
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLewis acid-assisted Ir(III) reductive elimination enables construction of seven-membered-ring sulfoxides†
Wu
Yang
,
Yingzi
Li
,
Jiefeng
Zhu
,
Wentan
Liu
,
Jie
Ke
and
Chuan
He
 *
*
Shenzhen Grubbs Institute, Department of Chemistry, Guangdong Provincial Key Laboratory of Catalysis, Southern University of Science and Technology, Shenzhen 518055, Guangdong, China. E-mail: hec@sustech.edu.cn; Web: http://faculty.sustech.edu.cn/hec/en/
First published on 3rd September 2020
Abstract
Iridium has played an important role in the evolution of C–H activation chemistry over the last half century owing to its high reactivity towards stoichiometric C–H bond cleavage; however, the use of Ir(III) complexes in catalytic C–H functionalization/C–C bond formation appears to have fallen off significantly. The main problem lies in the reductive elimination step, as iridium has a tendency to form stable and catalytically inactive Ir(III) species. Herein, with a rationally designed Lewis acid assisted oxidatively induced strategy, the sluggish Ir(III) reductive elimination is successfully facilitated, enabling the facile C–C bond formation. The X-ray crystal structure of a silver salt adduct of iridacycle and DFT calculations demonstrate that the sulfoxide group acts as a key bridge connecting the Ir(III) metal centre with the silver Lewis acid, which facilitates the reductive elimination of the Ir(III) metallacycle. Further identification of oxidants was carried out by performing stoichiometric reactions, which enables the development of catalytic construction of various highly functionalized seven-membered-ring sulfoxides, that are of great interest in medicinal chemistry and materials science.
Introduction
The development of new catalytic transformations based on converting carbon–hydrogen bonds into carbon–carbon bonds is one of the most attractive goals of modern synthetic chemistry.1–3 Over the last decade, the extraordinary advances achieved by transition metal catalysis in the field of C–H bond activation have revolutionized the rules for assembling molecules, enabling the rapid construction and late-stage diversification of functional molecules in a more efficient and straightforward manner.4–8 Generally, transition metals can react with carbon–hydrogen bonds in a certain way to produce carbon–metal bonds, and the organometallic intermediates open up opportunities for further carbon–metal functionalization.9,10 In the realm of C–H bond activation and subsequent C–C bond formation, the second-row platinum group metals, particularly palladium salts, unambiguously stand out as privileged catalysts owing to their outstanding reactivity and controllable reaction pathway.11–15 In contrast, the use of third-row transition metals in catalytic elaboration of C–C bonds from C–H bonds appears to have fallen off significantly.16–31 Iridium complexes have been shown to be highly active for stoichiometric C–H bond activation, while the relatively stable metallacyclic Ir(III) intermediates usually hamper their catalytic turnovers in the Ir(III)-catalyzed C–C bond forming reactions (Scheme 1a).32–34 The main problem lies in the reductive elimination step furnishing C–C bond formation, which is identified as the bottleneck in the catalytic cycle.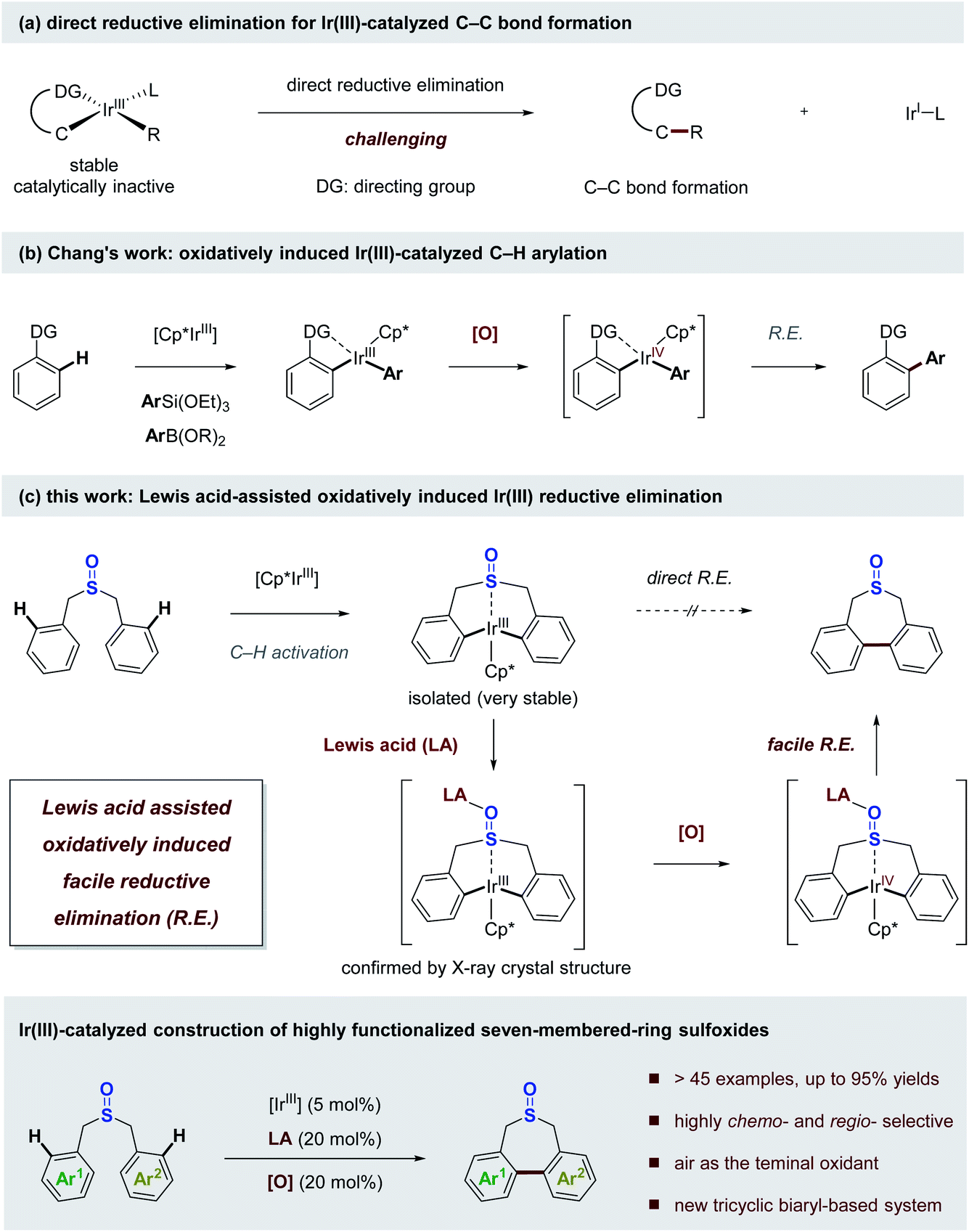 | ||
| Scheme 1 Ir(III) reductive elimination. | ||
Given the intrinsic outstanding reactivity in C–H bond activation, iridium would undoubtedly hold great potential for enabling catalytic C–H bond functionalization, in particular C–C bond forming reactions, if the sluggish reductive elimination of Ir(III) intermediates can be accelerated deliberately in the catalytic cycle, which may realize otherwise impossible reactions with many exciting opportunities. Recently, Chang and co-workers elegantly demonstrated that selectively changing the oxidation states of Ir(III) intermediates enables the facile oxidatively induced reductive elimination pathway, which operates successfully in the Ir(III)-catalyzed C–H arylation reaction with arylsilanes or arylboronic esters as the arylating reagents (Scheme 1b).35,36 The alternative high-valent catalytic pathway, where the reductive elimination is facilitated by making the metal centre electron-poor with a high oxidation state, has also been appreciated in palladium catalysis.37–41
As part of an overarching goal to develop new modes for catalytic conversion of carbon–hydrogen bonds into carbon–carbon bonds, we questioned whether some native functional groups could tune the electronic environment around the Ir(III) metal centre via coordination assisted by Lewis acids, which would increase the driving force for the reductive elimination step.42 Here we report the realization of this idea via a Lewis acid assisted oxidatively induced reductive elimination, which enables the facile Ir(III)-catalyzed C–C bond forming reaction (Scheme 1c). The sulfoxide functional group plays a crucial role in this transformation, allowing the selective C–H bond activation and acts as a key bridge connecting the Ir(III) metal centre with the Lewis acid, which facilitates the reductive elimination of the stable Ir(III) metallacycle intermediate. Based on the designed strategy, a novel Ir(III)-catalyzed C–H functionalization/C–C bond forming reaction is developed, which enables the facile construction of a wide range of highly functionalized seven-membered-ring sulfoxides in a chemo- and regio-selective manner with air as the terminal oxidant.
Results and discussion
At the outset of this study, we were interested in identifying effective ligands for Ir(III)-catalyzed C–H functionalization. One interesting study reported by Ison and co-workers showed that a DMSO (dimethyl sulfoxide) coordinated Cp*Ir acetate complex enabled direct C–H activation of benzene, and only DMSO was found to be effective for this transformation.43 Thus, we questioned whether some tailored sulfoxides could be used as ligands in Ir(III)-catalyzed C–H functionalization, or some sulfoxides tethered with proximal C–H bond sites could undergo facile C–H activation allowing the direct functionalization of themselves, which might access novel sulfoxide compounds of untapped potential. The studies began with the investigation of stoichiometric reactions between Ir(III) salts and various sulfoxides. When 2 equivalents of dibenzyl sulfoxide 1a were treated with a stoichiometric amount of the Cp*Ir(III)Cl2 dimer in the presence of a silver salt and NaOAc in TFE (trifluoroethanol), we observed a new organoiridium complex (Ir-I) where C–H activation had taken place twice at each of the phenyl groups proximal to the sulfoxide group (Scheme 2).44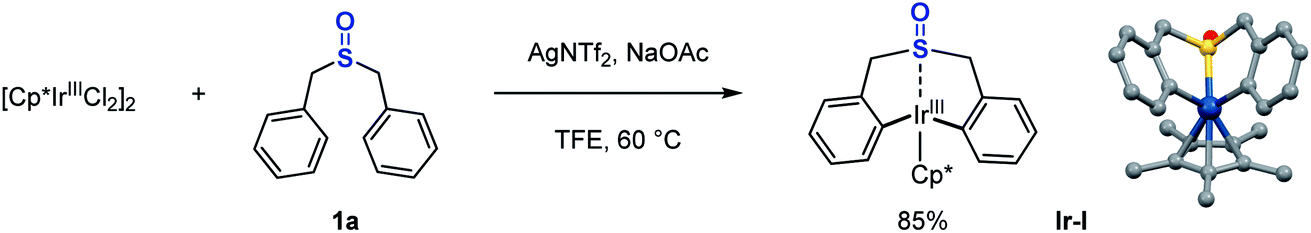 | ||
| Scheme 2 Stoichiometric reaction of dibenzyl sulfoxide with Ir(III). Crystal structure of iridacycle (Ir-I): blue, Ir; gray, C; red, O; yellow, S. | ||
Inspired by this sulfoxide-steered Ir(III)-mediated C–H bond activation mode, we questioned whether we could conduct the direct reductive elimination of this sulfoxide iridacycle (Ir-I), that might enable the C–C bond formation and construction of a seven-membered-ring sulfoxide. After many efforts, it turned out that this Ir(III) species (Ir-I) is quite stable. No reaction took place even at 90 °C under oxidant-free conditions (Scheme 3, entry 1). Preliminary theoretical study disclosed that the activation free energy for direct reductive elimination of the Ir(III) intermediate (Ir-I) is enormous 45.8 kcal mol−1, which indicated that the direct C–C bond reductive elimination of the Ir(III) intermediate (Ir-I) would not occur under mild conditions (see the ESI Fig. S17† for details). We next carried out reactions under oxidative conditions. We were delighted to observe the desired reductive elimination product, the seven-membered-ring sulfoxide 2a, when heating under an air atmosphere, albeit in low yields (Scheme 3, entries 2–4). A wide range of commonly used oxidants, such as AgOAc, Cu(OAc)2, PhI(OAc)2, Ce(IV) salts, V2O5, oxone, and MnO2 were ineffective in the transformation at room temperature (Scheme 3, entries 5–12); only Mn(OAc)3·2H2O produced 37% yield (Scheme 3, entry 13). Further optimization around oxidants did not result in any obvious improvements. Theoretically, making the metal centre electron-poor should increase the driving force for reductive elimination. Since the sulfoxide group coordinated to the Ir(III) metal centre, if we could find a method to reduce the electron density around the metal centre through this sulfoxide bridge, we may accelerate this challenging Ir(III) reductive elimination. With this idea in mind, several Lewis acid (LA) additives were employed in the reaction, which could reasonably coordinate to the O atom of the sulfoxide and pull electron density away from the Ir(III) metal centre. In the presence of magnesium or silver Lewis acids, the conversion of iridacycle (Ir-I) dramatically improved even with 20 mol% manganese(III) oxidant at room temperature (Scheme 3, entries 14–18). Notably, even without manganese(III) species, Mg(NTf2)2 alone could also produce the reductive elimination product in 14% yield in air (Scheme 3, entry 19). DFT calculations for this process indicated that assisted by a Lewis acid, the facile oxidatively induced reductive elimination of the C–C bond from Ir(IV) species occurs with only a small activation energy of 16.9 kcal mol−1 (see the ESI Fig. S17† for details).
 | ||
| Scheme 3 Reductive elimination of the sulfoxide iridacycle. Conditions: Ir-I (0.05 mmol), oxidant (entries 5–13, 1.2 equiv.; entries 14–18, 0.2 equiv. [Mn] in air), additive (0.2 equiv.), TFE (0.5 mL), 24 h; NMR yields; CAN = ceric ammonium nitrate; NTf2 = bis(trifluoromethanesulfonyl)imide; TFA = trifluoroacetate. | ||
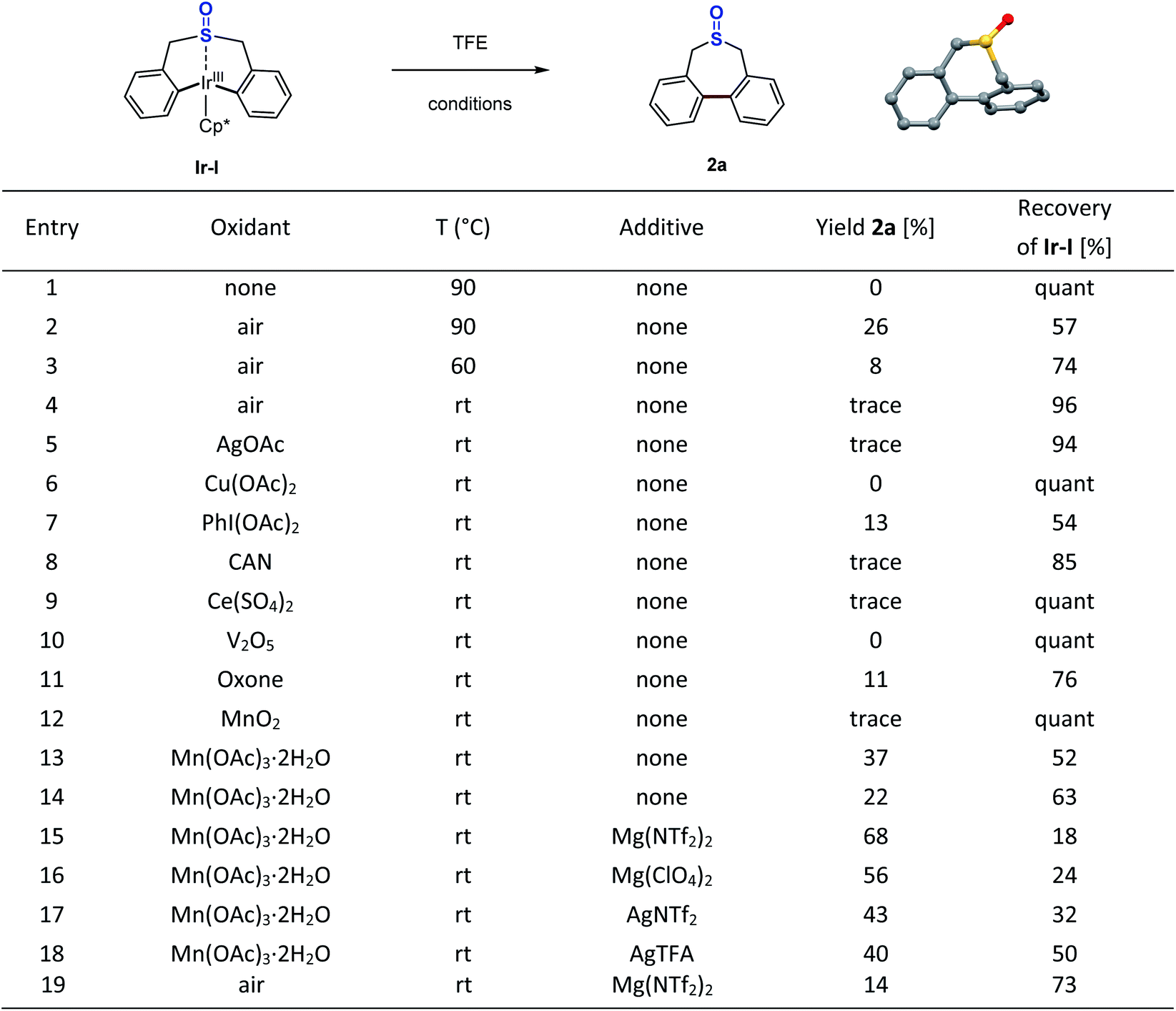
To further confirm the effect of the Lewis acid, stoichiometric reactions of the iridacycle (Ir-I) with Mg(NTf2)2 and several silver Lewis acids were investigated by NMR spectroscopy. The 1H NMR spectra all show a significant down-field chemical shift of the benzylic protons (Scheme 4), which strongly suggests that the coordination of Lewis acids pulls electron density away from the sulfoxide iridacycle (Ir-I). To our delight, we also obtained one X-ray crystal structure of the silver Lewis acid adduct of iridacycle (Ir-II), clearly displaying that one molecule of AgTFA coordinated to the O atom of sulfoxide, while another molecule of AgTFA had π-interactions with the two phenyl groups (Scheme 5). Both effects significantly reduced the electron density around the Ir(III) metal centre, allowing the facile Lewis acid assisted oxidatively induced reductive elimination.
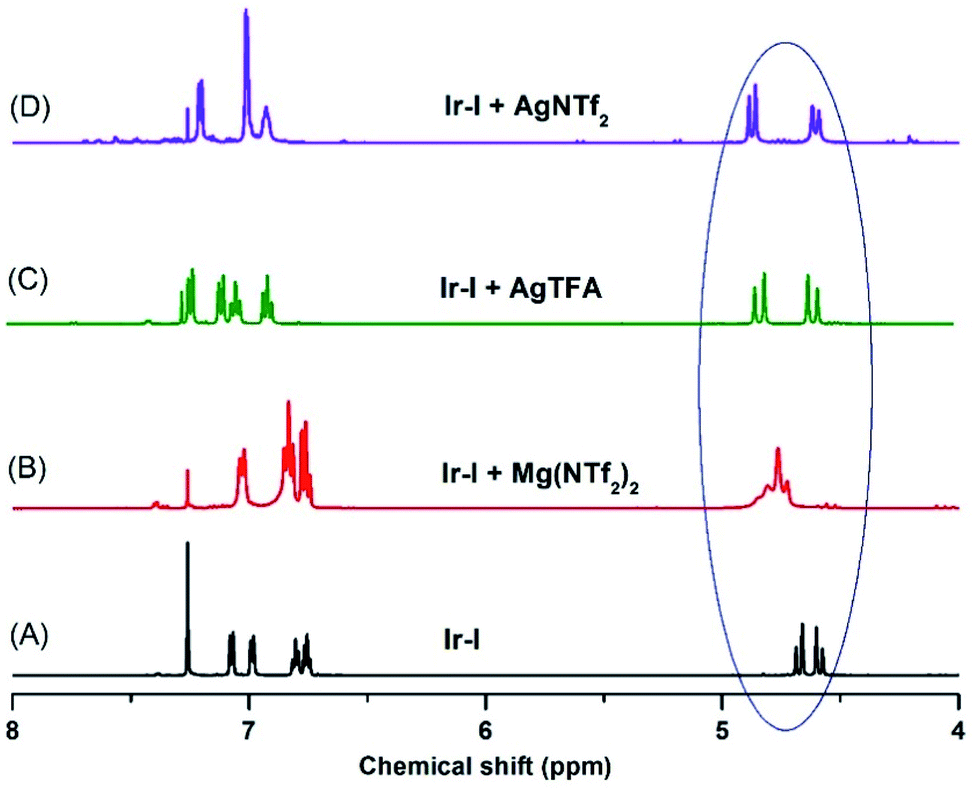 | ||
| Scheme 4 1H NMR studies of the Lewis acid effects. | ||
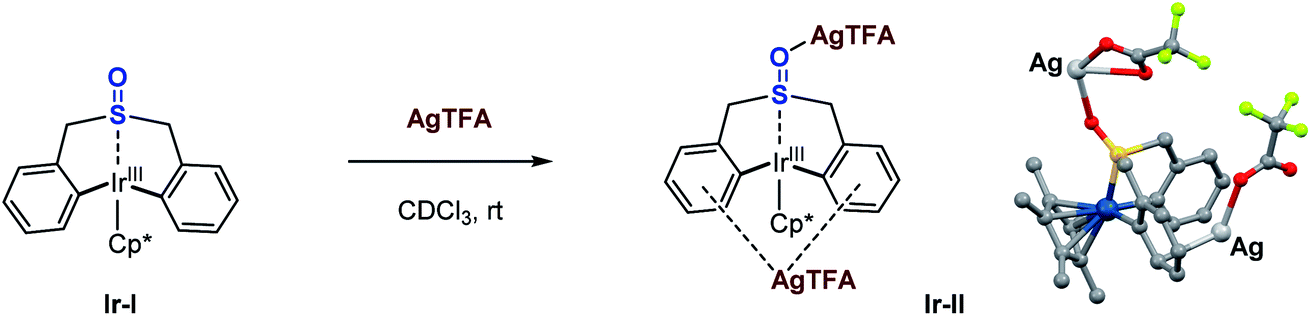 | ||
| Scheme 5 Silver Lewis acid adduct of the iridacycle. Crystal structure of Ir-II: blue, Ir; gray, C; red, O; green, F; yellow, S; light grey, Ag (marked). | ||
To support the above experimental results, electrostatic potential (ESP) calculations were used to clarify the role of the Lewis acid. The ESP surfaces showed that the negative charge is mainly located on the central iridium atom and sulfoxide group in Ir-I; while with the silver Lewis acid, the centre of the negative charge is located on the Lewis acid in Ir-II (Scheme 6). These results clearly suggest that the Lewis acid reduced the electron density around the iridium metal centre.
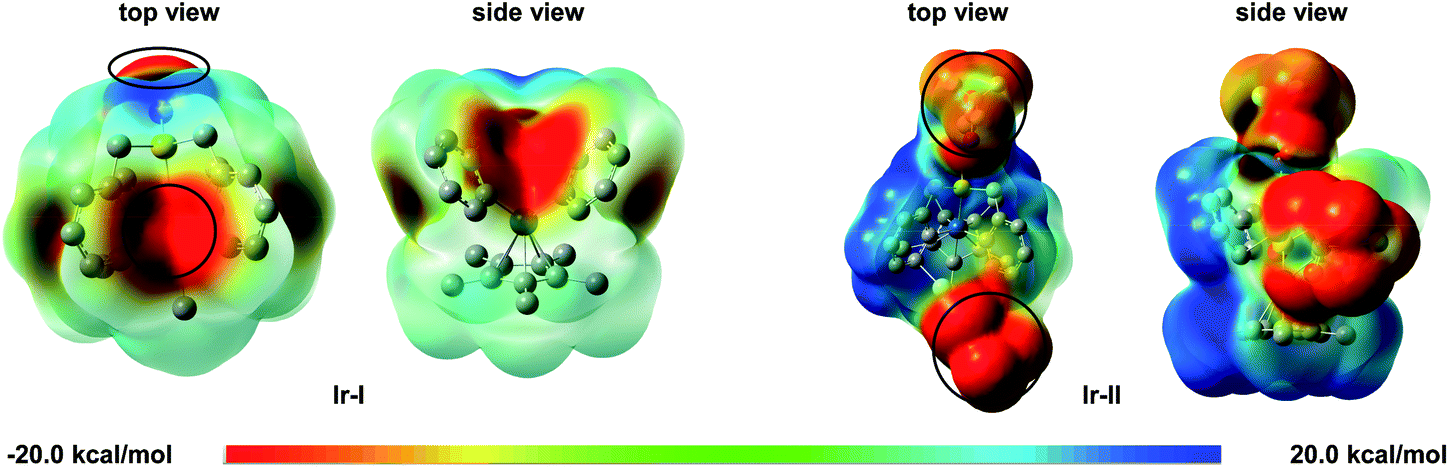 | ||
| Scheme 6 Electrostatic potential surfaces of iridacycle Ir-I and Ir-II (red indicating negative potential and blue indicating positive potential). | ||
With the successful stoichiometric reactions in hand, we next embarked on catalytic studies to establish the synthetic methodology for the construction of seven-membered-ring sulfoxides. Since the sulfoxide iridacycle is the key intermediate, we first used 10 mol% isolated iridacycle (Ir-I) as the catalyst in the reaction to test the turnover. In the presence of 20 mol% Mn(OAc)3·2H2O, 38% yield of desired product 2a was obtained (Scheme 7, entry 1). Based on the insights into the role of the Lewis acid in the stoichiometric reaction, adding 20 mol% magnesium or silver Lewis acids in the catalytic reaction enabled full conversion of 1a into 2a (Scheme 7, entries 2–4). Further examination of the catalyst precursors showed that the use of the 5 mol% Cp*Ir(III)Cl2 dimer along with 20 mol% Mn(OAc)3·2H2O and AgNTf2 provided the corresponding seven-membered-ring sulfoxide 2a in 93% yield (Scheme 7, entry 6). AgNTf2 was chosen owing to its ability to grab the chloride in the catalyst precursor, as well as to act as a Lewis acid. Without Mn(OAc)3·2H2O, almost no reaction occurred (Scheme 7, entry 5). It is noteworthy that this reaction could proceed smoothly even using a 1 mol% Cp*Ir(III)Cl2 dimer catalyst precursor (Scheme 7, entry 7). However, the Ir(I) catalyst precursor showed no reactivity in this process (Scheme 7, entry 8). In addition, Cp*RhCl2, (p-cymene)RuCl2 and Pd(OAc)2 catalysts were also tested in the reaction, as these catalytic systems usually display high reactivity in some sulfoxide directed C–H functionalization reactions.45–53 However, for this typical transformation, no yield of 2a could be obtained (see the ESI Table S6† for details).
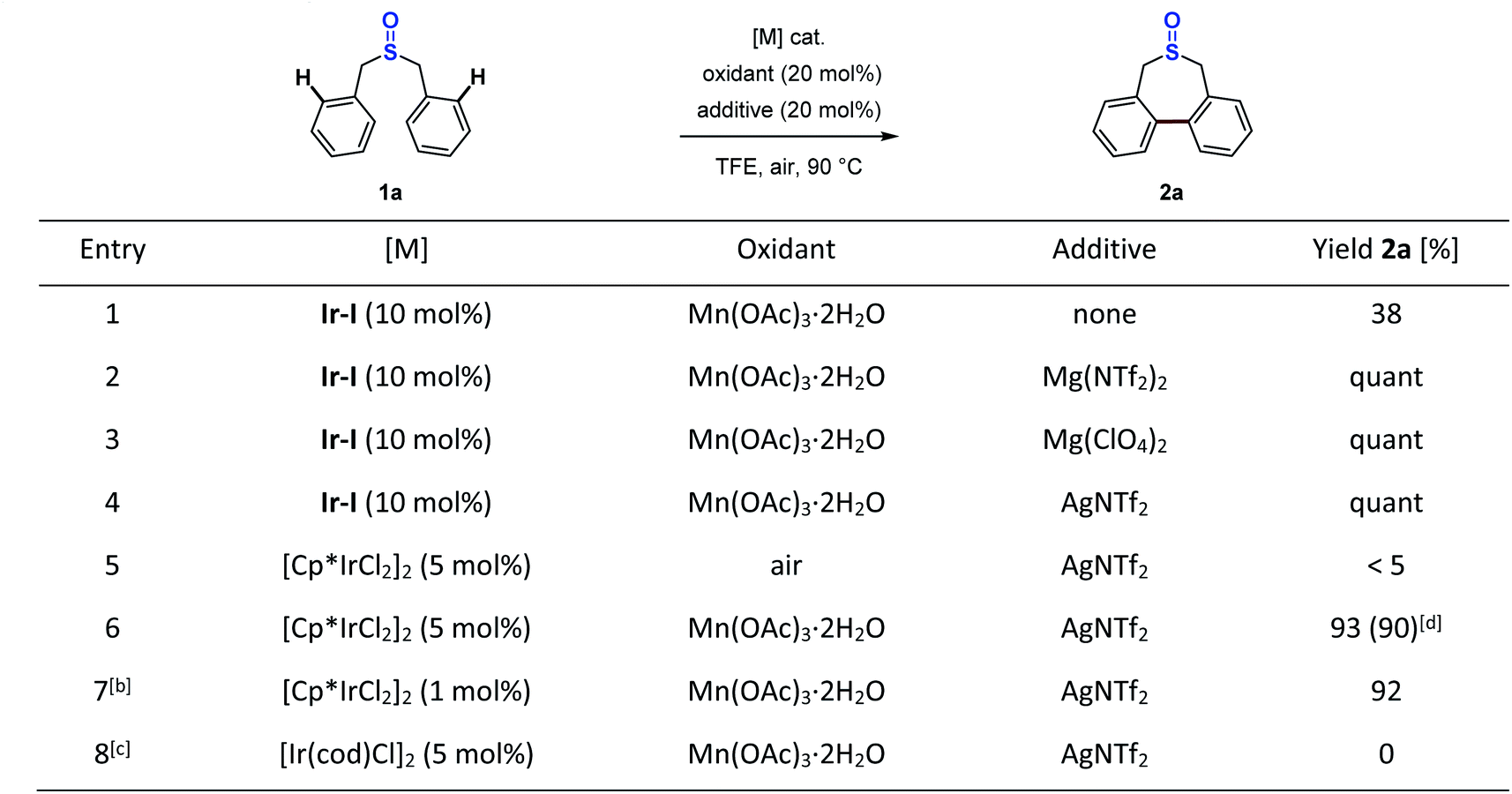 | ||
| Scheme 7 Development of the catalytic reaction. Conditions: 1a (0.1 mmol), oxidant (20 mol%), additive (20 mol%), TFE (0.5 mL); NMR yields. [b] 1a (0.2 mmol), Mn(OAc)3·2H2O (5 mol%), AgNTf2 (4 mol%), TFE (1.0 mL). [c] AgNTf2 (10 mol%). [d] Isolated yield in parentheses. | ||
In assessing the scope of the catalytic reaction, we found that a wide range of readily available symmetric sulfoxides containing benzyl groups were suitable substrates for the construction of seven-membered-rings (Scheme 8). Benzyl groups with substituents at the para, meta and ortho positions of the aromatic ring all provided the desired products in good yields (2b–2p). Both electron-withdrawing and electron-donating groups, such as methyl, tert-butyl, OCF3, CF3 and ester groups (2b, 2g, 2i–2m, and 2o), on the aromatic ring were well tolerated, and halogen substituents (2c–2f, 2n, and 2p) could also be introduced to produce the corresponding products without any problem including the iodo group (2f). Extended aromatic or olefin groups (2h, 2l, 2q, and 2r) as well as heterocycle thiophene (2s) could also be transformed into this seven-membered-ring sulfoxide scaffold. Moreover, di-p-tolyl sulfite could undergo similar transformation in 43% yield (2t). It is noteworthy that only a few reports demonstrated the construction of this type of seven-membered-ring sulfoxide scaffold, and the substrate was restricted to electron-rich aromatic rings.54,55
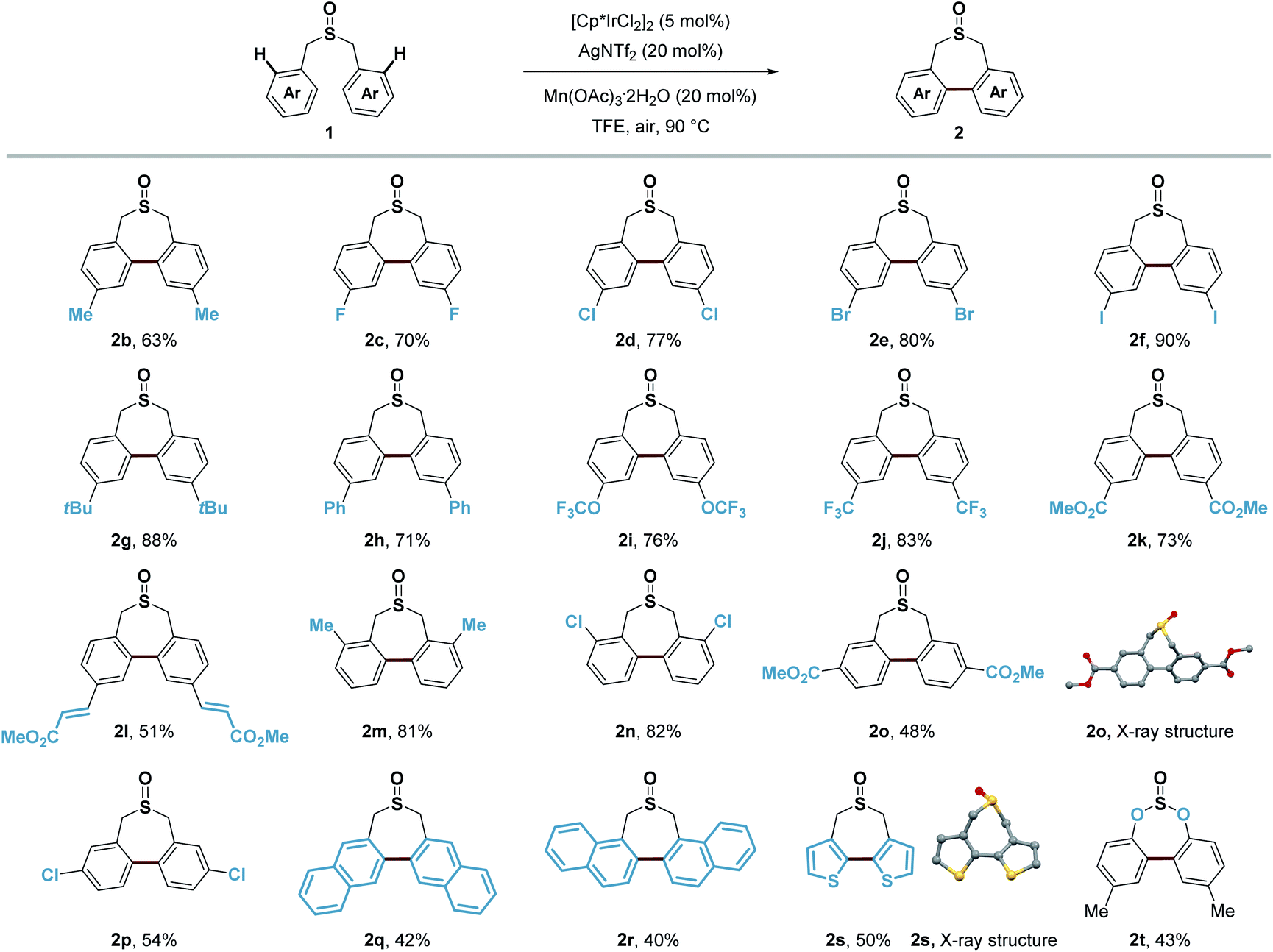 | ||
| Scheme 8 Substrate scope of symmetric sulfoxides. Isolated yields. | ||
Besides symmetric sulfoxides, the reaction was also readily extended to a variety of non-symmetric sulfoxides bearing diverse functional groups in good to excellent yields (Scheme 9). Methyl, halogen substituents, ester, OTBS, OCF3, SCF3, and alkenyl groups at different substituted positions all worked well giving the novel non-symmetric seven-membered-ring sulfoxides in 48–95% yields, which provided readily manipulatable functional groups suitable for downstream modification of varying substitution patterns (4a–4s). It is noteworthy that normally the over-oxidation of sulfoxides to sulfones as well as the oxidation of benzylic C–H bonds is an inevitable problem in many oxidative coupling reactions. Herein, with this Lewis acid assisted oxidatively induced strategy, we can selectively conduct Ir(III) reductive elimination achieving C–C bond formation under mild oxidative conditions without touching the sulfoxide group and benzylic C–H bonds. To further illustrate the utility of this transformation, we examined this Ir(III)-catalyzed C–H functionalization/C–C bond forming reaction employing the core structures of several bioactive molecules or pharmaceuticals, such as (+)-α-tocopherol (5a), lithocholic acid (5b), dehydrocholic acid (5c), lyrica (pregabalin) (5d), diclofenac (5e), naproxen (5f), isoxepac (5g) and indomethacin (5h). We were delighted to find that the corresponding seven-membered-ring sulfoxide products could be obtained in good yields, irrespective of existing diverse functional groups and complex molecular structures. This process transforms readily available sulfoxides into their highly functionalized seven-membered-ring derivatives, that have shown indication of applications in medicinal chemistry.56
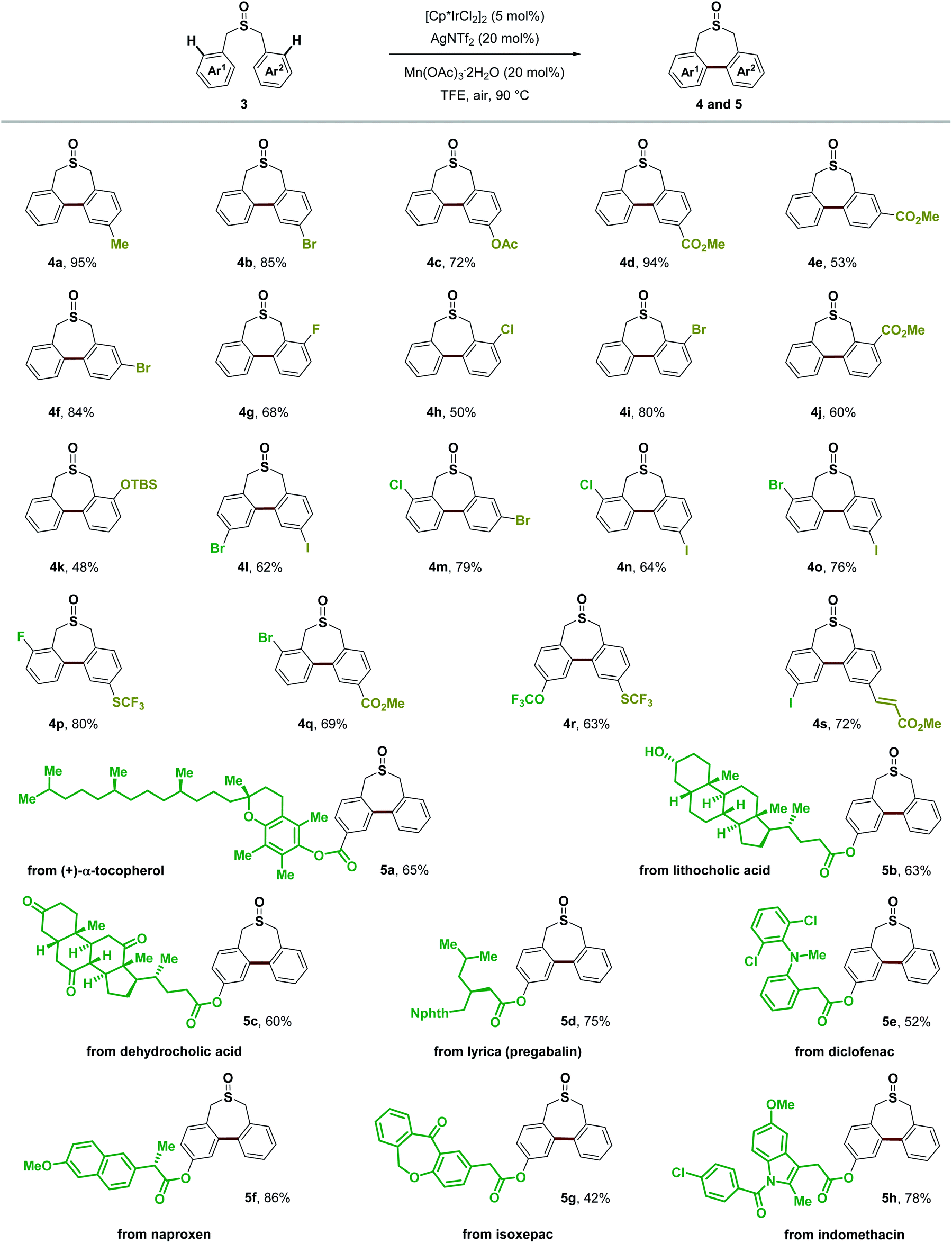 | ||
| Scheme 9 Substrate scope of non-symmetric sulfoxides. Isolated yields, due to the conformation of non-symmetric biaryl units within the seven-membered-ring sulfoxide, dr was observed, see the ESI† for details. | ||
In addition, we showed that the bithiophene-based seven-membered-ring sulfoxide product 2s could serve as a substrate for a selection of straightforward transformations to potentially useful building blocks in materials science (Scheme 10). Tricyclic bithiophene-based compounds where the 3,3′-positions of bithiophene are fastened by different bridging atoms have been demonstrated as superior building blocks to construct organic materials utilized in a range of diverse optoelectronic applications.57,58 Herein, by using this methodology, we developed a new tricyclic bithiophene-based system containing an additional sulfur atom within the seven-membered-ring. Simple reduction, oxidation or imidation of the sulfoxide of 2s generated the corresponding seven-membered-ring sulfide 6a, sulfone 6c, and sulfoximine 6b in good yields, respectively. Bromination of these compounds successfully delivered the dibromide derivatives 6d–6f. All of these compounds could presumably be used as novel monomers for the access of new donor–acceptor conjugated polymers of untapped potential, which could be highly attractive to organic material chemists. So far, we have done some preliminary studies which show that copolymerization of the electron-deficient sulfone bridged bithiophene 6f with some precedented building blocks, provided new high-performance polymer solar cells (PSCs) with a relatively high power conversion efficiency (PCE). The results of these investigations will be reported in due course.
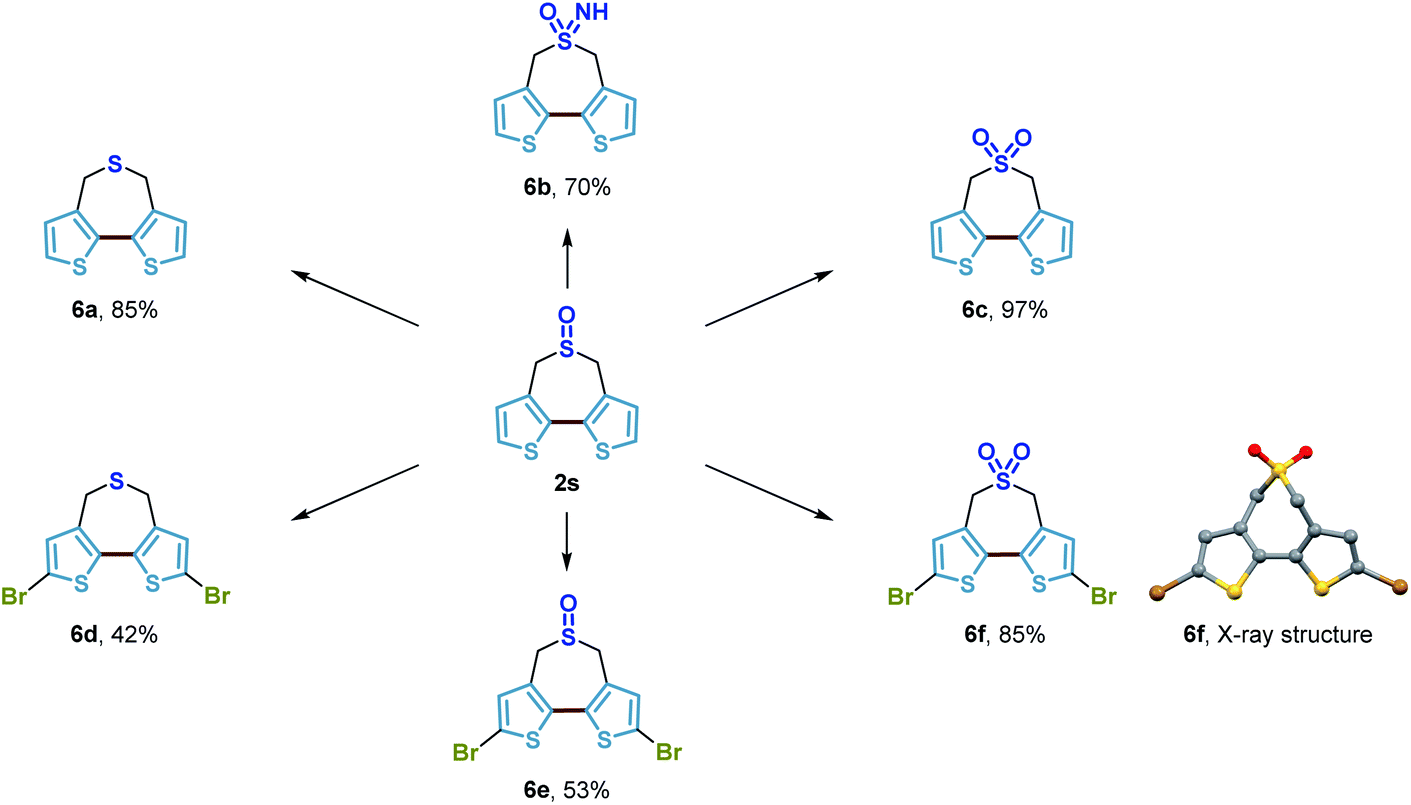 | ||
| Scheme 10 Derivatizations of the bithiophene-based seven-membered-ring sulfoxide. | ||
Finally, we investigated the kinetic parameters of this catalytic reaction. Two types of kinetic isotope effect (KIE) experiments were explored using 1a and d10-1a (Scheme 11). Both parallel competitive reactions and same-flask intermolecular competitive reactions exhibited significant isotope effects, suggesting that C–H bond cleavage occurs as part of the turnover-limiting step.59 Therefore, in the presence of the Lewis acid and manganese oxidant, the previous sluggish reductive elimination is clearly no longer the turnover-limiting step.
 | ||
| Scheme 11 KIE studies of the catalytic reaction. | ||
Taken together, based on the stoichiometric reactions and mechanistic studies, a catalytic cycle of this Ir(III)-catalyzed reaction is proposed in Scheme 12. First, Cp*Ir acetate complex A is generated from the precursor Cp*IrCl2 dimer by reacting with Ag(I) and manganese(III) acetate. Coordination with sulfoxide 1a produces complex B, which undergoes C–H bond activation affording the cyclometallated Ir(III) intermediate C (Ir-III) and D (Ir-I) sequentially. Assisted by the Lewis acid, this iridacycle adduct E is proposed to carry out a one-electron oxidation using manganese(III) acetate giving the highly reactive Ir(IV) intermediate F. The one-electron reduction of manganese(III) to manganese(II) was observed by EPR spectroscopy (See the ESI Fig. S8† for details). Finally, this high-valent Ir species may proceed to complete the Lewis acid assisted oxidatively induced reductive elimination step, enabling the construction of seven-membered-ring sulfoxide product 2a, while, the plausible Ir(II) species generated from the reductive elimination may be oxidized by the manganese oxidant to regenerate the catalytically active Ir(III) species, closing the catalytic cycle.
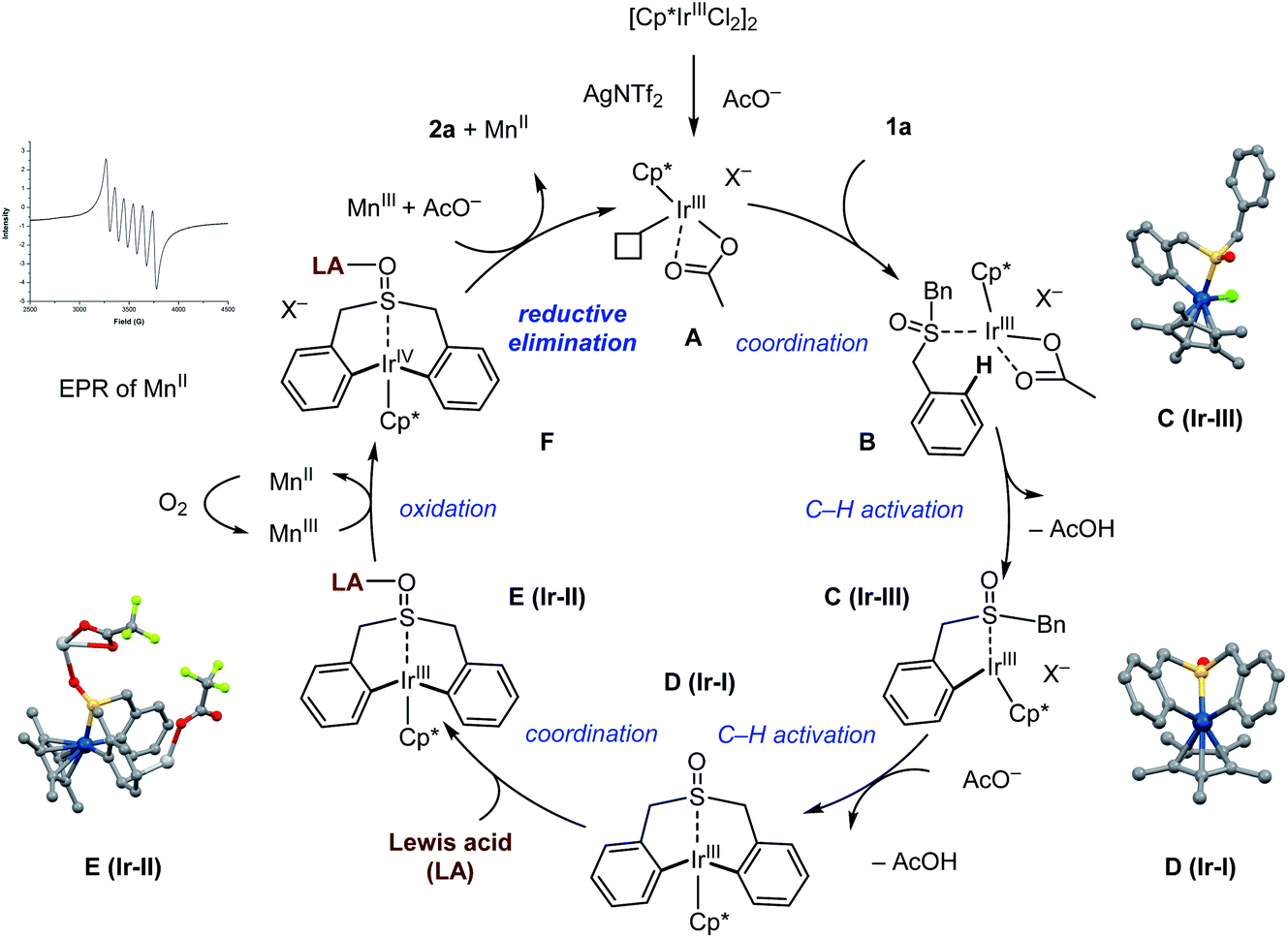 | ||
| Scheme 12 Catalytic cycle of the Ir(III)-catalyzed reaction. | ||
Conclusions
In summary, we have successfully facilitated Ir(III) reductive elimination by a rationally designed Lewis acid assisted oxidatively induced strategy, which enables the facile construction of versatile seven-membered-ring sulfoxides in good to excellent yields. The key X-ray crystal structure of the Lewis acid adduct of the iridacycle suggests that the sulfoxide group plays a crucial role in the transformation, acting as a bridge connecting the Ir(III) metal centre with the Lewis acid, which allows the facile reductive elimination of the stable and catalytically inactive Ir(III) metallacycle intermediate. This process transforms readily available sulfoxides into highly functionalized seven-membered-rings that we believe will prove attractive to practitioners of medicinal chemistry and materials science. Furthermore, this strategy by tuning the electronic density of the metal centre via the sulfoxide bridge and Lewis acid not only facilitates the challenging Ir(III) reductive elimination, but also provides insights into the elementary stages in general transition-metal-catalyzed coupling reactions, leads to a practical and powerful complement to classical condition development techniques, and offers an opportunity to explore previously inaccessible reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Thousand Talents Program for Young Scholars, Shenzhen Science and Technology Innovation Committee (JCYJ20190809142809370), the Shenzhen Nobel Prize Scientists Laboratory Project (C17783101), and the Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002). We also thank Dr Xiao-Yong Chang for assistance in solving the X-ray structures and Dr Yin-Hua Yang from the MCPC (Materials Characterization and Preparation Center) for assistance in the EPR experiments.Notes and references
- G. Dyker, Handbook of C–H Transformations: Applications in Organic Synthesis, Wiley-VCH, Weinheim, 2005, vol. 2 Search PubMed.
- K. M. Engle, J.-Q. Yu, H. M. L. Davies, Z. Xi, S.-L. You and Z.-J. Shi, in Organic Chemistry – Breakthroughs and Perspectives, ed. K. Ding and L.-X. Dai, Wiley-VCH, Weinheim, 2012, pp. 279–333 Search PubMed.
- K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS.
- W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- A. D. Ryabov, Chem. Rev., 1990, 90, 403–424 CrossRef CAS.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS.
- X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS.
- O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS.
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS.
- C. He, W. G. Whitehurst and M. J. Gaunt, Chem, 2019, 5, 1031–1058 CAS.
- J. A. Labinger, Chem. Rev., 2017, 117, 8483–8496 CrossRef CAS.
- K. Ueura, T. Satoh and M. Miura, J. Org. Chem., 2007, 72, 5362–5367 CrossRef CAS.
- D. A. Frasco, C. P. Lilly, P. D. Boyle and E. A. Ison, ACS Catal., 2013, 3, 2421–2429 CrossRef CAS.
- M. Itoh, K. Hirano, T. Satoh, Y. Shibata, K. Tanaka and M. Miura, J. Org. Chem., 2013, 78, 1365–1370 CrossRef CAS.
- P. Gao, W. Guo, J. Xue, Y. Zhao, Y. Yuan, Y. Xia and Z. Shi, J. Am. Chem. Soc., 2015, 137, 12231–12240 CrossRef CAS.
- L. Huang, D. Hackenberger and L. J. Goossen, Angew. Chem., Int. Ed., 2015, 54, 12607–12611 CrossRef CAS.
- J. Kim, S.-W. Park, M.-H. Baik and S. Chang, J. Am. Chem. Soc., 2015, 137, 13448–13451 CrossRef CAS.
- K. Shin, S.-W. Park and S. Chang, J. Am. Chem. Soc., 2015, 137, 8584–8592 CrossRef CAS.
- Y. Xia, Z. Liu, S. Feng, Y. Zhang and J. Wang, J. Org. Chem., 2015, 80, 223–236 CrossRef CAS.
- Y.-X. Lao, S.-S. Zhang, X.-G. Liu, C.-Y. Jiang, J.-Q. Wu, Q. Li, Z.-S. Huang and H. Wang, Adv. Synth. Catal., 2016, 358, 2186–2191 CrossRef CAS.
- X.-Y. Chen and E. J. Sorensen, Chem. Sci., 2018, 9, 8951–8956 RSC.
- G. Li, J. Hu, R. Zeng, D.-Q. Shi and Y. Zhao, Org. Lett., 2018, 20, 2454–2458 CrossRef CAS.
- Y. Qiu, M. Stangier, T. H. Meyer, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14179–14183 CrossRef CAS.
- G. Tan, Q. You, J. Lan and J. You, Angew. Chem., Int. Ed., 2018, 57, 6309–6313 CrossRef CAS.
- G. Tan, Q. You and J. You, ACS Catal., 2018, 8, 8709–8714 CrossRef CAS.
- G. Tan, C. Ran and J. You, Org. Chem. Front., 2018, 5, 2930–2933 RSC.
- J. G. Kim, K. Shin and S. Chang, Top. Organomet. Chem., 2016, 55, 29–51 CrossRef CAS.
- J. Choi and A. S. Goldman, Top. Organomet. Chem., 2011, 34, 139–168 CrossRef CAS.
- J. Wencel-Delord, F. W. Patureau and F. Glorius, Top. Organomet. Chem., 2016, 55, 1–27 CAS.
- K. Shin, Y. Park, M.-H. Baik and S. Chang, Nat. Chem., 2018, 10, 218–224 CrossRef CAS.
- J. Kim, K. Shin, S. Jin, D. Kim and S. Chang, J. Am. Chem. Soc., 2019, 141, 4137–4146 CrossRef CAS.
- K. Muniz, Angew. Chem., Int. Ed., 2009, 48, 9412–9423 CrossRef CAS.
- P. Sehnal, R. J. K. Taylor and I. J. S. Fairlamb, Chem. Rev., 2010, 110, 824–889 CrossRef CAS.
- L.-M. Xu, B.-J. Li, Z. Yang and Z.-J. Shi, Chem. Soc. Rev., 2010, 39, 712–733 RSC.
- K. M. Engle, T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478–1491 CrossRef CAS.
- A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177–185 CrossRef CAS.
- Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 7734–7735 CrossRef CAS.
- D. A. Frasco, S. Mukherjee, R. D. Sommer, C. M. Perry, N. S. Lambic, K. A. Abboud, E. Jakubikova and E. A. Ison, Organometallics, 2016, 35, 2435–2445 CrossRef CAS.
- W. Liu, W. Yang, J. Zhu, Y. Guo, N. Wang, J. Ke, P. Yu and C. He, ACS Catal., 2020, 10, 7207–7215 CrossRef CAS.
- A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 9842–9860 CrossRef CAS.
- R. Samanta and A. P. Antonchick, Angew. Chem., Int. Ed., 2011, 50, 5217–5220 CrossRef CAS.
- T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139–2144 CrossRef CAS.
- C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871–13875 CrossRef CAS.
- K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188–1191 CrossRef CAS.
- B. Wang, Y. Liu, C. Lin, Y. Xu, Z. Liu and Y. Zhang, Org. Lett., 2014, 16, 4574–4577 CrossRef CAS.
- B. Wang, C. Shen, J. Yao, H. Yin and Y. Zhang, Org. Lett., 2014, 16, 46–49 CrossRef CAS.
- K. Padala and M. Jeganmohan, Chem. Commun., 2014, 50, 14573–14576 RSC.
- Y.-C. Zhu, Y. Li, B.-C. Zhang, F.-X. Zhang, Y.-N. Yang and X.-S. Wang, Angew. Chem., Int. Ed., 2018, 57, 5129–5133 CrossRef CAS.
- T. Takada, M. Arisawa, M. Gyoten, R. Hamada, H. Tohma and Y. Kita, J. Org. Chem., 1998, 63, 7698–7706 CrossRef CAS.
- S. Sun, J. Yang, F. Li, Z. Lv, W. Li, H. Lou and L. Liu, Tetrahedron Lett., 2014, 55, 6899–6902 CrossRef CAS.
- K. Sindelar, J. Holubek, M. Ryska, I. Koruna and M. Protiva, Collect. Czech. Chem. Commun., 1986, 51, 2848–2868 CrossRef CAS.
- J.-S. Wu, S.-W. Cheng, Y.-J. Cheng and C.-S. Hsu, Chem. Soc. Rev., 2015, 44, 1113–1154 RSC.
- K. Strakova, L. Assies, A. Goujon, F. Piazzolla, V. Humeniuk Heorhii and S. Matile, Chem. Rev., 2019, 119, 10977–11005 CrossRef.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1943599–1943605 and 1971161. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04180g |
| This journal is © The Royal Society of Chemistry 2020 |