
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective synthesis of highly oxygenated acyclic quaternary center-containing building blocks via palladium-catalyzed decarboxylative allylic alkylation of cyclic siloxyketones†
Aurapat
Ngamnithiporn‡
 a,
Toshihiko
Iwayama‡
ab,
Michael D.
Bartberger
c and
Brian M.
Stoltz
*a
a,
Toshihiko
Iwayama‡
ab,
Michael D.
Bartberger
c and
Brian M.
Stoltz
*a
aWarren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: stoltz@caltech.edu
bCentral Pharmaceutical Research Institute, Japan Tobacco Inc., 1-1, Murasaki-cho, Takatsuki, Osaka, 569-1125, Japan
c1200 Pharma LLC, 844 East Green Street, Suite 204, Pasadena, CA 91101, USA
First published on 15th September 2020
Abstract
The development of a palladium-catalyzed enantioselective decarboxylative allylic alkylation of cyclic siloxyketones to produce enantioenriched silicon-tethered heterocycles is reported. The reaction proceeds smoothly to provide products bearing a quaternary stereocenter in excellent yields (up to 91% yield) with high levels of enantioselectivity (up to 94% ee). We further utilized the unique reactivity of the siloxy functionality to access chiral, highly oxygenated acyclic quaternary building blocks. In addition, we subsequently demonstrated the utility of these compounds through the synthesis of a lactone bearing vicinal quaternary-trisubstituted stereocenters.
Introduction
Silicon-containing hetereocycles are an important class of organic molecules often used in the context of temporary functional groups in the synthesis of natural products and drug molecules.1 Oxasilacycles or cyclic siloxanes, in particular, are frequently employed in various stages of total syntheses,2 characterized not only for their high tolerance to a range of reaction conditions, but also for the selective and facile transformations available for this functionality.While a number of non-asymmetric methods have been reported for accessing cyclic siloxanes,3 methods to access enantioenriched cyclic siloxanes are limited, with a majority of examples requiring the use of chiral, enantioenriched starting materials.4 To the best of our knowledge, there are only two reports detailing the enantioselective synthesis of cyclic siloxanes.5 In 2001, Hoveyda, Schrock, and coworkers reported an enantioselective desymmetrization of trienes via asymmetric ring-closing metathesis utilizing their developed Mo-catalyst (Scheme 1a).5a An enantioenriched cyclic siloxane could be obtained in good yields with excellent enantioselectivity. Jacobsen later disclosed in 2007 a method for asymmetric transannular Diels–Alder Reactions (Scheme 1b).5b In the presence of a chiral oxazaborolidine catalyst, the fused cyclic siloxane was obtained with good enantioselectivity. Subsequent oxidative cleavage of this siloxane provided a precursor to a number of sesquiterpine natural products. Although these two reports showcase unique synthetic strategies to access cyclic siloxanes in an enantioselective fashion, the substrate scope in each report only contains one example of a cyclic siloxane. Furthermore, an enantioselective method to access cyclic siloxanes bearing all-carbon quaternary stereocenters has not been reported to date.
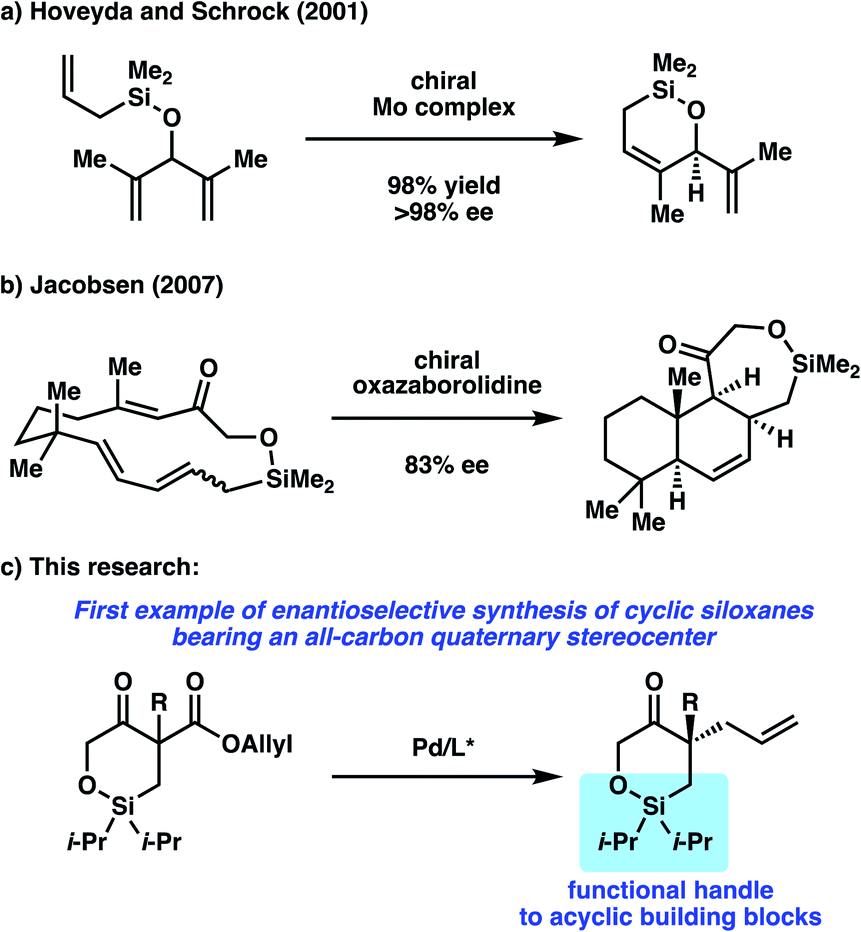 | ||
| Scheme 1 Enantioselective synthesis of cyclic siloxanes. | ||
To this end, we sought to target the synthesis of this useful motif as part of our on-going research interest in the development of asymmetric allylic alkylation methodologies.6,7 Namely, we envision performing an enantioselective decarboxylative allylic alkylation of a cyclic siloxy-β-ketoester to access a siloxyketone bearing a quaternary stereocenter (Scheme 1c).8 Furthermore, we believe that utilization of the silicon functionality in the alkylation products would allow for the synthesis of enantioenriched acyclic quaternary stereocenters, a motif that still poses a significant challenge in organic synthesis.9 Herein, we report our efforts toward the realization of these goals.
Results and discussion
As the synthesis of cyclic siloxy-β-ketoester substrates has not been previously reported, we initiated our study by first establishing a synthetic route to access the cyclic siloxy-β-ketoester substrates (Scheme 2). Our developed process begins with intermolecular O-silylation of alcohol 1 with silyl chloride 2, affording β-ketoester 3. Cyclization via intramolecular nucleophilic substitution and subsequent transesterification with allyl alcohol provided allyl β-ketoester 4 in 57% yield over 2 steps. Lastly, functionalization of 4 with various electrophiles delivered a variety of siloxy-β-ketoester substrates 5.10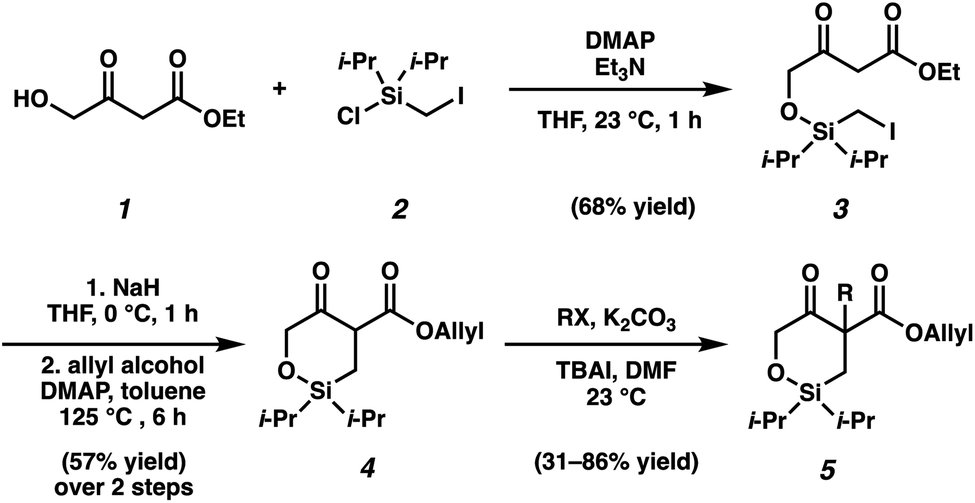 | ||
| Scheme 2 Siloxy-β-ketoester substrates synthesis. | ||
With the substrates in hand, our studies continued with an investigation of the palladium-catalyzed decarboxylative allylic alkylation (Table 1). We were pleased to find that treatment of 5a to commonly employed allylic alkylation conditions11 using Pd2(dba)3 and (S)-t-BuPHOX ligand (L1) in toluene proceeded smoothly with >95% conversion and provided the desired cyclic siloxane 6a in a moderate 75% ee (Table 1, entry 1). An examination of additional ligands revealed that switching to the more electron-deficient PHOX ligand L2 delivered the product in an excellent 91% ee (entry 2), while the use of Trost-type bisphosphine ligand L3 (ref. 12) provided 6a in a slightly diminished 89% ee (entry 3). Continuing with the optimal ligand L2, a survey of different solvents revealed that while consistently high levels of enantioselectivity could be achieved (entries 4–7), toluene remained the optimal solvent. Finally, by performing the reaction at 0 °C, the highest ee of 94% was obtained with no loss in reactivity.
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | % conv.b | % eec |
| a Conditions: 5a (0.1 mmol), Pd2(dba)3 (5 mol%), ligand (12.5 mol%) in solvent (3 mL). b Conversion determined by 1H NMR of crude reaction mixture using 1,3,5-trimethoxybenzene as a standard. c %ee determined by chiral SFC analysis of the isolated product. d Reaction performed at 0 °C. | ||||
| 1 | L1 | Toluene | >95 | 75 |
| 2 | L2 | Toluene | >95 | 91 |
| 3 | L3 | Toluene | >95 | −89 |
| 4 | L2 | THF | >95 | 87 |
| 5 | L2 | MTBE | >95 | 87 |
| 6 | L2 | 1,4-Dioxane | >95 | 89 |
| 7 | L2 | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexanes/toluene :1 hexanes/toluene |
>95 | 89 |
| 8 | L2 | Toluene | >95 | 94 |
|
||||

Having identified the optimal reaction conditions, we next explored the substrate scope of this transformation on 0.2 mmol scale (Table 2). With the benzyl group at the α-quaternary position, the enantioenriched cyclic siloxane product (6a) was isolated in 91% yield with 94% ee. An investigation of the electronic nature of this benzyl group demonstrates that both electron-withdrawing and electron-donating groups (6b–6e) were well tolerated (90–93% ee). A sterically hindered ortho-substituted benzyl substrate fares well under our optimized reaction conditions, providing product 6f in 83% yield with 92% ee. A significant drop in reactivity, however, was observed when a methyl-substituted substrate was employed (5g, ca. only 30% conversion). Pleasingly, we were able to improve the reactivity of this substrate by increasing the temperature to 23 °C, delivering product 6g in 79% yield with 89% ee. Additionally, cyclic siloxane starting materials bearing ester (5h) and Boc-protected alkyl amine (5i) functionalities delivered products 6h and 6g in 94% ee and 90% ee, respectively.
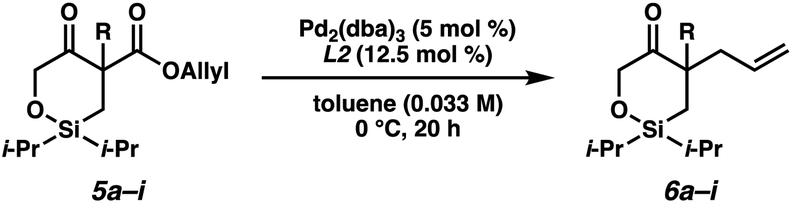
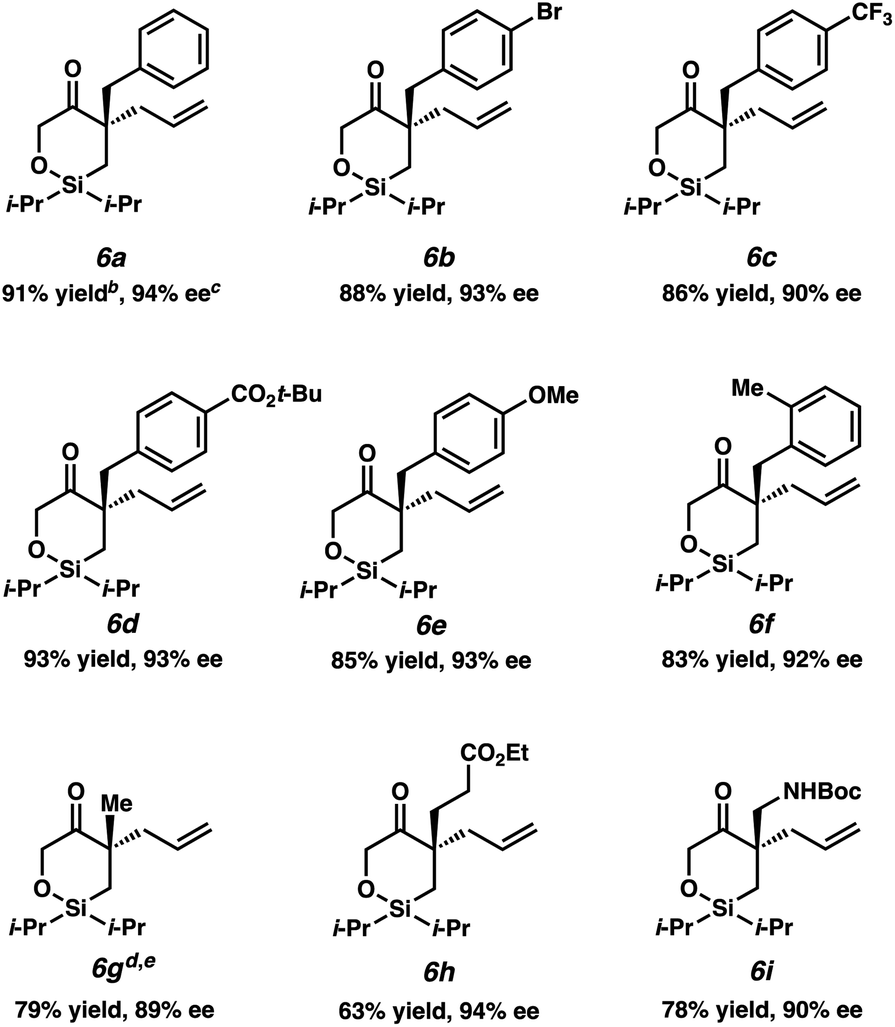
Despite several attempts at procuring X-ray quality crystals for determination of the absolute stereochemistry of the products, these efforts proved fruitless, presumably due to the non-polar nature and flexibility of these molecules. Thus, vibrational circular dichroism (VCD) was utilized for the determination of absolute configuration of systems 6a and 6g as single enantiomers. Sufficient agreement between the computed and measured VCD spectra were achieved such that the absolute configurations of 6a and 6g could be comfortably assigned as (R) and (S), respectively, as depicted in Table 2.13 Additionally, the measured optical rotation of 6g was found to be consistent with that computed for the Boltzmann-weighted conformational ensemble of the (S) enantiomer. With these configurations assigned, the absolute configurations for all other products have been inferred by analogy.
The next stage in our study was the utilization of the silyl fragment as a functional handle to access acyclic chiral building blocks (Scheme 3). Unfortunately, the direct ring-opening of cyclic siloxane 6a proved to be challenging as both neutral (TBAF) and oxidative (Tamao oxidation) conditions failed to deliver the desired acyclic products. However, we were pleased to find that with prior hydoboration/oxidation and concomitant ketone reduction, facile ring opening could be achieved. The diol 7 was isolated in 58% yield as a 93:7 mixture of diastereomers.14 Siloxane ring opening can then be achieved with TBAF either in the absence or presence of NaBO3 as an oxidant to deliver triol 8 or tetraol 9 in 77% yield and 51% yield, respectively.
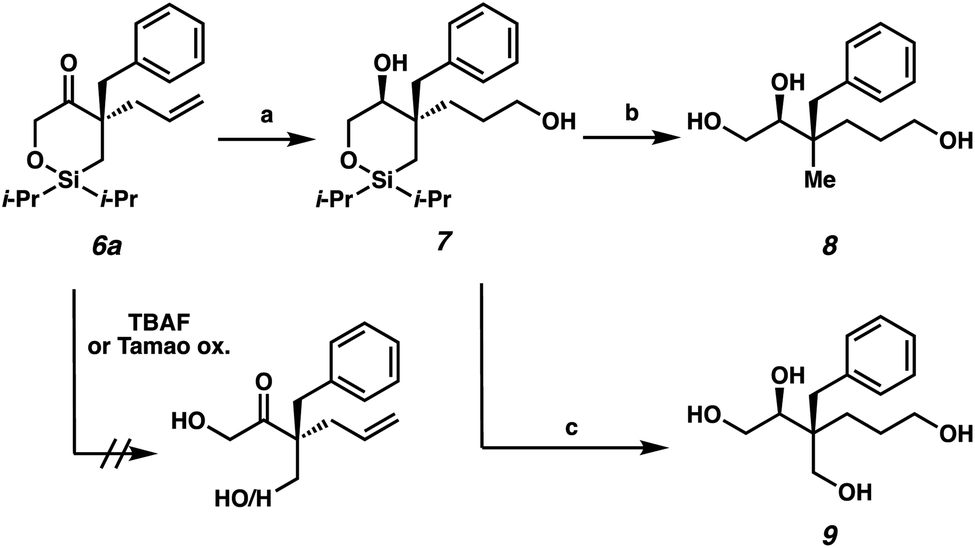 | ||
| Scheme 3 Synthesis of highly oxygenated, chiral, acyclic quaternary stereocenters: (a) BH3·SMe2, THF, 23 °C, 18 h; H2O, 23 °C, 10 min; NaBO3·4H2O, 23 °C, 4 h, 58% yield, dr = 93:7; (b) TBAF, DMF, 80 °C, 18 h, 77% yield; (c) TBAF, NaBO3·4H2O, THF, 60 °C, 18 h, 51% yield. | ||
We also sought to further demonstrate the utility of these highly oxygenated acyclic building blocks via the synthesis of a densely functionalized lactone (Scheme 4). First, selective benzylation of triol 8 provided monobenzylated product 10via a stannylene acetal intermediate.15 Subsequently, δ-lactone 11, bearing vicinal quaternary-trisubstituted stereocenters, could be generated from TEMPO oxidation in 39% yield over 3 steps.
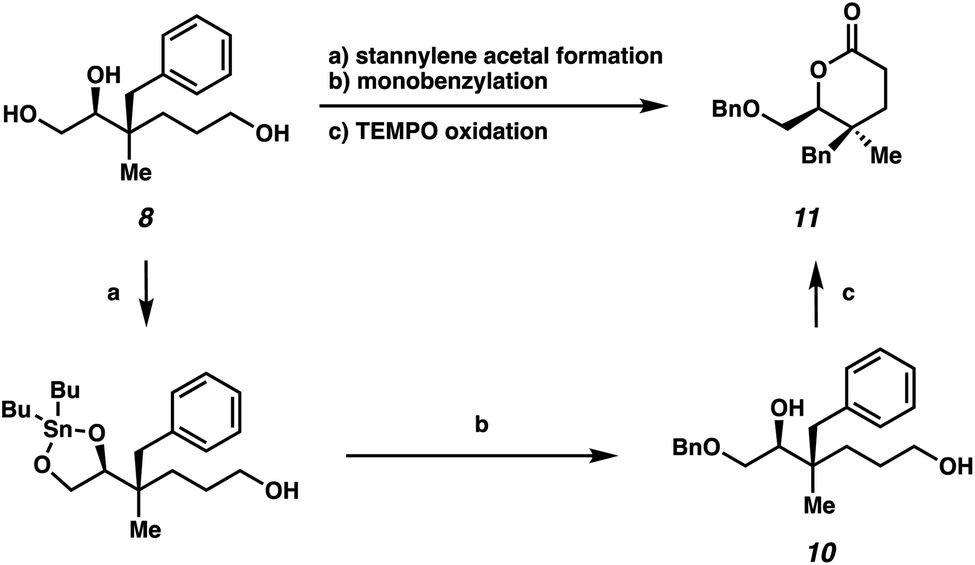 | ||
| Scheme 4 Synthesis of lactone bearing vicinal quaternary-trisubstituted stereocenters: (a) Bu2SnO, MeOH, reflux, 18 h; (b) BnBr, TBAB, toluene, reflux, 20 h, 60% yield over 2 steps; (c) TEMPO, PIDA, CH2Cl2, 23 °C, 18 h, 65% yield. | ||
Conclusions
In summary, we have developed the first enantioselective synthesis of cyclic siloxanes bearing an all-carbon quaternary stereocenter. Through Pd-catalyzed decarboxylative allylic alkylation, enantioenriched cyclic siloxanes can be constructed in good yield (up to 91% yield) and with high enantiomeric excess (up to 94% ee). The reaction conditions exhibit a high tolerance toward a range of functional groups, and the alkylation products can be further derivatized to obtain stereochemically rich acyclic building blocks. Efforts to further extend the substrate scope of this transformation, as well as applications in natural product synthesis, are currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The NIH-NIGMS (R01GM080269) and Caltech are thanked for support of our research program. A. N. thanks the Royal Thai Government Scholarship program. T. I. thanks Japan Tobacco Inc. for funding. We thank Dr Makoto Shiozaki for helpful discussion and Dr Mona Shahgholi for mass spectrometry assistant. Dr Scott Virgil is acknowledged for instrumentation and SFC assistance.Notes and references
- (a) M. Bols and T. Skrydstrup, Chem. Rev., 1995, 95, 1253–1277 Search PubMed; (b) S. Bracegirdle and E. A. Anderson, Chem. Soc. Rev., 2010, 39, 4114–4129 Search PubMed.
- For selected examples, see (a) A. Shvartsbart and A. B. Smith III, J. Am. Chem. Soc., 2015, 137, 3510–3519 Search PubMed; (b) P. Lu, A. Mailyan, Z. Gu, D. M. Guptill, H. Wang, H. M. L. Davies and A. Zakarian, J. Am. Chem. Soc., 2014, 136, 17738–17749 Search PubMed; (c) E. Lee, H. Y. Song, J. W. Kang, D.-S. Kim, C.-K. Jung and J. M. Joo, J. Am. Chem. Soc., 2002, 124, 384–385 Search PubMed.
- For a C–H oxygenation approach, see (a) C. Huang, N. Ghavtadze, B. Godoi and V. Gevorgyan, Chem.–Eur. J., 2012, 18, 9789–9792 Search PubMed. For a Heck reaction approach, see (b) M. Parasram, V. O. Iaroshenko and V. Gevorgyan, J. Am. Chem. Soc., 2014, 136, 17926–17929 Search PubMed . For other approaches such as cycloaddition and hydrosilylation, see ref. 1a and b.
- For examples of enantiospecific methods to access chiral siloxanes, see (a) B. M. Trost, Z. T. Ball and K. M. Laemmerhold, J. Am. Chem. Soc., 2005, 127, 10028–10038 Search PubMed; (b) A. G. Gallagher, H. Tian, O. A. Torres-Herrera, S. Yin, A. Xie, D. M. Lange, J. K. Wilson, L. G. Mueller, M. R. Gau, P. J. Carroll and D. Martinez-Solorio, Org. Lett., 2019, 21, 8646–8651 Search PubMed.
- (a) S. L. Aeilts, D. R. Cefalo, P. J. Bonitatebus Jr, J. H. Houser, A. H. Hoveyda and R. R. Schrock, Angew. Chem., Int. Ed., 2001, 40, 1452–1456 Search PubMed; (b) E. P. Balskus and E. N. Jacobsen, Science, 2007, 317, 1736–1740 Search PubMed.
- For our recent examples, see (a) C. I. Jette, Z. J. Tong, R. G. Hadt and B. M. Stoltz, Angew. Chem., Int. Ed., 2020, 59, 2033–2038 Search PubMed; (b) Z. P. Sercel, A. W. Sun and B. M. Stoltz, Org. Lett., 2019, 21, 9158–9161 Search PubMed; (c) E. J. Alexy, T. J. Fulton, H. Zhang and B. M. Stoltz, Chem. Sci., 2019, 10, 5996–6000 Search PubMed; (d) A. W. Sun, S. N. Hess and B. M. Stoltz, Chem. Sci., 2019, 10, 788–792 Search PubMed; (e) A. Ngamnithiporn, C. I. Jette, S. Bachman, S. C. Virgil and B. M. Stoltz, Chem. Sci., 2018, 9, 2547–2551 Search PubMed; (f) E. J. Alexy, H. Zhang and B. M. Stoltz, J. Am. Chem. Soc., 2018, 140, 10109–10112 Search PubMedFor a review, see (g) Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 Search PubMed.
- For reviews on the development of Pd-catalyzed decarboxylative allylic alkylation, see (a) J. D. Weaver, A. Recio, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 68 Search PubMed; (b) J. James, M. Jackson and P. J. Guiry, Adv. Synth. Catal., 2019, 361, 3016–3049 Search PubMed; (c) J. A. Tunge, Isr. J. Chem., 2020, 60, 351–359 Search PubMed.
- For asymmetric allylic alkylation of silicon-containing starting materials, see (a) J.-P. Chen, C.-H. Ding, W. Liu, X.-L. Hou and L.-X. Dai, J. Am. Chem. Soc., 2010, 132, 15493–15495 Search PubMed; (b) M. M. Yamano, R. R. Knapp, A. Ngamnithiporn, M. Ramirez, K. N. Houk, B. M. Stoltz and N. K. Garg, Angew. Chem., Int. Ed., 2019, 58, 5653–5657 Search PubMed.
- For recent reviews, see (a) B. Trost and J. E. Schultz, Synthesis, 2019, 51, 1–30 Search PubMed; (b) J. Feng, M. Holmes and M. J. Krische, Chem. Rev., 2017, 117, 12564–12580 Search PubMed.
- Attempts to access 5- and 7-membered siloxyketones via this synthetic route proved challenging. No reaction was observed in the α-alkylation of 5-membered allyl-β-ketoester substrate.
- D. C. Behenna, J. T. Mohr, N. H. Sherden, S. C. Marinescu, A. M. Harned, K. Tani, M. Seto, S. Ma, Z. Novák, M. R. Krout, R. M. McFadden, J. L. Roizen, J. A. Enquist Jr, D. E. White, S. R. Levine, K. V. Petrova, A. Iwashita, S. C. Virgil and B. M. Stoltz, Chem.–Eur. J., 2011, 17, 14199–14223 Search PubMed.
- (a) E. J. Alexy, S. C. Virgil, M. D. Bartberger and B. M. Stoltz, Org. Lett., 2017, 19, 5007–5009 Search PubMed; (b) B. M. Trost, J. Xu and T. Schmidt, J. Am. Chem. Soc., 2009, 131, 18343–18357 Search PubMed.
- See ESI† for the computational protocols, computed and measured VCD spectra, and computed and measured optical rotations..
- Diastereoselective reduction has been previously observed in similar systems, see (a) B. M. Trost, R. Radinov and E. M. Grenzer, J. Am. Chem. Soc., 1997, 119, 7879–7880 Search PubMed; (b) C.-K. Sha, R.-T. Chiu, C.-F. Yang, N.-T. Yao, W.-H. Tseng, F.-L. Liao and S.-L. Wang, J. Am. Chem. Soc., 1997, 119, 4130–4135 Search PubMed.
- A. B. C. Simas, K. C. Pais and A. A. T. da Silva, J. Org. Chem., 2003, 68, 5426–5428 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H NMR, 13C NMR, and IR spectra, SFC traces of racemic and chiral compounds, VCD data or other electronic format. See DOI: 10.1039/d0sc04383d |
| ‡ A. N. and T. I. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |