
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox-controlled chalcogen and pnictogen bonding: the case of a sulfonium/stibonium dication as a preanionophore for chloride anion transport†
Gyeongjin
Park
and
François P.
Gabbaï
 *
*
Department of Chemistry, Texas A&M University, College Station, Texas 77843-3255, USA. E-mail: francois@tamu.edu
First published on 10th September 2020
Abstract
Our interest in the chemistry of tunable chalcogen and pnictogen bond donors as Lewis acidic platforms for the complexation and transport of anions has led us to investigate examples of such compounds that can be activated by redox events. Here, we describe the synthesis of [o-MePhS(C6H4)SbPh3]2+ ([3]2+) and [o-MePhS(C6H4)Sb(p-Tol)3]2+ ([4]2+), two dicationic stibonium/sulfonium bifunctional Lewis acids which were obtained by methylation of the phenylthioether derivatives [o-PhS(C6H4)SbPh3]+ ([1]+) and [o-PhS(C6H4)Sb(p-Tol)3]+ ([2]+), respectively. An evaluation of the chloride anion transport properties of these derivatives using chloride-loaded POPC unilamellar vesicles shows that the activity of the monocations [1]+ and [2]+ greatly exceeds that of the dications [3]2+ and [4]2+, a phenomenon that we assign to the higher lipophilicity of the monocationic compounds. Harnessing this large transport activity differential, we show that [4]2+ can be used as a prechloridophore that is readily activated by reduction of the sulfonium moiety. Indeed, [4]2+ reacts with GSH to afford [2]+ as an active transporter. This activation, which has been monitored in aqueous solution, can also be carried out in situ, in the presence of the chloride-loaded POPC unilamellar vesicles.
Introduction
Promoting chloride anion transport across phospholipid bilayers is an active research area of relevance to the treatment of diseases such as cystic fibrosis and cancer.1–6 Given the complex requirements imposed by these therapeutic applications, many efforts have focussed on understanding the parameters that govern anion transport. Advances in the chemistry of organic transporters that interact with the anion via hydrogen bonds7–13 have identified hydrophobicity as one of the most important parameters since it controls the ability of a transporter to partition in phospholipid membranes while also enhancing anion binding.14–20 It follows that hydrophobicity modulation using an external stimulus could provide access to transporters whose activity can be turned-on under specific conditions. In a recent incarnation of this idea, Manna and co-workers have shown that the water-soluble sulfonium-based preanionophore A is converted into an active, membrane soluble transporter B upon reaction with glutathione (GSH), a tripeptide found in high concentration in cancer cells.21Advances in the field of hydrogen-bond donor anionophores have inspired parallel efforts based on chloridophilic main group compounds.22–27 Owing to their elevated and well-documented anion affinity, antimony derivatives have drawn attention. In a recent study, Matile and co-workers showed that SbPh(C6F5)2 is an active chloride anion transporter that interacts with the anion via pnictogen-bonding.24 Based on the understanding that such interactions are greatly strengthened by oxidation of the antimony center,28,29 our group concomitantly introduced a family of stibonium cations (C)30,31 whose anion transport activity is controlled both by the Lewis acidity of the antimony centre and the lipophilic exterior projected by the aryl substituents.25
Our interest in main group-based anion receptors32,33 has also led us to consider the use of sulfonium cations for anion capture via chalcogen-bonding,34–36 as in the case of D which forms an isolable fluoride anion complex.37 This early investigation also revealed that the sulfonium unit of this derivative is easily reduced by phenyl thiolate, leading to the release of the anionic guest. Considering this result and the recent transport activity turn-on strategy reported by Manna, we have now decided to target mixed sulfonium/stibonium dications aiming to achieve redox-controlled chloride anion transport (Fig. 1).
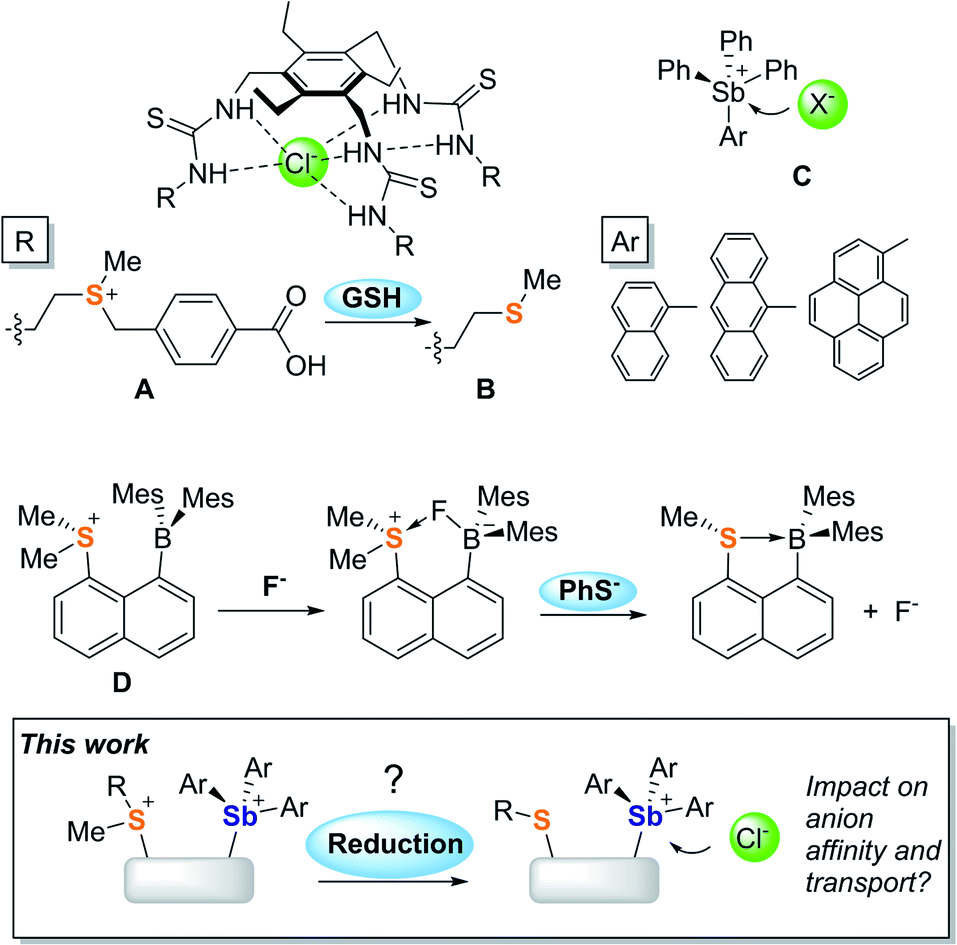 | ||
| Fig. 1 Important precedents and objectives of the current study. | ||
Results and discussion
It occurred to us that such dications could be easily constructed using an ortho-phenylene backbone. With this in mind, we targeted two examples of such derivatives that differ by the nature of the substituents appended to the antimony center. To access these compounds we first carried out the reaction of o-(PhS)C6H4Li with either Ph3SbBr2 or (p-Tol)3SbBr2 in Et2O/THF at −78 °C. These reactions afforded the corresponding bromide derivatives 1-Br and 2-Br which were converted into the corresponding stibonium salts [1][BF4] and [2][BF4] by treatment with AgBF4 (Scheme 1, Fig. S1–S12†). Next, these tetrafluoroborate salts were treated with [Me3O][BF4] in toluene/CH2Cl2. Methylation occurred upon heating of the solution to 90 °C in a sealed tube for 12 hours. The resulting dications [3]2+ and [4]2+ were isolated as air-stable tetrafluoroborate salts (Scheme 1, Fig. S13–S15 and S20–S22†). The 1H NMR spectra show a new resonance at 3.4 ppm for [3]2+ and 3.51 ppm for [4]2 confirming the presence of a sulfur-bound methyl group. The appearance of downfield phenylene resonances in the 7.93–8.21 ppm range for [3]2+ and 7.81–8.27 ppm range for [4]2+ speaks to the dicationic nature of these new derivatives.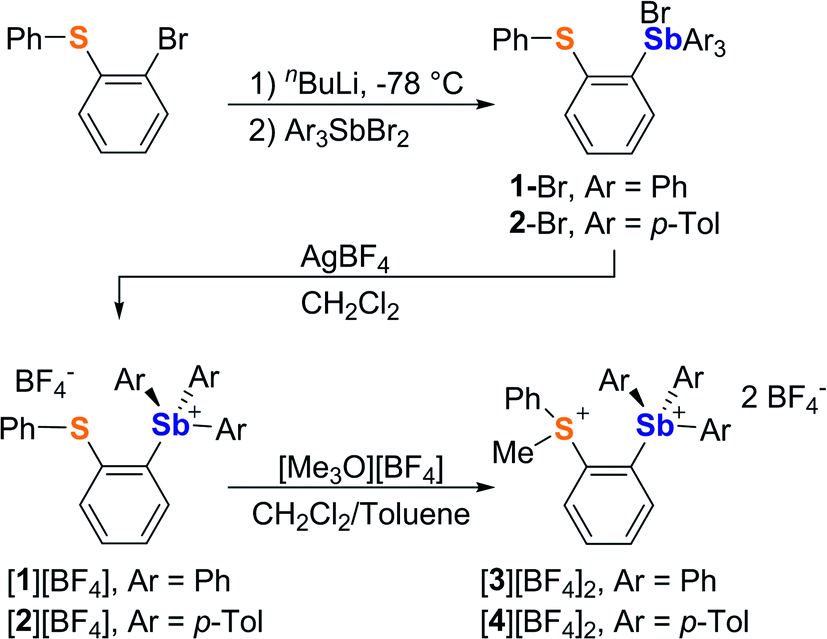 | ||
| Scheme 1 Synthesis of [1][BF4], [2][BF4], [3][BF4]2 and [4][BF4]2. | ||
In the solid-state, the antimony center of [1][BF4] and [2][BF4] adopts a slightly distorted tetrahedral geometry (Fig. 2).38 The sulfur and antimony atoms are separated by 3.2967(13) Å for [1]+ and 3.2999(5) Å for [2]+ a distance that falls within the sum of van der Waals radius of the two elements (ΣvdWR(Sb,S) = 4.33 Å).39 The short S–Sb distances in [1]+ and [2]+ are consistent with a backbone-enforced S → Sb interaction which can be visualized using Natural Bond Orbital (NBO) calculations. The output of these calculations shows the presence of donor–acceptor interactions of lp(S) → σ*(Sb–C) parentage. Deletion calculations indicate that these S → Sb interactions contribute Edel = 6.8 kcal mol−1 and 6.7 kcal mol−1 to the stability of [1]+ and [2]+, respectively. Although this interaction could dampen the electrophilic character of the stibonium centre, we note that [1]+ and [2]+ features a well-defined σ-hole located on the Sb(V) atoms and associated to maximum potential values VS,max of 90.5 kcal mol−1 for [1]+ and 87.1 kcal mol−1 for [2]+. A comparison with [Ph4Sb]+ for which VS,max = 93.9 kcal mol−1 indicates only moderate quenching of the σ-hole in [1]+ and [2]+. The lower VS,max value determined for [2]+ can be correlated to the electron releasing properties of the methyl groups of the p-tolyl substituents.
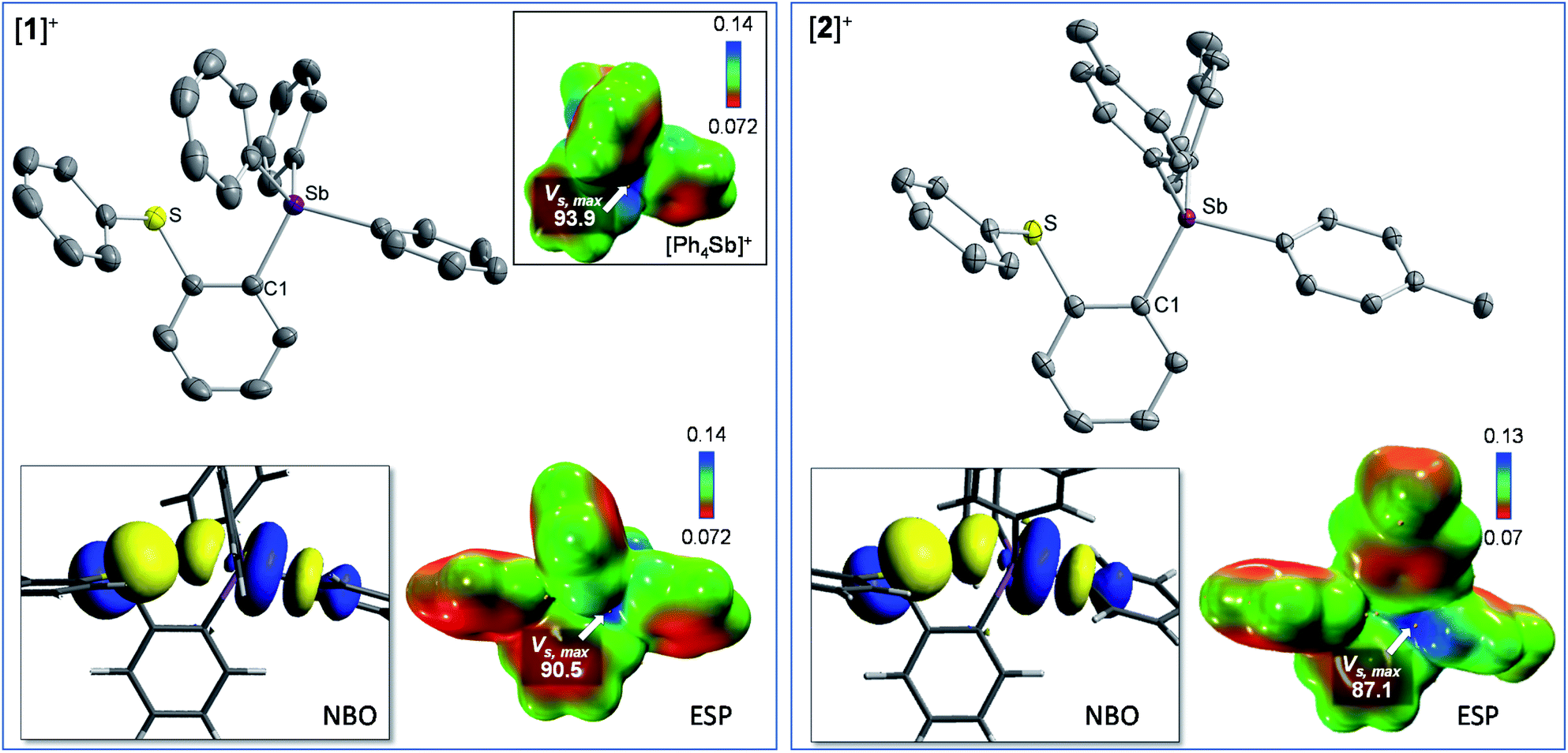 | ||
| Fig. 2 Crystal structures, representative NBOs involved in the S → Sb interactions, and electrostatic potential maps for [1]+ (left) and [2]+ (right). The inset in the left quadrant shows the ESP map of [Ph4Sb]+. For the crystal structures, the ellipsoids are drawn at the 50% probability level, and the hydrogen atoms are omitted for clarity. The ESP maps were obtained with a surface isovalue of 0.001 a.u. pretation, we note that the comput and the gradient scale values are given in a.u. The VS,max values, given in kcal mol−1, were obtained using the Multiwfn software. | ||
While crystals of [3][BF4]2 and [4][BF4]2 could not be obtained, we reproducibly grew single crystals of [3]2[μ2-F][BF4]3 upon exposing a solution of [3][BF4]2 in CH2Cl2 to vapors of pentane over two days (Fig. S16–S19†). The crystal structure of [3]2[μ2-F][BF4]3 confirms the formation of the dication [3]2+.38 In this structure, two of these dications are bridged by a fluoride anion40,41 connected to two symmetry-related trigonal bipyramidal antimony centers. The resulting Sb–F distance of 2.2849(7) Å and the Sb–F–Sb′ angle of 153.67(11)° are reminiscent of those found in polymeric (CH3)4SbF.42 The bridging fluoride anion occupies an apical site, trans from the o-sulfoniophenylene group as indicated by the C1–Sb–F angle of 172.28(8)° (Fig. 3). The S–Sb separation in [3]2+ (3.4361(10) Å) is elongated when compared to that observed in [1][BF4] (3.2967(13) Å), indicating a weakened S–Sb interaction.
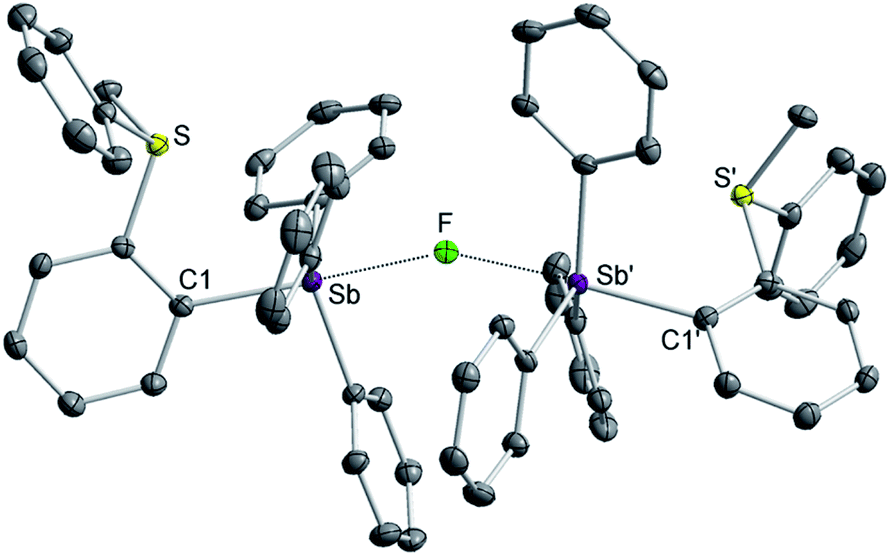 | ||
| Fig. 3 Solid-state structure of [3]2[μ2-F][BF4]3 Thermal ellipsoids are drawn at the 50% probability level. The hydrogen atoms and the free [BF4]− anions are omitted for clarity. Selected bond lengths (Å) and angles (deg): S–Sb = 3.4361(10), Sb–F = 2.2849(7), C1–Sb–F = 172.28(8), Sb–F–Sb′ = 153.67(11). | ||
Interestingly, the computed gas-phase structures of [3]2+ and [4]2+ also feature a lp(S) → σ*(Sb–C) donor–acceptor interaction whose stabilization energy (Edel = 2.4 kcal mol−1 for [3]2+ and Edel = 2.4 kcal mol−1 for [4]2+) is notably lower than in the corresponding monocations [1]+ and [2]+. This difference illustrates the reduced donicity of the sulfonium group in [3]2+ and [4]2+ when compared to the thioether group of [1]+ and [2]+. We have also compared the ESP maps of the monocations [1]+ and [2]+ to those of the dications [3]2+ or [4]2+. Owing to their dicationic nature and reduced S → Sb donor–acceptor bonding, the VS,max values of 153.8 kcal mol−1 for [3]2+ and 149.0 kcal mol−1 for [4]2+ are significantly larger than in the monocationic precursors [1]+ (VS,max = 90.5 kcal mol−1) and [2]+ (VS,max = 87.1 kcal mol−1) indicating the presence of deeper σ-hole at antimony (Fig. 4). It can thus be anticipated that [3]2+ and [4]2+ will display enhanced anion affinity compared to the monocations.
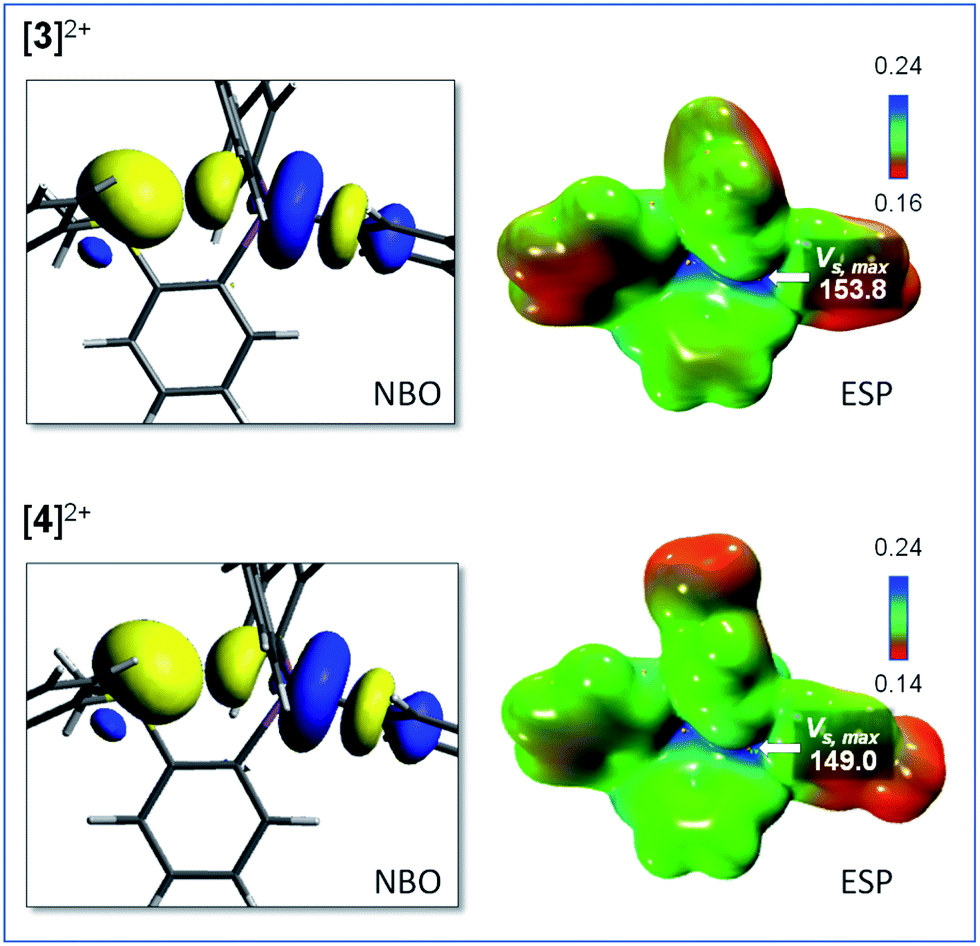 | ||
| Fig. 4 Representative NBOs involved in the S → Sb interactions and electrostatic potential maps for [3]2+ (top) and [4]2+ (bottom). The ESP maps were obtained with a surface isovalue of 0.001 a.u. and the gradient scale values are given in a.u. The VS,max values, given in kcal mol−1, were obtained using the Multiwfn software. | ||
To verify this assumption, we set out to investigate their chloride anion affinity (Fig. S28–S31†). UV-vis titration experiments carried out in MeCN by addition of TBACl indicate that the dications ([3]2+ and [4]2+) display chloride binding constants in excess of 106 M−1, in agreement with the high VS,max values defining their antimony-centred σ-hole. The optimized structures of the corresponding chloride complexes suggest that the chloride anion is concomitantly stabilized via pnictogen-bonding at the stibonium center and chalcogen-bonding43–50 at the sulfonium center (Scheme 2).35,37,51 The monocations [1]+ (KCl− = 4.4(±0.1) × 103 M−1) and [2]+ (KCl− = 2.34(±0.05) × 103 M−1) display significantly lower affinities for the chloride anion in line with their singly charged nature and less marked σ-hole. It follows that the computed ESP features of these derivatives and, in particular, the value of VS,max at the antimony atom, are proper descriptors of the Lewis acidity of these compounds. The lower chloride anion binding constant measured for [2]+ is again a reflection of the electron releasing properties of the methyl groups of the p-tolyl substituents which reduce the Lewis acidity of the antimony center.
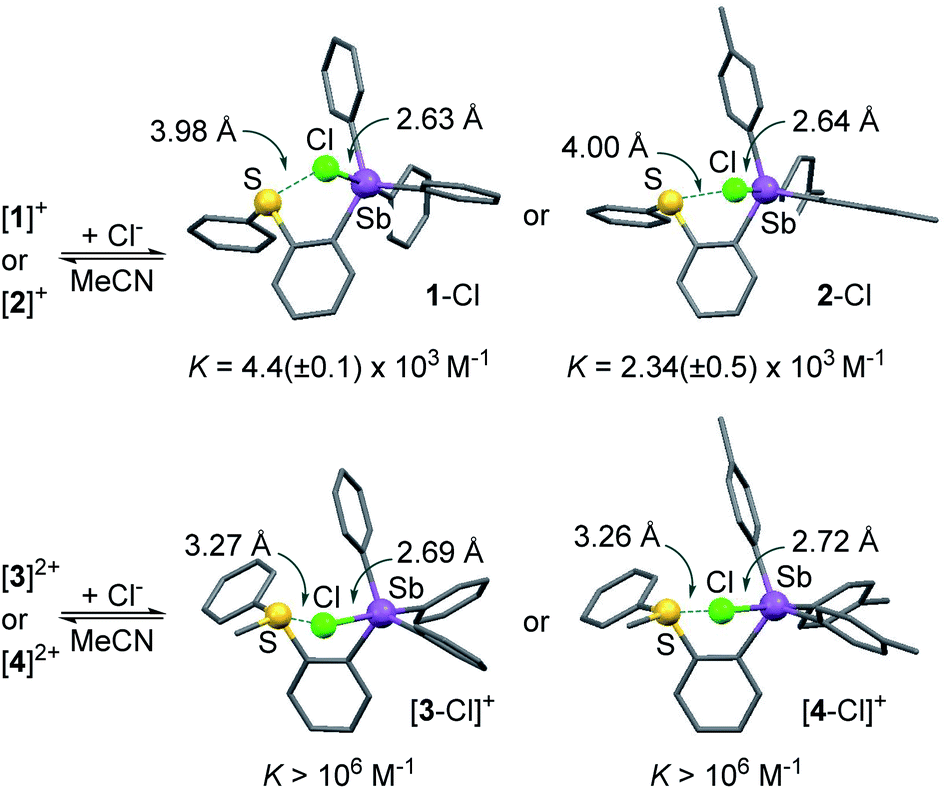 | ||
| Scheme 2 Chloride anion complexation by [1]+, [2]+, [3]2+ and [4]2+. The structures of the chloride complexes were obtained computationally using DFT methods. | ||
Given that we have previously correlated the anion transport properties of the stibonium cations to their anion affinity,25 we became eager to assay the chloride anion transport properties of [1]+, [2]+, [3]2+ and [4]2+. To this end, we prepared POPC-based large unilamellar vesicles (POPC = 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) loaded with a KCl solution buffered at pH 7.2. Chloride efflux was coupled with the addition of the potassium ion transporter valinomycin and monitored with an ion-selective electrode for 300 s after the addition of the transporter (Fig. 5). Since the stibonium salts were injected as DMSO solutions, we first tested the effect of pure DMSO on the chloride anion efflux. We found that this solvent does not mediate any significant transport. Next, we turned our attention to [1]+ and [2]+ and found that these cations promote rapid chloride efflux, with the activity of [2]+ outperforming that of [1]+ as indicated by the EC50 values of 4.7 mol% for [1]+ and 0.6 mol% for [2]+ (Fig. S37 and S38†). We also note that at t = 300 s, the % efflux value obtained with [2]+ (84%) is almost twice as high as that obtained with [1]+. We propose that the higher chloride anion transport activity of [2]+ results from the presence of a Sb(p-Tol)3 moiety that is more hydrophobic than the SbPh3 moiety of [1]+. Since [2]+ is less Lewis acidic that [1]+, we conclude that hydrophobicity is a more influential determinant. In support of this interpretation, we note that the computed n-octanol/water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kow)25 of [2]+ (7.68) is higher than that of [1]+ (logKow = 6.12) as shown in Table S9.† A similar correlation between hydrophobicity and transport activity was observed for stibonium cations of type C.25 We have also assayed [Ph4Sb]+ and found that it mediated moderate transport, which points to the importance of the hydrophobic phenylthioether functionality in [1]+ and [2]+ (Fig. 5). Ph2S was entirely inactive, ruling out the role of the sulfur moiety as being responsible for the anion transport activity of these cations.
Kow)25 of [2]+ (7.68) is higher than that of [1]+ (logKow = 6.12) as shown in Table S9.† A similar correlation between hydrophobicity and transport activity was observed for stibonium cations of type C.25 We have also assayed [Ph4Sb]+ and found that it mediated moderate transport, which points to the importance of the hydrophobic phenylthioether functionality in [1]+ and [2]+ (Fig. 5). Ph2S was entirely inactive, ruling out the role of the sulfur moiety as being responsible for the anion transport activity of these cations.
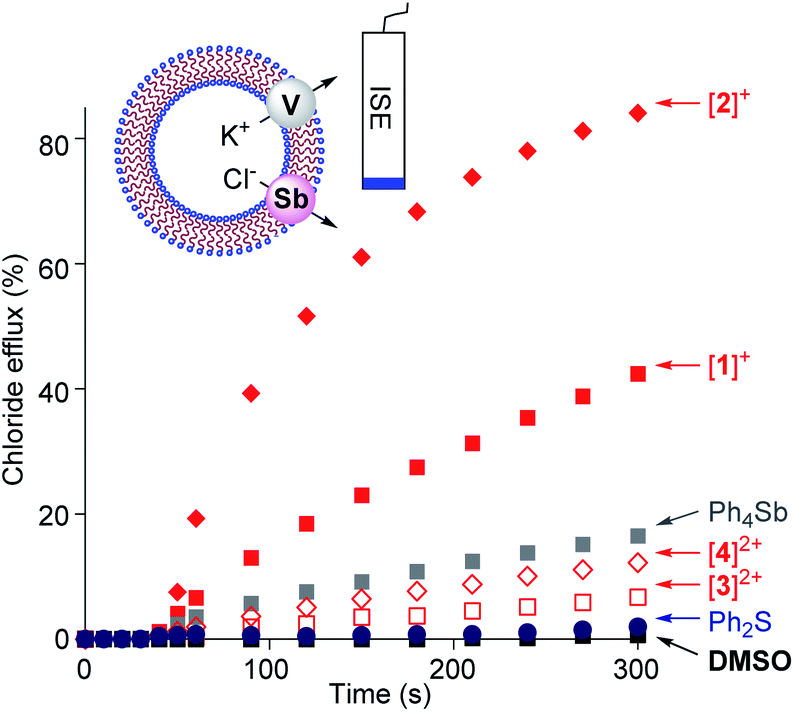 | ||
| Fig. 5 The valinomycin-coupled chloride efflux from POPC vesicles triggered by addition of a DMSO solution (7 μM) containing [1]+, [2]2+,[3]+, [4]2+, Ph4Sb+ or Ph2S (2 mol% in DMSO with respect to the lipid concentration) in the presence of valinomycin. POPC concentration: 0.7 mM. | ||
When compared to [1]+ and [2]+, the sulfonium-stibonium dications [3]2+ and [4]2+ both appear to be significantly less active. Their lower activity is reflected by the initial efflux rates Kini = 0.02% s−1 for [3]2+ and 0.04% s−1 for [4]2+ which are significantly smaller than those of the [1]+ (Kini = 0.26% s−1) and [2]+ (Kini = 1.03% s−1) (see Fig. S33–S36†). We invoke hydrophilicity as the main culprit for the poor performance of these dications. Indeed, the computed n-octanol/water partition coefficient of [3]2+ (logKow = −0.46) and [4]2+ (logKow = 1.26) are six order of magnitude lower than those of [1]+ and [2]+. We suspect that the high hydrophilicity of the dications limits their ability to partition in the vesicle membranes, thus explaining their low transport activity. Such arguments have been previously advanced to rationalize the poor performance of organic, hydrogen-bond donor transporters.52,53 Another factor that could explain the low transport activity of [3]2+ and [4]2+ is their possibly excessive chloride anion affinity, which may impede dynamic capture and release of the anion.50,54,55 A similar argument could be used to explain the better performance of [2]+ when compared to [1]+.
Using the best transporter identified in these studies ([2]+), we have carried out several additional experiments aimed at confirming the mechanism of transport. A chloride transport assay carried with DPPC-based vesicles (DPPC = 1,2-dipalmitoyl-sn-glycero-3-phosphtidycholine) at 25 °C and 45 °C suggests that transport occurs via a carrier type mechanism (Fig. S39†). A Hill analysis of the concentration-dependent chloride anion transport data obtained with [2]+ affords n = 1.3 in agreement with a carrier type mechanism in which one molecule of [2]+ transports one chloride anion (Fig. S38†). Further mechanistic insights were gained by combination of [2]+ with POPC LUVs loaded with carboxyfluorescein (Fig. S40†). Given that no leakage was observed, the anion transport phenomenon observed is unlikely to results from a transporter-induced destabilization of the phospholipid membrane. Finally, we have also confirmed that valinomycin is needed to observe anion transport, thus confirming the electrogenic nature of the transport mechanism (Fig. S32†).
Encouraged by the superior transport activity of [2]+, we became eager to establish whether the reduction of the sulfonium moiety in [4]2+, could be used to turn-on chloride anion transport. After observing that [4]2+ is slowly reduced by DMSO (Fig. S41†), we tested the effect of GSH and NADH, two ubiquitous biological reducing agents. While the reaction with NADH gave rise to a complex mixture of products, GSH swiftly reduced the sulfonium center of [4]2+ in DMSO to afford the corresponding monocation [2]+, as confirmed by NMR spectroscopy (Fig. S42†). This reaction is mostly complete in less than 10 minutes when 3 equivalents of GSH are used. Reduction of [4]2+ also occurs in aqueous solution (D2O:DMSO-d6, 7.9:2.1, v/v) as shown in Fig. 6. The reaction is slower than in DMSO; yet it reaches completion after 90 min at 37 °C, suggesting the possibility of in situ activation of the transporter.
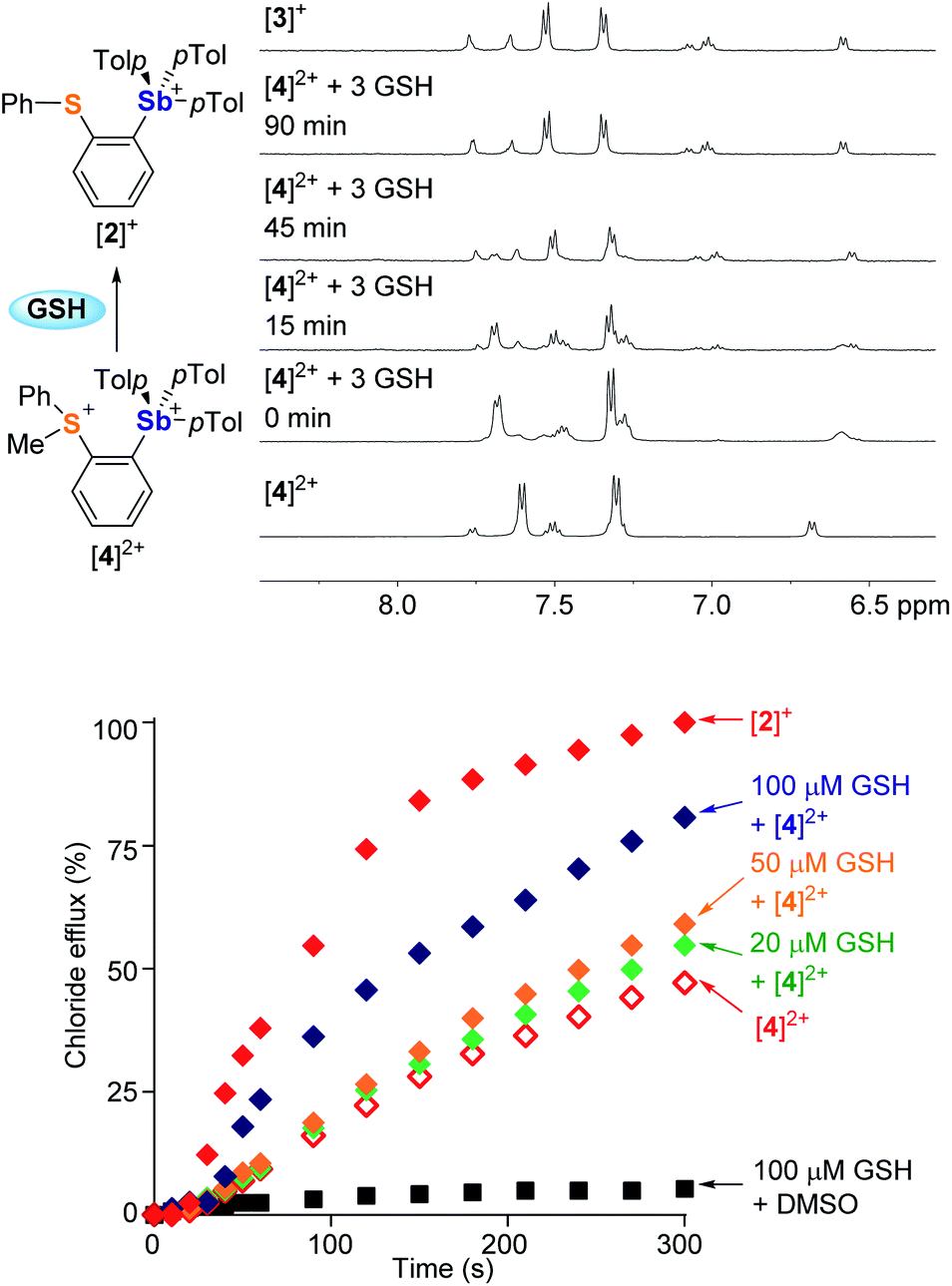 | ||
| Fig. 6 Top: reduction of [4]2+ (5 mM) by GSH (3 equiv.) at 37 °C and selected 1H NMR resonances showing the progress of the reaction in D2O:DMSO-d6 (7.9:2.1, v/v) at pH 7.6 (0.3 M phosphate buffer). Bottom: chloride efflux data of [4]2+ (14 μM), [4]2+ (14 μM)/GSH (20 μM, 50 μM, and 100 μM), and [2]+ after 20 min. Incubation in the external buffer (pH 7.4) containing the POPC vesicles loaded with a chloride cargo at 37 °C. | ||
To test this last idea, solutions containing KCl-loaded POPC vesicles (pH 7.4) were combined with [4]2+ (14 μM) and various amounts of GSH (0 μM, 20 μM, 50 μM or 100 μM). These solutions were incubated at 37 °C for 20 minutes, at which time valinomycin was added to initiate transport. The resulting transport data shows that the extent of chloride efflux correlates with the concentration of GSH (Fig. 6). Given that GSH alone did not mediate any transport, we conclude that the increased chloride efflux results from the in situ reduction of [4]2+, affording the monocation [2]+ as an active transporter.
Conclusion
The results presented herein demonstrate that sulfonium moieties provide a convenient trigger for controlling the activity of antimony-based anion transporters. This trigger is readily activated by the addition of GSH, which reduces the sulfonium centre into the corresponding thioether, leaving the antimony(V) centre intact. This selective reduction can be harnessed to manipulate the lipophilicity of the transporter as well as its overall anion affinity, leading to a drastic enhancement of the chloride anion transport activity. While this approach bears a precedent in the elegant work of Manna, we note that activation of [4]2+ by GSH is significantly faster than that of A which takes several days and necessitates the reduction of not one but three sulfonium centers.21 We propose that the susceptibility of dications such as [4]2+ towards reduction is enhanced by their dicationic nature and the destabilization resulting from the positioning of two adjacent cationic main group moieties. Finally, we note that a parallel exists between these results and the recent work of Gale who showed that gold-protected anion transporters can be activated by reduction of the noble metal.56Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the TAMU College of Science FY19 Strategic Transformative Research Program, the National Science Foundation (CHE-1856453), the Welch Foundation award number (A-1423), and Texas A&M University (Arthur E. Martell Chair of Chemistry).Notes and references
- P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC.
- P. A. Gale, R. Perez-Tomas and R. Quesada, Acc. Chem. Res., 2013, 46, 2801–2813 CrossRef CAS.
- S. V. Shinde and P. Talukdar, Angew. Chem., Int. Ed., 2017, 56, 4238–4242 CrossRef CAS.
- N. Busschaert, S. H. Park, K. H. Baek, Y. P. Choi, J. Park, E. N. W. Howe, J. R. Hiscock, L. E. Karagiannidis, I. Marques, V. Felix, W. Namkung, J. L. Sessler, P. A. Gale and I. Shin, Nat. Chem., 2017, 9, 667–675 CrossRef CAS.
- T. Saha, A. Gautam, A. Mukherjee, M. Lahiri and P. Talukdar, J. Am. Chem. Soc., 2016, 138, 16443–16451 CrossRef CAS.
- S. K. Ko, S. K. Kim, A. Share, V. M. Lynch, J. Park, W. Namkung, W. Van Rossom, N. Busschaert, P. A. Gale, J. L. Sessler and I. Shin, Nat. Chem., 2014, 6, 885–892 CrossRef CAS.
- X. Wu, E. N. W. Howe and P. A. Gale, Acc. Chem. Res., 2018, 51, 1870–1879 CrossRef CAS.
- A. Vargas Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791–2800 CrossRef CAS.
- S. Matile, A. Vargas Jentzsch, J. Montenegro and A. Fin, Chem. Soc. Rev., 2011, 40, 2453–2474 RSC.
- S. Hussain, P. R. Brotherhood, L. W. Judd and A. P. Davis, J. Am. Chem. Soc., 2011, 133, 1614–1617 CrossRef CAS.
- P. A. Gale, Chem. Commun., 2011, 47, 82–86 RSC.
- J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843–3862 RSC.
- A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC.
- S. J. Edwards, I. Marques, C. M. Dias, R. A. Tromans, N. R. Lees, V. Félix, H. Valkenier and A. P. Davis, Chem.–Eur. J., 2016, 22, 2004–2011 CrossRef CAS.
- L. E. Karagiannidis, J. R. Hiscock and P. A. Gale, Supramol. Chem., 2013, 25, 626–630 CrossRef CAS.
- M. J. Spooner and P. A. Gale, Chem. Commun., 2015, 51, 4883–4886 RSC.
- H. Valkenier, C. J. E. Haynes, J. Herniman, P. A. Gale and A. P. Davis, Chem. Sci., 2014, 5, 1128–1134 RSC.
- V. Saggiomo, S. Otto, I. Marques, V. Felix, T. Torroba and R. Quesada, Chem. Commun., 2012, 48, 5274–5276 RSC.
- C. J. E. Haynes, S. J. Moore, J. R. Hiscock, I. Marques, P. J. Costa, V. Félix and P. A. Gale, Chem. Sci., 2012, 3, 1436–1444 RSC.
- N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernández, R. Pérez-Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS.
- N. Akhtar, N. Pradhan, A. Saha, V. Kumar, O. Biswas, S. Dey, M. Shah, S. Kumar and D. Manna, Chem. Commun., 2019, 55, 8482–8485 RSC.
- S. Benz, M. Macchione, Q. Verolet, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 9093–9096 CrossRef CAS.
- A. Vargas Jentzsch, D. Emery, J. Mareda, S. K. Nayak, P. Metrangolo, G. Resnati, N. Sakai and S. Matile, Nat. Commun., 2012, 3, 905 CrossRef.
- L. M. Lee, M. Tsemperouli, A. I. Poblador-Bahamonde, S. Benz, N. Sakai, K. Sugihara and S. Matile, J. Am. Chem. Soc., 2019, 141, 810–814 CrossRef CAS.
- G. Park, D. J. Brock, J. P. Pellois and F. P. Gabbaï, Chem, 2019, 5, 2215–2227 CAS.
- L. E. Bickerton, A. J. Sterling, P. D. Beer, F. Duarte and M. J. Langton, Chem. Sci., 2020, 11, 4722–4729 RSC.
- G. Park and F. P. Gabbaï, Angew. Chem., Int. Ed., 2020, 59, 5298–5302 CrossRef CAS.
- M. Yang, D. Tofan, C. H. Chen, K. M. Jack and F. P. Gabbaï, Angew. Chem., Int. Ed., 2018, 57, 13868–13872 CrossRef CAS.
- D. Tofan and F. P. Gabbaï, Chem. Sci., 2016, 7, 6768–6778 RSC.
- M. Hirai, M. Myahkostupov, F. N. Castellano and F. P. Gabbaï, Organometallics, 2016, 35, 1854–1860 CrossRef CAS.
- I. S. Ke, M. Myahkostupov, F. N. Castellano and F. P. Gabbaï, J. Am. Chem. Soc., 2012, 134, 15309–15311 CrossRef CAS.
- A. J. Plajer, J. Zhu, P. Proehm, A. D. Bond, U. F. Keyser and D. S. Wright, J. Am. Chem. Soc., 2019, 141, 8807–8815 CrossRef CAS.
- A. J. Plajer, J. Zhu, P. Pröhm, F. J. Rizzuto, U. F. Keyser and D. S. Wright, J. Am. Chem. Soc., 2020, 142, 1029–1037 CrossRef CAS.
- Y. Kim, H. Zhao and F. P. Gabbaï, Angew. Chem., Int. Ed., 2009, 48, 4957–4960 CrossRef CAS.
- H. Y. Zhao and F. P. Gabbaï, Nat. Chem., 2010, 2, 984–990 CrossRef CAS.
- C. R. Wade, H. Zhao and F. P. Gabbaï, Chem. Commun., 2010, 6380–6381 RSC.
- H. Zhao and F. P. Gabbai, Org. Lett., 2011, 13, 1444–1446 CrossRef CAS.
- CCDC 2008416–2008418, 2021695 and 2021710 contain the supplementary crystallographic data for this paper.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- M. Hirai and F. P. Gabbaï, Angew. Chem., Int. Ed., 2015, 54, 1205–1209 CrossRef CAS.
- C.-H. Chen and F. P. Gabbaï, Angew. Chem., Int. Ed., 2017, 56, 1799–1804 CrossRef CAS.
- W. Schwarz and H. J. Guder, Z. Anorg. Allg. Chem., 1978, 444, 105–111 CrossRef CAS.
- A. F. Cozzolino, P. J. W. Elder and I. Vargas-Baca, Coord. Chem. Rev., 2011, 255, 1426–1438 CrossRef CAS.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC.
- R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, Chem. Rev., 2018, 118, 2010–2041 CrossRef CAS.
- J. Y. C. Lim and P. D. Beer, Chem, 2018, 4, 731–783 CAS.
- L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS.
- K. Strakova, L. Assies, A. Goujon, F. Piazzolla, H. V. Humeniuk and S. Matile, Chem. Rev., 2019, 119, 10977–11005 CrossRef.
- N. Biot and D. Bonifazi, Coord. Chem. Rev., 2020, 413, 213243 CrossRef CAS.
- B. Zhou and F. P. Gabbaï, Chem. Sci., 2020, 11, 7495–7500 RSC.
- Y. Kim, M. Kim and F. P. Gabbaï, Org. Lett., 2010, 12, 600–602 CrossRef CAS.
- M. J. Spooner, H. Li, I. Marques, P. M. R. Costa, X. Wu, E. N. W. Howe, N. Busschaert, S. J. Moore, M. E. Light, D. N. Sheppard, V. Félix and P. A. Gale, Chem. Sci., 2019, 10, 1976–1985 RSC.
- I. Marques, P. M. R. Costa, M. Q. Miranda, N. Busschaert, E. N. W. Howe, H. J. Clarke, C. J. E. Haynes, I. L. Kirby, A. M. Rodilla, R. Pérez-Tomás, P. A. Gale and V. Félix, Phys. Chem. Chem. Phys., 2018, 20, 20796–20811 RSC.
- J. P. Behr, M. Kirch and J. M. Lehn, J. Am. Chem. Soc., 1985, 107, 241–246 CrossRef CAS.
- A. Vargas Jentzsch, D. Emery, J. Mareda, P. Metrangolo, G. Resnati and S. Matile, Angew. Chem., Int. Ed., 2011, 50, 11675–11678 CrossRef CAS.
- M. Fares, X. Wu, D. Ramesh, W. Lewis, P. Keller, E. Howe, R. Pérez-Tomás and P. A. Gale, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202006392.
Footnote |
| † Electronic supplementary information (ESI) available. Experimental and computational details. NMR spectra of novel compounds. Anion binding and transport data. CCDC 2008416, 2008417, 2008418, 2021695 and 2021710. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04417b |
| This journal is © The Royal Society of Chemistry 2020 |