
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective oxidation of alkyl and aryl glyceryl monoethers catalysed by an engineered and immobilised glycerol dehydrogenase†‡
Susana
Velasco-Lozano
 a,
Maite
Roca
b,
Alejandro
Leal-Duaso
a,
José A.
Mayoral
ac,
Elisabet
Pires
*ac,
Vicent
Moliner
*b and
Fernando
López-Gallego
*ade
a,
Maite
Roca
b,
Alejandro
Leal-Duaso
a,
José A.
Mayoral
ac,
Elisabet
Pires
*ac,
Vicent
Moliner
*b and
Fernando
López-Gallego
*ade
aCatálisis Heterogénea en Síntesis Orgánicas Selectivas, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH-CSIC), University of Zaragoza, Pedro Cerbuna, 12, 50009, Zaragoza, Spain. E-mail: flopez@cicbiomagune.es; epires@unizar.es
bDepartament de Química Física i Analítica, Universitat Jaume I, 12071 Castelló, Spain. E-mail: moliner@uji.es
cDepto. de Química Orgánica, Facultad de Ciencias, University of Zaragoza, Pedro Cerbuna, 12, 50009, Zaragoza, Spain
dHeterogeneous Biocatalysis Laboratory, Center for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramón 182, 20014, Donostia San Sebastián, Spain
eIKERBASQUE, Basque Foundation for Science, María Díaz de Haro 3, 48013 Bilbao, Spain
First published on 5th October 2020
Abstract
Enzymes acting over glyceryl ethers are scarce in living cells, and consequently biocatalytic transformations of these molecules are rare despite their interest for industrial chemistry. In this work, we have engineered and immobilised a glycerol dehydrogenase from Bacillus stearothermophilus (BsGlyDH) to accept a battery of alkyl/aryl glyceryl monoethers and catalyse their enantioselective oxidation to yield the corresponding 3-alkoxy/aryloxy-1-hydroxyacetones. QM/MM computational studies decipher the key role of D123 in the oxidation catalytic mechanism, and reveal that this enzyme is highly enantioselective towards S-isomers (ee > 99%). Through structure-guided site-selective mutagenesis, we find that the mutation L252A sculpts the active site to accommodate a productive configuration of 3-monoalkyl glycerols. This mutation enhances the kcat 163-fold towards 3-ethoxypropan-1,2-diol, resulting in a specific activity similar to the one found for the wild-type towards glycerol. Furthermore, we immobilised the L252A variant to intensify the process, demonstrating the reusability and increasing the operational stability of the resulting heterogeneous biocatalyst. Finally, we manage to integrate this immobilised enzyme into a one-pot chemoenzymatic process to convert glycidol and ethanol into 3-ethoxy-1-hydroxyacetone and (R)-3-ethoxypropan-1,2-diol, without affecting the oxidation activity. These results thus expand the uses of engineered glycerol dehydrogenases in applied biocatalysis for the kinetic resolution of glycerol ethers and the manufacturing of substituted hydroxyacetones.
Introduction
Enzymes acting over glyceryl ether substrates are scarce in cell metabolisms. Among the low diversity of biochemical reactions involving glyceryl ethers, we highlight lignin biosynthesis,1 the metabolic synthesis of ether lipids2 and plasmalogens3–5 that play important physiological roles.6Despite the low abundance of glycerol derived alkyl ethers in nature, these molecules are enormously attractive for the chemical industry to manufacture both commodities7 (i.e. fuels and solvents)8 and specialised products (i.e. cosmetics and drugs).9–11 In particular, monoalkylated glycerol ethers are considered among the most promising targets to valorise the surplus of glycerol from biorefinery activities.12–14 The chemical structure of many approved and commercial drugs incorporates a glycerol skeleton with at least one alkoxy or aryloxy substituent (Scheme 1A). Some of these drugs are administered as pure enantiomers which encourages medical chemists to develop enantioselective synthetic schemes. Herein, biocatalysis bursts as a key enabling technology in chemical manufacturing due to the exquisite regio- and enantioselectivity of enzymes.
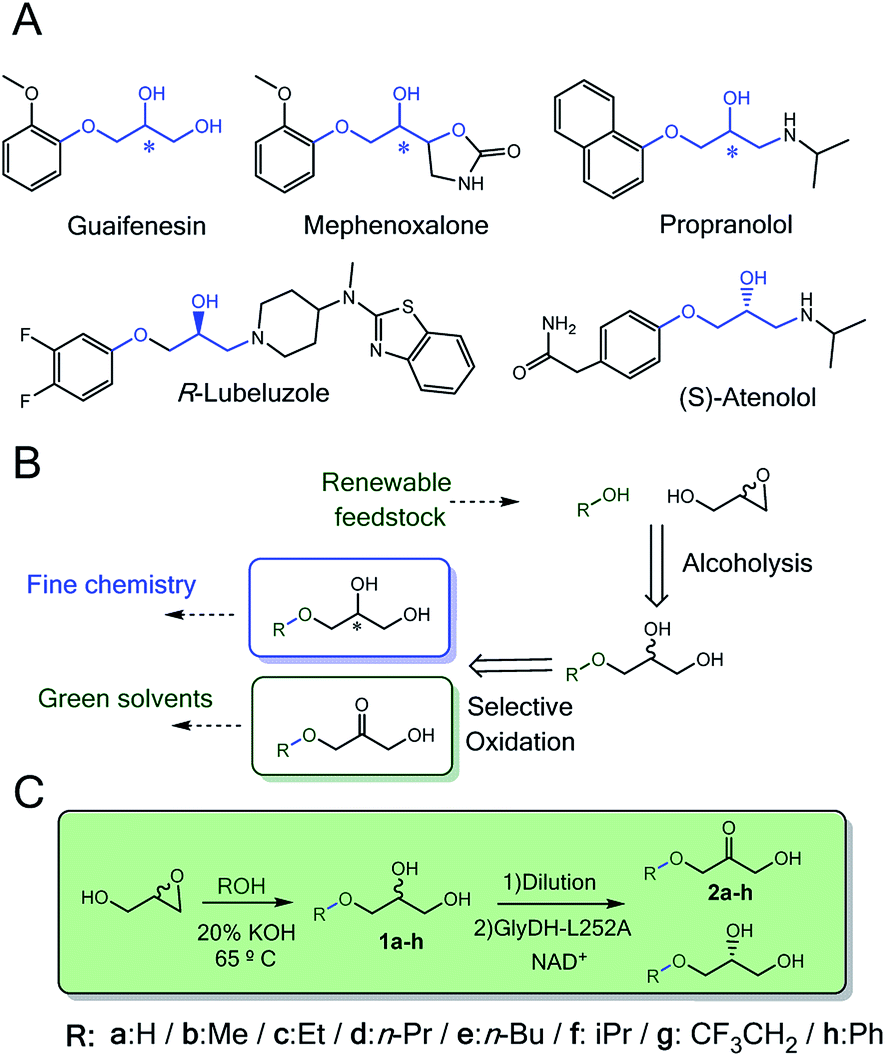 | ||
| Scheme 1 (A) Drugs containing alkyl/aryl-glycerol scaffolds (in blue). (B) Retrosynthesis pathway towards enantiopure alkyl/aryl glyceryl monoethers and substituted hydroxyketones from renewable starting materials. (C). Two-step chemo-enzymatic route to access (R)-3-(alkoxy/aryloxy)propan-1,2-diol and their corresponding 3-alkoxy/aryloxy-1-hydroxyacetone. | ||
In the last decade, the pharmaceutical industry is successfully integrating more biocatalytic steps in their drug synthetic processes; new enzyme routes for the manufacturing of sitagliptin15 and islatravir16 illustrate this trend.
Synthesis of monoalkyl/aryl glycerols have been attained through many different chemical methodologies, including both stoichiometric and catalytic ones.17 Recently, Leal-Duaso et al. reported a straightforward methodology to synthesise glyceryl monoethers from glycidol and several alcohols using KOH as catalyst (Scheme 1B).18 Nevertheless, the reaction yields a racemic product that makes the isolation of the target enantiomer a non-viable task for practical purposes. To access one of the enantiomers, a kinetic resolution is demanded. In this context, lipases19,20 are efficient catalysts to selectively acetylate only one enantiomer of glycerol derivatives, so the unreactive enantiomer is easily purified. Although alcohol dehydrogenases have also been applied for kinetic resolution of sec-alcohols,21 they have never been applied with glycerol derivatives. The enantioselective oxidation of monoalkyl glycerols will provide a double opportunity to access enantiomerically pure alkyl glycerols as building blocks for drug synthesis and manufacture of 3-alkoxy/aryloxy-1-hydroxyacetone as a new family of bio-based solvents to be explored (Scheme 1B).8 Moreover, enzymatic synthesis of these hydroxyacetone monoethers is unprecedented but highly interesting as key building blocks for the biosynthesis of non-natural deoxysugars22 and cosmetics.23
The best candidates to catalyse such selective oxidation would be the glycerol dehydrogenases (GlyDH) due to their natural substrate specificity. Biotechnological applications of these enzymes however are mainly limited to the whole cell production of dihydroxyacetone24 and valuable diols through metabolic engineering.25 Besides, this family of enzymes has also been widely applied in biosensing as the active phase of glycerol sensors.26 Nevertheless, their substrate scope is extremely narrow, restricted mainly to glycerol in the oxidation direction. Among the few exceptions, the wild-type GlyDHs from Klebsiella genus are able to oxidise a substrate bulkier than glycerol, and that promiscuity increases when replacing the catalytic Zn2+ by other divalent metals.27 Remarkably, the GlyDH from Enterobacter (Klebsiella) aerogenes oxidises the R-phenylglycol to 2-hydroxyacetophenone in a regio- and enantioselective manner.28
Hundreds of examples endorse the improvement of enzyme properties (activity, stability and selectivity) and the widening of their substrate scope through protein engineering. As safe path for enzyme optimization, the scientific community exploits, either separately or synergistically, random and extensive mutagenesis campaigns through directed evolution29 and minimalist mutations through site-directed mutagenesis guided by structural knowledge.22,30,31 In the particular case of GlyDH, one of the few documented successes is the combination of DNA-shuffling and site-directed mutagenesis to expand the GlyDH substrate scope to other diols. This variant was 2.6 times more active towards 1,3-butanediol than the wild-type.32 Despite these efforts, GlyDHs have never been challenged against bulkier substrates like glycerol monoethers.27
Besides re-shaping the active site of enzymes to improve their intrinsic functional features, the engineering journey must include the enzyme immobilisation to fabricate robust biocatalysts that are easily to separate from the reaction media and readily to integrate into different bioreactor configurations. In the last years, the immobilization of engineered enzyme variants has allowed their application under industrially relevant conditions.16,33–35 To that aim, selective immobilization protocols that assure the activity and increase the stability of the enzymes upon the attachment to the solid carrier are pretended.
In this work, we rationally engineer GlyDH from Bacillus stearothermophilus (Wt-BsGlyDH) to selectively oxidise alkyl/aryl glycerols to 3-alkoxy/aryloxy-1-hydroxyacetones. Before starting the engineering campaign, we shined light on the uncertainties found in its catalytic mechanism. Besides explaining how this enzyme positions the substrate into its active site, Quantum Mechanics/Molecular Mechanics (QM/MM) computational studies also reveal its enantiopreference towards the S-isomer of the alkylated glycerol. This information was instrumental to further design mutations at the active site that allowed the enzyme accommodating bulkier glycerol monoethers. From the molecular design, we walked towards the process development by immobilizing the most active engineered variant. The resulting heterogeneous biocatalyst eases the product separation and increases the enzyme operational stability. Finally, the heterogeneous biocatalyst herein presented was coupled to a one-pot sequential chemo-enzymatic process to synthesise enantiopure glycerol monoethers from glycidol and ethanol without intermediate purification steps (Scheme 1C).
Materials and methods
Chemicals
Glycerol, hydrogen peroxide, dihydroxyacetone (DHA), flavin-adenine-dinucleotide sodium salt (FAD+), acetic anhydride and N-methylimidazole, kanamycin sulfate from Streptomyces kanamyceticus and ampicillin, catalase from bovine liver (Hl-CAT) were acquired from Sigma-Aldrich (St. Louis, IL, USA). Nicotinamide adenine dinucleotide reduced disodium salt (NADH), nicotinamide adenine dinucleotide sodium salt (NAD+) and isopropyl β-D-thiogalactopyranoside (IPTG), were purchased from GERBU Biotechnik GmbH (Wieblingen, Germany). Glyceryl monoethers and diethers were synthesised as previously described (Table S1‡).18,36 Cobalt-activated agarose microbeads 4BCL (AG-Co2+) (particle size; 50–15 μm, pore size; 112 nm and 15 μmol of Co2+ per g carrier) were purchased from ABT technologies (Madrid, Spain). Precision plus protein™ standards, micro Bio-spin™ chromatographic columns and Bradford reagent were acquired from BIORAD. All other reagents and solvents were analytical grade or superior.Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 528 g for 30 min at 4 °C and the pellet was discarded. The supernatant containing cell extracts with the His-tagged proteins (except Tt-NOX) were collected and employed for further purification and/or immobilization.
528 g for 30 min at 4 °C and the pellet was discarded. The supernatant containing cell extracts with the His-tagged proteins (except Tt-NOX) were collected and employed for further purification and/or immobilization.
GlyDH activity (colorimetric assay). (1a, rac-1b, rac-1c, rac-1d, rac-1e, rac-1f, rac-1g, rac-1h, 1b-bis and 1c-bis) and 1 mM NAD+ in sodium phosphate or sodium bicarbonate buffer 100 mM at pH 7 or 9, respectively; were incubated with 5 μL of enzymatic solution or suspension at 30 °C. The increase of the absorbance was monitored at 340 nm. One unit of activity was defined as the amount of enzyme that was required to reduce 1 μmol of NAD+ per minute at the assayed conditions.
NOX activity (colorimetric assay). 200 μL of a reaction mixture containing 0.2 mM NADH and 150 μM FAD+ in phosphate buffer pH 7 were incubated with 5 μL of enzymatic solution or suspension at 30 °C. The decrease in the absorbance was monitored at 340 nm. One unit of activity was defined as the amount of enzyme that was required to oxidise 1 μmol of NADH per minute at the assayed conditions.
CAT activity (colorimetric assay). The activity was determined by recording the decrease in the absorbance at 240 nm of 200 μL of a reaction mixture containing 35 mM hydrogen peroxide in sodium phosphate buffer 100 mM pH 7 at 30 °C. The reaction was initiated by adding 5 μL of the enzymatic solution or suspension to the reaction mixture. One unit of CAT activity was defined as the amount of enzyme required for the disproportionation of one μmol of hydrogen peroxide per minute at the assessed conditions.
:1 mol ratio in respect to glycidol). Then the mixture was heated at 65 °C and 0.3 mL of rac-glycidol (4.35 mmol) were added dropwise for 15 min. After 2 hours, when rac-glycidol has completely been converted to rac-1c, the temperature was cooled down to 25 °C, KOH neutralised with 0.3 M HCl and the salts filtered off.
Heterogeneous biocatalyst GlyDH-L252A co-immobilised with Lp-NOX on AG-Co2+ (100 mg biocatalyst) was placed inside a 1.5 mL Bio-spin™ chromatographic column. Then, 500 μL of reaction mixture consisted in 20 mM of the rac-1c substrate (pure, raw from chemosynthesis with ethanol and raw from chemosynthesis without ethanol), 1 mM NAD+, 150 μM FAD+ in 100 mM sodium phosphate buffer at pH 8 and were added and incubated a 30 °C in a vertical rotating shaker at 40 rpm. After 24 and 48 h, columns were spin down and the recovered reaction mixtures were analysed as described in the chromatographic methods.
000 mU of Hl-CAT in 100 mM sodium phosphate buffer at pH 7 and incubated at 30 °C in a vertical rotating shaker at 40 rpm. After each reaction cycle (24 h), columns were spin down and washed one time with one volume of 25 mM sodium phosphate buffer at pH 7 before starting the next reaction cycle. Recovered reaction mixtures were analysed as described in the chromatographic methods.
Gas chromatography (GC). Prior GC analysis, samples were derivatised as described elsewhere.42 Briefly, 30 μL of aqueous reaction simple were placed in a 1.5 mL Eppendorf tube, followed by the addition of 30 μL of N-methylimidazole and 225 μL of acetic anhydride and incubated by 10 min at room temperature. Afterwards, 300 μL of distilled water was added and allowed to cool down. Later, liquid–liquid extraction of acetylated compounds was done by the addition of 300 μL of dichloromethane containing 2 mM eicosane as external standard discarding the aqueous phase. 30–50 mg of anhydrous MgSO4 were added to dry samples before GC analysis. Gas chromatography analyses were carried out in a Hewlett Packard 7890 Series II gas chromatograph using a column of phenyl silicone 5.5% (Zebron ZB-5HT Inferno 30 m × 0.25 mm × 0.25 μm), helium as carrier gas, and equipped with a flame ionization detector (FID). Injector at 250 °C, FID at 280 °C. Separation of 1c derivatives were done by the following temperature program: initial temperature at 60 °C, maintained 2 min, ramp to 240 °C at a rate of 20 °C min−1 and finally maintained 2 min. Retention times for acetylated compounds were: rac-1c: 7.88 min, 3-ethoxy-1-hydroxyacetone: 6.75 min and eicosane: 12.2 min. The samples were additionally analysed using a Hewlett Packard 6890 Series II Gas chromatograph using a column of phenyl silicone 5.5% (Zebron ZB-5HT Inferno 30 m × 0.25 mm × 0.25 μm), coupled to an Agilent 5973 inert Mass Spectrometer with Electronic Impact ionization.
:15).43 Retention times were: R-1h: 11.9 min, S-1h: 23.4 min and 1-phenoxy-3-hydroxyacetone (2h): 5.4 min.
Results and discussion
Engineering of GlyDH and mechanistic insights
Inspired by previous works,46,47 we sketched a retrosynthetic pathway to access enantiopure alkyl/aryl glyceryl monoethers starting from inexpensive epoxides such as glycidol, which can be readily synthesized from glycerol (Scheme 1B). Once racemic glycerol ethers are chemically synthesized under basic conditions,18 we propose their sequential enzymatic regio- and enantioselective oxidation. Unfortunately, none oxidoreductase has been described to efficiently conduct the selective oxidation towards this type of monoglyceryl ethers. GlyDH is one of the potential enzymes to catalyse that reaction, since it efficiently oxidises glycerol to dihydroxyacetone.48 Nevertheless, its oxidation activity is practically restricted to its natural substrate: glycerol. To go beyond glycerol, we tested two GlyDHs from different bacterial sources for the selective oxidation of monoalkylglycerols. We detected some oxidative activity towards a battery of pure monoalkylglyceryl ethers using both wild-type GlyDH from Cellulomonas sp. (CsGlyDH) and from Bacillus stearothermophilus (BsGlyDH) (Table S3‡). These results agree with the residual activity previously reported for CsGlyDH towards 1b.49 Remarkably, the relative activity of the enzyme from Bacillus was higher than the one from Cellulomonas towards a wider scope of glycerol ethers. These insights motivated us to engineer BsGlyDH through a structure-guided approach.The reported crystal structures (PDB ID: 1JQ5 and 1JQA)48 of BsGlyDH show that O1 and O2 atoms of glycerol are coordinated to the catalytic Zn2+ atom, which explains the exquisite specificity of this enzyme for 1,2-diols as substrates. In contrast, the O3 seems to be neutral in the catalytic mechanism but it is confined into the cavity formed by V131, Y142 and L252, supporting the fact, that glycerol is the preferential substrate (Fig. 1). Despite the available structural information, the precise catalytic mechanism of hydride transfer from C2 atom of glycerol to C4 atom in NAD+ remains unclear (Fig. 2). Then, before sculpting the active site to accommodate bulkier substituents in O3, we decided to explore the free energy profiles to unveil the BsGlyDH mechanism at atomistic level (see ESI‡). In the literature, a water molecule is suggested to act as a base to abstract the proton from the C2 hydroxyl group and form the alkoxide intermediate to subsequently proceed with the hydride transfer from C2 atom of glycerol to NAD+ cofactor.48 However, this mechanism has been demonstrated neither through experimental nor computational evidences. There are other short-chain alcohol dehydrogenases/reductases (SDRs) from Drosophila melanogaster and Drosophila lebanonensis50 that contain highly conserved active site residues (Ser and Tyr) that act as strong base for the proton abstraction. Moreover, in mannitol 2-dehydrogenase from Pseudomonas fluorescens,51 a Lys residue functions as the catalytic base that facilitates hydride transfer to NAD+ by proton abstraction from alcohol. Classical MD simulations of the fully solvated substrate–enzyme complex keep the Zn2+ ion five coordinate complex, as in the initial X-ray structure of Ruzheinikov et al.,48 including the coordination to O2 atom. Nevertheless, two different conformations of glycerol bound to the active center are identified (Fig. 2 and S2–S4‡). In one conformation the proton from O2 atom of glycerol establishes hydrogen bond interaction with a water molecule while in the other conformation; this proton establishes a hydrogen bond with D123 residue. This fact led us to propose two different reaction mechanisms (Fig. 2). Mechanism 1 involves the proton transfer from O2 atom of glycerol to a water molecule followed by a hydride transfer from C2 atom of glycerol to C4 atom of NAD+ as proposed in the literature.48 Contrariwise, mechanism 2 leads to a proton transfer between the substrate and the acid residue before the hydride transfer from glycerol to NAD+. To decipher the most likely mechanism, we generated the full free energy landscapes for the two proposed mechanisms by means of multiscale QM/MM simulations. In particular, free energy surfaces were computed for every chemical step as potentials of mean force (PMFs) at M06-2X/6-31+G(d,p):PM3/MM level of theory (see ESI Fig. S5 and S6‡).
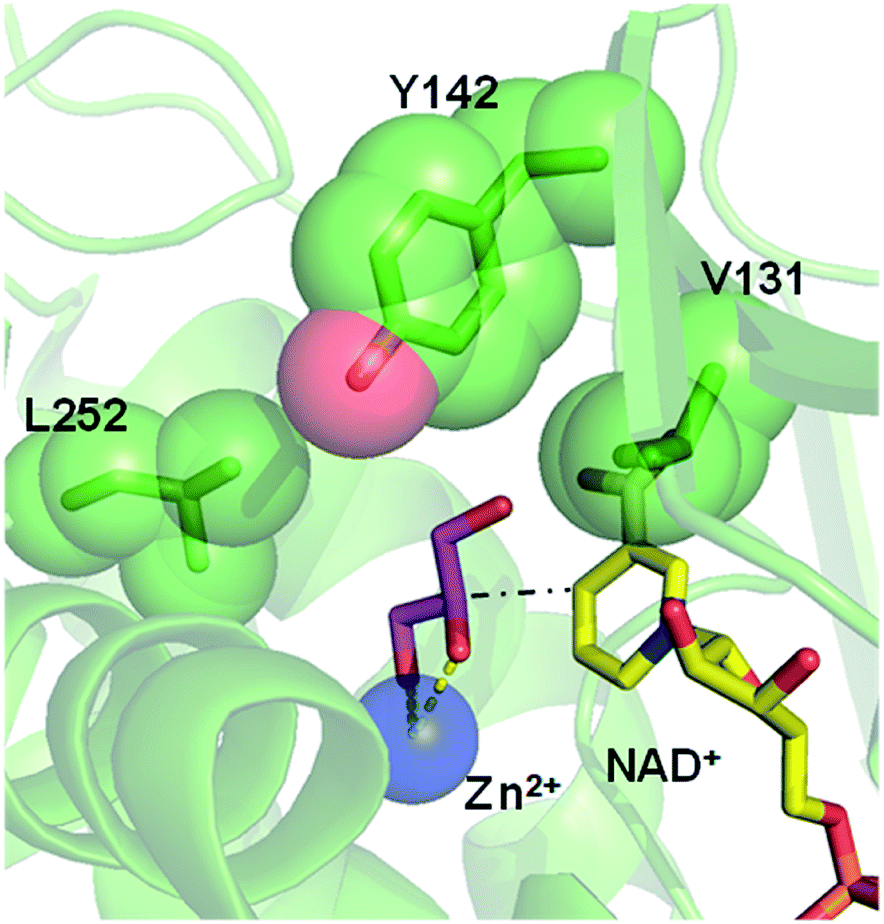 | ||
| Fig. 1 Overlap of crystal structures of GlyDH from Bacillus stearothermophilus binding its native substrate glycerol (pink sticks/PDB ID: 1JQA), the catalytic Zn2+ (blue sphere/PDB ID: 1JQ5) and the NAD+ (yellow sticks/PDB ID: 1JQ5). Residues V131, Y142 and L252 that form the substrate cavity are highlighted as green spheres. Black dash line represents the hydride transfer from C2 atom of glycerol to C4 atom of NAD+ (see Fig. 2). Yellow dash lines represent the coordination of O1 and O2 atoms from glycerol with the Zn2+ atom. | ||
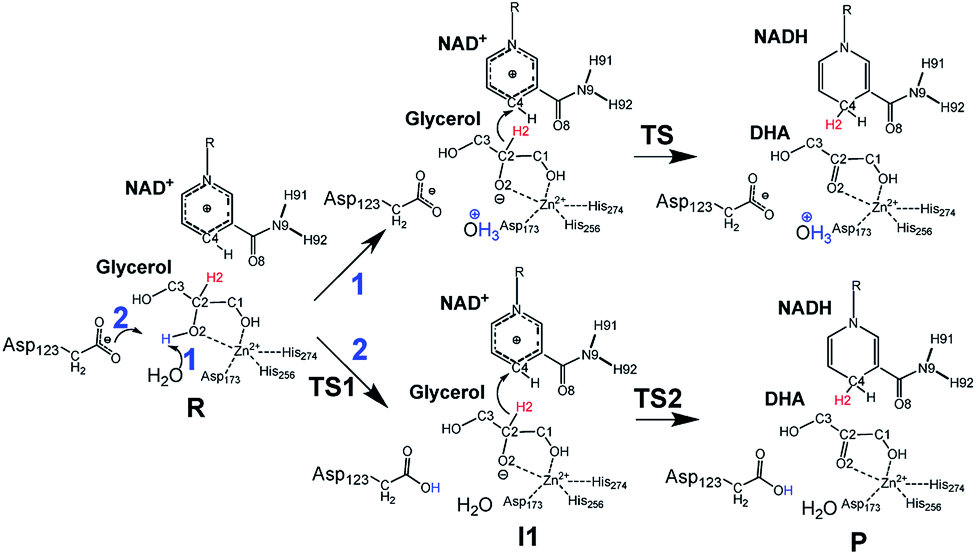 | ||
| Fig. 2 Proposed glycerol oxidation mechanisms for Wt-BsGlyDH. (1) Proton transfer through water molecule. (2) Proton transfer through the Asp123. R: glycerol/TS: transition state for mechanism 1/TS1 and TS2 transition state for mechanism 2/I1: alkoxide intermediate/P: dihydroxyacetone. | ||
According to the results, mechanism 1 can be discarded due to the resulting overall high free energy barrier (57.6 kcal mol−1), indicating that water is unable to act as base for the proton subtraction from the O2 hydroxyl group (for further details see ESI, Fig. S5 and S6A‡). Furthermore, our M06-2X/6-31+G(d,p):PM3/MM free energy surfaces and M06-2X/6-31+G(d,p)/MM potential energy surfaces confirms that mechanism 2 takes place in a stepwise manner (Fig. S5 and S6B‡), consisting in the proton abstraction of the alcohol by D123 residue followed by the hydride transfer from glycerol to NAD+. This second step, that is the rate-limiting one, renders an activation free energy (17.1 kcal mol−1) that agrees with the values that can be derived from the experimental rate constants (16.9 kcal mol−1).§ To experimentally confirm the mechanism 2, we constructed the mutant D123N that keeps the hydrogen bond interactions between glycerol and the amide group of D123N without affecting the size but precluding the formation of the alkoxy intermediate, as the amide group is unable to subtract the proton. D123N mutant indeed abolished the oxidative activity (Fig. S7‡), which agrees with the analysis of the geometries derived from classical MD simulations that show no alternative base in the active site. Hence, we propose the carboxylate of D123 as the base needed for abstracting the proton to form the alkoxy intermediate that precedes the hydride transfer to the nicotinamide cofactor (Fig. 2). For the first time, we present both experimental and computational data that unambiguously unveil the key role of D123 in the catalytic mechanism of a glycerol dehydrogenase (Fig. 2, mechanism 2, Fig. S5‡). This residue is highly conserved in GlyDHs and glycerol 1-phosphate dehydrogenases (Fig. S8 and S9‡), which supports its important role in the catalytic mechanism. Unlike mannitol dehydrogenases52 (medium chain dehydrogenases) and short-chain dehydrogenases,50 where Lys and Tyr residues acts as general base, respectively, BsGlyDH uses the carboxylate of D123 to subtract the proton from the secondary alcohol of glycerol.53–56 This conserved residue is not found in iron-containing polyol dehydrogenases, like 1,2-propanodiol dehydrogenase.57
Once the oxidation mechanism has been elucidated, we rationally selected three positions to sculpt the active site of BsGlyDH to better accommodate bulkier glycerol derivatives. We performed an alanine scanning in the positions V131, Y142 and L252 since they sterically confine the C3 hydroxyl group within the enzyme binding pocket according to the X-ray structure (Fig. 1). We also constructed the three possible combinations of double mutants and the triple one. By replacing these bulkier amino acids by alanine, we were able to expand the solvent accessible area of the active site up to 108 Å2 (Table S4‡). Nevertheless, there was no correlation between the size of the binding pocket and the activity towards the glyceryl ethers (Fig. S10‡). All mutations dramatically reduced the enzyme activity (<10%) towards the native substrate 1a (glycerol) except BsGlyDH-V131A (Fig. 3). On the other hand, the mutation Y142A conducted to inactive mutants towards any tested substrate regardless they were single, double or triple variants. Pleasantly, the variant BsGlyDH-L252A presented a remarkable activity towards 1c, even 30% higher than the oxidative activity of the Wt-GlyDH towards glycerol. For all those active mutants, we studied their substrate scope using a palette of alkyl/aryl glyceryl monoethers (Table S1‡). BsGlyDH-L252A was the most active variant towards all alkylated glycerol derivatives, while negligible activity was detected for the 1,3-dimethoxy (1b-bis) and 1,3-diethoxy (1c-bis) glyceryl derivatives. The absence of activity towards the glyceryl 1,3-dialkylethers agrees with the fact that GlyDH requires one free primary hydroxyl group to be coordinated with the catalytic Zn2+. Intriguingly, widening the active site did not enhance the oxidative activity towards aryl derivatives. In fact, we found out that both wild-type and the L252A variant were similarly active towards 1h.
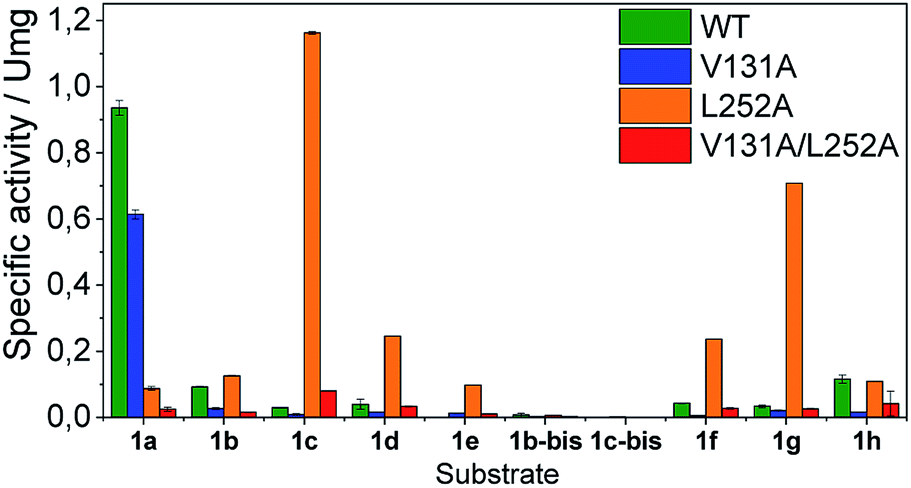 | ||
| Fig. 3 Specific activity of wild-type and several mutants of BsGlyDH towards alkyl/acylglyceryl ethers. In all cases, reaction mixtures consisted in 100 mM substrate, 1 mM NAD+ in 100 mM sodium phosphate buffer at pH 7 at 30 °C, 1b-bis and 1c-bis are the corresponding dialkyl-ether with methyl and ethyl substituents, respectively. | ||
The second best substrate was 1g bearing a 2,2,2-trifluorethyl substituent, which supports that the 22 Å2 larger active site also possesses the suitable shape to accommodate CH2CF3 moieties (Table S4‡). Surprisingly, when the aliphatic chain was either smaller or larger than 2 carbons, the enzyme activity dramatically decayed (Fig. 3). The specific activity results were supported by kinetic studies (Table 1, Fig. S11 and S12‡), which additionally demonstrate that shortening the apolar side chain at position 252 dramatically increases the KM towards glycerol without significantly affecting the kcat. In contrast, mutation L252A significantly increases (6 times) the BsGlyDH-L252A catalytic efficiency towards 1c due to the kcat enhancement (163 times). This variant showed similar trends towards all alkyl glyceryl derivatives except 1e, for which the kcat was significantly lower compared to other substituents (Table 1). BsGlyDH-L252A presented similar KM values towards NAD+ under the same conditions using both glycerol and 1c as substrates, which indicates that the mutation clearly affects the binding of the polyol rather than the cofactor (Fig. S13C–F‡). Using glycerol as substrate at pH 7, we also observed that both wild-type and mutant enzymes were inhibited by NAD+ at high concentration (>10 mM) (Fig. S13A–D‡). In the specific case of BsGlyDH-L252A, inhibition negligibly occurred when using 1c as substrate under neutral conditions (Fig. S13E and F‡), but it was noticeable at pH 9 (Fig. S13G and H‡). Remarkably, the catalytic efficiency of BsGlyDH-L252A was 40 times higher under alkaline conditions than under pH 7, and its optimal pH was shifted 1 unit towards basic values compared to the wild-type enzyme (Fig. S14‡). These kinetics studies reveal an important role of pH on the glycerol dehydrogenase performance that affects both binding (overall for the cofactor) and reaction rate constant.58Fig. 4, S15 and S16‡ show the free energies profiles and the hydride transfer transition states (named TS2, see Fig. 2) resulted from QM/MM simulations, for the oxidation reaction of different substrates catalysed by either the Wt-BsGlyDH or the BsGlyDH-L252A mutant. Energy barriers follow the same trend observed for the experimental kinetics shown in Table 1. This mutant presents an energy barrier of 16.2 and 16.5 kcal mol−1 for both 1c (Fig. 4A) and glycerol (Fig. S16‡) oxidations, respectively. These values are slightly smaller than the barrier previously obtained with the wild-type enzyme using glycerol as substrate (17.1 kcal mol−1) (Fig. S5‡). More interestingly, the free energy profile of Wt-BsGlyDH shows an energy barrier 2.6 kcal mol−1 higher than BsGlyDH-L252A towards the oxidation of 1c under the same conditions (Fig. 4A), in accordance with the experimental free energy barriers derived from the kcat values (Tables 1, S5‡). We suggest that the higher efficiency of engineered BsGlyDH-L252A results from a less sterically impeded 1c conformation within the mutated active site, which leads to transition states with lower energy barriers than within native one. QM/MM MD simulations revealed weak but stabilizing interactions energies between 1c and the residues at positions 131, 142 and 252 of BsGlyDH-L252A during the reactant state. Their synergistic Lennard-Jonnes interactions significantly contribute to the 1c binding (Fig. S17 and S18‡). Therefore, the insertion of Ala at position 252 but keeping the native residues at positions V131 and Y142 seems to optimise the binding pocket to improve the stability of the transition states with the ethyl glyceryl monoether. Fig. 4B shows indeed a rotation (125 degrees) for the CH3- of ethoxy substituent of 1c within the less hindered L252A active site. Similar results were found for the rational site-directed mutagenesis of a shuffled GlyDH, where the replacement of one aspartic that clashed with 1,3-butanodiol enhanced the oxidative activity up to 2.6 times compared to the wild-type variant.32
| Substrate | K M (mM) | V max (U mg−1) | k cat (s−1) | k cat/KM (M−1 s−1) | ||||
|---|---|---|---|---|---|---|---|---|
| Wt | L252A | Wt | L252A | Wt | L252A | Wt | L252A | |
| a Activity assay: 1 mM NAD+ in 100 mM sodium phosphate buffer pH 7 at 30 °C. n.a: not assessed because it did not reach the substrate saturation. | ||||||||
| 1a | 50 ± 3 | 1880 ± 80 | 1.40 ± 0.04 | 1.70 ± 0.05 | 7.4 ± 0.2 | 9.0 ± 0.3 | 148 | 5 |
| 1b | 45 ± 4 | 977 ± 131 | 0.160 ± 0.003 | 1.25 ± 0.06 | 0.84 ± 0.02 | 6.6 ± 0.3 | 19 | 7 |
| 1c | 13.4 ± 0.4 | 371 ± 71 | 0.036 ± 0.001 | 6.0 ± 0.5 | 0.190 ± 0.003 | 31 ± 3 | 14 | 85 |
| 1d | 30 ± 1 | 608 ± 102 | 0.052 ± 0.003 | 2.2 ± 0.2 | 0.27 ± 0.02 | 12 ± 1 | 9 | 19 |
| 1e | n.a | 162 ± 24 | n.a | 0.24 ± 0.02 | n.a | 1.3 ± 0.1 | n.a | 8 |
| 1f | 31.2 ± 0.3 | 707 ± 77 | 0.057 ± 0.001 | 2.4 ± 0.1 | 0.300 ± 0.001 | 13 ± 1 | 10 | 18 |
| 1g | 78 ± 8 | 281 ± 24 | 0.06 ± 0.02 | 2.8 ± 0.1 | 0.32 ± 0.01 | 14 ± 1 | 4 | 51 |
| 1h | 7 ± 2 | n.a | 0.14 ± 0.01 | n.a | 0.7 ± 0.1 | n.a | 112 | n.a |
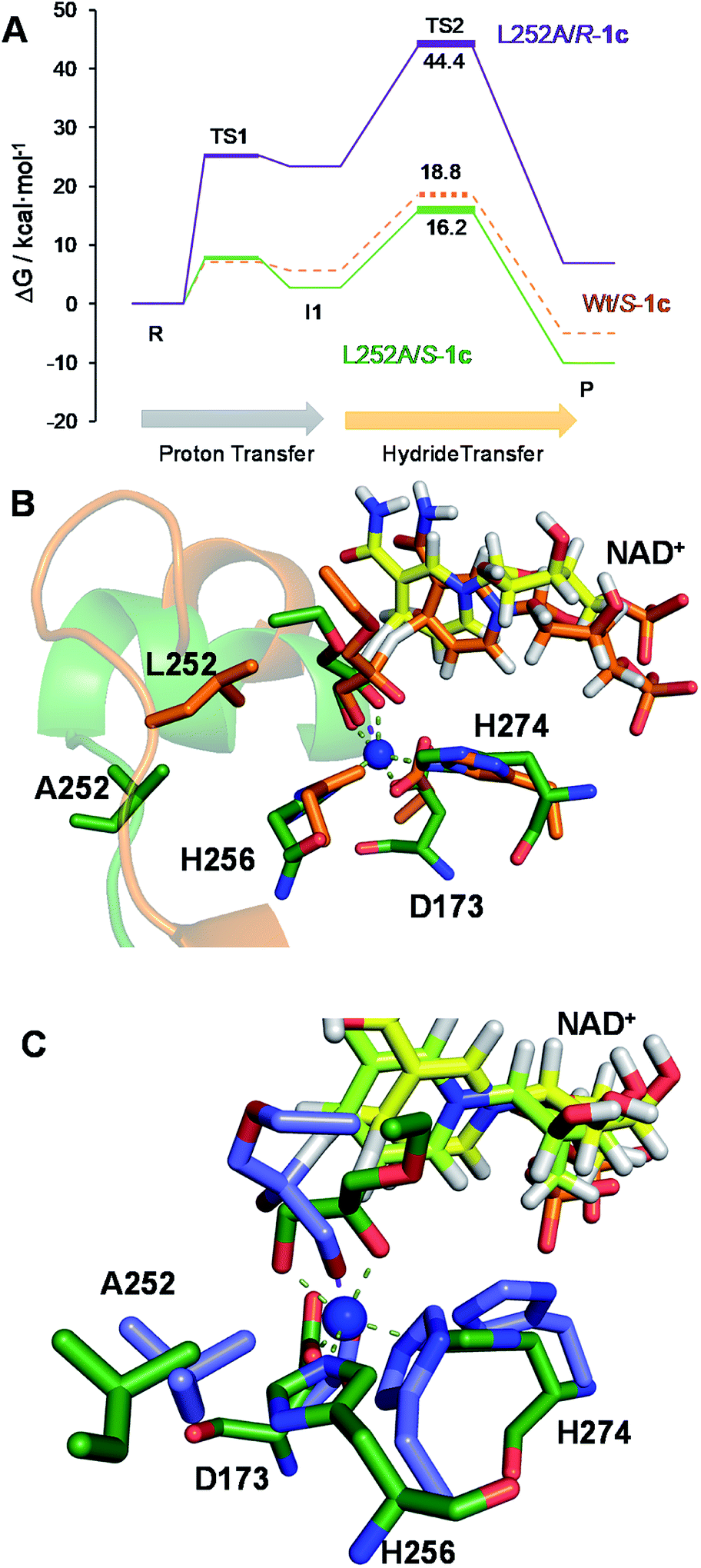 | ||
| Fig. 4 (A) Free energy profiles (M06-2X/6-31+G(d,p):PM3/MM PMFs + ZPE correction) following mechanism 2 for the S-1c oxidation catalysed by Wt-BsGlyDH (dashed orange line) and BsGlyDH-L252A (solid green line), and of R-1c oxidation catalysed by BsGlyDH-L252A (solid purple line). (B) Overlapped snapshot of the hydride transfer transition states (TS2) located in the oxidation of S-1c catalysed by the Wt-BsGlyDH (orange) and BsGlyDH-L252A (green). NAD+ bound to the complex of Wt and L252A mutant was coloured in orange and yellow, respectively. (C) Overlapped snapshots of the hydride transfer transition state (TS2) located in the oxidation of S-1c (green) and R-1c (purple) by BsGlyDH-L252A. NAD+ bound to the complex with S and R isomers was coloured in yellow and pale green, respectively. | ||
The excellent oxidative activity of BsGlyDH-L252A encouraged us to carry out the complete bioconversion of 1c and characterise the resulting product. Since this biotransformation is NAD+-dependent, we mixed BsGlyDH-L252A with an NADH oxidase from Thermus thermophilus HB27 (Tt-NOX) and the horse liver catalase (Hl-CAT) in one-pot, aiming at both in situ recycling the cofactor and removing the H2O2; a toxic by-product from the recycling system. Using the soluble enzymes, the reaction roughly reached 25% substrate conversion in 72 hours with enzyme and cofactor total turnover numbers of 3491 and 13, respectively (Table 2, entry 1). Despite that low conversion, we characterised the formed product through 13C and 1H NMR (Fig. S19–S22‡), and mass spectrometry (Fig. S23–S29‡). The analyses confirm that BsGlyDH-L252A oxidises the C2 of 1c as the wild-type GlyDH does for the glycerol. Remarkably, we did not detect the formation of the aldehyde, which indicates that the mutation at the active site alters the substrate specificity but not the enzyme regioselectivity. Although product yields were significantly lower than those ones obtained through chemical methods,59 the regioselectivity of this variant towards the C2 of the alkylglyceryl substrate thus enables the synthesis of the corresponding 3-alkoxy-1-hydroxyacetones under mild conditions using water as solvent. More importantly, the enzymatic route smoothly runs without protection/deprotection steps under acidic conditions that would provoke the decomposition of the product to methyl glyoxal, substantially decreasing the isolated yields.59
| Entry | Biocatalyst | Load (mg) | Conv./time (%)/(h) | TOFc (h−1) | TTNenzymed | TTNNAD+e |
|---|---|---|---|---|---|---|
|
a Reaction conditions; 18 mU of GlyDH-L252A (soluble or immobilised), 90 mU of soluble Tt-NOX and 4480 mU of soluble Hl-CAT mixed with 1.5 mL of 50 mM of rac-1c, 1 mM NAD+, 150 μM FAD+ in 100 mM sodium phosphate buffer at pH 7 at 30 °C.
b Reaction conditions; 144 mU of immobilised GlyDH-L252A, 720 mU of Tt-NOX and 36000 mU of Hl-CAT CAT mixed with 0.3 mL of 50 mM rac-1c, 1 mM NAD+, 150 μM FAD+ in 100 mM sodium phosphate buffer at pH 7 at 30 °C.
c Turnover frequency (TOF) was calculated as μmol of product per μmol of enzyme in 24 hours.
d TTNenzyme was calculated as μmol of product per μmol of enzyme.
e TTNNAD+ was calculated as μmol of product per μmol of cofactor.
|
||||||
| 1 | Soluble | 0.24a | 25 (72) | 617 | 25000 |
13 |
| 2 | AG-Co2+ | 0.186a | 50 (72) | 1433 | 64000 |
25 |
| 3 | 1.48b | 50 (24) | 68 | 1600 | 25 | |
Biotransformation of alkyl/aryl glyceryl ethers using immobilised GlyDH-L252A
In order to intensify the process and walk towards higher yields, we immobilised BsGlyDH-L252A on agarose porous microbeads activated with cobalt-chelates (AG-Co2+) to enhance its operational stability and increase the biocatalyst volumetric activity. Furthermore, the use of the immobilised enzyme will ease the product recovery and enable the enzyme reutilization once the reaction is completed. Since all BsGlyDH variants used in this work were tagged with 6× His polypeptide at their N-terminus for purification purposes, we harnessed that tag to site-selectively immobilise the BsGlyDH-L252A on AG-Co2+. This variant was quantitatively immobilised on this carrier; however, the specific activity of the immobilised enzyme was reduced to 19% compared to its free counterpart under colorimetric assay conditions (Table S6‡). The immobilised BsGlyDH-L252A performed the selective oxidation of 1c with a TOF 2.4-fold higher than the soluble enzyme, reaching a maximum yield of 50% after 72 h under batch conditions (Table 2, entry 2). This heterogeneous biocatalyst enabled an enzyme and cofactor total turnover numbers of 64 × 103 and 25, respectively, under the studied conditions.These data demonstrate that immobilised BsGlyDH-L252A mixed with soluble Tt-NOX and Hl-CAT outperforms the system with the three soluble enzymes. The apparent lower activity of the immobilised enzyme seems to be compensated by its higher robustness under operational conditions.60 The immobilization of BsGlyDH-L252A allowed us increasing 8 times the volumetric activity of the heterogeneous biocatalyst, reaching 50% conversion in only 24 h (Table 2, entry 3).
From the reaction time courses, we observed how the oxidation reaction slows down when the conversion approaches to 50% (Fig. 5A). Since we used 25 mM racemic mixture of 1c, that asymptotic behavior points out that BsGlyDH-L252A is extraordinarily selective for one of the two enantiomers. To unveil the enantioselectivity of this variant, we performed further QM/MM computational studies on the two isomers of 1c. The free energy barriers for the oxidation of S-1c (16.16 kcal mol−1) was significantly lower than that for the R-enantiomer (44.40 kcal mol−1) (Fig. 4A), indicating that the engineered BsGlyDH variant prefers oxidizing the S-enantiomer. Similar results were observed for the wild-type enzyme (Fig. S15 and Table S5‡), suggesting that the native active site also prefers the S-configuration of the substrate for the hydride transfer to the NAD+. Fig. 4C illustrates an unproductive position of R-1c as the secondary alcohol loses its coordination with the catalytic Zn2+. The same predicted enantiopreference for both engineered and native variants suggests that the L252A mutation aids the accommodation of larger glycerol-like substrates without affecting the regio- and stereoselectivity of the enzyme. This S-enantiopreference of BsGlyDH agrees with the enantioselectivity reported for archeal glycerol-1-phosphate dehydrogenase,61 suggesting a pro-R position of the NAD+ within the active site. GlyDHs from Enterobacter aerogenes and from Cellulomonas sp. selectively oxidise the R-isomer of 1,2-diols, kinetically resolving the corresponding S-enantiomers of phenylglycol (ee 99%, conv. 50%)58 and 1,2-propanodiol (ee 36%, conv. 26%),49 respectively. The opposite CIP priority between 1c and non alkoxylated 1,2-diols indicates that the diols are similarly coordinated and positioned within the enzyme active sites of those GlyDHs, and the hydride from the substrates is also transferred to Re-face of the NAD+. Emboldened by these computational insights, we challenged the immobilised BsGlyDH-L252A for the kinetic resolution of rac-1h (Figs. S29–S33‡). Fig. 5B shows that the reaction time course follows the classical kinetic resolution pattern where the S-enantiomer is fully oxidised to 3-phenoxy-1-hydroxyacetone, while the R-enantiomer remains nonreactive (Fig. S33‡). This result confirms the computational studies and thus demonstrates that BsGlyDH-L252A is an S-stereoselective polyol dehydrogenase, enriching the reaction crudes with R-1h (ee > 99%) after 72 hours. Since we exploited BsGlyDHA-L252A to selectively oxidise alkyl and aryl glyceryl monoethers, we are able to readily prepare the pure R-enantiomer of glycerol monoethers in one-pot under mild conditions. To the best of our knowledge, the only reported biotransformations able to produce this enantiomer is based on asymmetric reduction of hydroxyacetone monoethers through whole cells of different yeasts, but with significantly lower enantiopurity (ee = 73–88%).62 Beside the substrate specificity and exquisite enantioselectivity of BsGlyDH-L252A, its immobilization on solid carriers enables to reutilise this biocatalyst in consecutives operational cycles. Fig. 6 shows that the immobilised enzyme retains its activity during 6 operational cycles (a total of 6 days of operation). Glycerol dehydrogenases have been immobilised on a plethora of carriers through a variety of chemistries to stabilise them for further applications (mainly in the field of biosensors).63 Recently, Xu et al. co-aggregated and cross-linked a wild-type glycerol dehydrogenase together with a NADH oxidase (enzyme sources were not reported) for the transformation of glycerol into dihydroxyacetone.64 This system was able to produce up to 10 mM of product and proven reusable for 10 cycles maintaining 60% of its initial activity. In this work, we achieved up to 33 mM of 3-ethoxy-1-hydroxyacetone using 100 mM of substrate, yet the conversion was limited to 33%. A lower conversion than 50% suggests product inhibition issues, only manifested when using high substrate concentrations (>25 mM). In fact, BsGlyDH-L252A underwent a 50% activity decreasing when incubated at 100 mM dihydroxyacetone (Fig. S34‡), suggesting that BsGlyDH-L252 is also inhibited by hydroxyketones. Remarkably, the oxidation of rac-1c catalysed by the single mutant was inhibited by the ketone to a much lower extent than the glycerol oxidation performed by the wild-type enzyme. These results are aligned with other selective bio-oxidation catalysed by GlyDHs that also suffer inhibition issues.49,65 Such product inhibition can be suppressed through continuous product extraction to complete the kinetic resolutions at high substrate concentrations.65 According to these results, the application of immobilised GlyDH to the kinetic resolution of the alkyl glyceryl monoethers and the synthesis of 3-alkoxy-1-hydroxyacetone is one of the few examples in applied biocatalysis that use polyol dehydrogenases beyond the production of dihydroxyacetone. Nevertheless, this heterogeneous biocatalyst needs to be further optimised in order to overcome inhibition issues and increase its operational stability.
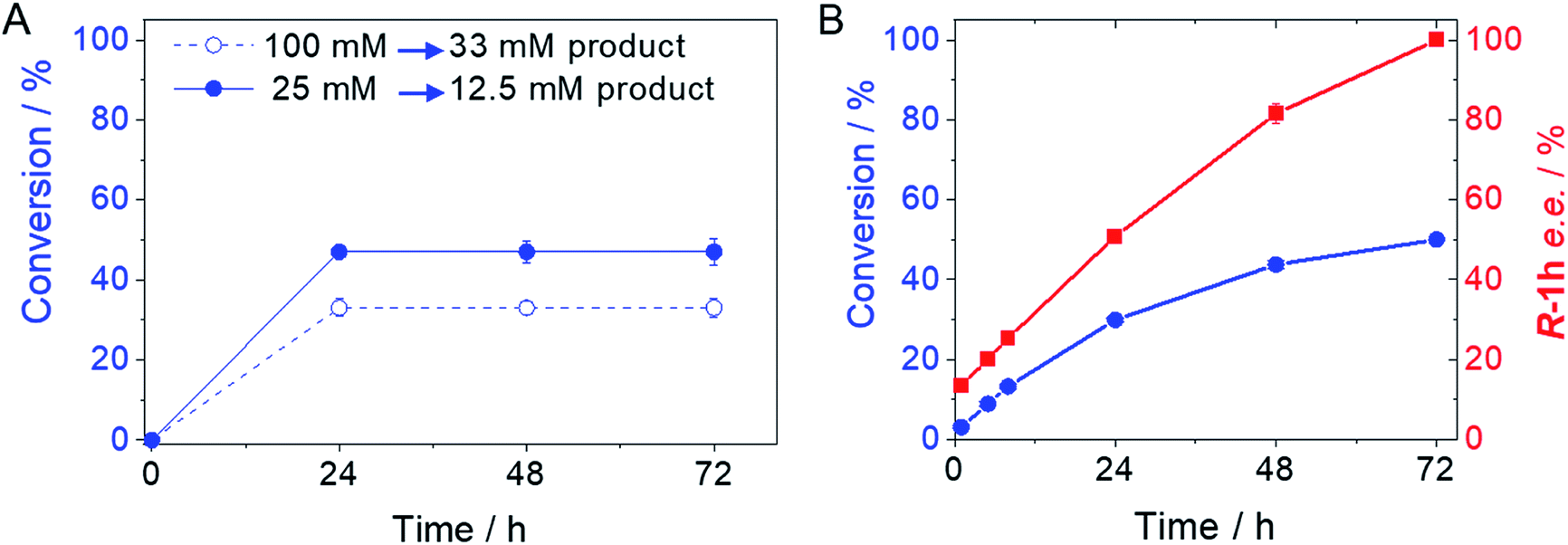 | ||
| Fig. 5 Time courses of the selective oxidation of rac-1c (A) and rac-1h (B) using BsGlyDH-L252A immobilised on AG-Co2+. For rac-1c, 144 mU of immobilised GlyDH-L252A, 720 mU of Tt-NOX and 36000 mU of Hl-CAT mixed with 0.3 mL of 25–100 mM substrate, 1 mM NAD+, 150 μM FAD+ in 100 mM sodium phosphate buffer at pH 7 at 30 °C. For rac-1h, 8.8 U of immobilised GlyDH-L252A were mixed with 1 mL of 2 mM substrate, 4 mM NAD+ in 100 mM sodium phosphate buffer at pH 7 at 30 °C. | ||
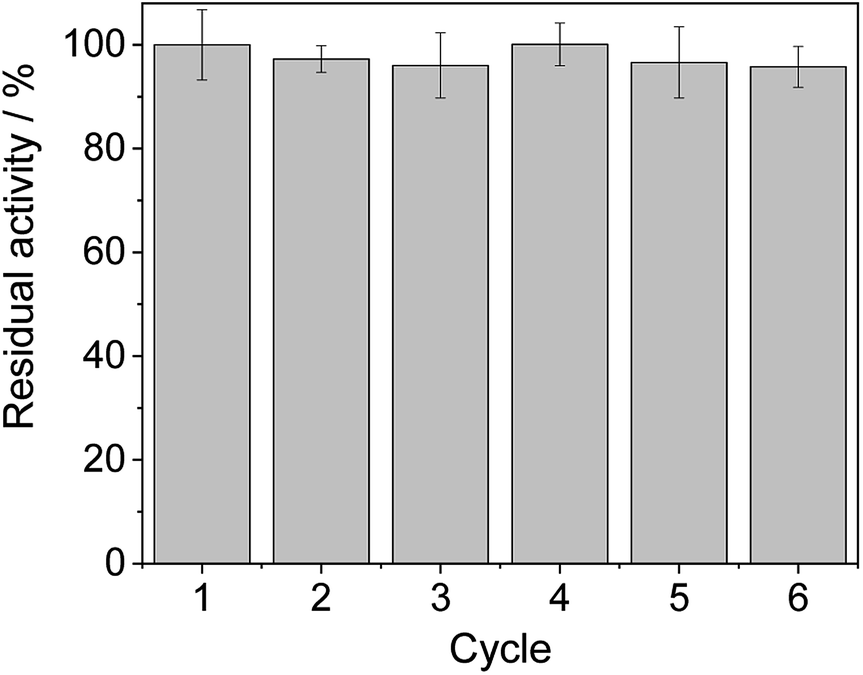 | ||
| Fig. 6 Recycling of heterogeneous biocatalyst BsGlyDH-L252A immobilised on AG-Co2+ (144 mU, 100 mg biocatalyst). Reaction mixture consisted in 300 μL of 25 mM of rac-1c, 1 mM NAD+, 150 μM FAD+, 720 mU of soluble Tt-NOX and 36000 mU of soluble Hl-CAT in 100 mM sodium phosphate buffer at pH 7 at 30 °C. Each cycle corresponds to 24 h at the described conditions. | ||
One-pot sequential chemo-enzymatic synthesis of 3-ethoxy-1-hydroxyacetone starting from ethanol and glycidol
To advance in the biorefinery concept, we addressed the synthesis of 3-ethoxy-1-hydroxyacetone using ethanol and racemic glycidol as raw materials (Scheme 1C). To that aim, we prepared a bi-functional heterogeneous biocatalyst through the selective co-immobilization of His-tagged GlyDH-L252A and the water-forming NAD+ oxidase from Lactobacillus pentosus (Lp-NOX) on AG-Co2+ porous microbeads. We swapped Tt-NOX by Lp-NOX, because the latter is a water-forming oxidase which makes unnecessary the use of CAT for the in situ removal of H2O2, simplifying the overall process. Furthermore, co-immobilised LpNOX allowed 14% higher conversion during the oxidation of 1c, using 2.2 times lower biocatalyst load (16% mass load including the carrier) than using the pair TtNOX/CAT immobilized on agarose porous beads functionalized with aldehyde groups (36% mass load) (Fig. S35‡).Firstly, we performed the alcoholysis of glycidol with an excess of ethanol and using KOH as basic catalyst.18 After the chemical step and using the same pot, the reaction crude was neutralised, cooled down to 25 °C and properly mixed with the reaction mixture (1 mM NAD+, 150 μM FAD+ in 100 mM sodium phosphate buffer at pH 8), diluting the substrate concentration to 20 mM. Finally, the bi-enzyme heterogeneous biocatalyst was directly added to the reaction crude to trigger the sequential enzymatic step. Fig. 7 shows that the diluted and neutralised crude coming from the chemical reaction can be readily used for the enzymatic selective oxidation without affecting the reaction efficiency. In all the cases, we obtained similar conversions, even in presence of the excess of ethanol needed for the chemical alcoholysis. This batch system was scaled up to 50 mL (120 mg of 1c) of reaction mixture reaching similar conversion (Fig. S36‡). Although desired, a concurrent process is impeded due to the basic conditions demanded by the epoxide ring opening that completely inactivates the enzymes participating in the selective oxidation. However, we herein demonstrate that the sequential chemo-enzymatic cascade in one-pot successfully works without any purification step. The alcoholysis/selective oxidation (kinetic resolution) herein presented, contributes to expand the repertoire of chemo-enzymatic processes for manufacturing of enantiomerically pure secondary alcohols as pharmaceutical66,67 and polymer building blocks.68–70
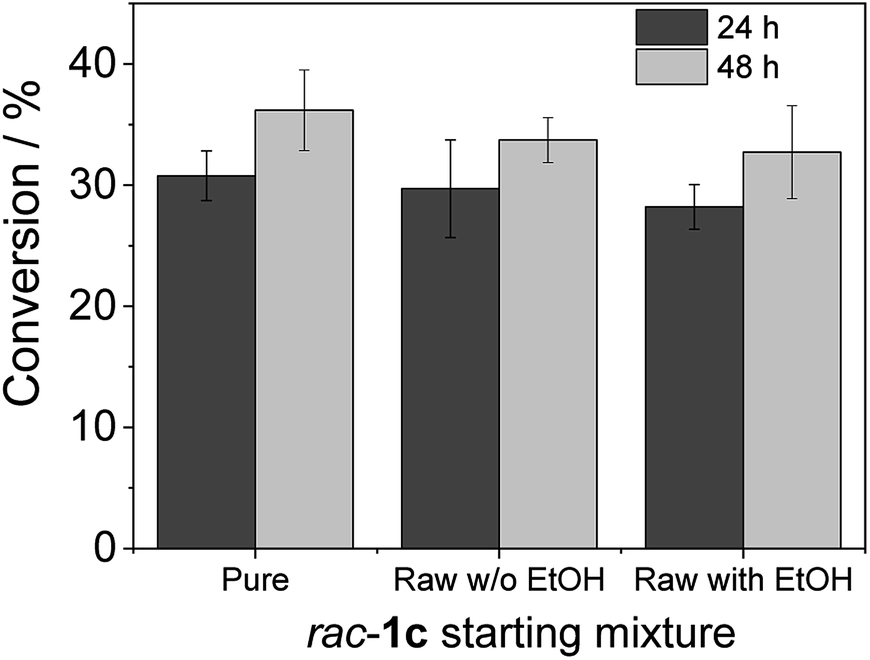 | ||
| Fig. 7 Chemo-enzymatic synthesis of 3-alkoxy-1-hydroxyacetone. Heterogeneous biocatalyst GlyDH-L252A co-immobilised with Lp-NOX on AG-Co2+ (100 mg biocatalyst). For the enzymatic step, the reaction mixture consisted in 500 μL of 20 mM of substrate (pure, raw from chemo synthesis with ethanol and raw from chemo synthesis without ethanol), 1 mM NAD+, 150 μM FAD+ in 100 mM sodium phosphate buffer at pH 8 at 30 °C. | ||
Conclusions
In this work, we have deciphered the oxidative catalytic mechanism of a GlyDH from Bacillus stearothermophilus towards polyols substrates through QM/MM molecular dynamics simulations. Our results show how the reaction takes place by means of a stepwise mechanism that consists in a proton transfer from the hydroxyl group of C2 atom of the alcohol to D123 residue to form the alkoxide intermediate followed by the hydride transfer from C2 atom of the alcohol to NAD+. Based on these mechanistic insights, we rationally engineered the BsGlyDH to regio- and enatioselectively oxidise glycerol monoethers. We found that a single mutation in its active site (L252A) provides enough space to accommodate bulkier glycerol derivatives in productive configurations. This variant was able to selectively oxidise a battery of glyceryl monoethers with different alkyl/aryl substituents. Besides the activity towards these substrates, we also report the exquisite enantiopreference of BsGlyDH towards S-isomers of substituted glycerol derivatives. After being immobilised on porous carriers, the best engineered variant serves as an efficient heterogeneous biocatalyst for the kinetic resolution of glyceryl monoethers and the production of the 3-alkoxy-1-hydroxyacetones. Herein, we have also contributed to enlarge the toolbox of the one-pot chemo-enzymatic processes through coupling the base-catalysed ethanolysis of glycidol with the enzymatic selective oxidation of 3-ethoxy-propan-1,2-diol in a sequential mode. All the results presented here support an interdisciplinary path that goes from the molecular design of enzymes to the process engineering and development for the manufacturing of products, like 3-alkoxy-1-hydroxyacetones. These compounds have been synthesised before neither through biocatalytic methods nor following a one-pot sequential reaction starting from bio-based materials (ethanol and glycidol). So far, the kinetic resolution of these glycerol derivatives has an impact in applied biocatalysis, but moving this system towards dynamic kinetic resolution to achieve 100% yield is a future challenge. Furthermore, we foresee that new engineering campaigns will expand the GlyDH substrate scope and change its enantiopreference. Likewise, the immobilization of GlyDH must be optimised to further increase the operational stability of this heterogeneous biocatalyst.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Ministerio de Ciencia, Innovación y Universidades (Spanish government) funded FLG (RTI2018-094398-B-I00 and RED2018-102403-T), VM (PGC2018-094852-B-C21), MR (RYC-2014-16592), and EP and JAM (RTI2018-093431-B-I00). Generalitat Valenciana funded VM (AICO/2019/195). Universitat Jaume I has funded VM (UJI-A2019-04) and MR (UJI-B2019-43). Gobierno de Aragón co-funded by FEDER 2014–2020 “Construyendo Europa desde Aragón” have funded EP, JAM, FLG, ALD and SV (Group E37_20R). IKERBASQUE and ARAID foundations have funded the contribution of FLG. The Mexican Council of Science and Technology (CONACyT) has funded SV (2017/2019-postdoctoral fellowship). Ministerio de Educación, Cultura y Deporte has funded ALD (FPU014/04338). The authors acknowledge the computational resources of the Servei d’Informàtica of Universitat Jaume I. This work was performed under the Maria de Maeztu Units of Excellence Programme – Grant No. MDM-2017-0720 Ministerio de Ciencia, Innovación y Universidades.References
- R. Whetten and R. Sederoff, Plant Cell, 1995, 7, 1001–1013 CrossRef CAS.
- H. Goldfine and T. A. Langworthy, Trends Biochem. Sci., 1988, 13, 217–221 CrossRef CAS.
- T. C. Lee, B. Malone and F. Snyder, J. Biol. Chem., 1986, 261, 5373–5377 CAS.
- C. O. Rock and F. Snyder, J. Biol. Chem., 1974, 249, 5382–5387 CAS.
- H. Taguchi and W. L. F. Armarego, Med. Res. Rev., 1998, 18, 43–89 CrossRef CAS.
- K. Watschinger and E. R. Werner, IUBMB Life, 2013, 65, 366–372 CrossRef CAS.
- J. F. Izquierdo, M. Montiel, I. Palés, P. R. Outón, M. Galán, L. Jutglar, M. Villarrubia, M. Izquierdo, M. P. Hermo and X. Ariza, Renewable Sustainable Energy Rev., 2012, 16, 6717–6724 CrossRef CAS.
- J. I. García, H. García-Marín and E. Pires, Green Chem., 2014, 16, 1007–1033 RSC.
- A. A. Bredikhin, Z. A. Bredikhina, D. V. Zakharychev and A. V. Pashagin, Tetrahedron: Asymmetry, 2007, 18, 1239–1244 CrossRef CAS.
- D. Cauwet and C. Dubief, France Pat., EP0555155A1, 1992.
- C. Goedl, T. Sawangwan, M. Mueller, A. Schwarz and B. Nidetzky, Angew. Chem., Int. Ed., 2008, 47, 10086–10089 CrossRef CAS.
- J. R. M. Almeida, L. C. L. Fávaro and B. F. Quirino, Biotechnol. Biofuels, 2012, 5, 48 CrossRef CAS.
- Z. Gholami, A. Z. Abdullah and K.-T. Lee, Renewable Sustainable Energy Rev., 2014, 39, 327–341 CrossRef CAS.
- A. Leal-Duaso, P. Pérez, J. A. Mayoral, E. Pires and J. I. García, Phys. Chem. Chem. Phys., 2017, 19, 28302–28312 RSC.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS.
- M. Sutter, E. D. Silva, N. Duguet, Y. Raoul, E. Métay and M. Lemaire, Chem. Rev., 2015, 115, 8609–8651 CrossRef CAS.
- A. Leal-Duaso, M. Caballero, A. Urriolabeitia, J. A. Mayoral, J. I. García and E. Pires, Green Chem., 2017, 19, 4176–4185 RSC.
- A. C. O. Machado, A. A. T. da Silva, C. P. Borges, A. B. C. Simas and D. M. G. Freire, J. Mol. Catal. B: Enzym., 2011, 69, 42–46 CrossRef CAS.
- C. Solarte, M. Balcells, M. Torres, N. Sala and R. Canela-Garayoa, RSC Adv., 2014, 4, 34623–34631 RSC.
- J. Dong, E. Fernández-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS.
- D. Güclü, A. Szekrenyi, X. Garrabou, M. Kickstein, S. Junker, P. Clapés and W.-D. Fessner, ACS Catal., 2016, 6, 1848–1852 CrossRef.
- P. Buehle and T. Rudolph, Germany Pat., PCT/EP2011/006006, 2012.
- J. Yan, J. Xu, M. Cao, Z. Li, C. Xu, X. Wang, C. Yang, P. Xu, C. Gao and C. Ma, Microb. Cell Fact., 2018, 17, 158 CrossRef CAS.
- N. E. Altaras and D. C. Cameron, Appl. Environ. Microbiol., 1999, 65, 1180–1185 CrossRef CAS.
- A. Mahadevan and S. Fernando, Biosens. Bioelectron., 2017, 92, 417–424 CrossRef CAS.
- S. Wang, J. Wang, X. Zhou, Y. Guo and B. Fang, Biotechnol. Bioprocess Eng., 2013, 18, 796–800 CrossRef CAS.
- Z.-B. Sun, Z.-J. Zhang, F.-L. Li, Y. Nie, H.-L. Yu and J.-H. Xu, ChemCatChem, 2019, 11, 3802–3807 CrossRef CAS.
- O. F. Brandenberg, K. Chen and F. H. Arnold, J. Am. Chem. Soc., 2019, 141, 8989–8995 CrossRef CAS.
- X. Garrabou, L. Gómez, J. Joglar, S. Gil, T. Parella, J. Bujons and P. Clapés, Chem.–Eur. J., 2010, 16, 10691–10706 CrossRef CAS.
- S. Junker, R. Roldan, H. J. Joosten, P. Clapés and W. D. Fessner, Angew. Chem., Int. Ed., 2018, 57, 10153–10157 CrossRef CAS.
- H. Zhang, G. T. Lountos, C. B. Ching and R. Jiang, Appl. Microbiol. Biotechnol., 2010, 88, 117–124 CrossRef CAS.
- J. Löwe, A. A. Ingram and H. Gröger, Bioorg. Med. Chem., 2018, 26, 1387–1392 CrossRef.
- W. Finnigan, J. Citoler, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2020 DOI:10.1021/acs.oprd.0c00075.
- J. M. Bolivar and F. López-Gallego, Current Opinion in Green and Sustainable Chemistry, 2020, 25, 100349 CrossRef.
- A. Leal-Duaso, P. Pérez, J. A. Mayoral, J. I. García and E. Pires, ACS Sustainable Chem. Eng., 2019, 7, 13004–13014 CrossRef CAS.
- J. Rocha-Martin, A. Acosta, J. Berenguer, J. M. Guisan and F. Lopez-Gallego, Bioresour. Technol., 2014, 170, 445–453 CrossRef CAS.
- C. Nowak, B. Beer, A. Pick, T. Roth, P. Lommes and V. Sieber, Front. Microbiol., 2015, 6, 957 Search PubMed.
- J. Sambrook, E. F. Fritsch and T. Maniatis, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, 1989 Search PubMed.
- J. Rocha-Martin, D. E. Vega, Z. Cabrera, J. M. Bolivar, R. Fernandez-Lafuente, J. Berenguer and J. M. Guisan, Process Biochem., 2009, 44, 1004–1012 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- J. Wu, M.-H. Li, J.-P. Lin and D.-Z. Wei, J. Chromatogr. Sci., 2011, 49, 375–378 CAS.
- B. Wang, B. Wu and B. He, RSC Adv., 2019, 9, 13757–13764 RSC.
- C. E. A. F. Schafmeister, W. F. Ross and V. Romanovsky, Universtity of California, San Francisco, 1995.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, V. W. D. Cruzeiro III, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, Y. Huang, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2018, University of California, San Francisco, 2018 Search PubMed.
- F. Hollmann, I. W. C. E. Arends, K. Buehler, A. Schallmey and B. Bühler, Green Chem., 2011, 13, 226–265 RSC.
- R. Bai, H. Zhang, F. Mei, S. Wang, T. Li, Y. Gu and G. Li, Green Chem., 2013, 15, 2929–2934 RSC.
- S. N. Ruzheinikov, J. Burke, S. Sedelnikova, P. J. Baker, R. Taylor, P. A. Bullough, N. M. Muir, M. G. Gore and D. W. Rice, Structure, 2001, 9, 789–802 CrossRef CAS.
- L. G. Lee and G. M. Whitesides, J. Org. Chem., 1986, 51, 25–36 CrossRef CAS.
- O. A. B. S. M. Gani, O. A. Adekoya, L. Giurato, F. Spyrakis, P. Cozzini, S. Guccione, J.-O. Winberg and I. Sylte, Biophys. J., 2008, 94, 1412–1427 CrossRef CAS.
- M. Klimacek and B. Nidetzky, Biochem. J., 2009, 425, 455–463 CrossRef.
- K. L. Kavanagh, M. Klimacek, B. Nidetzky and D. K. Wilson, J. Biol. Chem., 2002, 277, 43433–43442 CrossRef CAS.
- T.-S. Kim, S. K. S. Patel, C. Selvaraj, W.-S. Jung, C.-H. Pan, Y. C. Kang and J.-K. Lee, Sci. Rep., 2016, 6, 33438 CrossRef CAS.
- J. Choe, D. Guerra, P. A. M. Michels and W. G. J. Hol, J. Mol. Biol., 2003, 329, 335–349 CrossRef CAS.
- C. Selvaraj, G. Krishnasamy, S. S. Jagtap, S. K. S. Patel, S. S. Dhiman, T.-S. Kim, S. K. Singh and J.-K. Lee, Biochem. Eng. J., 2016, 114, 244–256 CrossRef CAS.
- J. E. Lucas and J. B. Siegel, Protein Sci., 2015, 24, 936–945 CrossRef CAS.
- C. Montella, L. Bellsolell, R. Pérez-Luque, J. Badía, L. Baldoma, M. Coll and J. Aguilar, J. Bacteriol., 2005, 187, 4957 CrossRef CAS.
- A. Liese, M. Karutz, J. Kamphuis, C. Wandrey and U. Kragl, Biotechnol. Bioeng., 1996, 51, 544–550 CrossRef CAS.
- D. Güclü, M. Rale and W.-D. Fessner, Eur. J. Org. Chem., 2015, 2015, 2960–2964 CrossRef.
- R. C. Rodrigues, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- V. Carbone, L. R. Schofield, Y. Zhang, C. Sang, D. Dey, I. M. Hannus, W. F. Martin, A. J. Sutherland-Smith and R. S. Ronimus, J. Biol. Chem., 2015, 290, 21690–21704 CrossRef CAS.
- T. Tsujigami, T. Sugai and H. Ohta, Tetrahedron: Asymmetry, 2001, 12, 2543–2549 CrossRef CAS.
- G. S. Kumar, Y. Wee, I. Lee, H. J. Sun, X. Zhao, S. Xia, S. Kim, J. Lee, P. Wang and J. Kim, Chem. Eng. J., 2015, 276, 283–288 CrossRef CAS.
- M.-Q. Xu, F.-L. Li, W.-Q. Yu, R.-F. Li and Y.-W. Zhang, Int. J. Biol. Macromol., 2020, 144, 1013–1021 CrossRef CAS.
- A. Liese, M. Karutz, J. Kamphuis, C. Wandrey and U. Kragl, Biotechnol. Bioeng., 1996, 51, 544–550 CrossRef CAS.
- N. Ríos-Lombardía, C. Vidal, E. Liardo, F. Morís, J. García-Álvarez and J. González-Sabín, Angew. Chem., Int. Ed., 2016, 55, 8691–8695 CrossRef.
- A. Boffi, S. Cacchi, P. Ceci, R. Cirilli, G. Fabrizi, A. Prastaro, S. Niembro, A. Shafir and A. Vallribera, ChemCatChem, 2011, 3, 347–353 CrossRef CAS.
- S. T. Ahmed, N. G. H. Leferink and N. S. Scrutton, Mol. Catal., 2019, 467, 95–110 CrossRef CAS.
- N. Ríos-Lombardía, C. Vidal, M. Cocina, F. Morís, J. García-Álvarez and J. González-Sabín, Chem. Commun., 2015, 51, 10937–10940 RSC.
- E. Liardo, N. Ríos-Lombardía, F. Morís, F. Rebolledo and J. González-Sabín, ACS Catal., 2017, 7, 4768–4774 CrossRef CAS.
Footnotes |
| † We want to dedicate this work to the memory of Prof. José Ignacio García who germinated this work, encouraged us to face this challenge and acted as a priceless advisor, providing answers and constantly supporting us. Rest in peace. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04471g |
| § Turnover of native GlyDH towards glycerol: kcat of 237 min−1 that corresponds a free energy barrier of 16.9 kcal mol−1 at a temperature of 303 K. |
| This journal is © The Royal Society of Chemistry 2020 |