-catalyzed enyne functionalization in future target molecule syntheses. To find widespread application, some key challenges in this area need to be overcome. For example, our understanding of, and our ability to predict, the regioselectivity of a given process will aid in synthesis planning. With regard to boro-/hydro-functionalization, methods that expand the scope of effective aromatic enyne substrates are needed, as electron-poor and hindered aromatic enynes are often unsatisfactory substrates (see Schemes 5, 14 and 16). Ultimately, the ability to design more efficient and more powerful processes will be key in a future shaped by sustainable catalysis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Leverhulme Trust (PDRA to Q. D.; RPG-2016-360), the European Union Horizon 2020 (Fellowship to S. M.; Marie Sklodowska-Curie grant 798846-CuCAN), the Royal Government of Thailand (DPST scholarship to W. P.), AstraZeneca (CASE Studentship to F. J. T. T.), EPSRC (Studentship to F. J. T. T. and Established Career Fellowship to D. J. P.; EP/M005062/1), SCI (Scholarship to F. J. T. T.), and the University of Manchester (Lectureship to G. J. P. P.).
Notes and references
-
(a) S. Javanbakht and A. Shaabani, ACS Appl. Bio Mater., 2020, 3, 156–174 CrossRef CAS;
(b) D. Insuasty, J. Castillo, D. Becerra, H. Rojas and R. Abonia, Molecules, 2020, 25, 505 CrossRef;
(c) A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef;
(d) J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Nat. Rev. Drug Discovery, 2018, 17, 709–727 CrossRef.
- B. M. Trost, Angew. Chem., Int. Ed., 1995, 34, 259–281 CrossRef CAS.
- Cu is 4000 times more abundant in the Earth's crust than Pd, see W. M. Haynes, in CRC Handbook of Chemistry and Physics. Section 14: Geophysics, Astronomy, and Acoustics, CRC Press LLC, 97th edn, 2016, ch. Abundance of Elements in the Earth's Crust and in the Sea, p. 17 Search PubMed.
-
Copper is classified as a class 3 impurities in USP, with daily intake 30 times higher than palladium, https://www.usp.org/sites/default/files/usp/document/our-work/chemical-medicines/key-issues/c232-usp-39.pdf Search PubMed.
- For selected examples that illustrate the range of olefins that can be functionalized by copper catalysts, see
(a) M. K. Armstrong, M. B. Goodstein and G. Lalic, J. Am. Chem. Soc., 2018, 140, 10233–10241 CrossRef CAS;
(b) Y. Yang, I. B. Perry and S. L. Buchwald, J. Am. Chem. Soc., 2016, 138, 9787–9790 CrossRef CAS;
(c) A. P. Pulis, K. Yeung and D. J. Procter, Chem. Sci., 2017, 8, 5240–5247 RSC;
(d) G. J. P. Perry, T. Jia and D. J. Procter, ACS Catal., 2020, 10, 1485–1499 CrossRef CAS;
(e) S.-L. Shi, Z. L. Wong and S. L. Buchwald, Nature, 2016, 532, 353–356 CrossRef CAS;
(f) L. Fu, S. Greßies, P. Chen and G. Liu, Chin. J. Chem., 2020, 38, 91–100 CrossRef CAS;
(g) X.-Y. Dong, Y.-F. Zhang, C.-L. Ma, Q.-S. Gu, F.-L. Wang, Z.-L. Li, S.-P. Jiang and X.-Y. Liu, Nat. Chem., 2019, 11, 1158–1166 CrossRef CAS;
(h) T. Jia, M. J. Smith, A. P. Pulis, G. J. P. Perry and D. J. Procter, ACS Catal., 2019, 9, 6744–6750 CrossRef CAS;
(i) W. Xue and M. Oestreich, ACS Cent. Sci., 2020, 6, 1070–1081 CrossRef CAS;
(j) F. J. T. Talbot, Q. Dherbassy, S. Manna, C. Shi, S. Zhang, G. P. Howell, G. J. P. Perry and D. J. Procter, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202007251.
- For selected examples that illustrate the range of functional groups that are accessible by copper catalysis, see
(a) K. Yeung, R. E. Ruscoe, J. Rae, A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 11912–11916 CrossRef CAS;
(b) F. Meng, H. Jang, B. Jung and A. H. Hoveyda, Angew. Chem., Int. Ed., 2013, 52, 5046–5051 CrossRef CAS;
(c) J. Yun, Asian J. Org. Chem., 2013, 2, 1016–1025 CrossRef CAS;
(d) D. Fiorito, Y. Liu, C. Besnard and C. Mazet, J. Am. Chem. Soc., 2020, 142, 623–632 CrossRef CAS;
(e) T. Jia, Q. He, R. E. Ruscoe, A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2018, 57, 11305–11309 CrossRef CAS;
(f) J. Lee, S. Radomkit, S. Torker, J. del Pozo and A. H. Hoveyda, Nat. Chem., 2018, 10, 99–108 CrossRef CAS;
(g) Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681–684 CrossRef CAS.
-
(a) B. M. Trost and J. T. Masters, Chem. Soc. Rev., 2016, 45, 2212–2238 RSC;
(b) E.-i. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979–2018 CrossRef CAS.
-
(a) C. Aubert, O. Buisine and M. Malacria, Chem. Rev., 2002, 102, 813–834 CrossRef CAS;
(b) Y. Hu, M. Bai, Y. Yang and Q. Zhou, Org. Chem. Front., 2017, 4, 2256–2275 RSC;
(c) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef.
-
(a) E. Soriano and I. Fernández, Chem. Soc. Rev., 2014, 43, 3041–3105 RSC;
(b) J. Ye and S. Ma, Org. Chem. Front., 2014, 1, 1210–1224 RSC;
(c)
H. Hopf, in Modern Allene Chemistry, John Wiley & Sons, Ltd, 2008, ch. Allenic Hydrocarbons – Preparation and Use in Organic Synthesis, vol. 185–241 Search PubMed;
(d) S. Ma, Chem. Rev., 2005, 105, 2829–2872 CrossRef;
(e) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595–5698 CrossRef CAS;
(f) K. Lauder, A. Toscani, N. Scalacci and D. Castagnolo, Chem. Rev., 2017, 117, 14091–14200 CrossRef CAS;
(g) H. Qian, D. Huang, Y. Bi and G. Yan, Adv. Synth. Catal., 2019, 361, 3240–3280 CrossRef CAS;
(h)
N. Wachter-Jurcsak and K. A. Conlon, in The Chemistry of Dienes and Polyenes, John Wiley & Sons, Ltd, 2003, ch. Synthetic Applications of Dienes and Polyenes, Excluding Cycloadditions, pp. 693–738 Search PubMed.
-
(a)
N. Krause and A. Hoffmann-Röder, in Modern Allene Chemistry, John Wiley & Sons, Ltd, 2008, ch. Allenic Natural Products and Pharmaceuticals, pp. 997–1040 Search PubMed;
(b)
K. M. Brummond and H. Chen, in Modern Allene Chemistry, John Wiley & Sons, Ltd, 2008, ch. Allenes in Natural Product Synthesis, pp. 1041–1089 Search PubMed;
(c) A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196–1216 CrossRef.
- K. Takahashi, T. Ishiyama and N. Miyaura, Chem. Lett., 2000, 29, 982–983 CrossRef.
- H. Ito, H. Yamanaka, J.-i. Tateiwa and A. Hosomi, Tetrahedron Lett., 2000, 41, 6821–6825 CrossRef CAS.
- D. Hemming, R. Fritzemeier, S. A. Westcott, W. L. Santos and P. G. Steel, Chem. Soc. Rev., 2018, 47, 7477–7494 RSC.
- Y. Sasaki, Y. Horita, C. Zhong, M. Sawamura and H. Ito, Angew. Chem., Int. Ed., 2011, 50, 2778–2782 CrossRef CAS.
-
(a) J. Li, C. Liu, J. He, S. Xu, X. Zhao, Y. Zhu and S. Cao, Org. Chem. Front., 2020, 7, 1495–1501 RSC; For a recent report see
(b) Z. Kuang, H. Chen, J. Qiu, Z. Ou, Y. Lan and Q. Song, Chem, 2020, 6, 2347–2363 CrossRef CAS.
- F. Meng, F. Haeffner and A. H. Hoveyda, J. Am. Chem. Soc., 2014, 136, 11304–11307 CrossRef CAS.
-
(a) T.-M. Huang, R.-H. Hsu, C.-S. Yang, J.-T. Chen, G.-H. Lee and Y. Wang, Organometallics, 1994, 13, 3657–3663 CrossRef CAS;
(b) J.-T. Chen, Y.-K. Chen, J.-B. Chu, G.-H. Lee and Y. Wang, Organometallics, 1997, 16, 1476–1483 CrossRef CAS;
(c) Y. Wakatsuki, H. Yamazaki, Y. Maruyama and I. Shimizu, J. Chem. Soc., Chem. Commun., 1991, 261–263 RSC;
(d) P. W. Blosser, D. G. Schimpff, J. C. Gallucci and A. Wojcicki, Organometallics, 1993, 12, 1993–1995 CrossRef CAS.
-
(a) X.-C. Gan, Q. Zhang, X.-S. Jia and L. Yin, Org. Lett., 2018, 20, 1070–1073 CrossRef CAS;
(b) X.-C. Gan and L. Yin, Org. Lett., 2019, 21, 931–936 CrossRef CAS.
- S. Manna, Q. Dherbassy, G. J. P. Perry and D. J. Procter, Angew. Chem., Int. Ed., 2020, 59, 4879–4882 CrossRef CAS.
- C. Yang, Z.-L. Liu, D.-T. Dai, Q. Li, W.-W. Ma, M. Zhao and Y.-H. Xu, Org. Lett., 2020, 22, 1360–1367 CrossRef CAS.
- Y. Liao, X. Yin, X. Wang, W. Yu, D. Fang, L. Hu, M. Wang and J. Liao, Angew. Chem., Int. Ed., 2020, 59, 1176–1180 CrossRef CAS.
- F.-F. Meng, Q.-Y. Xue, B. Jiang, M. Zhao, J.-H. Xie, Y.-H. Xu and T.-P. Loh, Org. Lett., 2019, 21, 2932–2936 CrossRef CAS.
- C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS.
- W. S. Mahoney, D. M. Brestensky and J. M. Stryker, J. Am. Chem. Soc., 1988, 110, 291–293 CrossRef CAS.
- D. H. Appella, Y. Moritani, R. Shintani, E. M. Ferreira and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 9473–9474 CrossRef CAS.
- B. H. Lipshutz and J. M. Servesko, Angew. Chem., Int. Ed., 2003, 42, 4789–4792 CrossRef CAS.
- Y. Yang, I. B. Perry, G. Lu, P. Liu and S. L. Buchwald, Science, 2016, 353, 144–150 CrossRef CAS.
- Y. Zhou, L. Zhou, L. T. Jesikiewicz, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2020, 142, 9908–9914 CrossRef CAS.
- Y. Huang, J. del Pozo, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2018, 140, 2643–2655 CrossRef CAS.
- L. Bayeh-Romero and S. L. Buchwald, J. Am. Chem. Soc., 2019, 141, 13788–13794 CrossRef CAS.
- S. Yu, H. L. Sang, S.-Q. Zhang, X. Hong and S. Ge, Commun. Chem., 2018, 1, 64 CrossRef.
-
H. Togo, in Advanced Free Radical Reactions for Organic Synthesis, Elsevier Science, 2004, ch. Intermolecular Radical Addition Reactions, pp. 123–156 Search PubMed.
-
(a) Z.-L. Li, G.-C. Fang, Q.-S. Gu and X.-Y. Liu, Chem. Soc. Rev., 2020, 49, 32–48 RSC;
(b) Q.-S. Gu, Z.-L. Li and X.-Y. Liu, Acc. Chem. Res., 2020, 53, 170–181 CrossRef CAS;
(c) F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS;
(d) A. J. Clark, Chem. Soc. Rev., 2002, 31, 1–11 RSC.
-
(a) F. Wang, D. Wang, Y. Zhou, L. Liang, R. Lu, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2018, 57, 7140–7145 CrossRef CAS; For recent reports see
(b) P.-H. Li, Y. Wei and M. Shi, Org. Biomol. Chem., 2020, 18, 7127–7138 RSC;
(c) H. Shen, H. Xiao, L. Zhu and C. Li, Synlett, 2020, 31, 41–44 CrossRef CAS.
- Y. Lan, X.-H. Chang, P. Fan, C.-C. Shan, Z.-B. Liu, T.-P. Loh and Y.-H. Xu, ACS Catal., 2017, 7, 7120–7125 CrossRef CAS.
- X. Zhu, W. Deng, M.-F. Chiou, C. Ye, W. Jian, Y. Zeng, Y. Jiao, L. Ge, Y. Li, X. Zhang and H. Bao, J. Am. Chem. Soc., 2019, 141, 548–559 CrossRef CAS.
- C. Ye, Y. Li, X. Zhu, S. Hu, D. Yuan and H. Bao, Chem. Sci., 2019, 10, 3632–3636 RSC.
-
(a) M. Jacobson, R. E. Redfern, W. A. Jones and M. H. Aldridge, Science, 1970, 170, 542–544 CrossRef CAS;
(b) G. Stork and M. Isobe, J. Am. Chem. Soc., 1975, 97, 4745–4746 CrossRef CAS.
- B. M. Trost and M. Lautens, J. Am. Chem. Soc., 1985, 107, 1781–1783 CrossRef CAS.
- X.-F. Wei, X.-W. Xie, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2017, 139, 4647–4650 CrossRef CAS.
- H. Li, M. Luo, G. Tao and S. Qin, Catalysts, 2018, 8, 359 CrossRef.
-
(a) P. Liu, Y. Fukui, P. Tian, Z.-T. He, C.-Y. Sun, N.-Y. Wu and G.-Q. Lin, J. Am. Chem. Soc., 2013, 135, 11700–11703 CrossRef CAS; For a recent report see
(b) C.-Y. He, Y.-X. Tan, X. Wang, R. Ding, Y.-F. Wang, F. Wang, D. Gao, P. Tian and G.-Q. Lin, Nat. Commun., 2020, 11, 4293 CrossRef CAS.
- C.-Y. He, L.-B. Xie, R. Ding, P. Tian and G.-Q. Lin, Tetrahedron, 2019, 75, 1682–1688 CrossRef CAS.
-
(a) J. Zhang, H. Zhang, D. Shi, H. Jin and Y. Liu, Eur. J. Org. Chem., 2016, 5545–5558 CrossRef CAS; For a recent report see
(b) T.-S. Zhang, W.-J. Hao, P.-J. Cai, G. Li, S.-J. Tu and B. Jiang, Front. Chem., 2020, 8, 234 CrossRef CAS.
- Y. Liu, J.-L. Zhang, M.-B. Zhou, R.-J. Song and J.-H. Li, Chem. Commun., 2014, 50, 14412–14414 RSC.
- Z. Meng, L. Souillart, B. Monks, N. Huwyler, J. Herrmann, R. Müller and A. Fürstner, J. Org. Chem., 2018, 83, 6977–6994 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Fabien J. T.
Talbot
,
Watcharapon
Prasitwatcharakorn
,
Gregory J. P.
Perry
,
Fabien J. T.
Talbot
,
Watcharapon
Prasitwatcharakorn
,
Gregory J. P.
Perry