
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLDL-mimetic lipid nanoparticles prepared by surface KAT ligation for in vivo MRI of atherosclerosis†
Alessandro
Fracassi
 a,
Jianbo
Cao
b,
Naoko
Yoshizawa-Sugata
c,
Éva
Tóth
d,
Corey
Archer
e,
Olivier
Gröninger
f,
Emanuela
Ricciotti
g,
Soon Yew
Tang
g,
Stephan
Handschin
h,
Jean-Pascal
Bourgeois
i,
Ankita
Ray
a,
Korinne
Liosi
a,
Sean
Oriana
a,
Wendelin
Stark
f,
Hisao
Masai
c,
Rong
Zhou
*b and
Yoko
Yamakoshi
*a
a,
Jianbo
Cao
b,
Naoko
Yoshizawa-Sugata
c,
Éva
Tóth
d,
Corey
Archer
e,
Olivier
Gröninger
f,
Emanuela
Ricciotti
g,
Soon Yew
Tang
g,
Stephan
Handschin
h,
Jean-Pascal
Bourgeois
i,
Ankita
Ray
a,
Korinne
Liosi
a,
Sean
Oriana
a,
Wendelin
Stark
f,
Hisao
Masai
c,
Rong
Zhou
*b and
Yoko
Yamakoshi
*a
aLaboratorium für Organische Chemie, ETH Zürich, Vladimir-Prelog-Weg 3, CH-8093 Zürich, Switzerland. E-mail: yamakoshi@org.chem.ethz.ch
bDepartment of Radiology, Institute for Translational Medicine and Therapeutics, University of Pennsylvania, John Morgan 198, 3620 Hamilton Walk, Philadelphia, PA19104, USA. E-mail: rongzhou@pennmedicine.upenn.edu
cDepartment of Genome Medicine, Tokyo Metropolitan Institute of Medical Science, 2-1-6 Kamikitazawa, Setagaya, Tokyo 156-8506, Japan
dCentre de Biophysique Moléculaire, CNRS UPR 4301, Université dOrléans, Rue Charles Sadron, 45071 Orléans, Cedex 2, France
eInstitut für Geochemie und Petrologie, ETH Zürich, Clausiusstrasse 25, CH-8092 Zürich, Switzerland
fInstitute for Chemical and Bioengineering, ETH Zurich, Vladimir-Prelog-Weg 1, CH-8093 Zurich, Switzerland
gDepartment of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, 3400 Civic Center Boulevard, Philadelphia, PA19104, USA
hScientific Center for Optical and Electron Microscopy, ETH Zürich, Auguste-Piccard-Hof 1, Zürich, CH-8093, Switzerland
iUniversity of Applied Science and Arts Western Switzerland, Bd de Pérolles 80, CH-1700 Fribourg, Switzerland
First published on 7th October 2020
Abstract
Low-density lipoprotein (LDL)-mimetic lipid nanoparticles (LNPs), decorated with MRI contrast agents and fluorescent dyes, were prepared by the covalent attachment of apolipoprotein-mimetic peptide (P), Gd(III)-chelate (Gd), and sulforhodamine B (R) moieties on the LNP surface. The functionalized LNPs were prepared using the amide-forming potassium acyltrifluoroborate (KAT) ligation reaction. The KAT groups on the surface of LNPs were allowed to react with the corresponding hydroxylamine (HA) derivatives of P and Gd to provide bi-functionalized LNPs (PGd-LNP). The reaction proceeded with excellent yields, as observed by ICP-MS (for B and Gd amounts) and MALDI-TOF-MS data, and did not alter the morphology of the LNPs (mean diameter: ca. 50 nm), as shown by DLS and cryoTEM analyses. With the help of the efficient KAT ligation, a high payload of Gd(III)-chelate on the PGd-LNP surface (ca. 2800 Gd atoms per LNP) was successfully achieved and provided a high r1 relaxivity (r1 = 22.0 s−1 mM−1 at 1.4 T/60 MHz and 25 °C; r1 = 8.2 s−1 mM−1 at 9.4 T/400 MHz and 37 °C). This bi-functionalized PGd-LNP was administered to three atherosclerotic apoE−/− mice to reveal the clear enhancement of atherosclerotic plaques in the brachiocephalic artery (BA) by MRI, in good agreement with the high accumulation of Gd in the aortic arch as shown by ICP-MS. The parallel in vivo MRI and ex vivo studies of whole mouse cryo-imaging were performed using triply functionalized LNPs with P, Gd, and R (PGdR-LNP). The clear presence of atherosclerotic plaques in BA was observed by ex vivo bright field cryo-imaging, and they were also observed by high emission fluorescent imaging. These directly corresponded to the enhanced tissue in the in vivo MRI of the identical mouse.
Introduction
Nanoparticles (NPs) are one of the most promising vehicles for delivery of drugs and imaging probes, and increasing numbers of NPs have recently been approved by the FDA or are currently under clinical trial.1 Using the NP platform, larger amounts of imaging probes and drugs can be administered with a small volume in vivo.2 By engineering the NP surface with disease-specific ligands, the local concentration of imaging probes can be increased by promoted accumulation of NPs to highlight the diseased tissue. Especially in the case of MRI contrast agents (MRI-CAs), NP-based Gd(III)-chelates generally reveal higher r1 relaxivity in comparison to the small molecule MRI-CAs due to the decreased tumbling rate of Gd(III)-chelates, which are bound on the NP surface.2–5 For the development of such NP-based biomaterials, efficient and biocompatible surface functionalization reactions, which work under physiological conditions, are highly demanded.6,7 Such reactions should not cause either decomposition or aggregation of NPs, and ideally proceed in an orthogonal manner under diluted conditions to be applicable for the attachment of biomolecules (e.g. unprotected peptide) which are often available only as diluted solutions due to their limited supply and/or insolubility.In previous studies, NP surface functionalization was performed using chemical reactions such as Michael addition, Cu(I)-catalyzed azide–alkyne cycloaddition, and Cu-free strain-promoted click reaction.8–14 To complement to these reactions, in this study, we used a recently reported potassium acyltrifluoroborate (KAT) ligation.15,16 This amide-forming reaction between a KAT derivative and a hydroxylamine (HA) derivative was thought to be suitable for NP functionalization because of the following reasons: (1) the reaction can be performed under biorelevant conditions (in buffer, at near neutral pH, and at room temperature) with fast kinetics even under highly diluted conditions. (2) Only biocompatible reagents were involved in the reaction without providing any toxic side products.17 (3) The reaction works in an orthogonal manner and is applicable in the conjugation of biomolecules such as unprotected peptides.16 So far, KAT ligation has been mostly used in homogeneous reactions such as peptide–peptide conjugation and has not been used in non-homogeneous systems, except for our recent preliminary result of the surface functionalization of NPs with a fluorescent molecule.18
In this study, by the use of this KAT ligation, we prepared a low-density lipoprotein (LDL)-mimetic NP-type MRI-CA for the in vivo imaging of atherosclerotic plaques. In previous studies, we reported a natural LDL-based MRI-CA for efficient contrast enhancement of atheroplaques.19,20 The LDL, isolated from human blood, was modified with a synthesized Gd(III)-chelate derivative by intercalation into the lipid monolayer. The obtained LDL-based MRI-CA, with a payload of ca. 200 Gd(III)-chelates per particle, successfully visualized atherosclerotic plaques by in vivo MRI of apoE−/− mice. However, in that natural LDL system, there were several disadvantages, such as infection risk and limited supply.
To make this system easily accessible, we planned to develop a synthetic LDL-mimetic lipid nanoparticle (LNP) by functionalizing the LNP core with an apolipoprotein-mimetic peptide. LNPs are biocompatible, inexpensive, and relatively easy to produce and handle, and can mimic the lipid core of LDL. They have been intensively used as a delivery vehicle for drugs and imaging probes, which were often entrapped in their hydrophobic cores,21–23 and have also been used in recent studies on the delivery of proteins and nucleic acids (such as siRNA).24–26
Based on our preliminary results on LNP surface functionalization with fluorescein by KAT ligation,18 in the present study, we prepared a synthetic LDL-mimetic LNP. To mimic apolipoprotein in LDL (apoB100) with a large MW (514 kDa), a peptide motif with an LDL receptor-binding site27–29 was used. HA derivatives of this peptide (P), Gd(III)-chelate (Gd), and sulforhodamine B (R) were synthesized and subjected to the KAT ligation with the LNP core having KAT moieties (LNP-KAT) on the surface to provide covalently functionalized LNPs with peptide and imaging probes (PGd-LNP or PGdR-LNP) (Fig. 1). With the help of efficient KAT ligation, a high payload of MRI probes (ca. 2800 Gd(III)-chelates per particle) was successfully achieved providing a high r1 relaxivity (r1 = 22.0 or 8.2 s−1 mM−1 at 1.4 T/60 MHz (25 °C) or 9.4 T/400 MHz (37 °C)) and resulting in a strong enhancement of the atherosclerotic plaques in the MRI of the mouse model. The details of the preparation and characterization of the functionalized LNPs and results of in vivo MRI and ex vivo analyses are described below.
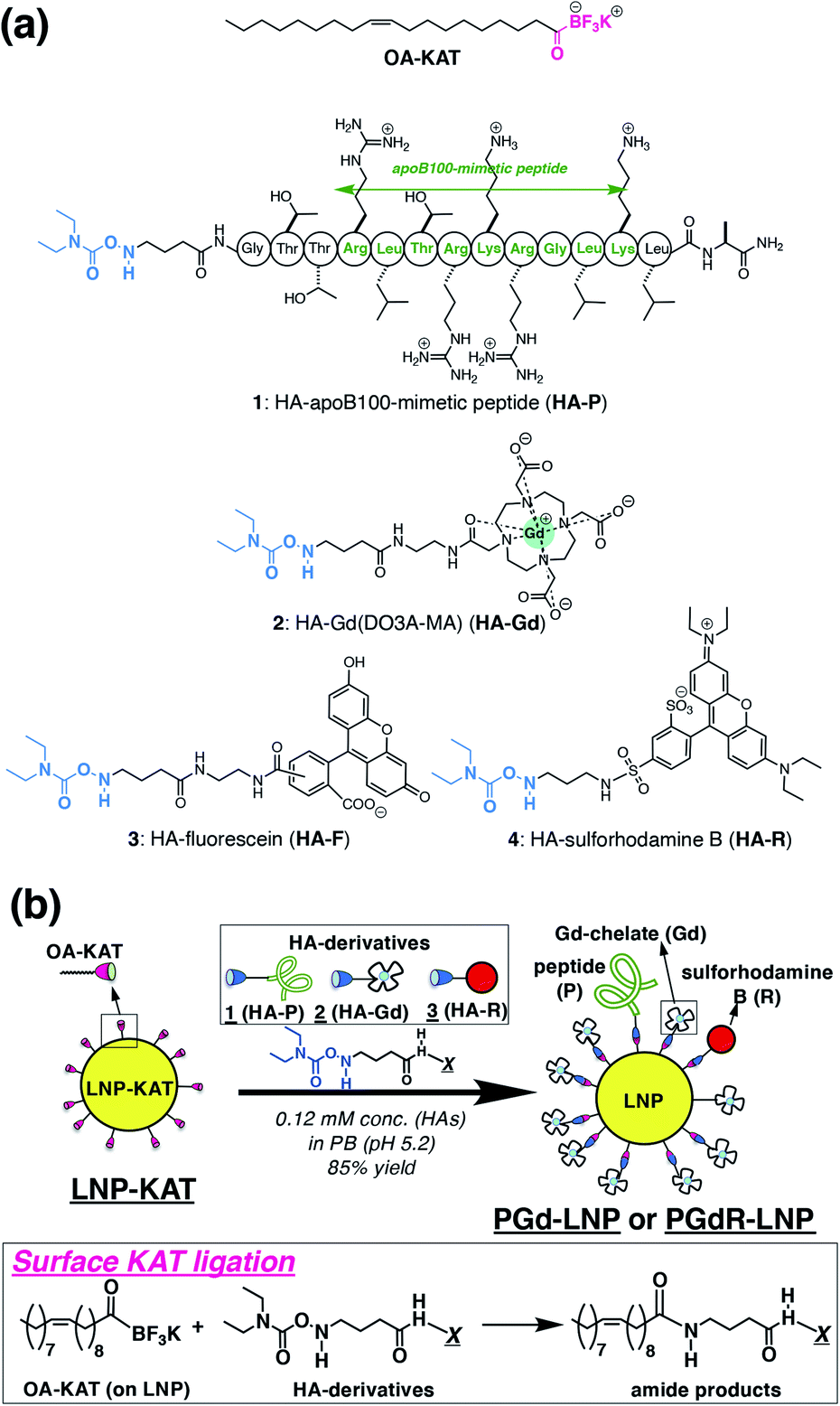 | ||
| Fig. 1 (a) Chemical structures of OA-KAT and hydroxylamine derivatives; HA-apoB100-mimetic peptide (HA-P (1)), HA-Gd(DO3A-MA) (HA-Gd (2)), and HA-sulforhodamine B (HA-R (3)). (b) Schematic illustration for surface KAT ligation of the LNP-KAT nanoparticle for the preparation of the multi-functionalized LNPs (PGd-LNP or PGdR-LNP). Reactions proceeded under diluted conditions (0.12 mM) in the presence of 1.2 equiv. (in total) of HA-derivatives in pH 5.2 phosphate buffer at room temperature. | ||
Results and discussion
Synthesis of HA derivatives of a peptide (P), an MRI-CA (Gd), and a fluorescent probe (R)
Three types of HA derivatives 1–3 were synthesized. HA-P (1), having a known binding motif to the LDL receptor in the apoB100 protein, was synthesized as an apolipoprotein-mimetic by a standard solid phase peptide synthesis (Scheme S1 in the ESI†). The synthesized N-Boc-HA-peptide on resin (S3) was treated with TFA and TIPS, for simultaneous deprotection of the N-Boc group of the HA moiety on the N-terminus, deprotection of the amino acid side chains on the peptide, and cleavage of the whole peptide from resin, to successfully provide an all-deprotected HA-P (1).30 The obtained 1 was purified by HPLC (Fig. S1 in the ESI†). The N-Boc precursor for HA-Gd(DO3A-monoamide) (or HA-Gd(DO3A-MA), HA-Gd (2)) was synthesized from cyclen via several synthetic intermediates (Scheme S3†). The N-Boc precursor of HA-sulforhodamine B (HA-R (3)) was synthesized as previously reported.31 These N-Boc HA derivatives (precursors for 2 and 3) were purified by HPLC and treated with TFA for the N-Boc deprotection immediately before subjecting them to surface KAT ligation with 5% or 10% LNP-KAT.Preparation of LNP-KAT and surface KAT ligation
The platform LNP-KAT particle was prepared from a mixture of three commercially available lipids (phophatidylcholine (PC), triolein (TO), and cholesteryl oleate (CO)) and one synthetic lipid (OA-KAT, 5 or 10 mol% of total lipids) as previously reported (Section 2.1 in the ESI†).18 Briefly, a mixture of the four lipids above (Fig. S31 in the ESI†) was treated under sonication and extrusion in pH 8.0 Tris–HCl buffer to form LNP-KAT (5 or 10%).32 The obtained LNP-KAT dispersion was washed by spin filtration (50 kDa MW cut-off) to remove the free lipid molecules – which were not incorporated into the NPs – and stored in pH 8.0 Tris–HCl buffer (with 10 mM of KF) as a stable dispersion (Fig. S36†).The surface KAT ligation of LNP-KAT and a mixture of HA derivatives (Fig. 1b) was carried out in pH 5.2 phosphate buffer due to its known faster reaction kinetics at lower pH.17 Particularly in this study, nanoparticle aggregation was suspected due to the electrostatic interaction of the negatively charged LNP-KAT surface and positively charged peptide (HA-P, Fig. 1a),33 and the reaction was carried out under diluted conditions (120 μM of HA reagents) with a minimum amount of HA reagents (in total 1.2 equiv. relative to the OA-KAT molecules incorporated into the LNP-KAT particle) at room temperature. For the MRI tests, a bi-functionalized PGd-LNP with peptide (P) and Gd(DO3AMA) (Gd) was prepared by the reaction of LNP-KAT (5%) with a mixture of HA-P (1) and HA-Gd (2). For both MRI and ex vivo cryo-fluorescence imaging, a triply functionalized PGdR-LNP was prepared by the addition of a mixture of 1, 2, and HA-R (3). The reaction was carried out simply mixing the HA derivatives with LNP-KAT (10%) in buffer at room temperature. After surface KAT ligation, each obtained LNP dispersion was thoroughly washed with PBS(−) by spin-filtration (MW cutoff: 50 kDa) to carefully remove the unreacted and non-specifically-bound HA derivatives 1–3 from the particle surface of PGd-LNP or PGdR-LNP before subjecting it to the following particle characterization and in vivo tests.
Reaction efficacy of surface KAT ligation
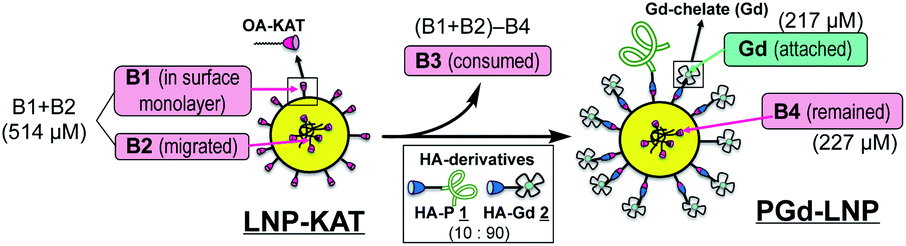
The reaction yield of surface KAT ligation was estimated by B and Gd quantification in LNP-KAT (5%) (starting material) and PGd-LNP (product) by ICP-MS analysis (Table 1). In the ligation reaction, a mixture of 1 and 2 was added in a mole ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90. ICP-MS analyses of the particles were carried out after digestion by HNO3. As shown in Table 1, the B contents in LNP-KAT (5%) and PGd-LNP were 514 and 227 μM suggesting that ca. 56% of the KAT group in LNP-KAT (B3 in the bottom figure) was consumed during the reaction. This result suggested that a sufficient number (more than half) of OA-KAT molecules were available for the reaction, presumably being embedded in the surface of the LNP-KAT particle with the help of amphiphilicity of the OA-KAT molecule, and efficiently exposed to the HA reactants in the external bulk solution. From the consumed B amount (B3 = (B1 + B2) − B4, 287 μM) and the gained Gd content (217 μM) in the reaction, taking into account that the HA reactant was a mixture of 1 and 2 (10:90), the conversion yield of surface KAT ligation was calculated to be 84% ((Gd × 100/90)/B3 × 100). The result indicated that the surface KAT ligation of the LNP-KAT (5%) particle proceeded in a sufficiently high yield under a diluted concentration (120 μM) and with a minimum amount of HA molecules (1.2 equiv.) without having side reactions. To confirm that these changes of B and Gd contents corresponded to the covalent reaction, the detection of ligation products was attempted by MALDI-MS as described below.
:90. ICP-MS analyses of the particles were carried out after digestion by HNO3. As shown in Table 1, the B contents in LNP-KAT (5%) and PGd-LNP were 514 and 227 μM suggesting that ca. 56% of the KAT group in LNP-KAT (B3 in the bottom figure) was consumed during the reaction. This result suggested that a sufficient number (more than half) of OA-KAT molecules were available for the reaction, presumably being embedded in the surface of the LNP-KAT particle with the help of amphiphilicity of the OA-KAT molecule, and efficiently exposed to the HA reactants in the external bulk solution. From the consumed B amount (B3 = (B1 + B2) − B4, 287 μM) and the gained Gd content (217 μM) in the reaction, taking into account that the HA reactant was a mixture of 1 and 2 (10:90), the conversion yield of surface KAT ligation was calculated to be 84% ((Gd × 100/90)/B3 × 100). The result indicated that the surface KAT ligation of the LNP-KAT (5%) particle proceeded in a sufficiently high yield under a diluted concentration (120 μM) and with a minimum amount of HA molecules (1.2 equiv.) without having side reactions. To confirm that these changes of B and Gd contents corresponded to the covalent reaction, the detection of ligation products was attempted by MALDI-MS as described below.
| Content of B [μM] | Content of Gd [μM] | Consumed B [%] | Reaction yield [%] | Conversion yield [%] | |
|---|---|---|---|---|---|
| a Total amount of B in the LNP-KAT (5%) before the reaction (B1 + B2). b Remaining amount of B in PGd-LNP after the reaction (B4). c Amount of Gd in the PGd-LNP after the reaction. d Calculated by (B1 + B2) − B4. e Reaction yield from Gd amount in PGd-LNP relative to the total amount of B in LNP-KAT (Gd/(B1 + B2) × 100). f Conversion yield from Gd amount in PGd-LNP relative to consumed B during the reaction (Gd/B3 × 100). | |||||
| LNP-KAT | 514a | — | — | — | — |
| PGd-LNP | 227 ± 20b | 217 ± 5.2c | 56 ± 3.8d | 47 ± 1.0e | 85 ± 6.8f |
|
|||||
Detection of ligation products in PGd-LNP by MALDI-MS
MALDI-MS analysis of PGd-LNP was employed for the detection of the ligation products (OA-peptide and OA-Gd(DO3A-MA)). An aliquot of PGd-LNP dispersion was analyzed using HCCA (α-cyano-4-hydroxycinnamic acid) as a matrix, and molecular ion peaks corresponding to the ligation products were clearly observed (Fig. 2a–c). Importantly, no trace of HA derivatives 1 and 2 was detected (Fig. 2d and e), indicating that there were no unreacted 1 and 2 that were physically adsorbed on the PGd-LNP surface. These results suggested that the ICP-MS results of the change in B and Gd contents shown in Table 1 corresponded to the covalent surface reaction.34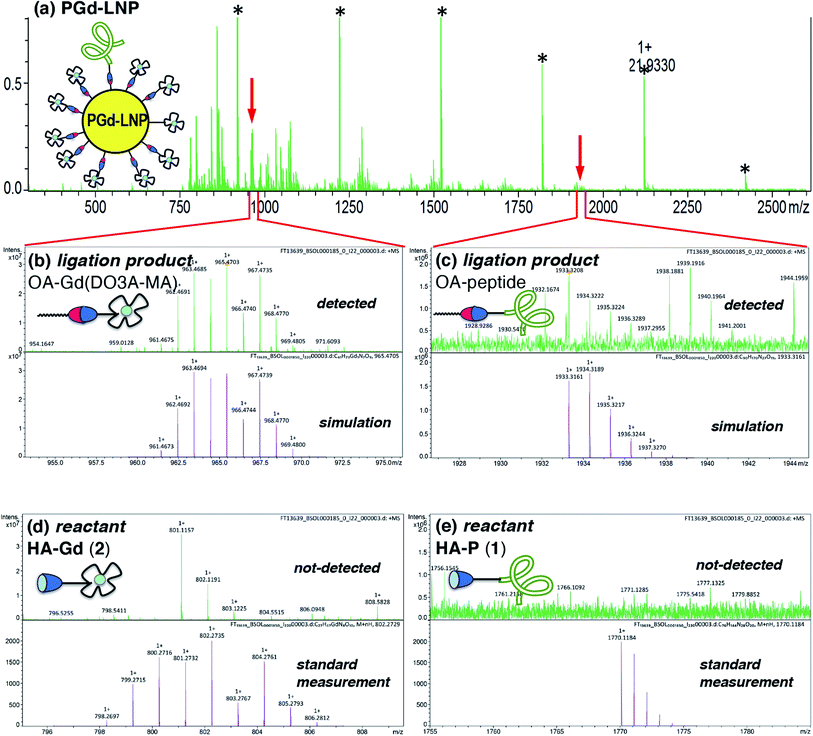 | ||
| Fig. 2 (a) HR-MALDI-MS spectrum of PGd-LNP (matrix: HCCA, *standard for calibration). (b and c) Expansion at the molecular ion peak of OA-Gd(DO3A-MA) (m/z: [M + H]+ found, 965.4703) (b) and OA-peptide (m/z: [M + H]+ found, 1933.3208) (c) with simulation. (d and e) Expansion around the molecular ion peaks of HA-Gd(DO3A-MA) 2 (d), and HA-peptide 1 (e) with standard measurements of 2 (m/z: [M + H]+ found, 802.2757) and 1 (m/z: [M + H]+ found, 1770.1185). | ||
Characterization of PGd-LNP by DLS and microscopy
The surface functionalization of NPs often alters their properties including charges and hydrophobicity or hydrophilicity, especially at the interface with the bulk water, causing damage to NPs such as aggregation and degradation.35 Since this damage is critical in biological tests, the sizes and morphologies of the particles were analyzed by dynamic light scattering (DLS) and microscopic methods. As shown in DLS, both nanoparticles before (LNP-KAT (5 or 10%)) and after the reaction (PGd-LNP (5%) or PGdR-LNP (10%)) revealed a single distribution of diameters with a mean of ca. 50 nm (Fig. 3), which is in a suitable size range for in vivo application.36 Importantly, no detectable aggregation was observed in all particles during the surface reaction and the LNPs were stably dispersed upon storage for at least 14 days at 4 °C in either pH 8.0 Tris–HCl buffer or pH 7.4 PBS(−) (Fig. S36†).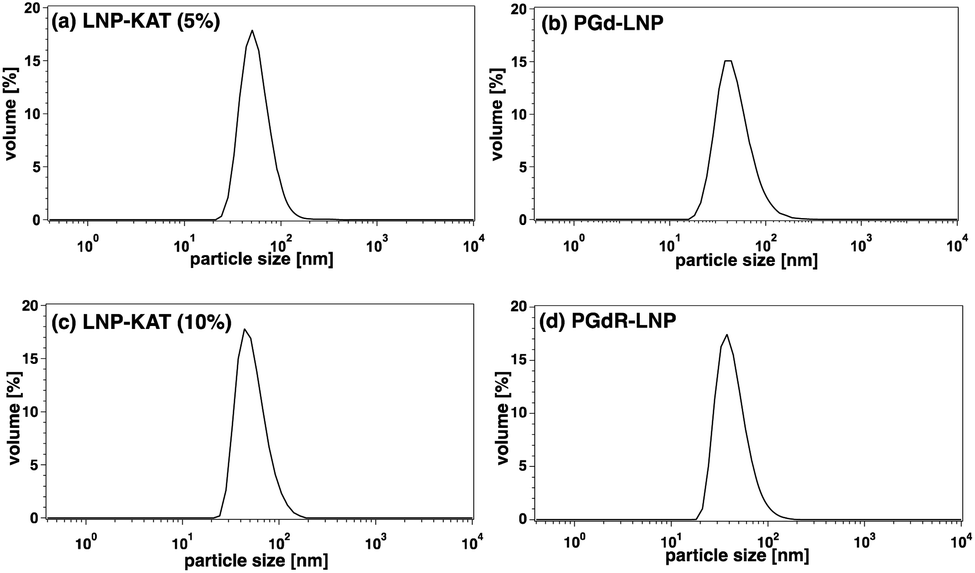 | ||
| Fig. 3 DLS of LNPs before and after the surface KAT ligation. (a) LNP-KAT (5%) with 5 mol% of OA-KAT (mean: 54.5 nm, width: 20.8 nm). (b) PGd-LNP prepared from LNP-KAT (5%) (mean: 45.7 nm, width: 19.3 nm). (c) LNP-KAT (10%) with 10 mol% of OA-KAT (mean: 51.7 nm, width: 20.6 nm). (d) PGdR-LNP prepared from LNP-KAT (10%) (mean: 42.6 nm, width: 16.8 nm). | ||
The dispersion status and the morphologies of the LNPs were further analyzed by AFM and cryoTEM (Fig. 4). For AFM imaging, an aliquot of LNP-KAT or PGd-LNP dispersion was deposited onto a mica surface and subsequently fixed with formalin. As shown in Fig. 4a and b, uniformly dispersed LNPs were observed by AFM without significant aggregation. The cryoTEM images were taken on the LNP samples, prepared by plunge freezing on grids in buffer. Measurements were performed at −180 °C without staining. As shown in Fig. 4c–f, all the LNP samples (LNP-KAT and PGd-LNP with 5 or 10% of KAT) exhibited well-dispersed spherical morphologies having ca. 30–70 nm diameter.37 Upon closer observation of individual LNPs (Fig. 4g), interestingly, 2 types of LNPs were found, one with a relatively darker solid morphology (marked with a purple square) and the other with a lighter inside and darker outer edge with ca. 5–10 nm thickness (marked with a blue square). These two types of NPs presumably corresponded to the one with a lipid monolayer and the other with a lipid bilayer as illustrated in Fig. 4h (see also Fig. S37†).38,39 This cryoTEM observation of a mixture of monolayer- and bilayer-type LNPs was in line with the results of the yield of surface KAT ligation – about half of KAT in LNP-KAT (5%) was consumed during the surface KAT ligation (see Table 1). The other half of B, remaining after surface KAT ligation, at least in part, corresponded to a migrated KAT moiety intercalated in the inner layer of the bilayer-type LNP-KAT (Fig. 4h).
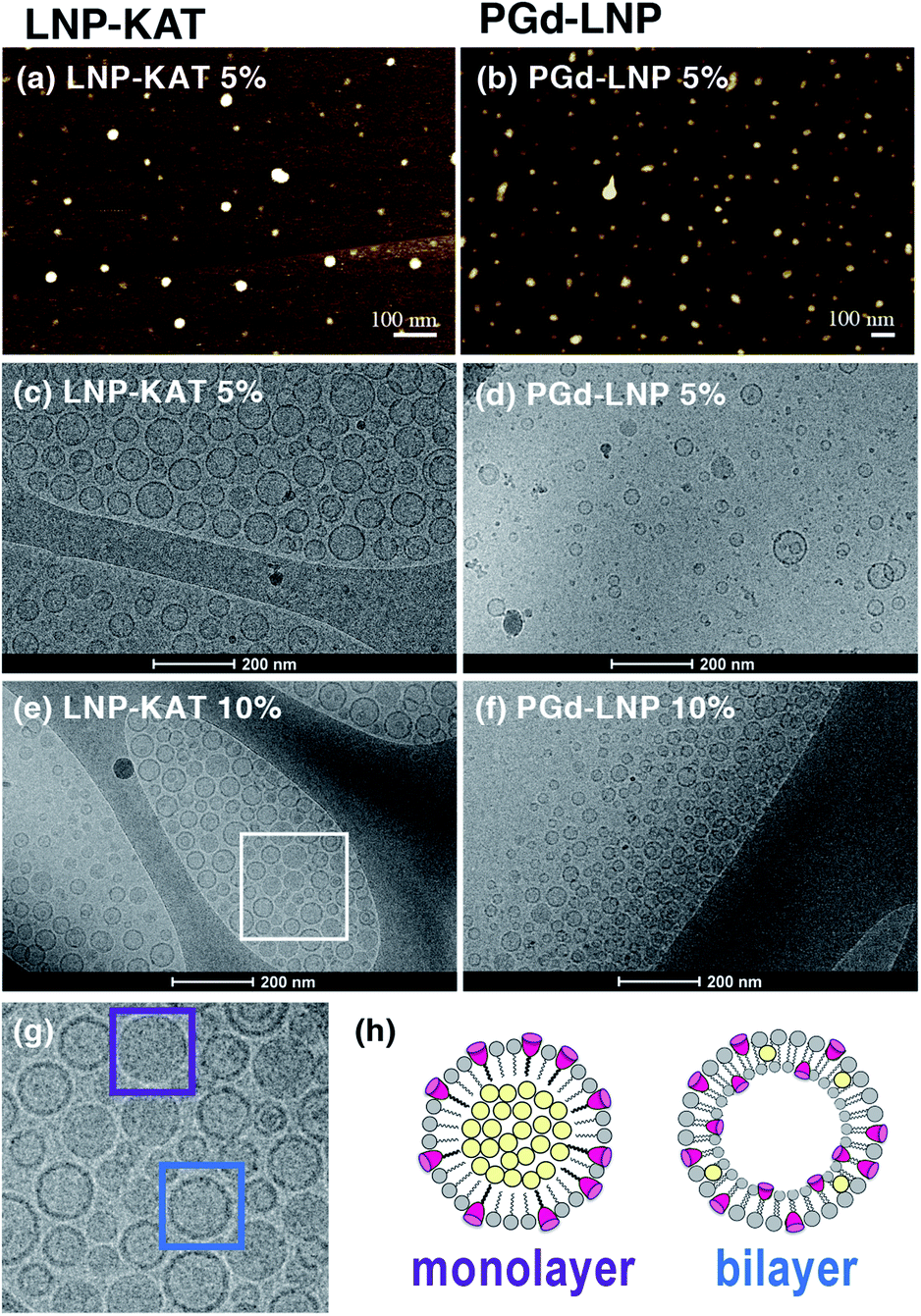 | ||
| Fig. 4 Microscopic images of LNPs. (a and b) AFM images of LNP-KAT (5%) (a) and PGd-LNP prepared from LNP-KAT (5%) (b). (c–f) CryoTEM images of LNP-KAT (5%) (c), PGd-LNP prepared from LNP-KAT (5%) (d), LNP-KAT (10%) (e), and PGd-LNP prepared from LNP-KAT (10%) (f). (g) Expansion of the cryoTEM image of LNP-KAT (10%) (from the area marked as a white square in (e)). (h) Illustration of LNP-KAT with monolayer and bilayer morphologies. The particles marked with purple and blue squares revealed a relatively dark solid morphology and a relatively light inside with a darker edge, respectively. | ||
Estimation of the Gd(III)-chelate payload in PGd-LNP
The Gd(III)-chelate payload per particle is an important factor affecting the r1 relaxivity and MRI contrast in vivo. The number of attached Gd(III)-chelates per PGd-LNP particle was estimated from (1) ICP-MS data of Gd concentration and (2) nanoparticle tracking analysis (NTA) data to count the number of particles (Tables 2 and S3†). From the results, Gd(III)-chelate payloads of PGd-LNP (5%) and PGd-LNP (10%) particles, prepared respectively from LNP-KAT (5%) and LNP-KAT (10%), were found to be ca. 2800 and 4600 per particle. Such large numbers of Gd(III)-chelate payload on PGd-LNP indicate the potential of these particles as an efficient delivery system of Gd(III)-chelates while providing high r1 relaxivity and corresponding strong contrast enhancement in MRI.Relaxivity of PGd-LNP
The proton longitudinal relaxation rate (1/T1) of PGd-LNP (5%) was measured to obtain r1 relaxivity. Under standard measurement conditions (1.4 T/60 MHz at 25 °C), the r1 relaxivity of PGd-LNP (5%) was 22.0 s−1 mM−1. This was much higher than those of small molecule MRI-CAs, such as ProHance® (Bracco Spa, Milano, Italy) with r1 = 4.0 s−1 mM−1 (1.4 T/60 MHz).40 It is known that the r1 relaxivity value is related to the rotational correlation time of the Gd(III)-chelate and a slowed rate causes higher r1 in comparison to the freely rotating small molecule Gd(III)-chelate especially at a lower magnitude.41 This is commonly observed by linking the Gd(III)-chelate molecules to macromolecules, polymers, and nanoparticles.3,4 The r1 value observed in the current PGd-LNP particle was also higher than that of most of the previously reported LNP-type Gd(III)-based MRI-CAs,42–44 including that of our previous natural LDL-based MRI-CA (r1 = 20.1 s−1 mM−1) with a Gd payload of 210 per particle.20 This was presumably, at least in part, due to the high payload of the Gd(III)-chelate (ca. 2800) per particle, which was successfully enabled by the fast kinetics of KAT ligation used in the current study. More importantly, the r1 of PGd-LNP measured at 9.4 T/400 MHz (at 37 °C) was 8.2 s−1 mM−1, showing significantly higher value than that of the small molecule ProHance® (r1 = 2.6 s−1 mM−1 at 9.4 T/400 MHz at 37 °C),46 although the NP effect providing a higher r1 value is generally weaker at higher magnetic field.45 These measurement conditions were the same as those for in vivo imaging in this study and sufficiently high contrast was expected in the in vivo imaging in the presence of PGd-LNP.In vivo MRI of an atherosclerotic mouse model injected with PGd-LNP
Based on the successful LNP functionalization with apoB100-mimetic peptide (P) and Gd(III)-chelate (Gd) providing a high r1 value, PGd-LNP (5%) was injected into an atherosclerotic mouse model for in vivo MRI. An apoE−/− mouse fed on a high fat diet for 2 months was used as a mouse model.47 After an initial baseline MRI measurement of three atherosclerotic apoE−/− mice (no. 1–3), the PGd-LNP (5%) dispersion, which was prepared from LNP-KAT (5%), was injected via the tail vein in a Gd(III)-chelate dose of 0.018–0.051 mmol kg−1 (Table 3).48 The MR images were acquired using a 9.4 T horizontal bore MR spectrometer and at 48 h post-injection, based on our previous results of atherosclerotic plaque imaging using a natural LDL-based MRI-CA.20 The thoracic aorta, including the aortic arch and its three branches (brachiocephalic artery (BA), left carotid artery (LC), and left subclavian artery (LS)), was visualized on coronal scout images, then multiple axial images were acquired.| No. | Mouse body weight | Dose of Gd [mmol kg−1] | Dry weight of tissue used [mg] | Gd content by ICP-MS [ppm] |
|---|---|---|---|---|
| 1 | 34 g | 0.018 | 0.8 | 3.66 |
| 2 | 49 g | 0.051 | 0.7 | 14.6 |
| 3 | 38 g | 0.042 | 0.9 | 4.26 |
As shown in Fig. 5, clear enhancements in the wall of the BA near its branching from the aortic arch were observed in all mice no. 1–3 (yellow arrow in Fig. 5c, d, g, h, k and l). These black blood (BB) images are useful for atherosclerotic plaque detection since the blood in the three arteries (BA, LC and LS, red arrows in Fig. 5) appear black due to the suppression of the blood signal, leading to clear delineation of the artery wall. The assignment of the arteries was confirmed by the comparison with white blood (WB) images, where the trachea appears dark and other arteries appear white (Fig. S39–S41 in the ESI†). In apoE−/− mice, atherosclerotic plaques are often formed at the branching point of BA from the aortic arch, where the MRI enhancement was detected in this study. Also, a similar enhancement was previously observed in this region in our previous reports with a natural LDL-based MRI-CA.19,20
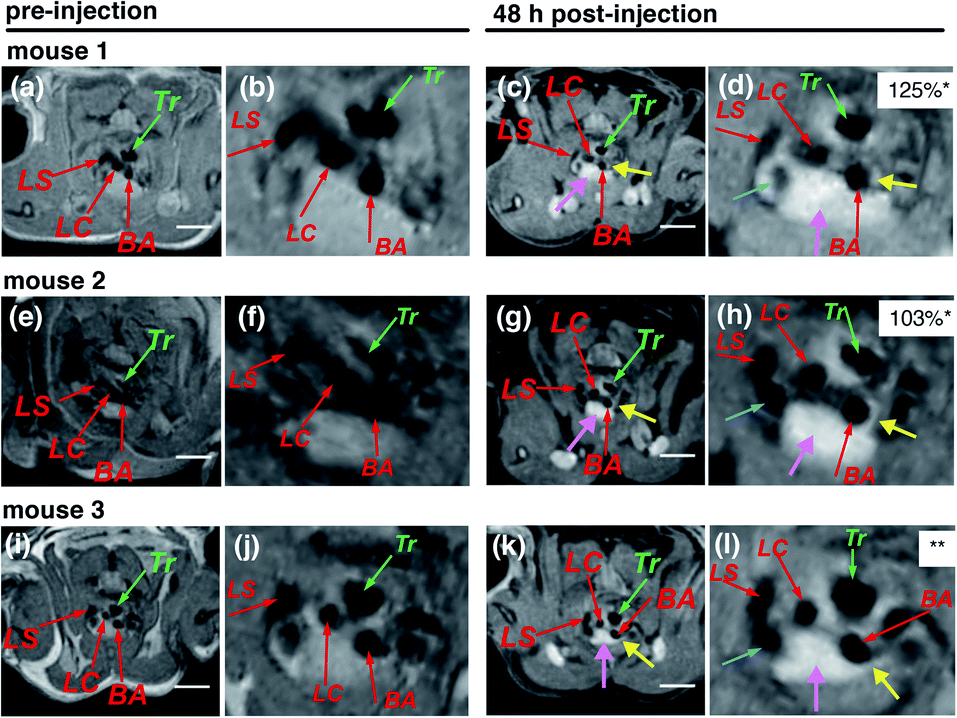 | ||
| Fig. 5 In vivo MR images of apoE−/− mice (no. 1–3) in the regions of arteries taken pre-injection (a, e and i) and 48 h post-injection of PGd-LNP (5%) with doses of 0.018, 0.051, and 0.042 mmol Gd(III)-chelate kg−1 (for no. 1, 2, and 3, respectively) (c, g and k) with their expanded images (b, f, j, d, h and l). Three branches of the aortic arch were identified by red arrows: BA = brachiocephalic artery; LC = left carotid artery; LS = left subclavian artery. Tr = trachea. Yellow arrows point to the enhancement of atherosclerotic plaques. Pink arrows point to the enhancement in the thymus. Green arrows point to the trachea. The enhancement surrounding the trachea is likely due to paratracheal lymph nodes, which contain macrophages taking up the PGd-LNP. Blue arrows point to the left cranial vena cava. *The normalized signal enhancement (% NSE), calculated using the following equation: % NSE = 100% × ((Iwallpost/Imusclepost)/(Iwallpre/Imusclepre) − 1). Iwallpre and Iwallpost are the signal intensity from the artery wall pre- and post-injection, respectively. Imusclepre and Imusclepost are defined similarly for the nearby skeletal muscle, which was used as reference tissue (see the ESI† for the details). **% NSE was not obtained due to the overlapping of the brachiocephalic artery wall with the enhanced thymus. White bars correspond to 5 mm. | ||
The normalized signal enhancement (% NSE) in mice no. 1 and 2 was quite high (125 and 103%). The % NSE of mouse no. 3 was not obtained due to the overlapping of the artery wall with the enhancement of the thymus (pink arrows) although a strong enhancement in the BA wall was apparent (Fig. 5i). These enhancements in BA were not observed in the MR images of the mice injected with small molecule ProHance® (Gd3+ dose = 0.085 mmol kg−1) (Fig. S39–S41 in the ESI,† showing % NSE of 53 or 54%) and are apparently higher than those in our previous natural LDL-based particle experiment providing % NSE of ca. 75%.20
Ex vivo ICP-MS analysis of the aorta of apoE−/− model mice
After MRI, the aortic arch with its three branches (BA, LC, and LS) just above the branching points were harvested from mice no. 1–3 and analyzed by ICP-MS for Gd contents. During dissection, plaques were clearly observed in all mice especially in the BA region. As shown in Table 3, significant accumulation of Gd3+ in the aorta was observed in all three apoE−/− mice. These data were comparable to or higher than our previous results on a natural LDL-based MRI-CA, showing the Gd content in the aortic arch (ca. 5.4 ppm).49 Mouse no. 2 with the highest Gd accumulation (14.6 ppm) in its aorta had the highest body weight and atherosclerotic plaque burden, which was observed during dissection. The increased accumulation of Gd in the aorta was because of selective accumulation of the high payload of Gd-chelate on the PGd-LNP (5%), which was facilitated by KAT ligation, with affinity to the atherosclerotic plaques in the in vivo system.In vivo MRI and ex vivo cryo-fluorescence imaging of apoE−/− injected with PGdR-LNP
Although the atherosclerotic plaque progression in the region of enhancement was obvious during dissection of the mice, it is important to directly correlate MRI enhancement and accumulation of nanoparticles by an ex vivo study to show the direct connection between LNP accumulation in plaques and MRI enhancement. For this purpose, together with P and Gd, we added a fluorescent probe (sulforhodamine B, R) to the LNPs to prepare triply labelled PGdR-LNP, enabling the ex vivo fluorescent imaging of the mouse tissues. To achieve a higher payload of Gd(III)-chelate, LNP-KAT (10%) was used as a starting material and the obtained PGdR-LNP (10%) was characterized by fluorescence spectroscopy and ICP-MS for sulforhodamine B and Gd content, respectively (Table S4†). Also, DLS showed no aggregation of NPs (Fig. S35†).An atherosclerotic apoE−/− mouse (no. 4), injected with PGdR-LNP, in Gd(III)-chelate doses of 0.109 mmol kg−1, was subjected to MRI. In this experiment, initially, MRI was compared at 24 and 48 h post-injection to optimize the MRI timing that provides the best enhancement. As shown in Fig. 6a, a clear enhancement of the BA branching region in the aortic arch was observed at both 24 and 48 h post-injection (yellow arrow). In comparison to the experiments of mice 1–3 that received PGd-LNP (5%) prepared from LNP-KAT (5%) (Fig. 5), a higher enhancement was observed in mouse 4 at 48 h post-injection of PGdR-LNP (10%) prepared from LNP-KAT (10%) (Fig. 6). By quantitative % NSE analysis, we found that the enhancement in BA was higher at 24 h post-injection than at 48 h (Fig. 6a), suggesting a partial washout of the contrast media between these time points. Based on this result, two weeks after the first MRI study, mouse no. 4 was injected again with PGdR-LNP (10%) and scanned by MRI at 24 h post-injection. After the MRI study, the mice were euthanized for ex vivo study by cryo-imaging of the whole mouse.
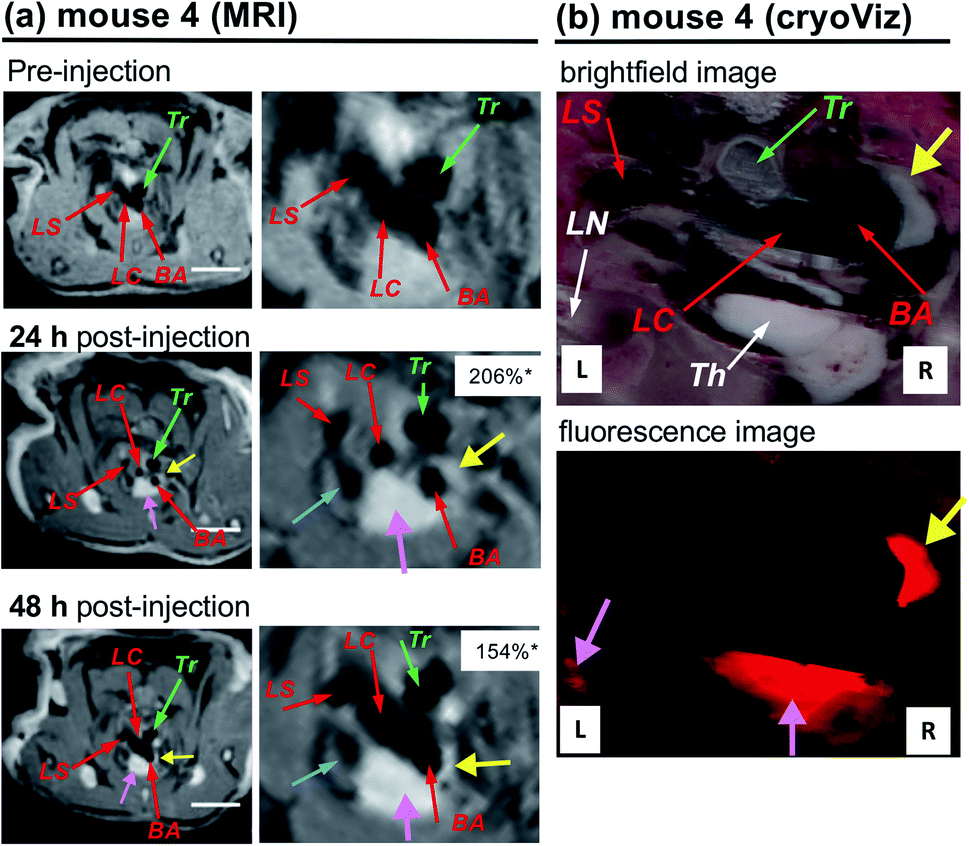 | ||
| Fig. 6 (a) In vivo MRI images of atheroma in the arteries of an apoE−/− mouse (no. 4) taken at pre-injection and at 24 and 48 h post-injection of PGdR-LNP (10%) (dose: 0.109 mmol Gd(III)-chelate kg−1) with their expanded images. (b) CryoViz images of mouse no. 4 at 24 h post-injection of PGdR-LNP (10%) with brightfield (upper) and fluorescence (lower) images. Three branches of the aortic arch were identified by red arrows: BA = brachiocephalic artery; LC = left carotid artery; LS = left subclavian artery. Yellow arrows point to the enhancement of atherosclerotic plaques and corresponding fluorescence emission. Pink arrows point to enhancement in the thymus (Th) and lymph node (LN). Green arrows point to the trachea (Tr). Blue arrows point to the left cranial vena cava. *The normalized signal enhancement (% NSE), calculated using the following equation: % NSE = 100% × ((Iwallpost/Imusclepost)/(Iwallpre/Imusclepre) − 1). Iwallpre and Iwallpost are the signal intensity from the artery wall pre- and post-injection, respectively. Imusclepre and Imusclepost are defined similarly from muscle (see the ESI† for the details). White bars correspond to 5 mm. | ||
In MR images of mouse no. 4 (Fig. 6a), a strong enhancement in the BA wall at the aortic arch branching point was clearly observed (yellow arrows) presumably due to the significant accumulation of PGdR-LNP at this location. The brightfield cryo-imaging of identical mice (Fig. 6b, upper) clearly indicates plaque deposition in the inner wall of BA (yellow arrow) which completely overlapped with the MRI contrast enhancement location. By fluorescent cryo-imaging (Fig. 6b lower), a strong sulforhodamine B fluorescence was observed in the plaque position (yellow arrow). This result confirmed that PGdR-LNP particles were accumulated in the plaque of BA enabling the in vivo MRI diagnosis of atherosclerosis with high enhancement.
Other LNP-type MRI contrast agents, especially HDL-based ones, have been previously reported with successful detection of atheroplaques in abdominal aorta.50–52 In this study, the use of an LDL-mimetic LNP contrast agent with a high payload of Gd(III)-chelate produced by efficient KAT ligation and cardiac-gated MRI to suppress the cardiac motion interference enabled the visualization of atherosclerotic plaques in the aortic arch by high-resolution MRI. Furthermore, we validated the MRI findings by confirming PGdR-LNP accumulation in enhanced anatomical structures using the state-of-the-art cryoViz technique.
Experimental
Synthesis
Details of the synthesis of OA-KAT and HA derivatives 1–3 are described in the ESI† with corresponding spectral data including all the synthetic intermediates. The HA-apoB100-mimetic peptide 1 was synthesized by a standard solid phase peptide synthesis (SPPS) and purified by reversed phase HPLC (Shiseido Capcell Pak C18 column, CH3CN–H2O in the presence of 0.1% TFA) to be obtained in a yield of 37% for the total peptide synthesis and characterized by MALDI-MS; HRMS (MALDI+) m/z: [M + H]+ calcd for C76H145N28O20+: 1770.1184; found, 1770.1185. HA-Gd(DO3A-MA) 2, and HA-sulforhodamine B 3 were synthesized by a common multistep organic synthesis.Preparation of the LNP-KAT particle
The lipid nanoparticle (LNP) with KAT moieties (LNP-KAT) was prepared in a similar way to that in our previous report8 using the same lipid components for LNP preparation as those reported by Halbert and co-workers29,53–56 and Forte and co-workers.57,58 Briefly, to a mixture of phosphatidyl choline (33.8 mg, 44.3 μmol), triolein (29.2 mg, 33.0 μmol) and cholesteryl oleate (9.4 mg, 16.5 μmol) in a mixture of CHCl3, MeOH, and acetone (2:1:1, v/v, 90 mL), a solution of OA-KAT (1.9 mg, 5 μmol) in a mixture of MeOH and acetone (1:1, v/v, 30 mL) was added. The solvents were slowly evaporated to provide a lipid thin film. Subsequently, the film was hydrated with 4 mL of Tris–HCl buffer (10 mM, pH 8.0, with KF (10 mM) and BHT (1 mg L−1)), sonicated, extruded with two stacked polycarbonate membrane filters (pore sizes: 0.05 and 0.1 μm) and filtered (0.45 μm). The filtrate was washed with Tris–HCl buffer by spin-filtration (50 kDa MW cut-off) to remove free lipid components, which were not incorporated in the particle, to yield 4 mL of LNP-KAT particle suspension in Tris–HCl buffer. The LNP-KAT was stored in Tris–HCl buffer at 4 °C until its use in the ligation reaction.
On-particle KAT ligation of LNP-KAT with HA derivatives
To a mixture of 10 mM KF in pH 5.8 phosphate buffer (10 mM) and 0.1 M HCl (400 μL), aqueous solutions of HA-peptide 1 (0.48 μmol) and HA-Gd(DO3A-MA) 2 (4.32 μmol) were added. Subsequently, the LNP-KAT particle suspension in Tris–HCl buffer was added and the reaction mixture (pH 5.2) was stirred overnight at room temperature. The resulting PGd-LNP particle was subjected to spin-filtration to provide 1.5 mL of suspension, which was washed with 5 mL of PBS (−) pH 7.4 three times to remove the unreacted 1 and 2 and subjected to particle characterization.Quantification of B and Gd contents in LNP-KAT and PGd-LNP
ICP-MS and ICP-OES were employed for the quantification of B and Gd contents in LNP-KAT and PGd-LNP for the particle characterization and estimation of on-surface KAT ligation yield. Each particle suspension was dried, digested in conc. HNO3, dried again, and dissolved in 2% HNO3 before being subjected to ICP-MS or ICP-OES analyses. Measurements were performed on an Element XR sector-field ICP-MS (ThermoFisher Scientific, Waltham, MA, USA) or an Ultima Expert ICP-OES (Horiba, Ltd., Kyoto, Japan).MALDI-MS analysis of PGd-LNP
The MALDI-MS analysis of PGd-LNP was performed on a Bruker SolariX-FTICR 9.4 T MS (Bruker Daltonics, Bremen, Germany) using α-cyano-4-hydroxycinnamic acid (HCCA) as a matrix.DLS analysis of PGd-LNP
The hydrodynamic size of the nanoparticles was estimated by dynamic light scattering (DLS) on a Malvern Nano-ZetaSizer (Malvern Instruments Ltd., Worcestershire, UK) in Tris–HCl buffer (10 mM, pH 8.0) or phosphate buffer (10 mM, pH 8.0) at 25 °C.AFM imaging of nanoparticles
Each nanoparticle suspension was deposited onto freshly cleaved mica (Ted Pella Inc., Redding, CA, USA) and fixed by the addition of 10% formalin. AFM imaging was performed on a Dimension Icon AFM (Bruker Nano Surfaces) in the Peak Force Tapping mode in liquid using an SNL 10-A cantilever (Bruker Co.).CryoTEM imaging of nanoparticles
The microscopic characterization of the particles in their hydrated state was carried out on a Tecnai F20 cryo-TEM (FEI/Thermo Fisher Scientific) using samples on 300-mesh lacey carbon-coated copper grids (Quantifoil Micro Tools GmbH, Jena, Germany). A mixture of liquid ethane/propane was used for sample vitrification. The grids were then transferred on a Gatan cryo-transfer holder (Gatan, Inc., Pleasanton, CA, USA) into a microscope and kept at −180 °C during observation. Micrographs were recorded under low dose conditions (<500 e− nm−2) using a Falcon II 4K Camera (FEI Thermo Fisher Scientific), operating the microscope at 200 kV acceleration voltage in bright field mode.Relaxivity measurement of PGd-LNP
Proton relaxivities at 60 MHz were measured on a Bruker WP80 NMR electromagnet adapted to variable field measurements and controlled by a Stelar SMARTracer PC-NMR console (Stelar s.r.l., Pavia, Italy). The temperature was monitored by a VTC91 temperature control unit and maintained by a gas flow. The temperature was determined by previous calibration with a Pt resistance temperature probe. Relaxivities at 400 MHz were measured on a Bruker AVANCE NMR spectrometer. The temperature was calculated according to previous calibration with ethylene glycol and methanol.59 The Gd concentration of the sample was 193 μM.NTA measurement of PGd-LNP
The nanoparticle tracking analysis (NTA) was recorded on a Zetaview® (Particle Metrix, Meerbusch, Germany) to obtain the number of particle dispersions. An aliquot (2 mL) of sample was injected into the sample cell and illuminated with a 405 nm laser sheet (45 mW). The 11 independent positions in the sample cell were imaged by a video camera mounted on a 10× microscope objective oriented 90° to the laser sheet, probing 0.4 nL of volume at each position. The aspect ratio of the camera is 640 pixels × 480 pixels, with an effective pixel size of 0.7 μm per pixel.ICP-MS analysis of the dissected aorta
All the animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania. The male apoE−/− mice were purchased from the Jackson Laboratory and fed with HFD (42% kcal from fat) for two months. To the apoE−/− model mice, PGd-LNP was injected. After 48 h, the mice were euthanized and perfused with PBS before harvesting the aortas for the determination of Gd content by ICP-MS, performed in the Toxicology Core Lab at the New Bolton Center of Veterinary Hospital of the University of Pennsylvania.In vivo imaging using the atherosclerotic mouse model
The apoE−/− model mice were used for in vivo MRI. Under anaesthesia, the PGd-LNP or PGdR-LNP particle was injected via a catheter placed in the tail vein of the mouse. All MRI was performed on a 9.4 T horizontal bore MR spectrometer (DirectDrive®, Agilent, Palo Alto, CA) equipped with a 12 cm (ID) gradient coil. The mouse was positioned prone in a quadrature volume radio frequency (RF) coil (ID = 3.5 cm, length = 8 cm, m2m imaging/Polarean) tuned to 1H resonance frequency (400 MHz). During imaging, the mouse was sedated with 0.8–1% of isoflurane mixed with air (flow rate = 1 L min−1). ECG, respiration, and core temperature of the mouse were monitored using an MRI-compatible vital sign monitoring system (SA Inc, Long Island, New York). The rectal temperature was maintained at 37 ± 0.2 °C by a feedback loop that turns on/off warm air directed into the magnet bore. Scout images were acquired to capture the aortic arch with three rising branches (LC, LS and BA). Then the image plane was placed to cut through the three aortic branching points. This image plane was used to acquire the “White Blood” (WB), where the blood signal in the aorta lumen is not suppressed, as well as “Black Blood” (BB) images, where the blood signal is suppressed (thus black). To obtain the WB image, an ECG-gated multi-slice gradient echo sequence (TR = 1 heartbeat, about 120 ms, TE = 2.4 ms) was employed with FOV = 26 × 25 mm, matrix size = 192 × 128, slice thickness = 0.8 mm. To obtain BB images, ECG-gated multi-slice FLASH (Fast imaging with Low Angle SHot) (TR = 76 ms, TE = 0.97 ms, FA = 40 degrees) was applied to the same slice. Due to the long interval between MRI sessions, we relied on the unique anatomy of the aortic arch, its branching points, and other thoracic arteries in the imaging plane for the comparison of pre- and post-injection images. For images acquired after PGd-LNP injection, BB was further combined with a fat suppression option in the MRI protocol although such option is not necessary for the detection of enhancement.Cryo-imaging analysis of the atherosclerotic mouse model after MRI
Immediately after euthanasia, the mouse was embedded in Tissue-Tek® Optimal Cutting Temperature (O. C. T.) solution (Sakura Finetek USA, Inc., Torrance, CA, USA) and frozen in liquid nitrogen before transfer to BioInVision Inc. (Cleveland, OH, USA). Concomitant cryosectioning and imaging was performed using an automated microtome-blockface episcopic imaging system (CryoViz™, BioInvision Inc.) that allows for microscopic, three-dimensional, high resolution brightfield and fluorescent imaging of macroscopic specimens. The mouse was sectioned from dorsal to ventral with a section thickness of 40 μm and an in-plane resolution of 10 μm × 10 μm, which were applied to the entire body.Conclusions
In this study, we developed a new method for the efficient covalent surface functionalization of LNPs using the amide-forming KAT ligation to synthesize multi-functional hybrid LNPs. Under neutral and diluted aqueous conditions, surface functionalization proceeded in a high yield with several types of bioactive molecules including a peptide (P), a Gd(III)-chelate (Gd), and a fluorescent dye (sulforhodamine B, R). The resulting PGd-LNP and PGdR-LNP particles showed no significant aggregation with a hydrodynamic diameter of ca. 50 nm. The KAT ligation with fast reaction rate enabled the LNP-KAT surface functionalization, by using a minimum amount (1.2 equiv.) and low concentration (0.12 mM) of HA derivatives, to provide PGd-LNP or PGdR-LNP with outstanding loadings of Gd(III)-chelate (ca. 2300 per NP). The obtained LNPs revealed a high relaxivity (r1 = 22.0 s−1 mM−1 at 1.4 T/60 MHz at 25 °C), which was correlated with the strong enhancement in in vivo images in the atherosclerotic plaques of BA in apoE−/−. Further ex vivo study by cryo-fluorescent imaging of a whole mouse injected with triply functionalized PGdR-LNP clearly suggested the strong accumulation of LNPs in atheroplaque in the BA region, completely overlapped with the observed MRI enhancement of the identical mice. This study demonstrated a powerful and versatile method to produce multifunctional LNPs with a sufficiently high payload of functional moieties on the surface on demand, such as peptides and imaging probes. By taking advantage of this efficient attachment of required functional groups by KAT ligation, further production of multi-functional LNPs that employ this LNP-KAT scaffold particle is currently in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication No. 85-23 Rev. 1985) and was approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania (Philadelphia, PA, USA). Authors thank Ms. Agnès Pallier of CNRS Orléans for performing relaxivity measurements. Authors thank Prof. Helm of EPFL for educational discussion. Authors thank Prof. Walde of ETH for intensive and fruitful discussion. Authors thank Prof. Leroux of ETH for his help in nanoparticle preparation and NTA measurements. Authors thank Prof. Spencer of ETH for his help in AFM imaging and Prof. Morbidelli of ETH for his help in DLS measurements. Authors thank Dr Pickup of the Small Animal Imaging Facility (SAIF), Department of Radiology, University of Pennsylvania for his expert assistance. Authors thank Mr Wirz and Dr Rubi of MoBiAS of ETH for their help in MALDI-MS measurements. The ScopeM facility in ETH is acknowledged for TEM measurements. Authors thank Prof. Lowell of Virginia Tech., and Dr Pattabiraman and Prof. Bode of ETH for fruitful discussion. This research was supported in part by an ETH Research Grant (ETH-25 11-1, ETH-45 19-1, YY), the Swiss National Foundation (200021_156097, 205321_173018, IZLJZ2_183660, YY), the Researcher's Developing Grant from the American Heart Association (0930140N, YY), and JSPS KAKENHI (Grant-in-Aid for Scientific Research [A], 6251004, HM).Notes and references
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS.
- M. A. Bruckman, X. Yu and N. F. Steinmetz, Nanotechnology, 2013, 24, 462001 CrossRef.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512–523 RSC.
- E. J. Werner, A. Datta, C. J. Jocher and K. N. Raymond, Angew. Chem., Int. Ed., 2008, 47, 8568–8580 CrossRef CAS.
- H. Li and T. J. Meade, J. Am. Chem. Soc., 2019, 141, 17025–17041 CrossRef CAS.
- N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2011, 44, 925–935 CrossRef CAS.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS.
- R. I. Jølck, L. N. Feldborg, S. Andersen, S. M. Moghimi and T. L. Andresen, Enfgineering Loposomes and Nanoparticles for biological tagering, in Biofunctionaliation of Polymers and their Applications, Springer-Verlag Berlin Heidelberg, 2011, pp. 251–280 Search PubMed.
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825–858 CrossRef CAS.
- E. Lallana, A. Sousa-Herves, F. Fernandez-Trillo, R. Riguera and E. Fernandez-Megia, Pharmaceutical Research, 2012, 29, 1–34 CrossRef CAS.
- N. W. Li and W. H. Binder, J. Mater. Chem., 2011, 21, 16717–16734 RSC.
- P. Shieh and C. R. Bertozzi, Org. Biomol. Chem., 2014, 12, 9307–9320 RSC.
- L. N. Feldborg, R. I. Jolck and T. L. Andresen, Bioconjugate Chem., 2012, 23, 2444–2450 CrossRef CAS.
- C. E. Carney, I. L. Lenov, C. J. Baker, K. W. MacRenaris, A. L. Eckermann, S. G. Sligar and T. J. Meade, Bioconjugate Chem., 2015, 26, 899–905 CrossRef CAS.
- A. M. Dumas, G. A. Molander and J. W. Bode, Angew. Chem., 2012, 51, 5683–5686 CrossRef CAS.
- H. Noda, G. Eros and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 5611–5614 CrossRef CAS.
- D. Mazunin, N. Broguiere, M. Zenobi-Wong and J. W. Bode, ACS Biomater. Sci. Eng., 2015, 1, 456–462 CrossRef CAS.
- S. Oriana, A. Fracassi, C. Archer and Y. Yamakoshi, Langmuir, 2018, 34, 13244–13251 CrossRef CAS.
- Y. Yamakoshi, H. Qiao, A. N. Lowell, M. Woods, B. Paulose, Y. Nakao, H. L. Zhang, T. Liu, S. Lund-Katz and R. Zhou, Chem. Commun., 2011, 47, 8835–8837 RSC.
- A. N. Lowell, H. Qiao, T. Liu, T. Ishikawa, H. L. Zhang, S. Oriana, M. Wang, E. Ricciotti, G. A. FitzGerald, R. Zhou and Y. Yamakoshi, Bioconjugate Chem., 2012, 23, 2313–2319 CrossRef CAS.
- W. J. M. Mulder, G. J. Strijkers, G. A. F. van Tilborg, A. W. Griffioen and K. Nicolay, NMR Biomed., 2006, 19, 142–164 CrossRef CAS.
- D. P. Cormode, P. C. Naha and Z. A. Fayad, Contrast Media Mol. Imaging, 2014, 9, 37–52 CrossRef CAS.
- P. Saulnier and J. P. Benoit, in Nanoparticulates as Drug carriers, ed. V. P. Torchilin, Imperial College Press, London, U.K., 2014, pp. 213–224 Search PubMed.
- J. Chang, X. H. Chen, Z. Glass, F. Gao, L. Q. Mao, M. Wang and Q. B. Xu, Acc. Chem. Res., 2019, 52, 665–675 CrossRef CAS.
- Q. Y. Lin, J. Chen, Z. H. Zhang and G. Zheng, Nanomedicine, 2014, 9, 105–120 CrossRef CAS.
- R. L. Ball, K. A. Hajj, J. Vizelman, P. Bajaj and K. A. Whitehead, Nano Lett., 2018, 18, 3814–3822 CrossRef CAS.
- T. J. Knott, R. J. Pease, L. M. Powell, S. C. Wallis, S. C. Rall Jr, T. L. Innerarity, B. Blackhart, W. H. Taylor, Y. Marcel and R. Milne, et al. , Nature, 1986, 323, 734–738 CrossRef CAS.
- C. Y. Yang, S. H. Chen, S. H. Gianturco, W. A. Bradley, J. T. Sparrow, M. Tanimura, W. H. Li, D. A. Sparrow, H. Deloof, M. Rosseneu, F. S. Lee, Z. W. Gu, A. M. Gotto and L. Chan, Nature, 1986, 323, 738–742 CrossRef CAS.
- G. Baillie, M. D. Owens and G. W. Halbert, J. Lipid Res., 2002, 43, 69–73 CAS.
- Since it is known that peptide is more stable on resin, the HA-peptide was kept on resin until right before use (S3, Scheme S1†).
- D. Mazunin, Formation and Functionalization of Hydrogels with the Potassium Acyltrifluoroborate (KAT) Ligation, PhD thesis, ETH Zurich, 2016.
- Details are described in the ESI.†.
- In the preliminary tests using higher concentration of peptide, unfavorable aggregation of nanoparticles was observed in the reaction mixture. This was presumably due to the coulombic interaction between the negatively charged BF3− moieties on the particle surface and positively charged apoB100-mimetic peptide side chain. This was successfully avoided by diluting the HA-peptide reactant in the reaction mixture. The reaction under highly diluted conditions is better when less soluble and more expensive biological molecules are used as HA derivatives in the surface KAT ligation.
- Please note that compounds 1 and 2 were detected clearly under the same MALDI-MS conditions as shown in Fig. S2 and S30.†.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS.
- A. Albanese, P. S. Tang and W. C. W. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS.
- The preparation of the grid for cryoTEM can yield thin films with slightly different thicknesses and therefore the numbers of particles on the grids may vary. Indeed, if the film is particularly thin, the particles tend to accumulate in proximity of the grid where the film is slightly thicker as observed in the case of the LNP.
- Since cryoTEM images do not always reflect the ratios of monolayer LNP and bilayer LNP of the entire population of LNPs, the ratio of these two types of LNPs could not be determined in this study.
- T. Fujimoto, Y. Ohsaki, M. Suzuki and J. Cheng, Chapter 13 – Imaging Lipid Droplets by Electron Microscopy, in Methods in Cell Biology, ed. H. Yang and P. Li, Lipid Droplets, Academic Press, 2013, vol. 116, pp. 227–251 Search PubMed.
- J. S. Haigh, Investigation in to the Effect of Spin Locking on Contrast Agent Relaxivity, PhD Dissertation, Portland State University, Portland, OR, 2015, p. 87.
- É. Tóth, L. Helm and A. Merbach, in The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. Merbach, L. Helm and É. Tóth, Wiley, Chichester, U.K., 2nd edn, 2013, p. 57 Search PubMed.
- C. S. Thaxton, J. S. Rink, P. C. Naha and D. P. Cormode, Adv. Drug Delivery Rev., 2016, 106, 116–131 CrossRef CAS.
- D. P. Cormode, R. Chandrasekar, A. Delshad, K. C. Briley-Saebo, C. Calcagno, A. Barazza, W. J. Mulder, E. A. Fisher and Z. A. Fayad, Bioconjugate Chem., 2009, 20, 937–943 CrossRef CAS.
- W. J. Mulder, G. J. Strijkers, G. A. van Tilborg, D. P. Cormode, Z. A. Fayad and K. Nicolay, Acc. Chem. Res., 2009, 42, 904–914 CrossRef CAS.
- P. Caravan, C. T. Farrar, L. Frullano and R. Uppal, Contrast Media Mol. Imaging, 2009, 4, 89–100 CrossRef CAS.
- Measured in this study.
- P. Libby, Nature, 2002, 420, 868–874 CrossRef CAS.
- The PGd-LNP particle dispersion was concentrated by spin-filtration right before the injection, so that the volume of injection to each mouse was below 250 μL.
- Unpublished data (5.4 ± 3.9 ppm in the aorta of apoE−/− mouse models and 0.32 ± 0.44 ppm in C56BL/6j mice).
- D. P. Cormode, K. C. Briley-Saebo, W. J. M. Mulder, J. G. S. Aguinaldo, A. Barazza, Y. Q. Ma, E. A. Fisher and Z. A. Fayad, Small, 2008, 4, 1437–1444 CrossRef CAS.
- J. C. Frias, Y. Ma, K. J. Williams, Z. A. Fayad and E. A. Fisher, Nano Lett., 2006, 6, 2220–2224 CrossRef CAS.
- J. C. Frias, K. J. Williams, E. A. Fisher and Z. A. Fayad, J. Am. Chem. Soc., 2004, 126, 16316–16317 CrossRef CAS.
- M. D. Owens, G. Baillie and G. W. Halbert, Int. J. Pharm., 2001, 228, 109–117 CrossRef CAS.
- S. Hayavi and G. W. Halbert, Biotechnol. Prog., 2005, 21, 1262–1268 CrossRef CAS.
- S. Hayavi, G. Baillie, M. D. Owens and G. W. Halbert, J. Pharm. Pharmacol., 2006, 58, 1337–1342 CrossRef CAS.
- P. X. Zhou, S. Hatziieremia, M. A. Elliott, L. Scobie, C. Crossan, A. M. Michie, T. L. Holyoake, G. W. Halbert and H. G. Jorgensen, J. Controlled Release, 2010, 148, 380–387 CrossRef CAS.
- M. Nikanjam, E. A. Blakely, K. A. Bjornstad, X. Shu, T. F. Budinger and T. M. Forte, Int. J. Pharm., 2007, 328, 86–94 CrossRef CAS.
- M. Nikanjam, A. R. Gibbs, C. A. Hunt, T. F. Budinger and T. M. Forte, J. Controlled Release, 2007, 124, 163–171 CrossRef CAS.
- D. S. Raiford, C. L. Fisk and E. D. Becker, Anal. Chem., 1979, 51, 2050–2051 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04106h |
| This journal is © The Royal Society of Chemistry 2020 |