
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric synthesis of 1,2-fluorohydrin: iridium catalyzed hydrogenation of fluorinated allylic alcohol†
Sudipta
Ponra‡
a,
Jianping
Yang‡
 a,
Haibo
Wu
a,
Wangchuk
Rabten
a and
Pher G.
Andersson
*ab
a,
Haibo
Wu
a,
Wangchuk
Rabten
a and
Pher G.
Andersson
*ab
aDepartment of Organic Chemistry, Stockholm University, Svante Arrhenius väg 16C, SE-10691 Stockholm, Sweden. E-mail: Pher.Andersson@su.se
bSchool of Chemistry and Physics, University of KwaZulu-Natal, Private Bag X54001, Durban, 4000, South Africa
First published on 25th September 2020
Abstract
We have developed a simple protocol for the preparation of 1,2-fluorohydrin by asymmetric hydrogenation of fluorinated allylic alcohols using an efficient azabicyclo thiazole-phosphine iridium complex. The iridium-catalyzed asymmetric synthesis of chiral 1,2-fluorohydrin molecules was carried out at ambient temperature with operational simplicity, and scalability. This method was compatible with various aromatic, aliphatic, and heterocyclic fluorinated compounds as well as a variety of polyfluorinated compounds, providing the corresponding products in excellent yields and enantioselectivities.
Introduction
Despite the limited number of fluorinated natural products, the introduction of a fluorine atom into an organic molecule can have an immense impact in small molecule drug discovery.1 Moreover, the stereocontrolled introduction of fluorine atoms into organic molecules can be expected to be the next chapter for the development of drugs possessing excellent bioactivity and bioavailability.1b,c,e,2 However, current synthetic methodologies lack a waste-free and easy stereoselective approach for the construction of complex bioactive fluorinated molecules. Fluorohydrins3 are well-known as an important subclass of organofluorine compounds. They serve as key intermediates in the synthesis of mono-fluorinated analogues for many bioactive compounds.4 They are also used as derivatizing agents in determining enantiomeric composition by 19F NMR spectroscopy.5 Classic examples of drug molecules containing the 1,2-fluorohydrin substructure are fludrocortisone (the first fluorine-containing marketed drug);6 anti-inflammatory corticosteroid difluprednate7a–c and antihepatitis C agent sofosbuvir.7d Along with steroid and nucleotide analogs a few alkaloid-derived fluorohydrins have also been reported.7e Nucleophilic opening of epoxides with various fluoride sources are one of the most reliable methods for fluorohydrin synthesis. The use of HF-based reagents (e.g., Olah's reagent) as a fluoride source is particularly notable.3b,8 Many of the existing methods for the synthesis of stereoselective fluorohydrins have economical or practical setbacks, often relating to the fluorinating agents. The lack of a general atom economical asymmetric process prompted us to devise a highly enantioselective synthetic method for the versatile generation of fluorohydrin molecules.The asymmetric hydrogenation of olefins using iridium complexes without a coordinating group or a weakly coordinating group is one of the most fundamental and atom economical processes in synthetic organic chemistry.9 For the trisubstituted olefins, the smallest substituent, which is hydrogen, determines which enantioface of the olefin preferentially coordinates to the Ir center. Since, H and F are both small substituents and almost of similar size, there is more than one possible orientation of the olefin for trisubstituted fluorinated allylic alcohols. This presents a significant challenge since it results in enantiomeric mixtures of the hydrogenated product.10 Selectivity together with the frequent occurrence of defluorination (de-F) during hydrogenation and the electron-withdrawing property of fluorine makes trisubstituted fluorinated olefins an even more difficult substrate for hydrogenation.11 Since olefins are inexpensive and widely available it led us to investigate the asymmetric hydrogenation of fluorinated allylic alcohols as a possible method to produce bio-relevant fluorohydrin compounds.12 Recently, we reported the successful development of an efficient catalytic system for the enantioselective synthesis of fluorine motifs based on the highly selective hydrogenation of alkenyl fluorides.10,12d,g Our Ir-N,P catalyst was moderately successful for the asymmetric synthesis of fluorohydrins by hydrogenation, however it was limited to specific substrates12a or gave poor enantioselectivity with a high extent of de-F.12b With these limitations in mind, we set out to develop an efficient catalytic system for the atom economical asymmetric synthesis of fluorohydrins by hydrogenation of fluorinated allylic alcohols (Scheme 1).
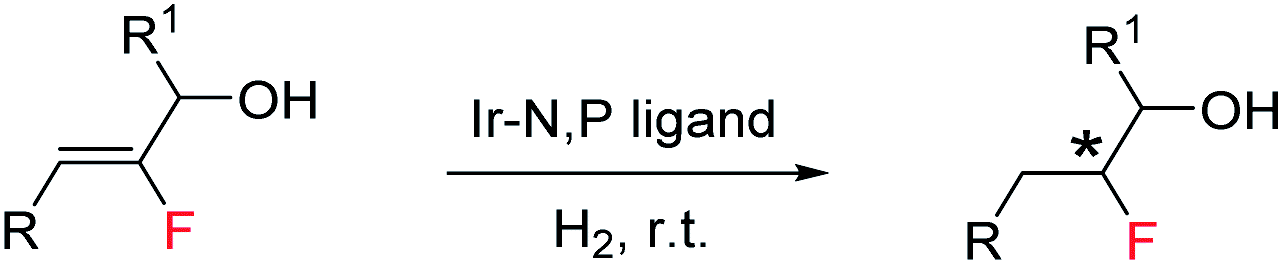 | ||
| Scheme 1 Atom economical synthesis of asymmetric 1,2-fluorohydrin. | ||
Results and discussion
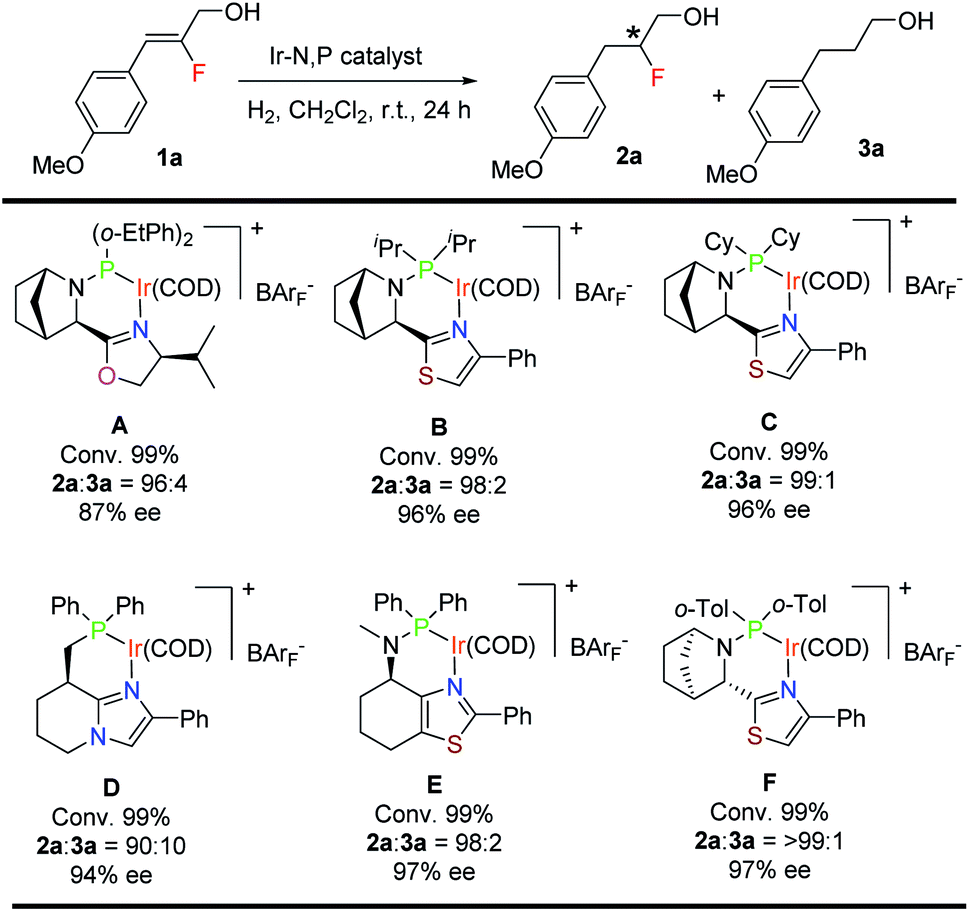
For the asymmetric hydrogenation investigation reported here, we selected (Z)-2-fluoro-3-(4-methoxyphenyl)prop-2-en-1-ol (1a) as the model substrate and bicyclic oxazoline-o-Et-phenylphosphine iridium complex12aA as the catalyst using the following reaction conditions: 1 mol% catalyst, CH2Cl2, and 10 bar H2. Substrate 1a gave 99% conversion of the starting material along with slight de-F (4%) and very good enantioselectivity (87% ee) (Table 1). Catalysts having a thiazole backbone (catalyst B and catalyst C) led to an improved enantioselectivity of 96% ee with very minute de-F (1–2%). Further optimization of the heterocycle skeleton of the N,P-iridium complex involved changing thiazole to imidazole (catalyst D) which provided a comparable result but with 10% de-F. Catalyst E which was superior in our previous study12a on asymmetric hydrogenation of fluorinated allylic alcohol gave 97% ee with 2% de-F (more details, see: ESI†). We finally evaluated the effect of the substituent on phosphorous and replaced the two cyclohexyl groups with ortho-tolyl groups (catalyst F) and obtained the best result for 1a: 99% conversion with excellent enantioselectivity (97%) and <1% of the de-F by-product.| a Reaction conditions: 0.05 mmol of 1a, 1 mol% catalyst, 0.5 mL CH2Cl2, 10 bar H2. Conversion was determined by 1H-NMR spectroscopy. Enantiomeric excess was determined by SFC. |
|---|
|
Having successfully found a new effective catalyst containing a bicyclic thiazole backbone with o-tolyl substituents on phosphorus F, we carried out further optimization of the solvent, catalyst loading and hydrogen pressure (Table 2). Solvent screening with CH2Cl2, DCE and PhCF3 proved that CH2Cl2 was the best in terms of reactivity, enantioselectivity and de-fluorination. When the H2 pressure decreased from 10 bar (entry 1) to 4 bar (entry 4) using 1.0 mol% of catalyst F for 24 hours, conversion was not affected but de-F increased slightly to 2%. A decrease in the catalyst loading at 10 bar of H2 pressure for substrate 1a from 1.0 mol% (entry 1) to 0.5 mol% (entry 5) also increased the de-F to 4%. Hence, for substrate 1a the optimal conditions for full conversion, good enantioselectivity and minimal de-F are a catalyst loading of 1.0 mol% (catalyst F) and 10 bar H2 pressure for 24 hours (entry 1). It should be noted that almost no de-F product (<1%) was observed as a side reaction which is a recurring problem in hydrogenations of alkenyl fluorides.11b
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | H2 (bar) | Catalyst (mol%) | Conversion (%) | de-F (%) | ee (%) |
| a Reaction conditions: 0.05 mmol of 1a, 0.5 mL solvent. Conversion was determined by 1H-NMR spectroscopy. Enantiomeric excess was determined by SFC. | ||||||
| 1 | CH2Cl2 | 10 | 1.0 | 99 | <1 | 97 |
| 2 | DCE | 10 | 1.0 | 99 | 3 | 97 |
| 3 | PhCF3 | 10 | 1.0 | 98 | 3 | 97 |
| 4 | CH2Cl2 | 4 | 1.0 | 99 | 2 | 97 |
| 5 | CH2Cl2 | 10 | 0.5 | 99 | 4 | 97 |
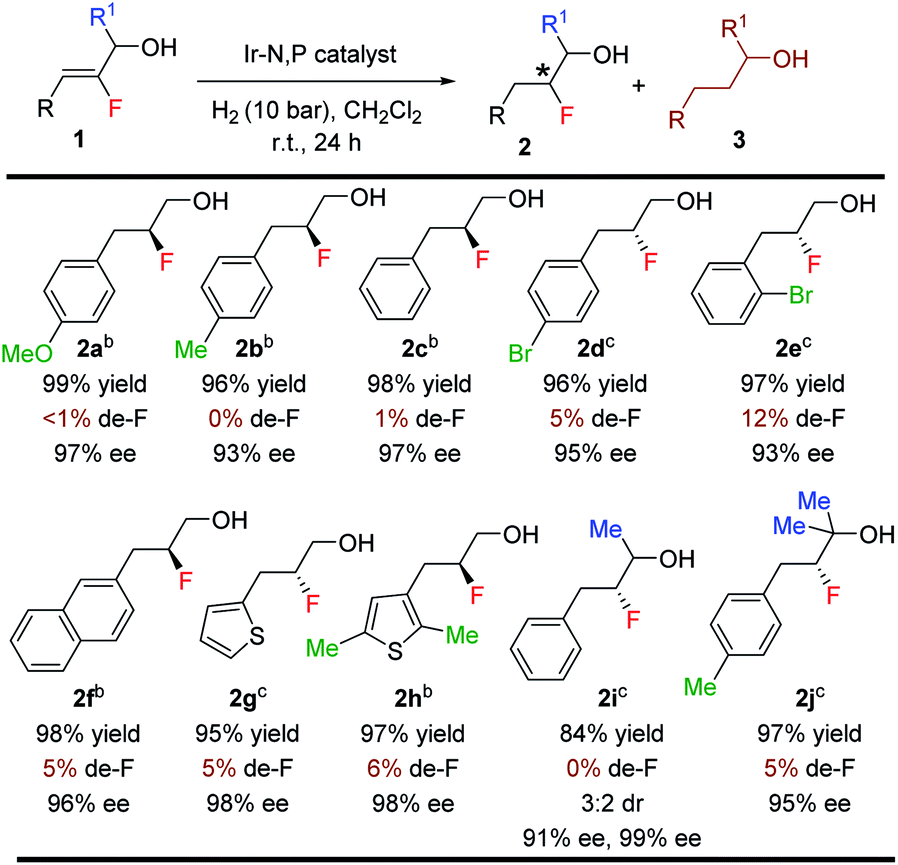
With the optimized reaction conditions established, we evaluated the hydrogenation of variously substituted (Z)-2-fluoro-3-phenylprop-2-en-1-ols 1 (Table 3). A variety of fluorinated allylic alcohols were successfully hydrogenated to generate the desired products 2a–2e in excellent yields and enantioselectivities. The (Z)-2-fluoro-3-phenylprop-2-en-1-ol substrates with either electron-donating or electron-withdrawing substituents at the para or ortho position of the aryl rings provided the desired products in high isolated yields (96–99%) and excellent enantioselectivities (93–97% ee). Replacing the phenyl group with 2-naphthyl or a heterocyclic (thienyl) substituent also yielded the desired products (2f–2h) in excellent yields (95–98%) and ees (96–98%). Next, we examined the secondary and tertiary vinyl-F alcohols and as expected, all gave satisfactory results. Again, very low de-F (0–12%) was observed in all examples, which underlines the effectiveness of this catalytic method.
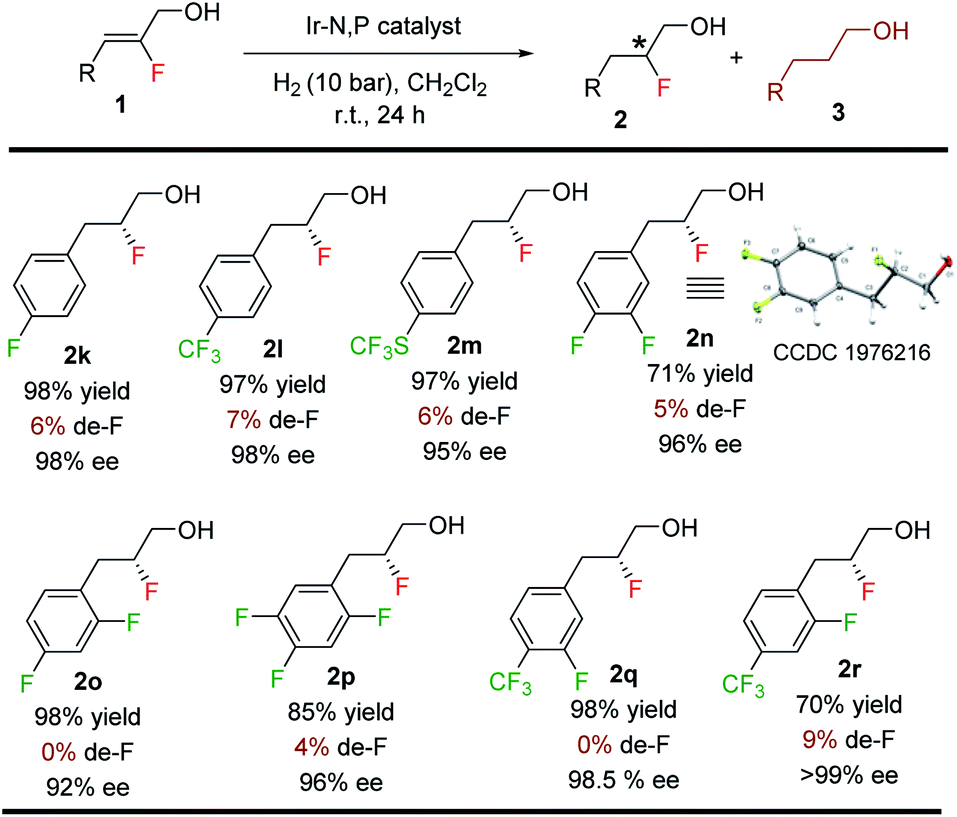
Since de-F during hydrogenation of alkenyl fluorides presents potential challenges,11 various polyfluorinated allylic alcohols were hydrogenated under standard conditions (Table 4). For all examples, irrespective of the position of the fluorine or the trifluoromethyl substituent on the aromatic ring, we observed only traces of de-F products (0–9%) along with high yields (70–99%) and enantioselectivities (95–>99% ee) for the polyfluorinated 1,2-fluorohydrins.
| a Reaction conditions: 0.25 mmol of substrate, 1 mol% of catalyst ent-F, 2.0 mL CH2Cl2. Yields are isolated hydrogenated product. Enantiomeric excess was determined by SFC or GC/MS using chiral stationary phases. |
|---|
|
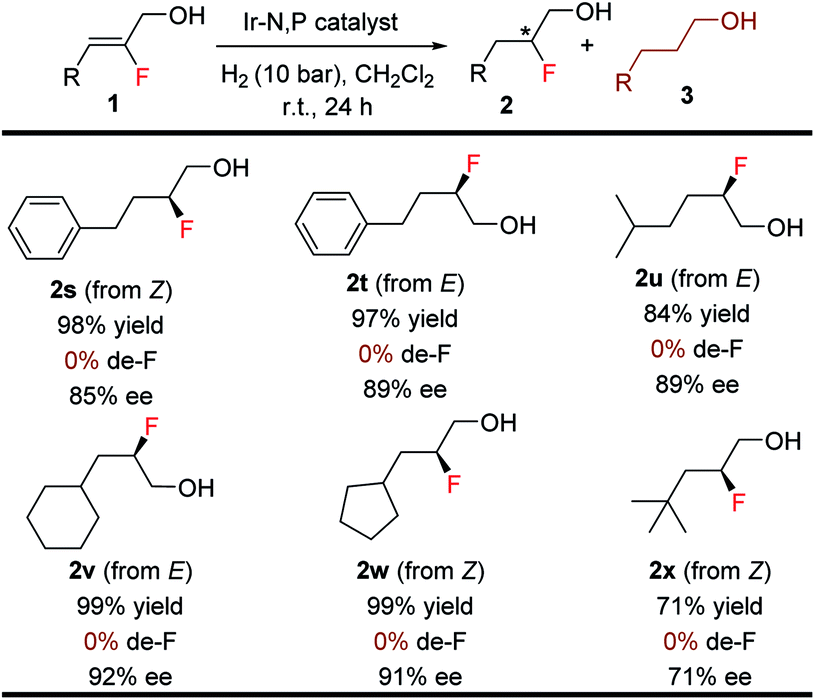
The efficacy of this stereoselective 1,2-fluorohydrin synthesis was investigated by evaluating various aliphatic fluorinated allylic alcohols (Table 5). Gratifyingly, a number of aliphatic fluorinated allylic alcohols (1s–1x) were also efficiently hydrogenated in good to excellent yields (71–99%) with high enantioselectivities (71–92% ee). Interestingly, both isomers (E & Z) of 2-fluoro-4-phenylbut-2-en-1-ol were hydrogenated with excellent results. Various acyclic and cyclic primary, secondary, and tertiary aliphatic substituents afforded the anticipated product (2s–2x) without generating any problematic de-F.
| a Reaction conditions: 0.25 mmol of substrate, 1 mol% catalyst F, 2.0 mL CH2Cl2. Yields are isolated hydrogenated product. Enantiomeric excess was determined by HPLC or GC/MS using chiral stationary phases. |
|---|
|
Notably, this method was amenable for (Z)-3-fluorooct-2-en-1-ol 1y. The hydrogenated product 2y was obtained in good ee (83%), where the chiral center containing fluorine is at the β position to the CH2OH group (Scheme 2).
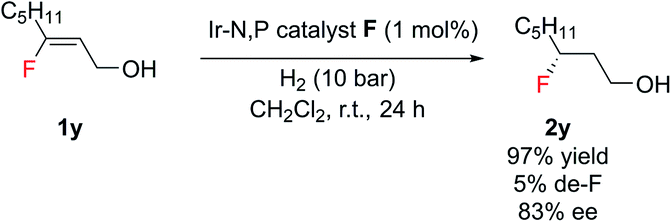 | ||
| Scheme 2 Hydrogenation of (Z)-3-fluorooct-2-en-1-ol. | ||
A preparative-scale production of chiral fluorohydrin under these reaction conditions was also carried out. Starting with 1.0 g of allylic alcohol 1c and using 0.5 mol% of catalyst F under 10 bar H2, the desired fluorohydrin 2c was obtained in excellent 97% yield with 97% ee (Scheme 3).
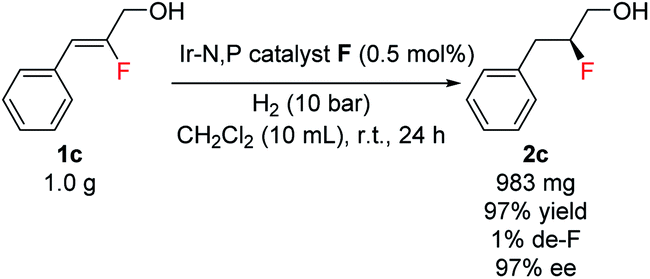 | ||
| Scheme 3 Gram-scale production of chiral fluorohydrin. | ||
The enantioenriched 1,2-fluorohydrins can be transformed into a variety of many useful chiral fluorinated derivatives: acid 4, aldehyde 5, and sulfamate 6 were all obtained without any loss in enantiopurity (Scheme 4). The synthetic intermediate (S)-2-fluoro-3-phenylpropyl sulfamate 6 could be a potential key precursor for the synthesis of dapoxetine13 analogs, a useful selective serotonin reuptake inhibitor (SSRI).
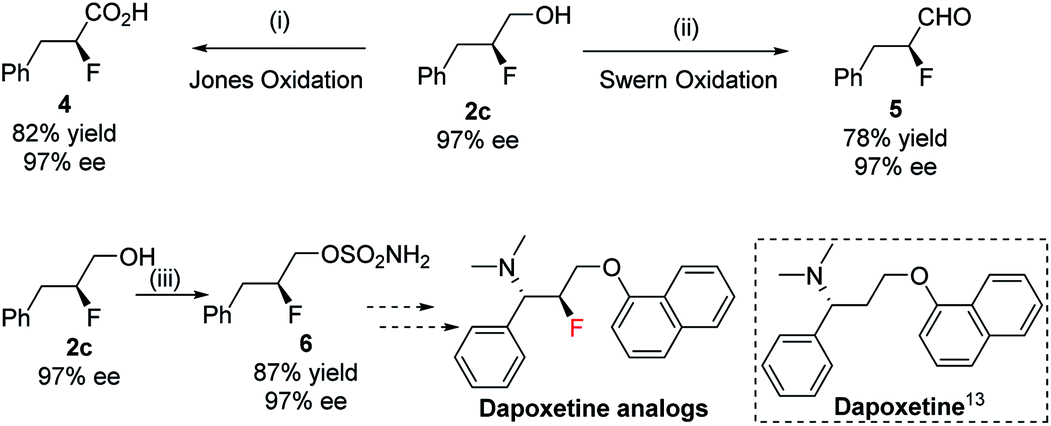 | ||
| Scheme 4 Synthesis of chiral fluorine molecule having different functional groups. Conditions: (i) CrO3, H2SO4, H2O, acetone, 1 h, 0 °C; (ii) (COCl)2, DMSO, TEA, 2 h, −78 to 0 °C; (iii) ClSO2NCO, HCOOH, 0 °C – r.t. | ||
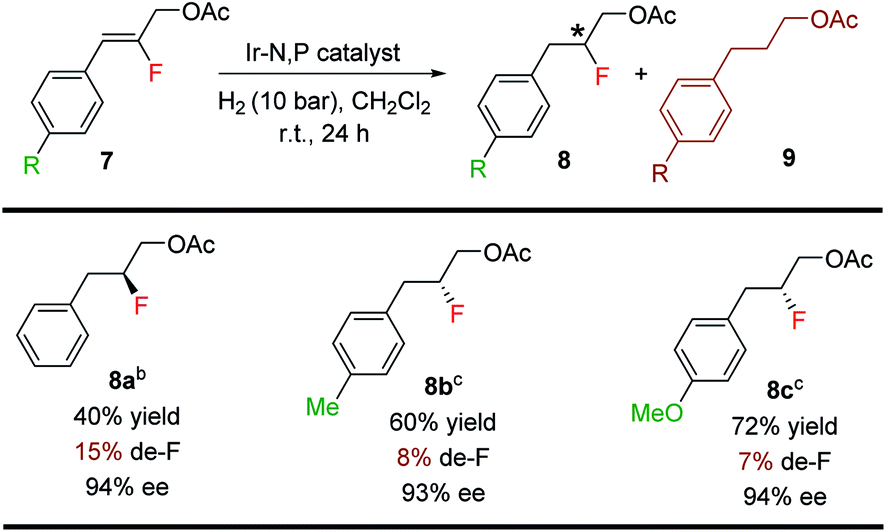
The Ir-complex (F) having the bicyclic thiazole backbone with o-tolyl substituents on phosphorus was also effective for fluorinated allylic acetates (8a–8c) (Table 6), giving good yields (40–72%) and enantioselectivities (93–94% ee), although at a higher level of defluorination.
Steric interactions between the olefin and the N,P-iridium catalyst controls the enantioselectivity of iridium-catalyzed asymmetric hydrogenations.14,15 A quadrant model was developed14 using catalyst F to explain the enantioselectivity and is similar to our earlier observations.10 The Ir-N,P ligand (F) was positioned as depicted in Scheme 5a, with the iridium atom at the center of the four quadrants (Scheme 5b). The olefin (fluorinated allylic alcohol) coordinates vertically trans to phosphorus (Scheme 5c). The phenyl group on the thiazole moiety points outwards, making quadrant iii sterically hindered. One of the o-tolyl groups on the phosphorus atom also points slightly outwards, making quadrant ii the semi-hindered quadrant. Since quadrants i and iv do not experience obstructions from the ligand, they are considered open quadrants. When the trisubstituted (Z)-fluoro alcohol 1c is placed trans to phosphorus, such that the smallest substituent (H) occupies the hindered quadrant iii and the other small substituent (F) occupies the semi-hindered quadrant ii, this results in a favored arrangement (Scheme 5d). Since the H and F atoms are of similar size, another sterically favorable possible arrangement is to place fluorine in the sterically hindered quadrant iii (Scheme 5e). The two other possible arrangements are sterically not favored (Scheme 5f and g). Thus, based on the quadrant model, both sterically favored orientations of fluoro alcohol 1c result in hydrogenation from the same face of the olefin. The predicted absolute configuration of hydrogenated product 2c is S, which is in agreement as measured by the single crystal X-ray structure of similar compound 2n.
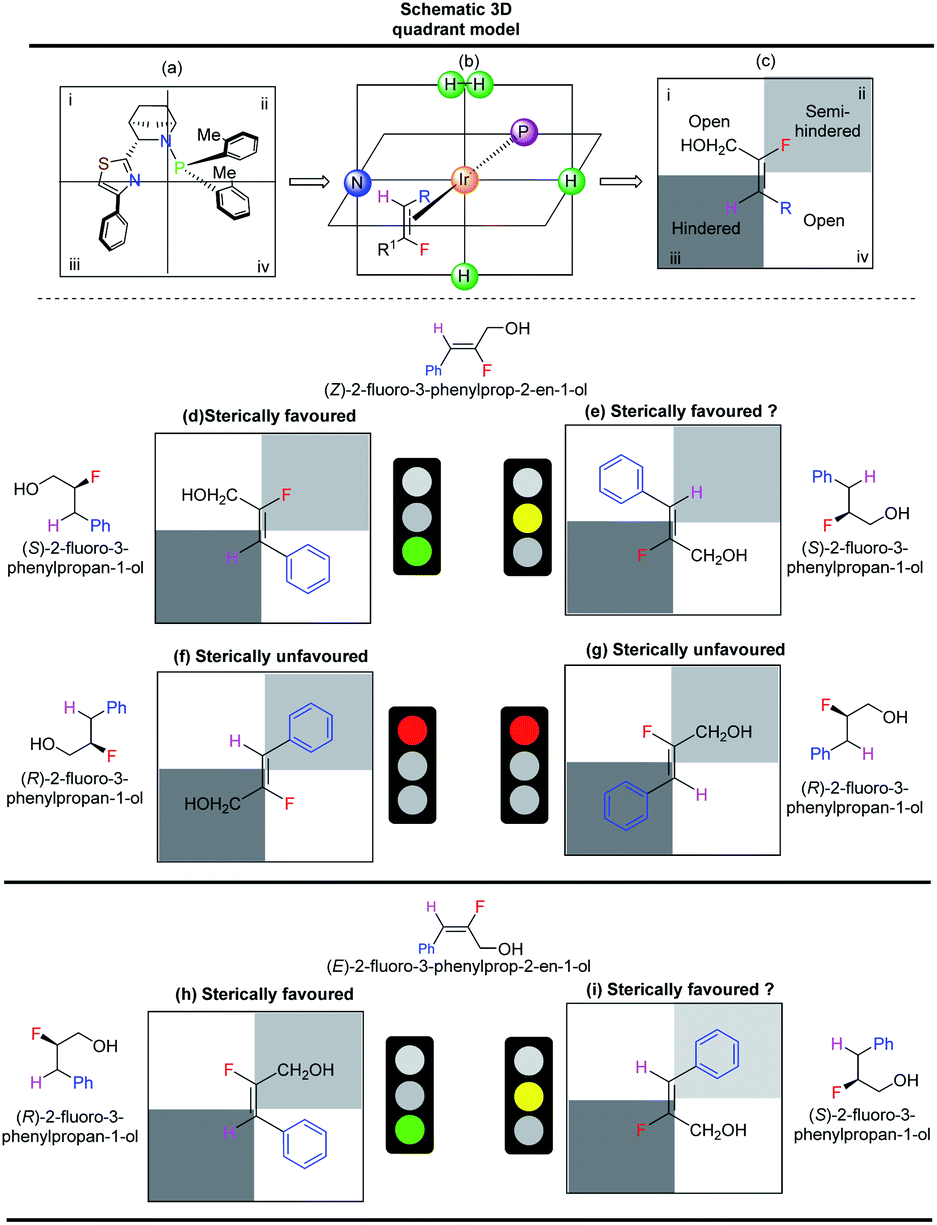 | ||
| Scheme 5 Determination of absolute configuration of olefin 1c using catalyst F. (a–c) General fluorinated allylic alcohol in the selectivity model of the ligand; (d–g) four different products obtained by different possible coordination of Z isomer; (h and i) two different products obtained by different possible coordination of E isomer. | ||
The quadrant model also explains the lower enantioselectivity of the E-isomer of aromatic allylic alcohol. For (E)-fluoro alcohol 1c′, the two sterically favored orientations (Scheme 5h and i) feature coordination from opposite faces, which results in the formation of a mixture of the R and S products.
The enantioselectivity predictions using the quadrant model are in good agreement with the experimental results (Scheme 6) as the (Z)-fluoro alcohol 1c provided product 2c with high enantioselectivity (97% ee) while the E isomer gave the corresponding product 2c in only 65% ee.
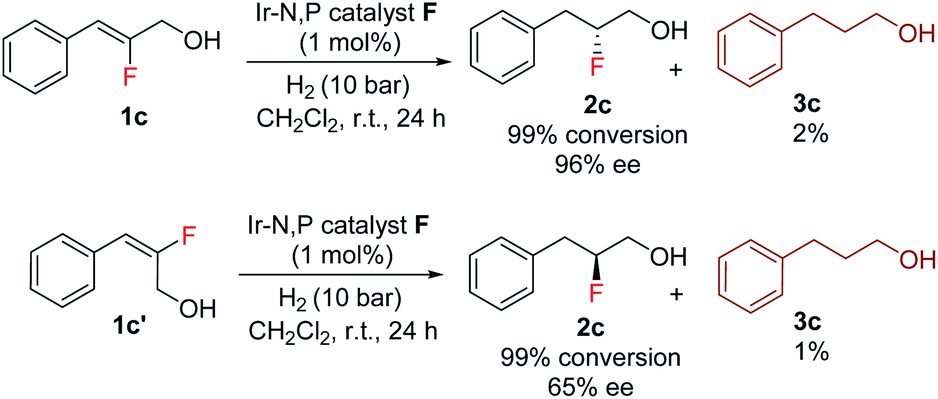 | ||
| Scheme 6 Hydrogenation of both the Z and E isomer of vinyl fluoride olefin 1c. | ||
Conclusions
In summary, we have developed a general and efficient catalyst for the stereoselective synthesis of various fluorohydrins, which complements previous catalytic asymmetric syntheses of 1,2 fluorohydrins. This method is straightforward and also suppresses the problem of defluorination. The catalyst has been used effectively on various aromatic and aliphatic trisubstituted fluorinated allylic alcohols, providing chiral 1,2 fluorohydrins in excellent yields and enantioselectivities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Swedish Research Council (VR), Stiftelsen Olle Engkvist Byggmästare, Knut and Alice Wallenberg Foundation (KAW 2018:0066) supported this work. We thank Dr Thishana Singh, School of Chemistry and Physics, University of Kwazulu-Natal, South Africa, for proofreading and editing the manuscript.Notes and references
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 Search PubMed; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 Search PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 Search PubMed; (d) S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 Search PubMed; (e) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 Search PubMed; (f) S. Dallongeville, N. Garnier, C. Rolando and C. Tokarski, Chem. Rev., 2016, 116, 2–79 Search PubMed.
- (a) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 Search PubMed; (b) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 Search PubMed.
- For reviews, see: (a) P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073–9174 Search PubMed; (b) G. Haufe, J. Fluorine Chem., 2004, 125, 875–894 Search PubMed.
- (a) R. M. Roe, V. Kallapur, R. J. Linderman and F. Viviani, Pestic. Biochem. Physiol., 2005, 83, 140–154 Search PubMed; (b) A. K. Podichetty, S. Wagner, S. Schröer, A. Faust, M. Schäfers, O. Schober, K. Kopka and G. Haufe, J. Med. Chem., 2009, 52, 3484–3495 Search PubMed; (c) M.-Y. Chang, N.-C. Lee, M.-F. Lee, Y.-P. Huang and C.-H. Lin, Tetrahedron Lett., 2010, 51, 5900–5903 Search PubMed; (d) A. J. Cresswell, S. G. Davies, J. A. Lee, P. M. Roberts, A. J. Russell, J. E. Thomson and M. J. Tyte, Org. Lett., 2010, 12, 2936–2939 Search PubMed; (e) A. J. Cresswell, S. G. Davies, J. A. Lee, M. J. Morris, P. M. Roberts and J. E. Thomson, J. Org. Chem., 2011, 76, 4617–4627 Search PubMed.
- (a) E. L. Eliel, S. H. Wilen and L. N. Mander, in Stereochemistry of Organic Compounds, John Wiley & Sons, 1994 Search PubMed; (b) F. Cuevas, P. Ballester and M. A. Pericàs, Org. Lett., 2005, 7, 5485–5487 Search PubMed; (c) S. Rodríguez-Escrich, D. Popa, C. Jimeno, A. Vidal-Ferran and M. A. Pericàs, Org. Lett., 2005, 7, 3829–3832 Search PubMed; (d) M. Apparu, Y. B. Tiba, P.-M. Léo, S. Hamman and C. Coulombeau, Tetrahedron: Asymmetry, 2000, 11, 2885–2898 Search PubMed; (e) Y. Takeuchi, M. Konishi, H. Hori, T. Takahashi, Y. Takeuchi, T. Kometani and K. L. Kirk, Chem. Commun., 1998, 365–366 Search PubMed; (f) S. Hamman, M. Barrelle, F. Tetaz and C. G. Beguin, J. Fluorine Chem., 1987, 37, 85–94 Search PubMed.
- F. Basolo and F. P. Dwyer, J. Am. Chem. Soc., 1954, 76, 1454–1455 Search PubMed.
- (a) L. Mulki and C. S. Foster, Drugs Today, 2011, 47, 327–333 Search PubMed; (b) K. N. Jamal and D. G. Callanan, Clin. Ophthalmol., 2009, 3, 381 Search PubMed; (c) M. Korenfeld, Cataract and Refractive Surgery Today, 2008, 105–107 Search PubMed; (d) A. Yancey, A. Armbruster and S. Tackett, J. Pharm. Technol., 2014, 31, 29–37 Search PubMed; (e) V. Chagnault, M.-P. Jouannetaud, J.-C. Jacquesy and J. Marrot, Tetrahedron, 2006, 62, 10248–10254 Search PubMed.
- (a) Q. Zhang and H. M. Nguyen, Chem. Sci., 2014, 5, 291–296 Search PubMed; (b) G. Haufe and S. Bruns, Adv. Synth. Catal., 2002, 344, 165–171 Search PubMed; (c) G. Haufe, S. Bruns and M. Runge, J. Fluorine Chem., 2001, 112, 55–61 Search PubMed; (d) M. Shimizu and Y. Nakahara, J. Fluorine Chem., 1999, 99, 95–97 Search PubMed; (e) R. Skupin and G. Haufe, J. Fluorine Chem., 1998, 92, 157–165 Search PubMed; for a study on the regioselectivity of the ring opening with different sources of nucleophilic fluoride, see: (f) A. Sattler and G. Haufe, J. Fluorine Chem., 1994, 69, 185–190 Search PubMed; for seminal contributions, see: (g) G. A. Olah, J. T. Welch, Y. D. Vankar, M. Nojima, I. Kerekes and J. A. Olah, J. Org. Chem., 1979, 44, 3872–3881 Search PubMed; (h) G. A. Olah and D. Meidar, Isr. J. Chem., 1978, 17, 148–149 Search PubMed.
- (a) O. Pàmies, P. G. Andersson and M. Diéguez, Chem.–Eur. J., 2010, 16, 14232–14240 Search PubMed; (b) A. Lightfoot, P. Schnider and A. Pfaltz, Angew. Chem., Int. Ed., 1998, 37, 2897–2899 Search PubMed; (c) X. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272–3296 Search PubMed.
- S. Ponra, J. Yang, S. Kerdphon and P. G. Andersson, Angew. Chem., Int. Ed., 2019, 58, 9282–9287 Search PubMed.
- (a) M. Hudlicky, Chemistry of Organic Fluorine Compounds, 2nd edn, Ellis Harwood, Chichester, 1992 Search PubMed; (b) D. M. Sedgwick and G. B. Hammond, J. Fluorine Chem., 2018, 207, 45–58 Search PubMed.
- (a) M. Engman, J. S. Diesen, A. Paptchikhine and P. G. Andersson, J. Am. Chem. Soc., 2007, 129, 4536–4537 Search PubMed; (b) P. Kaukoranta, M. Engman, C. Hedberg, J. Bergquist and P. G. Andersson, Adv. Synth. Catal., 2008, 350, 1168–1176 Search PubMed; (c) A. Stumpf, M. Reynolds, D. Sutherlin, S. Babu, E. Bappert, F. Spindler, M. Welch and J. Gaudino, Adv. Synth. Catal., 2011, 353, 3367–3372 Search PubMed; (d) S. Ponra, W. Rabten, J. Yang, H. Wu, S. Kerdphon and P. G. Andersson, J. Am. Chem. Soc., 2018, 140, 13878–13883 Search PubMed; (e) Z. Han, Y.-Q. Guan, G. Liu, R. Wang, X. Yin, Q. Zhao, H. Cong, X.-Q. Dong and X. Zhang, Org. Lett., 2018, 20, 6349–6353 Search PubMed; (f) Y.-Q. Guan, Z. Han, X. Li, C. You, X. Tan, H. Lv and X. Zhang, Chem. Sci., 2019, 10, 252–256 Search PubMed; (g) S. Kerdphon, S. Ponra, J. Yang, H. Wu, L. Eriksson and P. G. Andersson, ACS Catal., 2019, 9, 6169–6176 Search PubMed.
- S. Kang and H.-K. Lee, J. Org. Chem., 2010, 75, 237–240 Search PubMed.
- (a) T. L. Church, T. Rasmussen and P. G. Andersson, Organometallics, 2010, 29, 6769–6781 Search PubMed; (b) P. Brandt, C. Hedberg and P. G. Andersson, Chem.–Eur. J., 2003, 9, 339–347 Search PubMed; (c) Y. Fan, X. Cui, K. Burgess and M. B. Hall, J. Am. Chem. Soc., 2004, 126, 16688–16689 Search PubMed.
- C. Mazet, S. P. Smidt, M. Meuwly and A. Pfaltz, J. Am. Chem. Soc., 2004, 126, 14176–14181 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, crystallography reports, and analytical data (PDF). CCDC 1976216 crystallographic data for 2n (CIF). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04032k |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |