
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regioselective addition/annulation of ferrocenyl thioamides with 1,3-diynes via a sulfur-transfer rearrangement to construct extended π-conjugated ferrocenes with luminescent properties†
Lipeng
Yan
,
Jingbo
Lan
 *,
Hu
Cheng
,
Yihang
Li
,
Mangang
Zhang
,
Di
Wu
and
Jingsong
You
*
*,
Hu
Cheng
,
Yihang
Li
,
Mangang
Zhang
,
Di
Wu
and
Jingsong
You
*
Key Laboratory of Green Chemistry and Technology of Ministry of Education, College of Chemistry, Sichuan University, 29 Wangjiang Road, Chengdu 610064, People's Republic of China. E-mail: jingbolan@scu.edu.cn; jsyou@scu.edu.cn
First published on 16th September 2020
Abstract
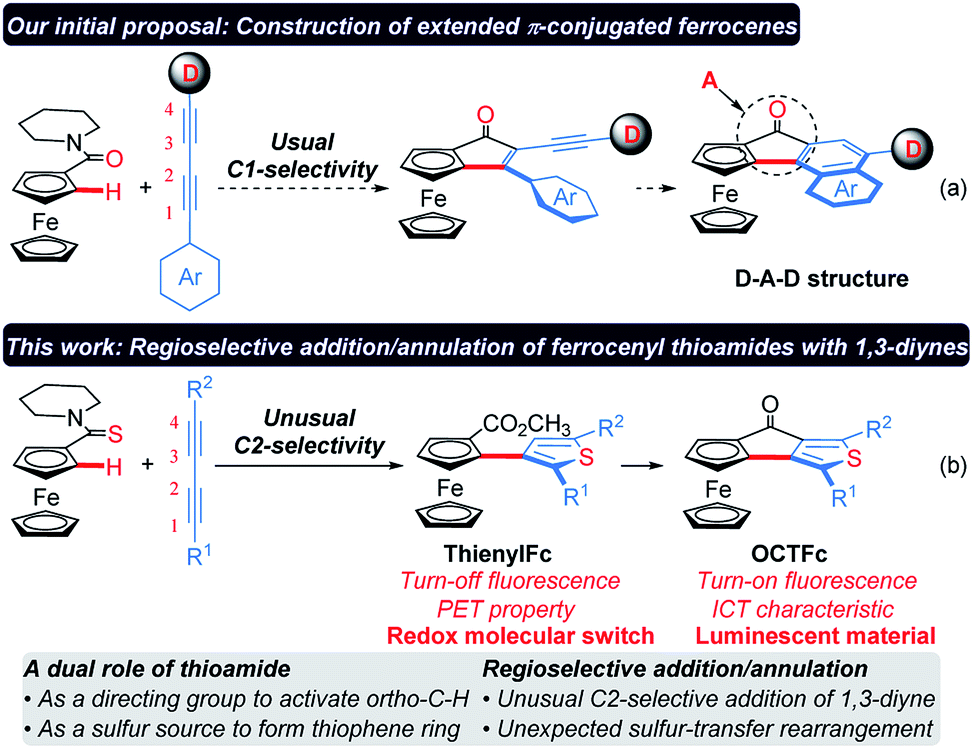
Herein a regioselective addition/annulation strategy of ferrocenyl (Fc) thioamides with alkynes to construct thienylferrocene (ThienylFc) structures, involving a rhodium-catalyzed C–H activation, an unusual C2-selective addition of 1,3-diyne, and an unexpected intramolecular sulfur-transfer rearrangement process is described. In this protocol, thioamide not only serves as a directing group to activate the ortho-C–H bond of the ferrocene, but also as a sulfur source to form the thiophene ring. The resulting carboxylic ester group after sulfur transfer can act as a linkage to construct extended π-conjugated ferrocenes (OCTFc) with luminescent properties. ThienylFc displays effective fluorescence quenching due to the photoinduced electron transfer (PET) from the Fc unit to the excited luminophore, which turns out to be a promising type of redox molecular switch. OCTFc exhibit relatively strong emission owing to their intramolecular charge transfer (ICT) characteristics. The ring-fused strategy is herein employed for the first time to construct luminescent materials based on ferrocenes, which provides inspiration for the development of novel organic optoelectronic materials, such as electroluminescent materials based on ferrocenes.
Introduction
Ferrocene (Fc) is an important building block for versatile ligands, medicines and optoelectronic materials because of its unique sandwich structure, strong π-donating ability, and chemical and thermal stabilities, as well as good reversibility in one-electron oxidation.1–3 Since the first synthesis of ferrocene was reported in 1951,4 its functionalization and relevant application have drawn increasing attention.1–3 In principle, ferrocene can serve as an electron donor (D) to construct luminescent materials with a D–A (A, electron acceptor) structure. However, D–A molecules containing an Fc unit usually show quenched fluorescence due to a photoinduced electron transfer (PET) process from the ferrocene to the excited electron acceptor. Ferrocene can be oxidized to an electron-deficient ferricenium cation (Fc+), which terminates the PET process, thus leading to an enhanced emission. Based on this mechanism, various redox-fluorescence switches derived from ferrocene have been developed recently.5–7Although the negative charge of the cyclopentadienyl (Cp) ligands endows ferrocene with electron-rich aromatic characteristics, many ferrocene derivatives are unavailable through direct electrophilic reactions, such as nitration, chlorination or bromination, because the ferrocene nucleus may be oxidized and/or destructed by nitrate ions, chlorine or bromine.8–11 Thus, Friedel–Crafts acylation and lithiation were proved to be the most used methods for the derivatization of ferrocene.12–17 However, it remains challenging to prepare 1,2-disubstituted ferrocenes because they usually require highly reactive organolithium reagents as strong bases to accomplish the ortho-metalation of mono-substituted ferrocenes,14–16 and thus inevitably suffers from poor functional group compatibility. Recently, chelating-assisted C–H bond functionalization has made significant advances,18–25 which provide new opportunities for the rapid construction of 1,2-disubstituted ferrocenes.26–36 1,3-Diynes are an important class of organic architectures and have been widely used in the construction of various five-membered heteroaromatic rings via the reaction with heteroatoms.37–40 However, it is still a challenging task to control the regioselectivity in the migratory insertion of 1,3-diyne into the carbon–metal bond.41 Herein, we present a regioselective addition/annulation sequence of ferrocenyl thioamides with alkynes to construct thienylferrocene (ThienylFc) structures, which includes a rhodium-catalyzed C–H activation, an unusual C2-selective addition of 1,3-diyne, and an unexpected intramolecular sulfur-transfer rearrangement (Scheme 1b). In this protocol, thioamide not only serves as a directing group to activate the ortho-C–H bond of the ferrocene, but also as a sulfur source to provide the indispensable heteroatom for the formation of the thiophene ring. In addition, these resulting ThienylFc structures can be conveniently transformed into extended π-conjugated ferrocenes (4-oxocyclopentathiophene-fused ferrocenes, OCTFc) with luminescent properties via an intramolecular Friedel–Crafts reaction.
 | ||
| Scheme 1 Design and synthesis of extended π-conjugated ferrocenes. D: electron donor. A: electron acceptor. PET: photoinduced electron transfer. ICT: intramolecular charge transfer. | ||
Results and discussion
Considering that the migratory insertion of 1,3-diyne into the carbon–metal bond usually occurs at the C1-position,41 we initially proposed to construct extended π-conjugated ferrocenes via sequential addition/annulation of ferrocenyl amide with 1,3-diyne and subsequent intramolecular annulation (Scheme 1a). Disappointingly, the reaction of ferrocenyl amide 1 with 1,3-diyne 2a did not deliver such an annulated product 3 (Scheme 2). Actually, the analysis of X-ray single crystal diffraction demonstrated the formation of alkyne addition product 4 (Scheme 2 and Fig. 1a), which was generated via the migratory insertion of 1,3-diyne into the carbon–metal bond at the C2-position rather than the usual C1-position.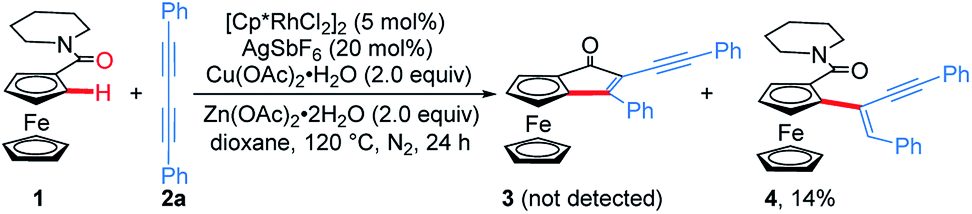 | ||
| Scheme 2 Rh-catalyzed ortho-C–H activation/addition of ferrocenyl amide 1 with 1,3-diyne 2a. | ||
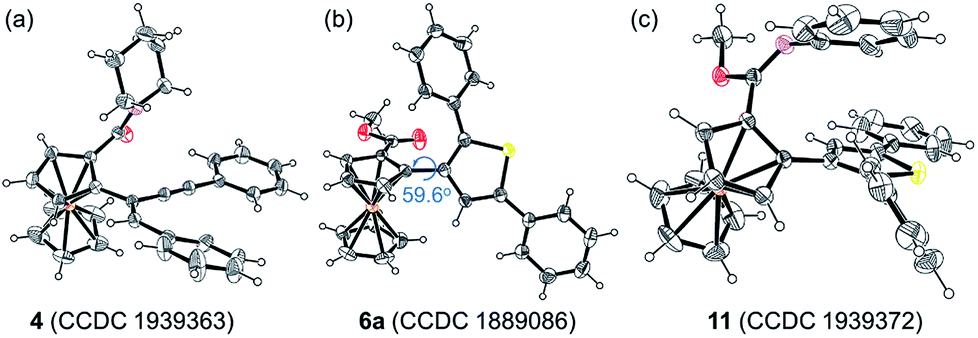 | ||
| Fig. 1 ORTEP diagrams of (a) 4, (b) 6a, and (c) 11. Displacement ellipsoids are drawn at the 30% probability level. | ||
Subsequently, ferrocenyl thioamide (5) was attempted instead of ferrocenyl amide (1). Unexpectedly, in the presence of 5 mol% of [Cp*RhCl2]2, 20 mol% of AgSbF6 and 2.0 equiv. of Cu(OAc)2·H2O in methanol at 80 °C for 24 h, the reaction of 5 with 2a gave ThienylFc (6a) in 46% yield (Scheme 3a and Table S1,† entry 1). The existence of a thiophene ring was confirmed clearly by the X-ray single crystal analysis of 6a (Fig. 1b). In addition, 6a could be conveniently converted into OCTFc 7a through an intramolecular Friedel–Crafts acylation (Scheme 3b).
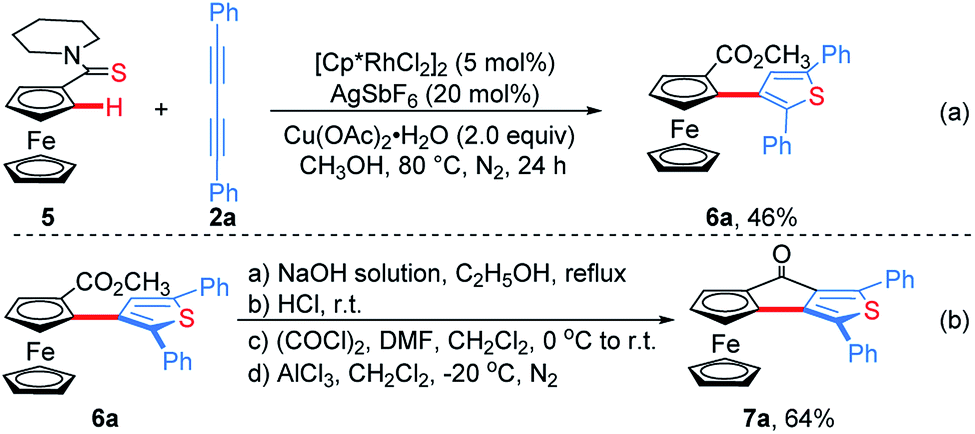 | ||
| Scheme 3 Synthesis of ThienylFc 6a and OCTFc 7a. | ||
The reaction conditions of 5 with 2a were further optimized. Screening of silver salts indicated that AgOTs was better than others, affording 6a in 61% yield (Table S1,† entries 2–5). Other oxidants were proven to be less effective than Cu(OAc)2·H2O (Table S1,† entries 6–9). Owing to the poor solubility of 1,3-diyne in methanol, the solvent volume was doubled, improving the yield to 79% (Table S1,† entry 10). Moreover, the mixed solvent of trichloroethanol and methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v) could further improve the solubility of 1,3-diyne, delivering 6a in an increased yield of 81% (Table S1,† entry 12). Shortening the reaction time and reducing the catalyst dosage both would decrease the yield of 6a (Table S1,† entries 13 and 14). [Cp*Rh(CH3CN)3](OTs)2 was also attempted, which gave 6a in 72% yield, demonstrating that the cationic Rh species is the real reactive catalyst of this addition/annulation of ferrocenyl thioamides with 1,3-diynes (Table S1,† entry 15).
:3, v/v) could further improve the solubility of 1,3-diyne, delivering 6a in an increased yield of 81% (Table S1,† entry 12). Shortening the reaction time and reducing the catalyst dosage both would decrease the yield of 6a (Table S1,† entries 13 and 14). [Cp*Rh(CH3CN)3](OTs)2 was also attempted, which gave 6a in 72% yield, demonstrating that the cationic Rh species is the real reactive catalyst of this addition/annulation of ferrocenyl thioamides with 1,3-diynes (Table S1,† entry 15).
We next examined the substrate scope of 1,3-diynes (Scheme 4). 1,4-Diphenyl-1,3-butadiynes with both electron-donating and electron-withdrawing substituents at the para- or meta-position of the phenyl ring afforded the desired products 6b–6j in good yields. 3,5-Dimethyl, 3,5-dimethoxyl and 3,4-dimethoxyl phenyl-substituted 1,3-butadiynes also worked well under the standard conditions, delivering 6k, 6l and 6m in 72%, 67% and 69% yields, respectively. The unsymmetrical aryl alkyl 1,3-butadiyne could undergo the addition/annulation process with complete regioselectivity confirmed with the 1H–1H NOESY spectrum, but giving a lower yield (6n). No product was obtained when 1,4-dialkyl-1,3-butadiynes were used as the substrate. 1,4-Di(furan-2-yl) and 1,4-di(thiophen-2-yl) substituted 1,3-butadiynes were tolerated by using methanol as solvent, giving 6o–6r in 45–68% yields. 1,4-Di(indol-5-yl)-1,3-butadiyne and 1,4-diferrocenyl-1,3-butadiyne afforded the corresponding thienylferrocenes 6s and 6t in moderate yields when using 1,2-dichloroethane (DCE) as the cosolvent. In addition, ThienylFc containing special functional groups such as triphenylamine and 9,9′-spirobifluorene were prepared when doubling the volume of mixed DCE/CH3OH solvent and increasing the temperature to 120 °C (6u and 6v).
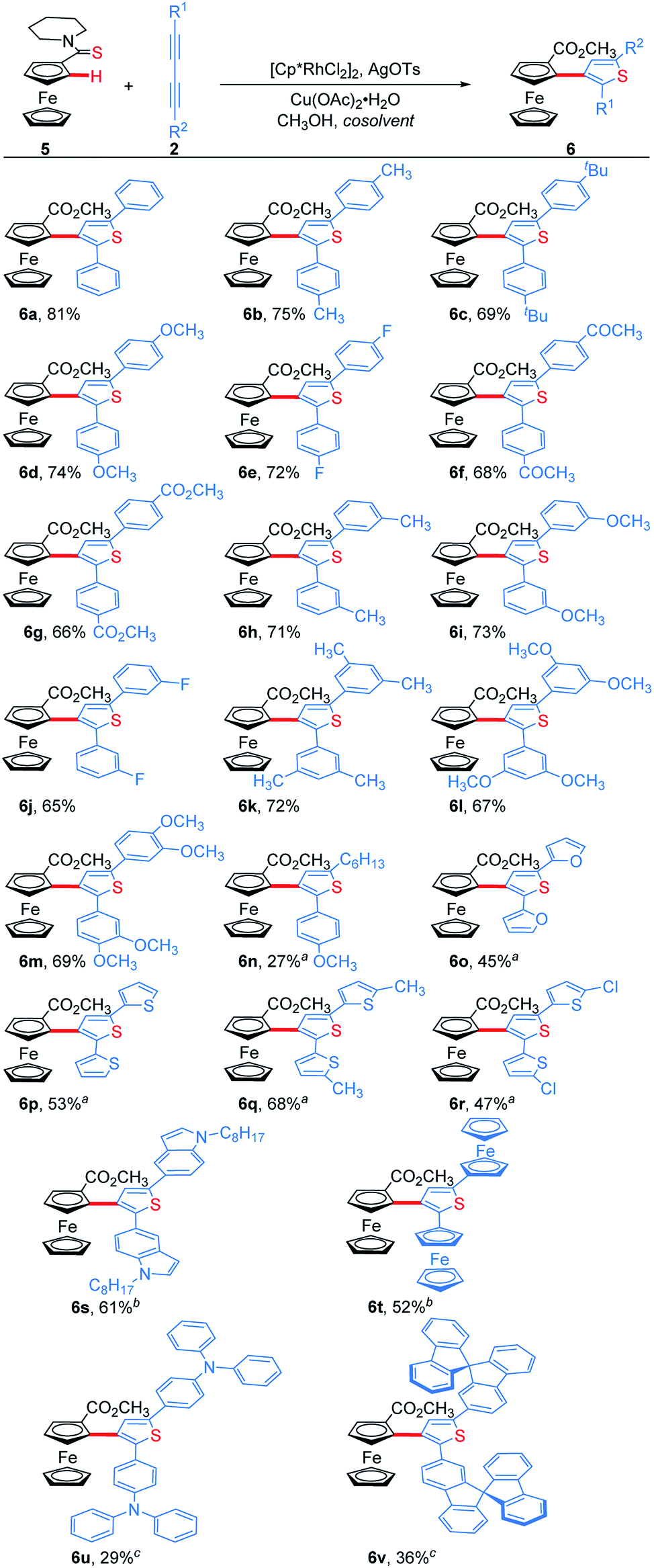 | ||
| Scheme 4 Regioselective addition/annulation of ferrocenyl thioamide with 1,3-diyne. Standard conditions: 5 (0.2 mmol), 2 (0.1 mmol), [Cp*RhCl2]2 (5 mol%), AgOTs (20 mol%), and Cu(OAc)2·H2O (2.0 equiv.) in trichloroethanol (0.5 mL)/methanol (1.5 mL) at 80 °C for 24 h under N2. Isolated yields. a Methanol (2.0 mL) as solvent. b 1,2-Dichloroethane (1.0 mL) and methanol (1.0 mL) as solvents. c 1,2-Dichloroethane (2.0 mL) and methanol (2.0 mL) as solvents at 120 °C. | ||
To get clearer insight into the pathway of the reaction relay, a series of control experiments were conducted (Scheme 5 and Section VII in the ESI†). In the absence of Cu(OAc)2·H2O, the reaction of an equivalent mole of 5 with 1,3-diyne 2a did not provide the desired product 6a, but 5 almost disappeared completely (Scheme 5a). Moreover, without Cu(OAc)2·H2O, 6a could not be obtained when employing other oxidants such as AgOAc, benzoquinone, O2 or air (Table S1,† entries 7–9 and 16). The analysis of HRMS indicated the formation of the thiopyran intermediate III ([M]+: 514.1286, found: 514.1289) (Scheme 6 and Fig. S2†), which could not be isolated from the reaction mixture due to its poor stability. Upon addition of 20 mol% of Cu(OAc)2·H2O, 6a was obtained in 43% yield (Scheme 5b), indicating that the sulfur-transfer reaction may need the participation of the copper ion. The coordination of Cu2+ with the sulfur atom or alkyne may facilitate the cleavage of the C–S bond of thioamide (Scheme 6). Considering that ferrocenyl amide (1) and methyl ferrocene carboxylate (8) could be detected under the standard reaction conditions (Scheme 4, 6a), the reactions of 1 and 8 with 2a were performed in the presence of external sulfur sources, such as S8, NaHS·H2O and Na2S·9H2O, respectively.42–44 However, 6a was not observed (Scheme 5c and d). The cycloaddition reaction of 4 did not occur in the presence of additional sulfur sources (Scheme 5e). Moreover, 6a was not detected in the reactions of 1, 5 and 8 with 2,5-diphenylthiophene (9), respectively (Scheme 5f–h). The above results demonstrate that the sulfur atom of the thiophene ring of 6a comes from the thioamide group via the intramolecular sulfur-transfer rearrangement, and moreover, the sulfur transfer is a successive process. Although thioamide has been employed as a directing group to activate ortho-C–H bonds,33,45,46 it remains undisclosed that a directing group containing sulfur simultaneously acts as a sulfur source to participate in the functionalization process.
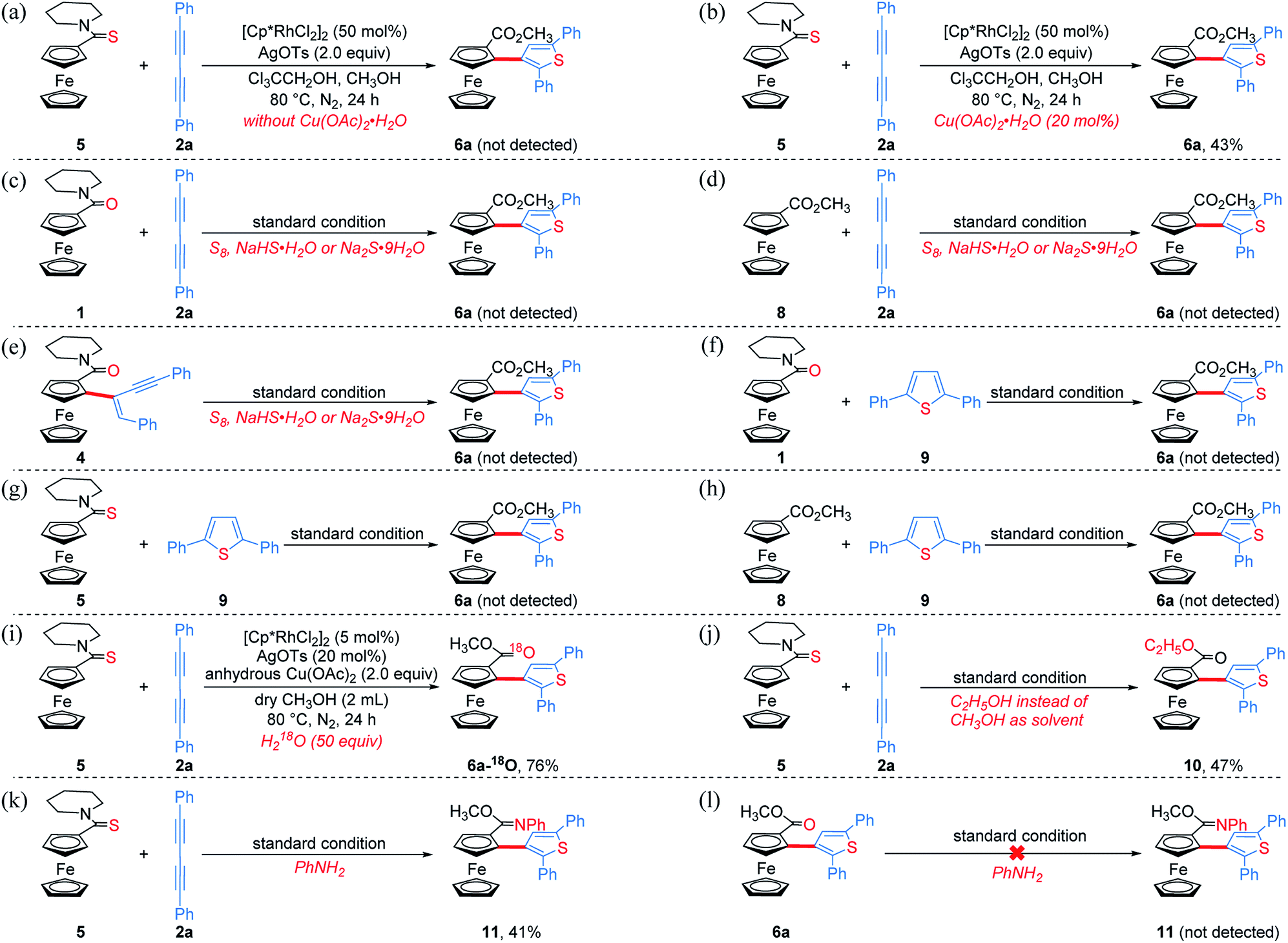 | ||
| Scheme 5 Control experiments and mechanistic investigations. | ||
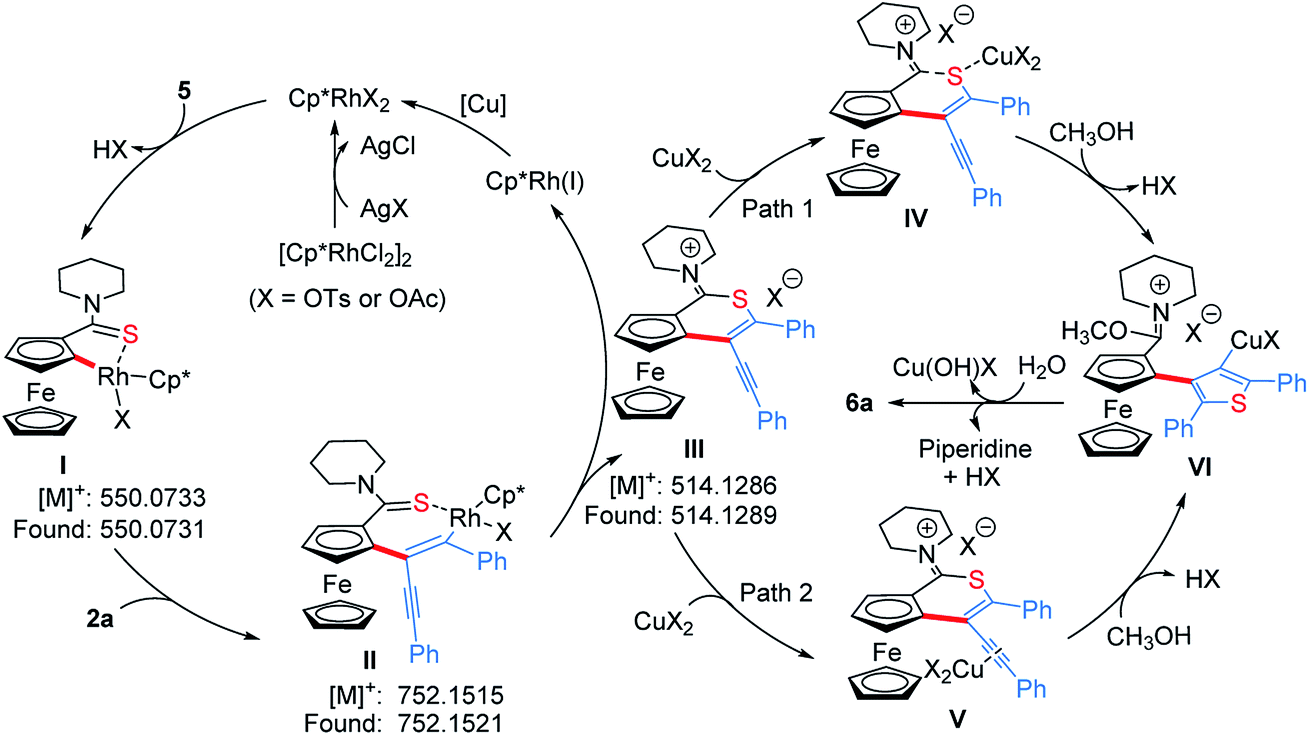 | ||
| Scheme 6 Proposed mechanistic pathway. | ||
Furthermore, 6a–18O and ethyl thienylferrocenyl carboxylate (10) were formed upon addition of H218O and ethanol into the reaction system, respectively, indicating that the carbonyl oxygen atom of 6a stems from water and the alkoxy originates from alcohol through the nucleophilic substitution (Scheme 5i, j and Fig. S1†).47 When adding aniline into this reaction system, N-phenyl carbimidate substituted ThienylFc 11 was obtained in 41% yield (Fig. 1c and Scheme 5k). However, the reaction of 6a and aniline could not deliver 11 under the standard conditions (Scheme 5l). The above observations further validate a successive nucleophilic substitution process.
Based on the above mechanistic studies, a plausible pathway was proposed in Scheme 6. Firstly, the thioamide-directed ortho-C–H activation of 5 forms the five-membered cyclic rhodium intermediate I, detected by ESI-HRMS ([M]+: 550.0733, found: 550.0731, Fig. S2†). 1,3-Diyne 2a inserts into the C–Rh bond to generate the seven-membered cyclic rhodium intermediate II ([M]+: 752.1515, found: 752.1521, Fig. S3†). The reductive elimination of II delivers thiopyran III ([M]+: 514.1286, found: 514.1289, Fig. S2†), and the released Rh(I) species is re-oxidized by copper salt to the reactive Rh(III). The coordination of the copper ion with sulfur or alkyne affords intermediate IV or V. Then, the nucleophilic attack of methanol to the imine cation and subsequent Cu(II)-assisted sulfur-transfer form intermediate VI. Finally, the hydrolysis of the imidate and thienylcopper fragments gives 6a.
To demonstrate the effectiveness and utility of our methodology, a gram scale experiment was performed, affording 6a in 72% yield (1.03 g, Scheme 7). Subsequently, ThienylFc structures 6u and 6v were transformed into the extended π-conjugated OCTFc 7b and 7c through the hydrolysis of methyl carboxylate in NaOH solution, acidification, nucleophile substitution, and intramolecular Friedel–Crafts acylation (Scheme 8).
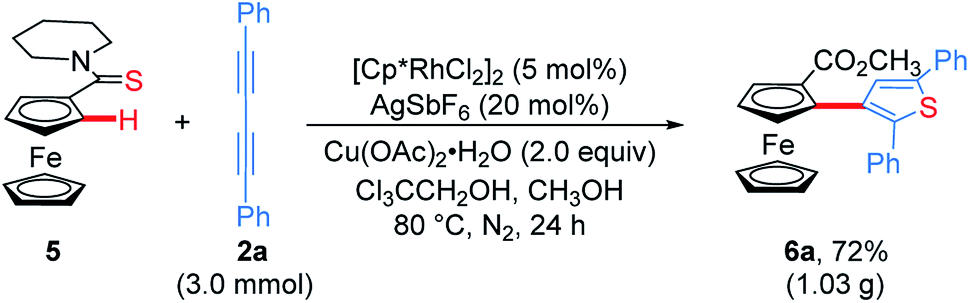 | ||
| Scheme 7 Gram-scale synthesis of ThienylFc 6a. | ||
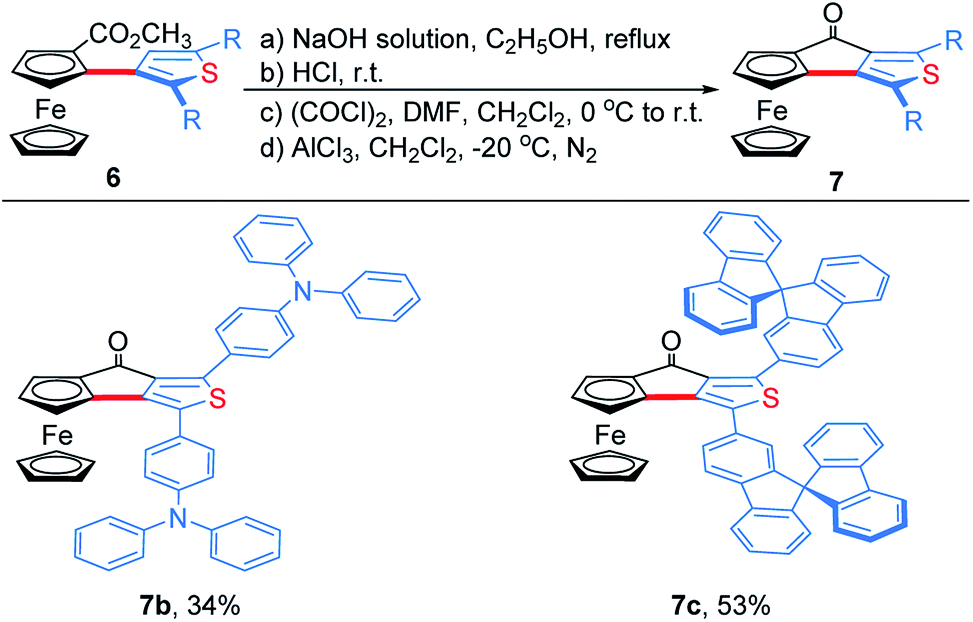 | ||
| Scheme 8 Synthesis of extended π-conjugated ferrocenes 7b and 7c. | ||
The photophysical properties of 6u, 6v, 7b and 7c were studied. The absorption spectra of 6u and 6v show peaks at 372 nm and 361 nm, respectively (Fig. S4†). The absorption bands of 7b and 7c are broadened and red-shifted due to their extended π-conjugation and intramolecular charge transfer (ICT) transitions (Fig. S4†). Density functional theory (DFT) computation demonstrates that the dihedral angles between Cp and thiophene rings are 55.8° and 46.6° for the ThienylFc structures 6u and 6v, respectively, which are roughly consistent with that in the single-crystal data of 6a (Fig. 1b, 2a and S5a†). The twisted conformation inhibits the ICT process. Therefore, 6u and 6v display effective fluorescence quenching due to the photoinduced electron transfer (PET) from the Fc unit to the excited luminophore (Fig. 2c and S6†).5–7 In sharp contrast, conformational twisting is not allowed in the OCTFc structure (Fig. 2b and S5b†). Thus, 7b and 7c exhibit relatively strong red-shifted emission owing to their ICT characteristics (Fig. 2c and S6†). The emission wavelengths of 7b and 7c are gradually red-shifted with an increasing solvent polarity, which are in accordance with the ICT effect (Fig. S7†).48 Although fused ferrocenes have been studied widely,49–53 the ring-fused strategy is herein employed for the first time to construct luminescent materials based on ferrocenes, which would give us inspiration for the development of novel organic optoelectronic materials, such as electroluminescent materials based on ferrocenes.
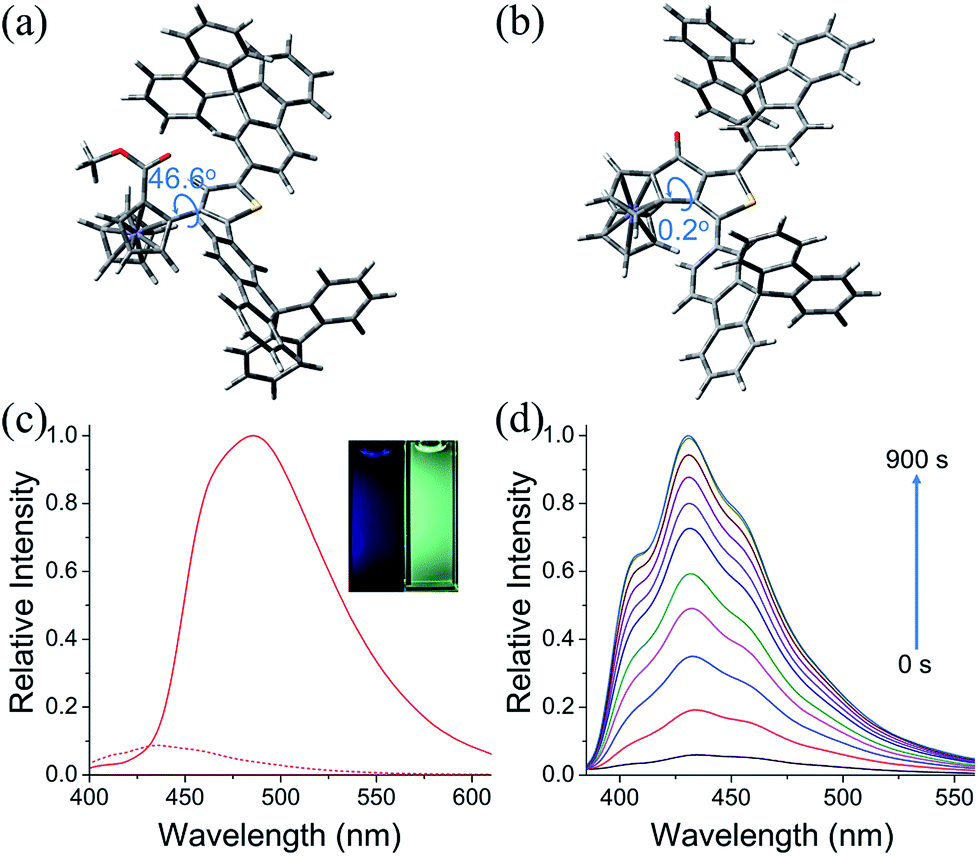 | ||
| Fig. 2 Calculated molecular conformations of (a) 6v and (b) 7c. Molecular optimization was performed by density functional theory (DFT) computation with Gaussian 09 at the B3LYP/6-31G* level. (c) Fluorescence spectra of 6v (dash line) and 7c (solid line) in CH3CN (1 × 10−5 M). Inset: fluorescence images of 6v (left) and 7c (right) in CH3CN (1 × 10−5 M) under UV light (365 nm). (d) Fluorescence spectra of 6v in CH3CN (5 × 10−6 M) containing n-Bu4NPF6 (0.1 M) when applying an oxidation potential of +0.48 V (vs. Fc/Fc+) during 0–900 s. | ||
The fluorescence on/off properties of 6v in an electro-redox process was investigated (Fig. 2d, S8 and S9†). To the CH3CN solution (5 × 10−6 M) of 6v with n-Bu4NPF6 (0.1 M) as the supporting electrolyte, an oxidation potential of +0.48 V (vs. Fc/Fc+) was applied. As shown in Fig. 2d, the fluorescence intensity of 6v exhibits an obviously upward trend with increasing time of electrochemical oxidation. Emission enhancement of approximately 17-fold is observed when applying the oxidation potential for 900 s. When applying a reduction potential of −0.62 V (vs. Fc/Fc+) to the oxidized solution of 6v, the emission intensity decreases gradually to the initial low fluorescence intensity. Moreover, the oxidation and reduction of 6v were carried out for several cycles without significant fatigue (Fig. S9†). These results show that 6v is a promising redox molecular switch.
Conclusions
In summary, we have developed a highly efficient strategy to construct ThienylFc structures via ortho-C–H activation/addition/annulation of ferrocenyl thioamides with 1,3-diynes. Mechanistic studies demonstrate that the formation of the thiophene ring is a successive process via the addition reaction of the sulfur atom of the thiocarbonyl group to the C1-position of 1,3-diyne and subsequent intramolecular sulfur-transfer rearrangement from the carbonyl group to the C4-position of 1,3-diyne. In this protocol, thioamide plays a dual role, firstly, as a directing group to activate ortho-C–H on ferrocene, and secondly, as a sulfur source to form the thiophene ring. The resulting ThienylFc can be conveniently transformed into extended π-conjugated ferrocenes with luminescent properties through the sequential hydrolysis and intramolecular Friedel–Crafts acylation. ThienylFc displays effective fluorescence quenching due to the PET process from the Fc unit to the excited luminophore, while OCTFc exhibits relatively strong emission owing to their ICT characteristics. In this work, the ring-fused strategy is employed for the first time to construct luminescent materials based on ferrocenes, which provides inspiration for the development of novel organic optoelectronic materials, such as electroluminescent materials based on ferrocenes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 21871193, 21772133, and 21672154). We also thank the Comprehensive Training Platform Specialized Laboratory, College of Chemistry, Sichuan University.References
- R. Gómez Arrayás, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674–7715 Search PubMed.
- R. Pietschnig, Chem. Soc. Rev., 2016, 45, 5216–5231 Search PubMed.
- M. Patra and G. Gasser, Nat. Rev. Chem., 2017, 1, 0066 Search PubMed.
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 Search PubMed.
- R. Martínez, I. Ratera, A. Tárraga, P. Molina and J. Veciana, Chem. Commun., 2006, 3809–3811 Search PubMed.
- R. Zhang, Z. Wang, Y. Wu, H. Fu and J. Yao, Org. Lett., 2008, 10, 3065–3068 Search PubMed.
- M. Li, Z. Guo, W. Zhu, F. Marken and T. D. James, Chem. Commun., 2015, 51, 1293–1296 Search PubMed.
- H. Grubert and K. L. Rinehart, Tetrahedron Lett., 1959, 1, 16–17 Search PubMed.
- F. L. Hedberg and H. Rosenberg, J. Am. Chem. Soc., 1973, 95, 870–875 Search PubMed.
- A. Zirakzadeh, A. Herlein, M. A. Groß, K. Mereiter, Y. Wang and W. Weissensteiner, Organometallics, 2015, 34, 3820–3832 Search PubMed.
- J. H. Olshansky, T. X. Ding, Y. V. Lee, S. R. Leone and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 15567–15575 Search PubMed.
- R. B. Woodward, M. Rosenblum and M. C. Whiting, J. Am. Chem. Soc., 1952, 74, 3458–3459 Search PubMed.
- M. Rosenblum and R. B. Woodward, J. Am. Chem. Soc., 1958, 80, 5443–5449 Search PubMed.
- F. Rebière, O. Riant, L. Ricard and H. B. Kagan, Angew. Chem., Int. Ed. Engl., 1993, 32, 568–570 Search PubMed.
- M. Tsukazaki, M. Tinkl, A. Roglans, B. J. Chapell, N. J. Taylor and V. Snieckus, J. Am. Chem. Soc., 1996, 118, 685–686 Search PubMed.
- C. Genet, S. J. Canipa, P. O'Brien and S. Taylor, J. Am. Chem. Soc., 2006, 128, 9336–9337 Search PubMed.
- C. Drolen, E. Conklin, S. J. Hetterich, A. Krishnamurthy, G. A. Andrade, J. L. Dimeglio, M. I. Martin, L. K. Tran, G. P. A. Yap, J. Rosenthal and E. R. Young, J. Am. Chem. Soc., 2018, 140, 10169–10178 Search PubMed.
- X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 Search PubMed.
- C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 Search PubMed.
- S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 Search PubMed.
- G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 Search PubMed.
- L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 Search PubMed.
- M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843–895 Search PubMed.
- T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 Search PubMed.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 Search PubMed.
- L. A. López and E. López, Dalton Trans., 2015, 44, 10128–10135 Search PubMed.
- D.-W. Gao, Q. Gu, C. Zheng and S.-L. You, Acc. Chem. Res., 2017, 50, 351–365 Search PubMed.
- D.-W. Gao, Y.-C. Shi, Q. Gu, Z.-L. Zhao and S.-L. You, J. Am. Chem. Soc., 2013, 135, 86–89 Search PubMed.
- C. Pi, Y. Li, X. Cui, H. Zhang, Y. Han and Y. Wu, Chem. Sci., 2013, 4, 2675–2679 Search PubMed.
- T. Shibata and T. Shizuno, Angew. Chem., Int. Ed., 2014, 53, 5410–5413 Search PubMed.
- D.-W. Gao, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2016, 138, 2544–2547 Search PubMed.
- Z.-J. Cai, C.-X. Liu, Q. Gu and S.-L. You, Angew. Chem., Int. Ed., 2018, 57, 1296–1299 Search PubMed.
- Y.-H. Liu, P.-X. Li, Q.-J. Yao, Z.-Z. Zhang, D.-Y. Huang, M. D. Le, H. Song, L. Liu and B.-F. Shi, Org. Lett., 2019, 21, 1895–1899 Search PubMed.
- Z.-J. Cai, C.-X. Liu, Q. Wang, Q. Gu and S.-L. You, Nat. Commun., 2019, 10, 4168 Search PubMed.
- S. R. Yetra, T. Rogge, S. Warratz, J. Struwe, W. Peng, P. Vana and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 7490–7494 Search PubMed.
- L. Liu, H. Song, Y.-H. Liu, L.-S. Wu and B.-F. Shi, ACS Catal., 2020, 10, 7117–7122 Search PubMed.
- W. Shi and A. Lei, Tetrahedron Lett., 2014, 55, 2763–2772 Search PubMed.
- V. Lavallo, G. D. Frey, B. Donnadieu, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 5224–5228 Search PubMed.
- C. Maeda, T. Yoneda, N. Aratani, M.-C. Yoon, J. M. Lim, D. Kim, N. Yoshioka and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 5691–5694 Search PubMed.
- D. J. Schipper, L. C. H. Moh, P. Müller and T. M. Swager, Angew. Chem., Int. Ed., 2014, 53, 5847–5851 Search PubMed.
- D.-G. Yu, F. de Azambuja, T. Gensch, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 9650–9654 Search PubMed.
- J. Tang and X. Zhao, RSC Adv., 2012, 2, 5488–5490 Search PubMed.
- H. Jiang, W. Zeng, Y. Li, W. Wu, L. Huang and W. Fu, J. Org. Chem., 2012, 77, 5179–5183 Search PubMed.
- G. Zhang, H. Yi, H. Chen, C. Bian, C. Liu and A. Lei, Org. Lett., 2014, 16, 6156–6159 Search PubMed.
- J. E. Spangler, Y. Kobayashi, P. Verma, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 11876–11879 Search PubMed.
- A. T. Tran and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 10530–10534 Search PubMed.
- J. Yin, M. Tan, D. Wu, R. Jiang, C. Li and J. You, Angew. Chem., Int. Ed., 2017, 56, 13094–13098 Search PubMed.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 Search PubMed.
- R. H. Mitchell, Y. Chen, N. Khalifa and P. Zhou, J. Am. Chem. Soc., 1998, 120, 1785–1794 Search PubMed.
- F. Pammer, Y. Sun, M. Pagels, D. Weismann, H. Sitzmann and W. R. Thiel, Angew. Chem., Int. Ed., 2008, 47, 3271–3274 Search PubMed.
- B. Hu, M. Meng, Z. Wang, W. Du, J. S. Fossey, X. Hu and W.-P. Deng, J. Am. Chem. Soc., 2010, 132, 17041–17044 Search PubMed.
- M. Toganoh, A. Sato and H. Furuta, Angew. Chem., Int. Ed., 2011, 50, 2752–2755 Search PubMed.
- T. Chen, D. Wang, L.-H. Gan, Y. Matsuo, J.-Y. Gu, H.-J. Yan, E. Nakamura and L.-J. Wan, J. Am. Chem. Soc., 2014, 136, 3184–3191 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. Details of experimental procedures, characterization and spectroscopic data of compounds. The X-ray single crystal diffraction data of compound 4, 6a and 11 have been deposited at the Cambridge Crystallographic Data Centre (CCDC) with independent accession codes 1939363, 1889086 and 1939372. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04597g |
| This journal is © The Royal Society of Chemistry 2020 |