
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-mediated formation of Cu/metal oxide hybrid nanocrystals with tunable number of interfaces†
Seyedeh Behnaz
Varandili
 ,
Dragos
Stoian
,
Jan
Vavra
,
James
Pankhurst
and
Raffaella
Buonsanti
*
,
Dragos
Stoian
,
Jan
Vavra
,
James
Pankhurst
and
Raffaella
Buonsanti
*
Laboratory of Nanochemistry for Energy (LNCE), Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne, CH-1950 Sion, Switzerland. E-mail: raffaella.buonsanti@epfl.ch
First published on 27th October 2020
Abstract
Combining domains of different chemical nature within the same hybrid material through the formation of heterojunctions provides the opportunity to exploit the properties of each individual component within the same nano-object; furthermore, new synergistic properties will often arise as a result of unique interface interactions. However, synthetic strategies enabling precise control over the final architecture of multicomponent objects still remain scarce for certain classes of materials. Herein, we report on the formation of Cu/MOx (M = Ce, Zn and Zr) hybrid nanocrystals with a tunable number of interfaces between the two domains. We demonstrate that the organic ligands employed during the synthesis play a key role in regulating the final configuration. Finally, we show that the synthesized nanocrystals serve as materials platforms to investigate the impact of the Cu/metal oxide interfaces in applications by focusing on the electrochemical CO2 reduction reaction as one representative example.
Introduction
Combining domains of different chemical nature within the same hybrid material through the formation of heterojunctions provides the opportunity to exploit the properties of each individual component within the same nano-object; furthermore, new synergistic properties will often arise as a result of unique interface interactions. In the past decade, colloidal chemistry has proven to be one of the most suitable synthetic methods to achieve tunability on hybrid materials at the nanoscale.1–10 Indeed, colloidal hybrid nanocrystals (HNCs) have been utilized as the key components in a variety of applications spanning from catalysis to plasmonics to biomedicine and energy conversion.1–10 Phase segregation, partial cation exchange, thermally induced coalescence and seeded growth are some of the strategies employed to access a huge library of HNCs.1–10 Despite the beautiful examples reported in the literature, some material combinations remain more challenging or underexplored than others.Non-noble metal/metal oxide HNCs are extremely interesting systems because of the unique synergistic interactions that arise at the interface between the two domains.9,11,12 Nevertheless, synthetic strategies that enable precise control over the final architecture of these NCs remain scarce.9,13–15 One of the challenges in these systems is the tendency of the non-noble metal domain to easily oxidize, which limits the reaction environment to organic solvent, and precludes the possibility of using the metal as a seed for the growth of the oxide domain, which is a common approach for HNCs.6,16–18
In this work, we choose Cu/CeO2 HNCs as one example of this class of materials because of the relevance of this interface for various applications, including thermal and electrochemical CO2 conversion.9,19,20 We report a ligand-mediated synthesis to access Cu/(CeO2)n, with n = 1, 2, 6 representing the average number of Cu/CeO2 heterojunctions in each particle. The CeO2 NCs are employed as nucleation seeds for the copper and different ligands are utilized to tune the number of junctions. We demonstrate that the final architecture is crucial when seeking property tunability by providing a representative example in the electrochemical CO2 reduction reaction. Finally, we show the generality of the synthetic concept that is applicable to construct other Cu/metal oxide heterojunctions.
Results and discussion
In a typical synthesis, 7 nm spherical CeO2 NCs (Fig. S1†) are dispersed in octadecene (ODE) and employed as seeds. A precursor solution containing copper acetate (Cu(OAc)) and the selected organic ligands (OLAM/OLAC, OLAM/OLAC/TOP or TOP only, where OLAM = oleylamine, OLAC = oleic acid, TOP = trioctylphosphine) in ODE is injected dropwise at 300 °C into the seed solution. OLAM and OLAC are common ligands in the synthesis of HNCs including one metal oxide domain, TOP was chosen as demonstrated to play an important role in the formation mechanism of copper NCs.9,21 The dropwise injection was important to avoid homogeneous nucleation of copper. Various synthetic parameters (i.e. temperature, ligand concentration, amount of seeds) were optimized to maximize the yield of HNCs (i.e. 100 × no HNCs/(no isolated seeds + no HNCs)).Structural characterization of the (Cu/CeO2)n HNCs
Fig. 1 gives an overview on the Cu/CeO2 HNCs synthesized with OLAC/OLAM (Fig. 1a and b), OLAM/OLAC/TOP (Fig. 1c and d) and TOP only (Fig. 1e and f). TEM images at lower magnifications are also provided in Fig. S2† to confirm the homogeneity of the samples. The HNC yield, as measured by statistical analysis in TEM images, was 80%, 92% and 90%, meaning that less than 20%, 8% and 10% of ceria seeds remain isolated, respectively. No homogeneously nucleated Cu NCs are identified. Representative X-ray diffraction (XRD) patterns of the HNCs are reported in Fig. S3.†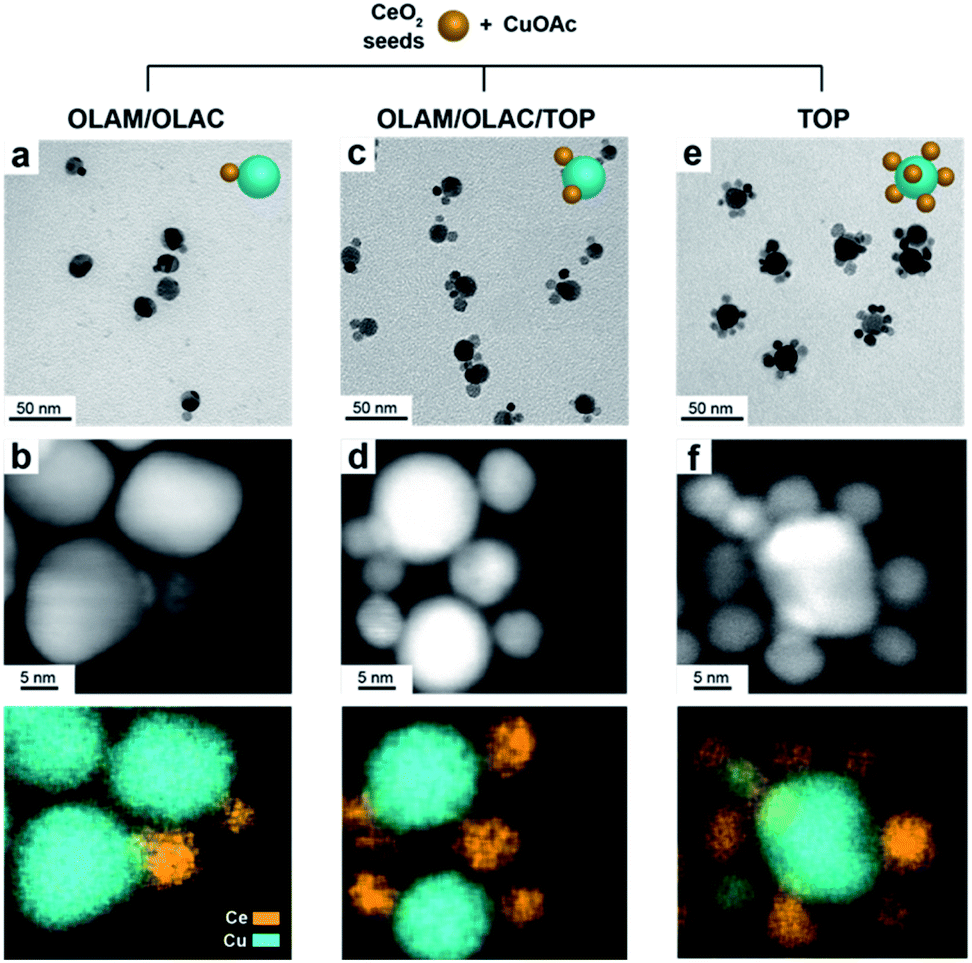 | ||
| Fig. 1 (a, c, e) Representative TEM and (b,d,f) HAADF-STEM images and corresponding EDXS elemental maps of (a, b) Cu/(CeO2)1, (c, d) Cu/(CeO2)2 and (e,f) Cu/(CeO2)6, synthesized in the presence of OLAC/OLAM, OLAC/OLAM/TOP and TOP only, respectively. | ||
Transmission electron microscopy (TEM), high-angle annular dark-field scanning TEM (HAADF-STEM) and the relative area-selective energy-dispersive X-ray spectroscopy (EDXS) maps evidence the systematic formation of HNCs consisting of one central domain of spherical Cu, with an average size of around 15 nm, and one or more CeO2 domain seeds of around 7 nm. For the rest of the manuscript, these HNCs will be referred to as Cu/(CeO2)n with n = 1,2,6 indicating the average number of heterojunctions in each NCs based on a statistical analysis of the stoichiometry distribution (Fig. 2). Concomitantly, elemental analysis performed by inductively coupled plasma-optical emission spectroscopy (ICP-OES) indicates decreasing Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ce ratio as n increases (3.6:1, 2.1:1 and 1.7:1 for Cu/(CeO2)1, Cu/(CeO2)2 and Cu/(CeO2)6, respectively).
:Ce ratio as n increases (3.6:1, 2.1:1 and 1.7:1 for Cu/(CeO2)1, Cu/(CeO2)2 and Cu/(CeO2)6, respectively).
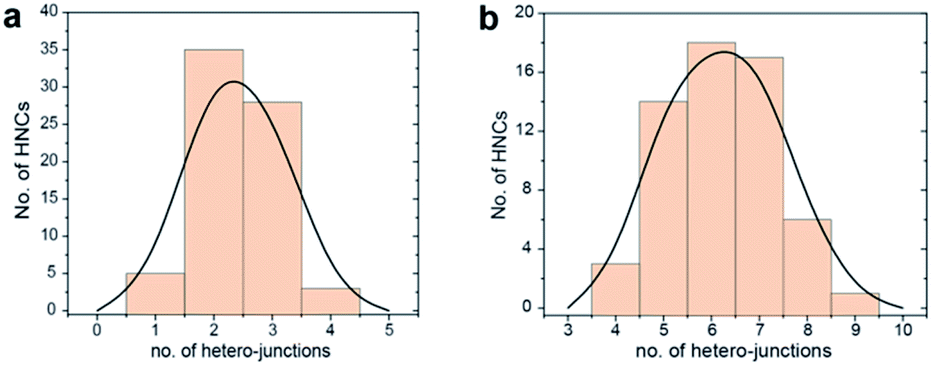 | ||
| Fig. 2 Statistical analysis of the number of hetero-junctions in: (a) Cu/(CeO2)2 and (b) Cu/(CeO2)6. This analysis was performed by counting at least 100 HNCs per sample. | ||
Clearly, the number of the ceria domains in each hybrid architecture depends on the employed ligand mixture. Specifically, OLAC/OLAM (Fig. 1a and b), OLAM/OLAC/TOP (Fig. 1c and d) and TOP only (Fig. 1e and f) lead to the formation of Cu/(CeO2)1, Cu/(CeO2)2 and Cu/(CeO2)6, respectively.
High-resolution TEM was performed to investigate the nature of the interface. Notably, the samples were ultrasonicated immediately before deposition on the grid as a proof that the two domains are permanently linked in one object. Fig. 3 shows that the Cu and CeO2 NCs are intimately connected, however no straightforward epitaxial relationship was observed, which is reasonable considering the high misfit between the lattice parameters of Cu and CeO2. X-Ray photoelectron spectroscopy (XPS) analysis provides an additional proof that the two domains are coupled as the electronic structure of the HNCs is different than the physical mixture of Cu and CeO2 NCs (Fig. S4†).
 | ||
| Fig. 3 HR-TEM characterization of: the HNCs (a, c, e) and the interface areas shown by white squares (b, d, e) for (a, b) Cu/(CeO2)1, (c, d) Cu/(CeO2)2 and (e, f) Cu/(CeO2)6, synthesized in the presence of OLAC/OLAM, OLAC/OLAM/TOP and TOP only, respectively. | ||
Investigation of the role of the ligands
Configurations of HNCs similar to Cu/(CeO2)2 and Cu/(CeO2)6 have been previously reported in the literature and were often referred to as flower- or clover-like structures.3,6,16,17,22–25 However, in these examples, the HNC core always consists of the NC seeds. Oppositely, in Cu/(CeO2)2 and Cu/(CeO2)6, the core is the nucleating Cu domain while the “petals” are the ceria seeds, which is both unique and surprising. As for the mechanism, the formation of abovementioned flower- or clover-like structures has been correlated to the polarity of the solvent, the change of seed size or structure (single crystal vs. multi-twinned) and the reaction time.3,6,16,17,22–25 In our system, the solvent and the seeds are the same for all of the HNCs so their influence on the heterojunction formation can be ruled out. Instead, the chemical nature of the ligands clearly plays a significant role.Ligands can impact NC nucleation and growth in two main ways: by modulating the surface chemistry and by acting as reactants in the synthesis (i.e. complexing agents or reductants).2,26
From the results above, it is clear that the presence of TOP in the reaction mixture induces the formation of multiple heterojunctions. Interestingly, no convincing evidence of the TOP acting as surface ligand on Cu/(CeO2)2 and Cu/(CeO2)6 HNCs was found by Fourier transform infrared spectroscopy (FT-IR) and XPS (Fig. S5†). Another possibility is that TOP alters the surface chemistry of the seeds by exchanging with the original OLAM ligand of the CeO2 NC seeds during the synthesis. However, no HNCs formed when TOP-coated CeO2 NCs were used as seeds in place of the OLAM-coated ones; instead, only homogeneous nucleation of Cu NCs occurred (Fig. S6†). These results suggest that TOP plays a key role as complexing agent for the formation of petal-like HNCs and must be involved in the Cu monomer formation as previously observed.21
Fig. 4 reports the NMR spectra of the precursor solutions (CuOAc + TOP) and (CuOAc + OLAM/OLAC). The data for the (CuOAc + TOP) mixture (Fig. 4a) indicate the formation of a [Cu(TOP)4][OAc] complex, where the acetate anion is non-coordinating. In the 31P{1H} NMR spectrum, the large shift of the free TOP signal to a more positive chemical shift, as well as the broadening of the resonance, are strong indicators of coordination to the Cu+ center.
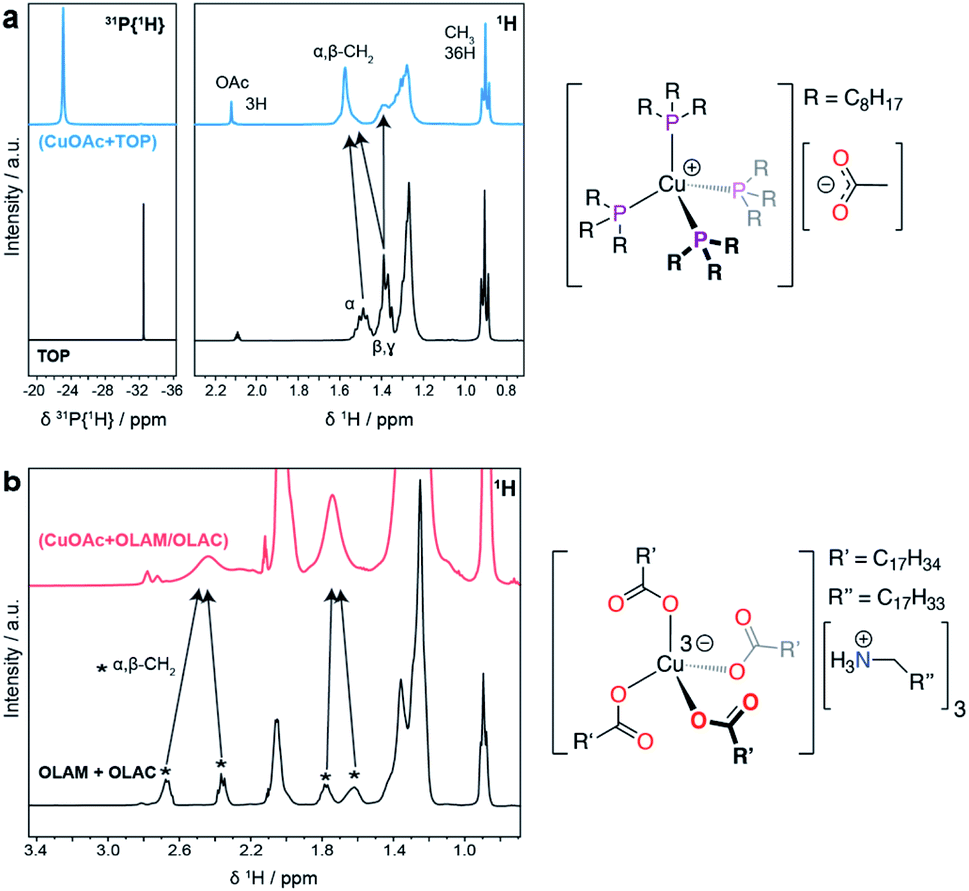 | ||
| Fig. 4 Characterization of the precursor solutions. (a) 31P{1H} NMR spectra and 1H NMR spectra of the Cu complex formed by a combination of CuOAc and TOP. (b) 1H NMR spectra of the Cu complex formed by a combination of CuOAc, OLAM and OLAC. The proposed molecular structures of the Cu complexes in each mixture are shown on the right of the corresponding spectra. | ||
All four coordinated TOP molecules are clearly symmetry equivalent, indicating a tetrahedral geometry. Likewise, in the 1H NMR spectrum, the free TOP alpha- and beta-CH2 resonances move to more positive chemical shift values after coordination to Cu+, and are also broadened due to the quadrupolar moment on Cu. A resonance for the OAc ligand was also observed, and its integration indicates the expected 1:4 ratio with TOP. No free TOP is observed in the precursor mixture.
In the case of the (CuOAc + OLAM/OLAC) mixture (Fig. 4b), there are a number of broad resonances in the 1H NMR spectrum that can be assigned to the α- and β-CH2 environments of either OLAM and/or OLAC. In a control experiment, OLAM and OLAC were mixed and the 1H NMR spectrum was measured; the spectrum was consistent with an oleylammonium oleate salt, which forms from the acid–base reaction between OLAM and OLAC, as expected. The 1H NMR spectrum of the precursor mixture compared quite well with this spectrum, albeit with an important difference: the well-resolved resonances for the α-CH2 protons in the [OLAM-H][OLAC] salt (2.67 and 2.36 ppm) appear as a single broad resonance in the precursor mixture (2.44 ppm). An equivalent observation was made for the β-CH2 protons (peaks at 1.78 and 1.62 ppm averaging to 1.74 ppm). This loss of resolution could be due to association of the ammonium carboxylate ions with Cu+. The speciation of copper ions in ionic solutions has previously been shown to be quite complex, where transition metals tend to coordinate to additional anions, forming anionic complexes.27 These complexes are then surrounded by associated cations, in mono- or multi-nuclear aggregates.28 We therefore tentatively propose that Cu+ is coordinated by four oleate ligands in a tetrahedral geometry, with three oleylammonium counter ions balancing the charge, i.e. [Cu(OLAC)4][(OLAM-H)3].
Discussion on the formation mechanism of the HNCs
Generally, two different mechanisms might take place to form the HNCs: an aggregative electrostatic self-assembly and heterogeneous nucleation. In the former, the Cu NCs would homogeneously self-nucleate in solution and successively assemble with the CeO2 seeds. Here, the ligands would modulate the surface passivation and charge to drive such a controlled attachment. In the heterogeneous nucleation, the CeO2 NCs would act as seeds for the nucleation of the Cu domain.To get further insights, we monitored the temporal evolution for Cu/(CeO2)1 (Fig. S7†) and Cu/(CeO2)6 (Fig. 5). In both cases, the TEM analysis suggests that the configuration of the HNCs, with either single or multiple heterojunctions, is established since the early stages of the synthesis and an increased reaction time contributes towards the growth of the Cu domains (Fig. 5a–c). No evidence of homogeneously nucleated Cu NCs was found at any time. As for the CeO2 seeds, statistical analysis on the TEM images indicates a sudden drop of isolated NCs at around 75 s (Fig. 5d), which corresponds to the time at which the HNCs are first observed in the TEM images. In order to exclude the impact of aggregation during drying of the TEM grid, dynamic light scattering (DLS) was performed as a complementary technique (Fig. 5e). Consistently with the TEM data, a continuous increase of particle size in solution is observed, which tracks well the increase in the Cu domain size. If aggregative growth of the HNCs occurred, a bimodal distribution should have been detected at some point, which was not the case. Therefore, as far as the current experimental evidences indicate, heterogeneous nucleation is the main mechanism behind the formation of the HNCs.
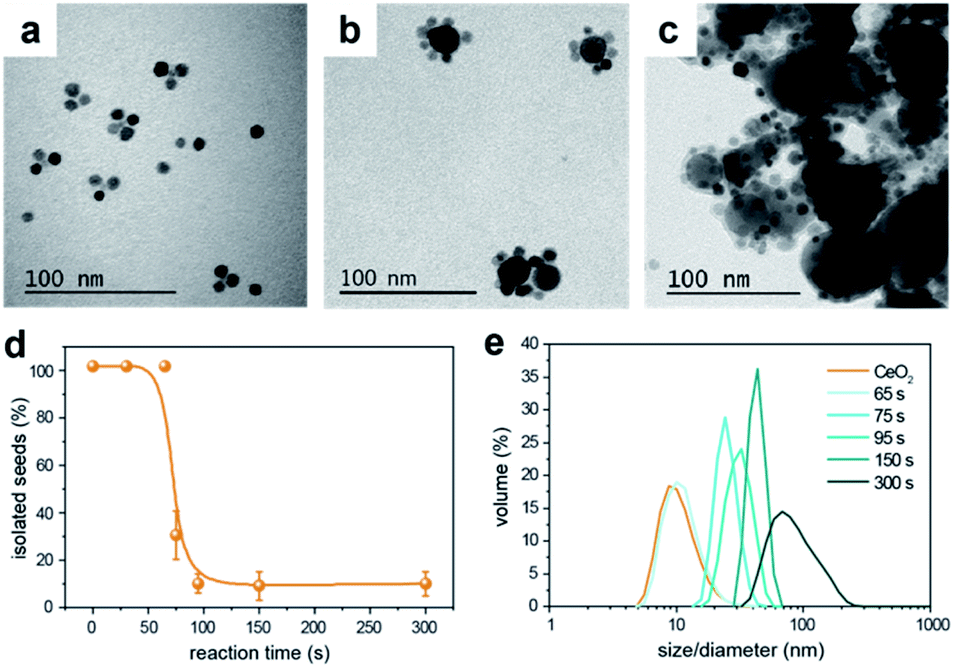 | ||
| Fig. 5 Temporal evolution of the HNCs. (a, b, c) Representative TEM images of samples collected at a reaction time of (a) 75 s, (b) 150 s and (c) 300 s; (d) percentage of isolated CeO2 seeds versus the reaction time obtained from statistical analysis on TEM aliquots; (e) DLS spectra of aliquots collected at different reaction times. All data are relative to the synthesis of Cu/(CeO2)6. | ||
If heterogeneous seeded-growth is taking place, then the nature of the complexes formed between the ligands and the Cu precursor must play a role in determining the final configuration of the HNCs. Based on the metal–ligand bond strengths and on previous calculations,21 the copper–phosphine complex is expected to be more stable than the copper complex forming in the OLAM/OLAC mixture, and so to decompose more slowly and/or only at high temperature to release the copper monomers in solution. As a matter of fact, when the two precursor solutions were left at room temperature for two days and monitored via UV-vis spectroscopy (Fig. S8†), the (CuOAc + OLAM/OLAC) mixture turned blue and a brown sediment was observed at the bottom of the vial; instead, no change was observed in the (CuOAc + TOP), in agreement with its higher redox stability. Furthermore, when the reaction temperature was decreased from 300 °C to 270 °C (Fig. S9†), neither HNCs or Cu NCs formed when utilizing (CuOAc + TOP), instead Cu/CeO2 HNCs were detected when (CuOAc + OLAM/OLAC) was employed. The above experimental evidences suggest that the organic ligands can regulate the copper monomer supersaturation by modulating the precursor stability. Specifically, we speculate that (CuOAc + TOP) and (CuOAc + OLAM/OLAC) result in low and high supersaturation, respectively, when they are delivered to the seed-containing reaction mixture. Low supersaturation is expected to induce a kinetic preference for high-energy products.2 In the HNCs, a significant lattice mismatch exists between ceria (a = b = c = 5.411 Å) and copper (a = b = c = 3.610 Å), and so the system will tend to minimize the free energy by reducing the number of interfaces between the two domains. Therefore, Cu/(CeO2)1 form, through the formation of a partially amorphous interface, when the OLAM/OLAC mixture is employed during the synthesis. Instead, the multi-junction HNCs, Cu/(CeO2)2 and Cu/(CeO2)6, are obtained when TOP is used instead.
Overall, the absence of isolated Cu domains at any time during the synthesis along with no TOP on the surface of the HNCs and instead involved in the formation of Cu-TOP, suggest that electrostatic self-assembly is unlikely. While still speculative at this time, the ligand-regulated monomer saturation offers an alternative explanation to the undoubted role played by the ligands in determining the number of heterojunctions in the HNCs (Fig. 6).
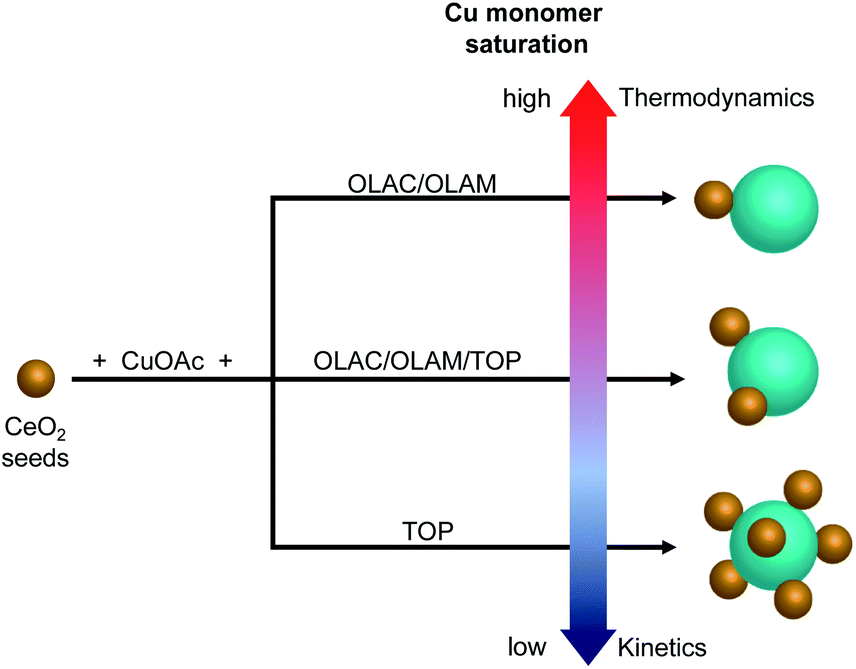 | ||
| Fig. 6 Schematic representation of the proposed pathway toward the formation of different (Cu/CeO2)n HNCs. | ||
Impact of the number of heterojunctions on CO2 reduction reaction (CO2RR)
To illustrate the importance of controlling the number of heterojunctions in the HNCs, the performance of the (Cu/CeO2)n hybrids as electro-catalysts for CO2RR were investigated. In a previous study, we found the Cu/CeO2 interface to promote CO2RR versus the competing hydrogen evolution reaction (HER) and to form methane as the major CO2RR product.9Operando X-ray absorption spectroscopy (XAS) and DFT calculations explained this behavior based on the promotion of the Ce4+ → Ce3+ reduction and the concomitant formation of oxygen vacancies. The latter accounts for improved stabilization of the key CO2RR intermediates (CHO* and H2CO*) at the Cu/CeO2 interface.9 Based on these results, understanding if an increased interfacial area further enhances these beneficial effects is interesting and crucial to develop better catalysts.Fig. 7a reports the faradaic efficiencies (FE) and CO2RR partial current densities (JCO2RR) normalized by the electrochemically active surface area (ECSA-Fig. S10†) for Cu–CeO2 physical mixture, Cu/(CeO2)1, Cu/(CeO2)2, and Cu/(CeO2)6 HNCs, respectively. Data for isolated Cu and CeO2 NCs of similar sizes of the HNC domains are reported in Fig. S11† for reference. First of all, the results evidence that the Cu/(CeO2)1 HNCs outperform the isolated Cu NCs and Cu–CeO2 physical mixture in terms of total FE towards CO2RR, which reaches up to 60%. The higher JCO2RR indicates that such increase in FE is related to the higher intrinsic activity of the HNCs. In agreement with our previous study, Cu/(CeO2)1 produce methane as the major product with FE up to 50%.9 This result highlights the role of the hetero-junction obtained by employing the colloidal synthesis method presented herein. Notably, as the number of heterojunctions increases, the CO2RR FE decreases to around 37% and 32% for Cu/(CeO2)2 and Cu/(CeO2)6, respectively. Among the CO2RR products, both ethylene and methane are suppressed while CO and formate are promoted. Concomitantly, the JCO2RR is lower for the two samples compared to the single junction HNCs. Overall the FEs for CO2RR of Cu/(CeO2)n≥2 HNCs are comparable to the isolated Cu NCs (Fig. S11†), however the ratio of C1/C2 products is increased which could be an indication of interrupted pathway toward C2 products. The lower JCO2RR indicates that the overall intrinsic activity towards CO2RR decreases as well.
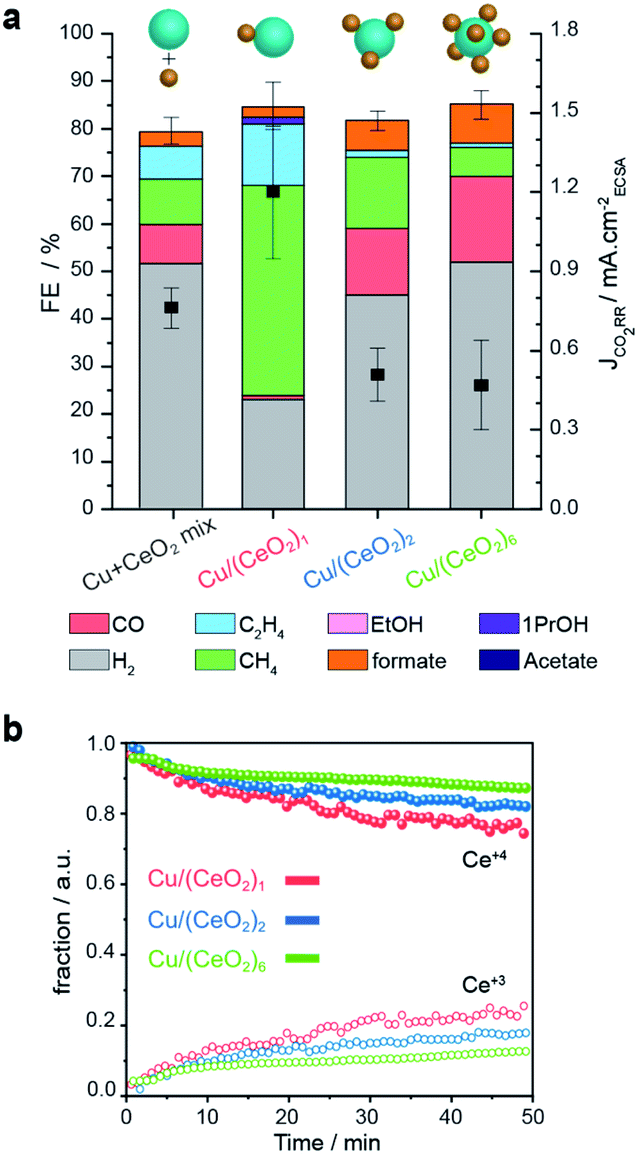 | ||
| Fig. 7 CO2RR performance of the Cu/(CeO2)n HNCs. (a) faradaic efficiencies and CO2RR partial current-densities for Cu/(CeO2)1, Cu/(CeO2)2 and Cu/(CeO2)6 and Cu–CeO2 physical mixture measured at −1.2 VRHE in 0.1 M KHCO3. It is worth mentioning that the total FE of the samples are less than 90%. Oxidation of some unaccounted formate and methanol at the platinum anode has been reported to contribute to the lack of 100% total FE.29,30 Also, some electrons may be used for the formation of oxygen vacancies in CeO2−x, thus, contributing to non-productive current.9 (b) Concentration profiles of Ce4+ (solid symbols) and Ce3+ (open symbols) derived from the multivariate analysis of Ce L3-edge XANES spectra during CO2RR. | ||
To gain further insight into the oxidation states of the Cu and Ce domains during CO2RR, we performed operando XAS measurements. Fig. 7b shows the concentration profiles of Ce4+ (solid symbols) and Ce3+ (open symbols) as a function of the reaction time at the operating potential. These values were extracted from a time-resolved map via multivariate spectral analysis (Fig. S12†). In all three samples, we observed the reduction Ce4+ → Ce3+ over time until reaching a steady state value. Instead, the Cu counterpart, partially oxidized at the beginning of the experiment, reduced very quickly and at a similar rate in all the samples as soon as the negative operating potential was applied; then, it remained stable in its metallic form during the catalysis (Fig. S12†). Importantly, we found that a larger fraction of Ce3+ forms in the Cu/(CeO2)1 HNCs compared to the Cu/(CeO2)n≥2 HNCs (roughly 25% > 15% > 10% as n increases from 1 to 6). Thus, the results from the operando XAS of Ce L3-edge confirm the positive correlation between Ce3+ and the CO2RR promotion. They also suggest that the presence of multiple metal oxides surrounding the Cu NCs inhibits the Ce4+ → Ce3+ transformation, which is opposite to what expected based on increased interfacial area. One possible explanation is that, as the number of heterojunctions increases, the constant number of the charges (supplied from applied negative potential, −1.2 VRHE) have to be distributed among a higher number of ceria NCs. As the loading and size of the Cu NCs in all the samples are constant, it's reasonable that the charge portion allotted to each ceria NCs would decrease.
Moreover, one must consider that increasing the number of ceria domains reduces the copper surface available to perform CO2RR. The CeO2 NCs themselves are mostly active for HER (Fig. S11†), therefore we hypothesize that the increased HER results from their increase weight fraction in the catalyst. As the protons are consumed by the ceria, less remain available for the proton coupling steps needed to convert CO to higher hydrocarbons. Thus, mostly CO increases among the CO2RR products while CH4 and C2H4 decrease. More detailed studies are planned for the future to further corroborate this hypothesis. For example, HNCs with a reverse configuration where the several Cu NCs nucleated on one CeO2 seed, would be an interesting system to study as the number of heterojunctions will increase without compromising the Cu surface available for the CO2RR to take place.
Generality of the synthetic method
Finally, having learned the crucial role of the organic ligands in controlling the number of heterojunctions in the Cu/CeO2 HNCs, we decided to investigate if the same synthesis approach could be applied to different systems. We selected ZrO2 and ZnO as examples, because Cu/ZrO2 and Cu/ZnO interfaces are of potential interest for both CO2 hydrogenation and CO2RR.31,32Fig. 8 depicts the Cu/ZnO (Fig. 8a–c) and Cu/ZrO2 (Fig. 8d–f) HNCs that were successfully obtained with a configuration resembling that of Cu/CeO2, where the central copper domain is decorated by a number of metal oxide seeds, and with the number of heterojunctions increasing as OLAM/OLAC, OLAM/OLAC/TOP or TOP only were used as the organic ligands during the synthesis. While some system specific differences emerge in the final HNCs (i.e. the faceted morphology of the Cu domain in Fig. 8d, which might stem from some effects induced by the crystallinity of ZrO2 seeds3,6,16,17,22–25,33,34), the results suggest that the method is indeed general. | ||
| Fig. 8 Generality of the synthetic approach. TEM images and corresponding EDXS elemental maps of Cu/ZnO HNCs (a–c) and Cu/ZrO2 HNCs (d–f) synthesized using (a, d) OLAM/OLAC, (b, e) OLAM/OLAC/TOP and (c, f) TOP only. | ||
Conclusions
In summary, we have successfully developed a seeded-growth approach to synthesize Cu/metal oxide HNCs with adjustable numbers of interfaces. Investigating the role of the ligands was key to understand that they affect the copper precursor stability and, thus, the monomer saturation, which in turn governs the growth regime and the final configuration of the HNCs. Finally, we also demonstrate that the achieved degree of architectural tunability is crucial to explore synergistic interface effects in catalysis. By testing Cu/(CeO2)n as electrocatalysts for CO2RR, we learned that optimal electrocatalytic properties are obtained as a trade-off between interfacial synergy and geometric configuration determining accessibility of the catalytic surfaces.Having tunable materials platforms, like the (Cu/CeO2)n HNCs reported in this study, becomes crucial to enable these correlations in catalysis and in other applications where these interfaces might be useful, such as plasmonic, biomedical, sensing and photocatalysis applications.3,31,32,35–37 Therefore, additional efforts in the community to understand the chemistry behind the formation of such tailored structures are important to further master the synthesis of other non-noble metal/metal oxide HNCs.
Experimental section
Chemicals
Copper(I) acetate (or CuOAc, 97%), trioctylphosphine ({CH3(CH2)7}3P, or TOP, 90%), oleic acid (C17H33CO2H or OLAC, 90%), oleylamine (C17H33NH2 or OLAM, 70%), 1-octadecene (C18H36 or ODE, 90%) were all purchased from Sigma-Aldrich and used as received.Synthesis
Characterization
Electrocatalytic measurements
A Biologic SP-300 was used as the potentiostat. Platinum foil was employed as the counter electrode and an Ag/AgCl reference electrode (leak free series from Innovative Instruments, Inc.) was used. Voltages were converted to the reversible hydrogen electrode (RHE) scale.
For gas product analysis, a gas chromatograph (GC, SRI instruments) equipped with a HayeSep D porous polymer column, thermal conductivity detector, and flame ionization detector was used. Ultrahigh purity N2 (99.999%) was used as a carrier gas. After passing through the cell, CO2 was flowed directly into the gas-sampling loop of the GC for online gaseous product analysis, which was carried out every 10 min. For all experiments, electrolysis was carried out for 80 min with gas analysis recorded at 10 min intervals. The liquid products were collected from the electrolyte after electrolysis and analyzed by the high-performance liquid chromatography (HPLC) on an UltiMate 3000 instrument from Thermo Scientific.
Operando X-ray absorption spectroscopy (XAS)
Operando XAS experiments were performed at the SuperXAS beamline at the Swiss Light Source synchrotron facility (Paul Scherrer Institute, Switzerland). The measurements were performed in fluorescence mode at an incident angle of about 45 degrees.We employed a dedicated set-up for the low energy Ce L3-edge as described in our previous work.9 The catalyst solution was drop-casted on a thin (2.5 × 2.5 × 0.5 mm3) glassy carbon (GC) support and a Kapton window allows the X-rays to pass through. Additional details for the data analysis are reported in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was primarily financed by the European Research Council under Starting Grant ERC-HYCAT with agreement number 715634. D. S. is supported by the Sandoz foundation. The authors thank Dr Mounir Mensi for the acquisition of the XPS data and Ona Segura Lecina for her contribution to NMR spectroscopy. The authors acknowledge the Paul Scherrer Institut, Villigen, Switzerland for provision of synchrotron radiation beamtime at beamline SuperXAS of the SLS and would like to thank Dr Adam Clark for assistance.Notes and references
- J. L. Fenton, B. C. Steimle and R. E. Schaak, Science, 2018, 360, 513–517 CrossRef CAS.
- A. N. Chen, M. M. Scanlan and S. E. Skrabalak, ACS Nano, 2017, 11, 12624–12631 CrossRef CAS.
- S. Peng, C. Lei, Y. Ren, R. E. Cook and Y. Sun, Angew. Chem., Int. Ed., 2011, 50, 3158–3163 CrossRef CAS.
- L. Amirav, F. Oba, S. Aloni and A. P. Alivisatos, Angew. Chem., Int. Ed., 2015, 54, 7007–7011 CrossRef CAS.
- U. Banin, Y. Ben-Shahar and K. Vinokurov, Chem. Mater., 2014, 26, 97–110 CrossRef CAS.
- J. Piella, A. Gónzalez-Febles, J. Patarroyo, J. Arbiol, N. G. Bastús and V. Puntes, Chem. Mater., 2019, 31, 7922–7932 CrossRef CAS.
- S. Najafishirtari, C. Guglieri, S. Marras, A. Scarpellini, R. Brescia, M. Prato, G. Righi, A. Franchini, R. Magri, L. Manna and M. Colombo, Appl. Catal., B., 2018, 237, 753–762 CrossRef CAS.
- M. Cargnello, Chem. Mater., 2019, 31, 576–596 CrossRef CAS.
- S. B. Varandili, J. Huang, E. Oveisi, G. L. De Gregorio, M. Mensi, M. Strach, J. Vavra, C. Gadiyar, A. Bhowmik and R. Buonsanti, ACS Catal., 2019, 9, 5035–5046 CrossRef CAS.
- A. Aitbekova, E. D. Goodman, L. Wu, A. Boubnov, A. S. Hoffman, A. Genc, H. Cheng, L. Casalena, S. R. Bare and M. Cargnello, Angew. Chem., 2019, 131, 17612–17618 CrossRef.
- M. Cargnello, V. V. T. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Science, 2013, 341, 771–773 CrossRef CAS.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- M. Casavola, A. Falqui, M. A. García, M. García-Hernández, C. Giannini, R. Cingolani and P. D. Cozzoli, Nano Lett., 2009, 9, 366–376 CrossRef CAS.
- B. Nakhjavan, M. N. Tahir, M. Panthöfer, H. Gao, T. Gasi, V. Ksenofontov, R. Branscheid, S. Weber, U. Kolb, L. M. Schreiber and W. Tremel, Chem. Commun., 2011, 47, 8898–8900 RSC.
- S. H. Choi, B. N. Hyon, I. P. Yong, K. An, G. K. Soon, Y. Jang, M. H. Park, J. Moon, S. S. Jae, C. S. In, K. M. Woo and T. Hyeon, J. Am. Chem. Soc., 2008, 130, 15573–15580 CrossRef CAS.
- W. C. Smith, J. R. Morse, C. R. M. Bria, R. E. Schaak and S. K. R. Williams, ACS Appl. Nano Mater., 2018, 1, 6435–6443 CrossRef CAS.
- E. Fantechi, A. G. Roca, B. Sepúlveda, P. Torruella, S. Estradé, F. Peiró, E. Coy, S. Jurga, N. G. Bastús, J. Nogués and V. Puntes, Chem. Mater., 2017, 29, 4022–4035 CrossRef CAS.
- J. M. Hodges, J. R. Morse, M. E. Williams and R. E. Schaak, J. Am. Chem. Soc., 2015, 137, 15493–15500 CrossRef CAS.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. Fernández Sanz and J. A. Rodriguez, Science, 2015, 345, 546–550 CrossRef.
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113–7119 CrossRef CAS.
- M. Strach, V. Mantella, J. R. Pankhurst, P. Iyengar, A. Loiudice, S. Das, C. Corminboeuf, W. van Beek and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 16312–16322 CrossRef CAS.
- W. Shi, H. Zeng, Y. Sahoo, T. Y. Ohulchanskyy, Y. Ding, Z. L. Wang, M. Swihart and P. N. Prasad, Nano Lett., 2006, 6, 875–881 CrossRef CAS.
- C. Wang, W. Tian, Y. Ding, Y. Q. Ma, Z. L. Wang, N. M. Markovic, V. R. Stamenkovic, H. Daimon and S. Sun, J. Am. Chem. Soc., 2010, 132, 6524–6529 CrossRef CAS.
- H. Yu, M. Chen, P. M. Rice, S. X. Wang, R. L. White and S. Sun, Nano Lett., 2005, 5, 379–382 CrossRef CAS.
- Y. Wei, R. Klajn, A. O. Pinchuk and B. A. Grzybowski, Small, 2008, 4, 1635–1639 CrossRef CAS.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS.
- J. M. Hartley, C. M. Ip, G. C. H. Forrest, K. Singh, S. J. Gurman, K. S. Ryder, A. P. Abbott and G. Frisch, Inorg. Chem., 2014, 53, 6280–6288 CrossRef CAS.
- P. De Vreese, N. R. Brooks, K. Van Hecke, L. Van Meervelt, E. Matthijs, K. Binnemans and R. Van Deun, Inorg. Chem., 2012, 51, 4972–4981 CrossRef CAS.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C.-T. Dinh, T. Burdyny, M. G. Kibria, C. P. O'Brien, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2018, 11, 2531–2539 RSC.
- Y. X. Chen, A. Miki, S. Ye, H. Sakai and M. Osawa, J. Am. Chem. Soc., 2003, 125, 3680–3681 CrossRef CAS.
- K. Larmier, W. C. Liao, S. Tada, E. Lam, R. Verel, A. Bansode, A. Urakawa, A. Comas-Vives and C. Copéret, Angew. Chem., Int. Ed., 2017, 56, 2318–2323 CrossRef CAS.
- D. Ren, J. Gao, L. Pan, Z. Wang, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Angew. Chem., Int. Ed., 2019, 58, 15036–15040 CrossRef CAS.
- Y. Xiong and Y. Xia, Adv. Mater., 2007, 19, 3385–3391 CrossRef CAS.
- Y. Yin and A. Paul, Nature, 2005, 437, 664–670 CrossRef CAS.
- P. Rai, R. Khan, S. Raj, S. M. Majhi, K. K. Park, Y. T. Yu, I. H. Lee and P. K. Sekhar, Nanoscale, 2014, 6, 581–588 RSC.
- S. Narayanan, B. N. Sathy, U. Mony, M. Koyakutty, S. V. Nair and D. Menon, ACS Appl. Mater. Interfaces, 2012, 4, 251–260 CrossRef CAS.
- X. Liu, J. Iocozzia, Y. Wang, X. Cui, Y. Chen, S. Zhao, Z. Li and Z. Lin, Energy Environ. Sci., 2017, 10, 402–434 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04739b |
| This journal is © The Royal Society of Chemistry 2020 |