
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe sporothriolides. A new biosynthetic family of fungal secondary metabolites†
Dong-Song
Tian
 a,
Eric
Kuhnert
a,
Jamal
Ouazzani
b,
Daniel
Wibberg
c,
Jörn
Kalinowski
c and
Russell J.
Cox
*a
a,
Eric
Kuhnert
a,
Jamal
Ouazzani
b,
Daniel
Wibberg
c,
Jörn
Kalinowski
c and
Russell J.
Cox
*a
aCentre of Biomolecular Drug Research (BMWZ), Institute for Organic Chemistry, Leibniz University Hannover, Schneiderberg 38, 30167, Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
bFrench National Center for Scientific Research (CNRS), Institute for the Chemistry of Natural Substances (ICSN), Avenue de la Terrasse, 91198, Gif-sur-Yvette, Cedex, France
cCenter for Biotechnology (CeBiTec), Bielefeld University, Universitätsstraße 27, 33615, Bielefeld, Germany
First published on 21st October 2020
Abstract
The biosynthetic gene cluster of the antifungal metabolite sporothriolide 1 was identified from three producing ascomycetes: Hypomontagnella monticulosa MUCL 54604, H. spongiphila CLL 205 and H. submonticulosa DAOMC 242471. A transformation protocol was established, and genes encoding a fatty acid synthase subunit and a citrate synthase were simultaneously knocked out which led to loss of sporothriolide and sporochartine production. In vitro reactions showed that the sporochartines are derived from non-enzymatic Diels–Alder cycloaddition of 1 and trienylfuranol A 7 during the fermentation and extraction process. Heterologous expression of the spo genes in Aspergillus oryzae then led to the production of intermediates and shunts and delineation of a new fungal biosynthetic pathway originating in fatty acid biosynthesis. Finally, a hydrolase was revealed by in vitro studies likely contributing towards self-resistance of the producer organism.
Gamma-lactone and alkyl citrate compounds derived from oxaloacetate are widespread natural products in fungi and often possess potent biological activities. Examples include sporothriolide 1,1,2 piliformic acid 2,3 tyromycin 34 and the cyclic maleidrides including byssochlamic acid 45,6 among others (Fig. 1). In some cases, for example those of 4 and squalestatin S1 5,7 detailed molecular studies have revealed that a dedicated polyketide synthase (PKS) produces a carbon skeleton that is then condensed with oxaloacetate by a citrate synthase (CS) to give an early alkyl citrate intermediate that is further oxidatively processed. In other cases, such as 1 and the sporochartines 6, the biosynthetic pathways are not yet clear.
 | ||
| Fig. 1 Structures of γ-lactone and alkyl citrate metabolites from fungi. Bold bonds show oxaloacetate-derived carbons where known. | ||
Sporochartines 6a–6d8,9 from the fungus Hypoxylon monticulosum CLL 205 (now referred to as Hypomontagnella spongiphila)10 possesses potent cytotoxicity (IC50: 7.2 to 21.5 μM) vs. human cancer cell lines and are proposed to be Diels Alder (DA) adducts of the furofurandione sporothriolide 1, itself a potent antifungal agent (EC50: 11.6 ± 0.8 μM),11 and trienylfuranol A 7,12 originally obtained from an endophytic fungus Hypoxylon submonticulosum DAOMC 242471 (now referred to as Hypomontagnella submonticulosa).13 Since the biosynthesis of sporothriolide 1 and related compounds is unknown, and biological DA reactions in fungi are currently of high interest,14 we decided to examine the biosynthesis of the sporochartines 6 in the Hypomontagnella spp. strains MUCL 54604 and CLL 205 (ref. 10 and 13) in detail.
Results
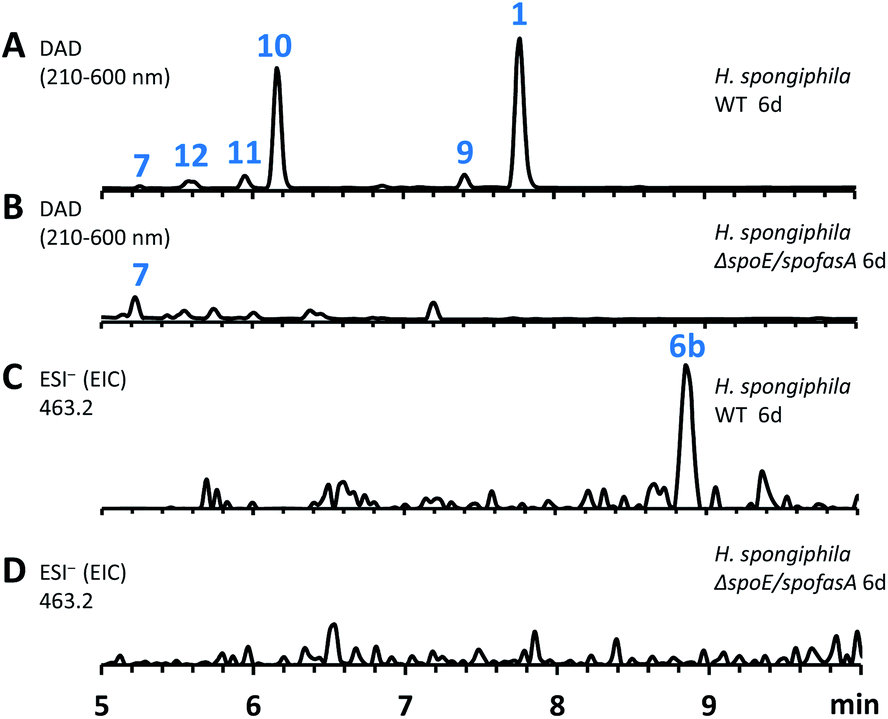
The three known producers of sporochartines, sporothriolide 1 and trienylfuranol A 7 (H. monticulosa MUCL 54604, H. spongiphila CLL-205 and H. submonticulosa DAOMC 242471) were grown in PDB liquid media and examined for production of metabolites by LCMS. Production time course experiments (see ESI Fig. S1.11–S1.14†) showed a high production of sporothriolide 1 in all three organisms (ca 180–238 mg L−1), as well as very low titers of sporochartine B 6b (ca 4–6 mg L−1). Compounds 1, 6b and 7, and cometabolites 8–12 (Fig. 2) were purified, characterised by 1D NMR and compared to literature (see ESI Tables S3.1–S3.8†). All three fungi are also capable of producing trienylfuranol A 7, but the H. submonticulosa strain yields higher amounts.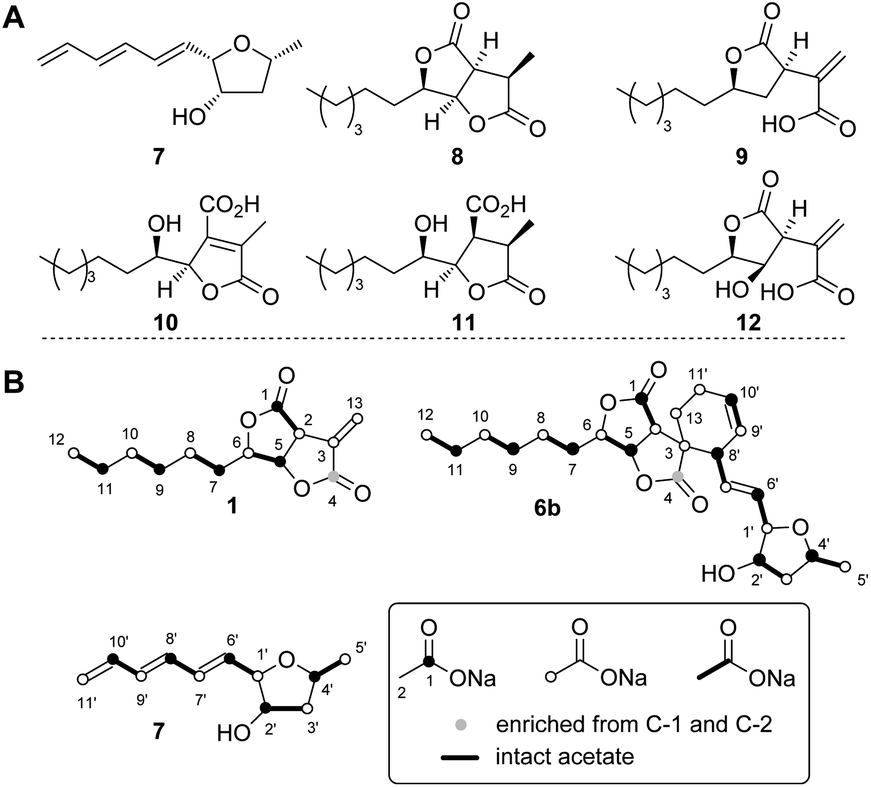 | ||
| Fig. 2 (A) Isolated compounds from H. spongiphila CLL 205. (B) Isotope labelled compounds from [1-13C], [2-13C] and [1,2-13C2]-acetate feeding experiments. | ||
The biosynthetic origin of the different carbon chains in 1, 6b and 7, was determined by isotopic labelling experiments with [1-13C], [2-13C] and [1,2-13C2] acetate (see ESI Tables S1.11–S1.13†). The observed labelling patterns of compounds 1 and 6b were consistent with a fatty acid or polyketide origin (see ESI Table S1.2†), coupled to a decarboxylated Krebs cycle intermediate such as oxaloacetate (Fig. 2). Carbons C-6 and C-1′ of 6b derived from C-2 of acetate were oxygenated indicating the involvement of an oxygenase during biosynthesis. Trienylfuranol A 7 was also shown to be derived from at least six intact acetates, but the loss of a C-1 acetate-derived carbon at C-11′, and oxygenation at C-1′ once again suggests the involvement of oxidative steps.
Genomic DNA from all three organisms was sequenced and assembled to afford high quality draft genomes (see ESI Table S1.1†).10 antiSMASH15 fungal version predicted 72, 69 and 53 secondary metabolite biosynthetic gene clusters (BGC) in the CLL, MUCL and DAOMC genomes, respectively (see ESI Fig. S1.1†).
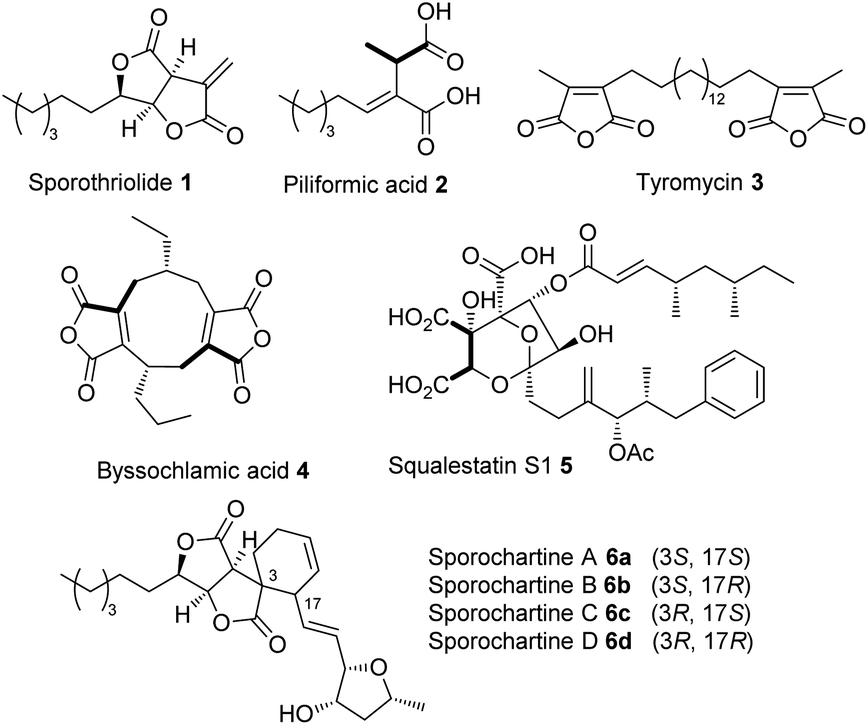
Initial searches for BGC containing PKS and CS encoding genes, as found for the biosynthesis of related compounds such as 4 and 5, were not productive. However, search for fungal fatty acid synthase (FAS) genes co-located with CS genes, as found for example in the BGC for the oryzines,16 rapidly identified closely related target BGC in all three organisms, which also encoded the expected oxygenase and decarboxylase catalysts (Fig. 3). BLASTp17 and PHYRE2 (ref. 18) platforms were used to annotate and predict the putative function of all proteins encoded by the BGC (see ESI Table S1.2†). In addition to the expected citrate synthase spoE, and two fungal FAS subunits (FASα and FASβ) spofasA and spofasB, the BGC was found to encode: a methylcitrate dehydratase (spoL); a decarboxylase (spoK) similar to the cis-aconitate decarboxylase;19 a dioxygenase (spoG); and two putative lactonases (spoH and spoJ). In addition, the clusters encoded two transporters (spoC and spoF), along with a transcriptional regulator (spoD), and spoI with unknown function (GenBank MT889334, see ESI Table S1.3†). The three clusters were highly homologous, containing the same genes in the same order and orientations (see ESI Fig. S1.3 and S1.4†).
 | ||
Fig. 3 Transcriptome analysis of the sporothriolide BGC from H. monticulosa MUCL 54604. Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2-fold changes are calculated between producing and non-producing conditions. NE, no expression. 2-fold changes are calculated between producing and non-producing conditions. NE, no expression. | ||
RNA from H. monticulosa MUCL 54604 was isolated separately under 1 and 6b producing (PDB medium) and non-producing (DPY medium) conditions and subjected to transcriptome sequencing (see ESI S1.1.3 for details†). Differential gene expression analysis of the predicted functional genes from spofasB to spofasA, showed a strong upregulation under producing condition (Fig. 3), while genes outside this region showed either low expression (see ESI Table S1.4†) or no change in transcription level.
The spoE (CS) and spofasA genes in H. spongiphila CLL 205 were then deleted simultaneously. This was achieved using the bipartite method of Nielsen and coworkers20 to insert a hygromycin resistance cassette. Forty-six hygromycin resistant transformants were generated and cultivated under sporothriolide 1 producing conditions and compared to the wild type (WT) strain (Fig. 4). One of these produced neither sporothriolide 1 nor the related sporochartines 6 or congeners 8–12. Polymerase chain reaction (PCR) analysis confirmed the incorporation of the hygromycin resistance gene at the target position and successful disruption of spoE and spofasA (see ESI Fig. S1.9 and S1.10†).
 | ||
| Fig. 4 HPLC analysis of crude extracts from H. spongiphila CLL 205 grown under producing conditions of 1: (A), Diode Array Detector (DAD) chromatogram of wild-type (WT); (B), DAD chromatogram of ΔspoE/spofasA; (C), extracted ion chromatogram (EIC of 6b, ESI−, 463.2, M + HCOO−) of WT extract; (D), EIC of ΔspoE/spofasA transformant showing absence of 6b. | ||
In this mutant the titre of trienylfuranol A 7 was increased, and this strain was therefore a suitable organism for the purification of 7 in usable amounts (e.g. 40 mg L−1). Sporochartines are formed as mixtures of diastereomers at the cyclohexene moiety and the observation that the endo DA product 6b always occurs in higher amounts than exo adduct 6a can be explained by the preference of DA reactions for endo product formation over exo. We therefore considered it possible that the proposed DA condensation between sporothriolide 1 and trienylfuranol A 7 could be non-enzymatic. In order to probe this question, we first added sporothriolide 1 to the supernatant obtained from the H. spongiphila ΔspoE/spofasA strain grown under producing conditions of 1 to accumulate 7. After 24 h of incubation 6a/6b were observed in the medium by HPLC-MS (Fig. 5B). In addition, 1 and trienylfuranol A 7 were also reacted under nitrogen in the dark in ethyl acetate (the standard extraction solvent). After 2 hours at room temperature, sporochartine A 6a (minor) and sporochartine B 6b (major) were observed by LCMS (Fig. 5D). At 40 °C (Fig. 5E), the reaction accelerated to give the same compounds in higher titre (see ESI S1.2.11 for details†).
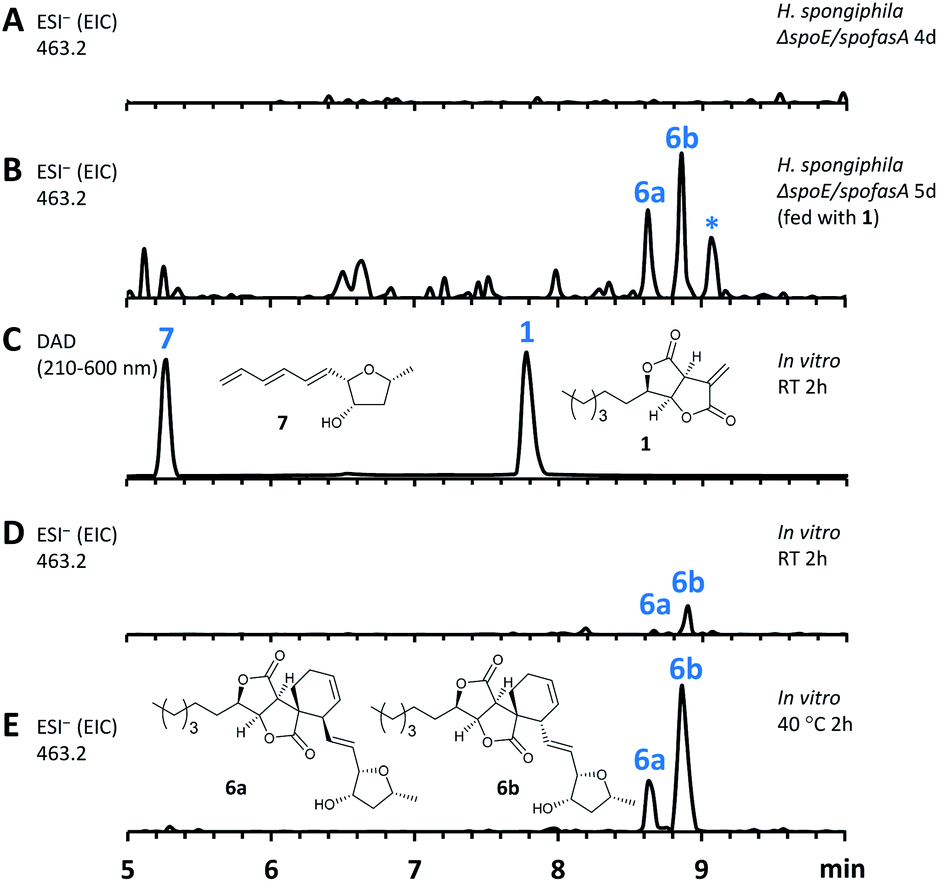 | ||
| Fig. 5 In vivo and in vitro assay for spontaneous sporochartine (6a and 6b) production. (A) Extracted ion chromatogram (EIC of 6a/6b, ESI−, 463.2, M + HCOO−) of crude extract from H. spongiphila CLL 205 ΔspoE/spofasA; (B), EIC of 6a/6b from H. spongiphila CLL 205 ΔspoE/spofasA fed with 1; (C) and (D) UV/vis spectrum and EIC (6a/6b) of in vitro reaction between 7 and 1 in ethyl acetate at RT, 2 h; (E), EIC (6a/6b) of in vitro reaction between 1 and 7 in ethyl acetate at 40 °C, 2 h. *Unrelated peak. | ||
Further analysis of the biosynthesis was performed by heterologous expression of combinations of the spo genes in Aspergillus oryzae NSAR1 (Table 1 and Fig. 6). All gene fragments were amplified from cDNA and cloned into fungal expression vectors (see ESI Fig. S1.8†). Expression of the FAS components alone, or with addition of the CS (spoE) and DH (spoL) did not result in the production of new compounds observable by LCMS (Table 1, entries 1–3).
| EXP | spo | Products | |||||||
|---|---|---|---|---|---|---|---|---|---|
| fasA | E | G | H | J | K | L | fasB | ||
| FASA | CS | DO | Lact | Lact | DC | DH | FASB | ||
| 1 | ✓ | — | — | — | — | — | — | ✓ | — |
| 2 | ✓ | ✓ | — | — | — | — | — | ✓ | — |
| 3 | ✓ | ✓ | — | — | — | — | ✓ | ✓ | — |
| 4 | ✓ | ✓ | — | — | — | ✓ | ✓ | ✓ | 20 |
| 5 | ✓ | ✓ | ✓ | — | — | ✓ | ✓ | ✓ | 9, 12, 14–15, 20, 22–23 |
| 6 | ✓ | ✓ | ✓ | ✓ | — | ✓ | ✓ | ✓ | 9, 12, 20, 22–23 |
| 7 | ✓ | ✓ | ✓ | — | ✓ | ✓ | ✓ | ✓ | 9, 12, 20, 22–23 |
| 8 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | 1, 9, 13–15 |
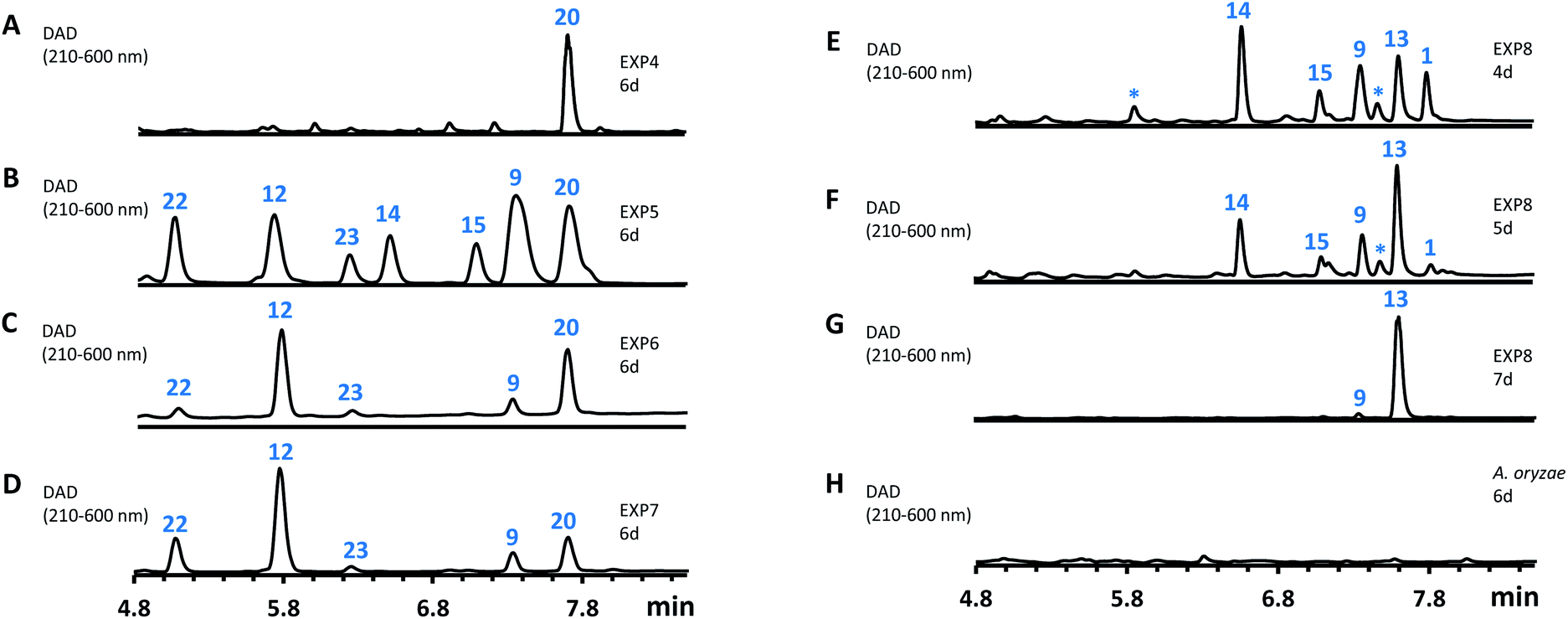 | ||
| Fig. 6 HPLC analysis of the expression of spo genes in A. oryzae. (A–H), diode array detector (DAD) traces of extracts of A. oryzae transformants. EXP refers to the gene combinations in Table 1. | ||
However, heterologous expression of all eight genes (Table 1, entry 8) led to the observation of a range of new metabolites in A. oryzae including sporothiolide 1 after 4 days (Fig. 6E). After longer fermentation up to 7 days these were converted to 13 (ca 8 mg L−1, Fig. 6G) which has the same molecular formula as sporothriolide (C13H18O4), but a slightly different retention time. Isolation and full NMR analysis of 13 showed this to be the monocyclic analog of 1, named dehydro-deoxysporothric acid, presumably either resulting from an eliminative ring opening of 1 itself, or being a biosynthetic precursor. However experiments with sporothriolide 1 itself showed that this compound is converted to the observed 13 after exposure to PDB, DPY or CMP fermentation media (pH 5.6–6.5) for short periods (see ESI Fig. S1.19†).
Further experiments were then conducted using reduced spo gene sets (Table 1, EXP 4–7). The results of EXP4 then indicated a biosynthetic pathway in which the fungal FAS proteins produce a dedicated decanoic acid 17, probably as a CoA thiolester, as these proteins possess a bifunctional malonyl-palmitoyl transferase (MPT) domain which loads malonyl CoA and off-loads an acyl CoA.21 Decanoyl CoA 17 is likely to be reacted with oxaloacetate by CS, in analogy to other known pathways,6 to give an octanyl citrate 18 which is then dehydrated to give octanyl aconitate 19 and decarboxylated to the first observable intermediate, octanyl itaconic acid 20, a known compound from the endophytic fungus Pestalotiopsis theae.22 The R absolute configuration at C-2 was confirmed by comparing the optical rotation with literature (see ESI Table S3.12†).22
The addition of the putative dioxygenase spoG to the previous set of genes in A. oryzae results in formation of a broad range of new compounds in addition to 20 (EXP 5, Fig. 6B). Four of the observed products (9, 12, 15, 23) were purified and their structures were deduced by NMR (see ESI Tables S3.5–S3.11†). Compound 23 was identified as the C-5 hydroxylated product of 20, which quickly converts in solution into the ring-closed shunt 16 (see ESI Fig. S3.59†). Compound 12 was shown to be sporothric acid, characterized by a lactone ring established between C-6 and the carboxylic acid group of the fatty acid chain.
Due to the presence of a hydroxyl group at C-5, we assume that 12 is derived from a double hydroxylated intermediate 22, which undergoes spontaneous lactonization and thus putatively represents a pathway intermediate. A main peak corresponding with the molecular formula for 22 was observed in the crude extract, but could not be purified to support its identity as the proposed intermediate. Compound 9 is structurally related to 12, but lacks the hydroxyl group at C-4. It is most likely a shunt metabolite derived from the putative monohydroxylated intermediate 21. Compound 15 is also monohydroxylated at C-6, but instead of the lactone ring it contains a maleic anhydride. Likewise 15 is probably also derived from the putative intermediate 21 through spontaneous reaction of the carboxylic acid moieties. Isolation of 14 was not successful, and LCMS chromatograms showed that 14 was partially converted to 10 in a short time (see ESI Fig. S3.46–S3.48†). As 14 shares the same molecular formula and very similar retention time with 10 its structure was deduced as the isomeric form of 10, which itself is likely formed by spontaneous lactonization between C-5 and the carboxylic acid moiety derived from oxaloacetate of the putative intermediate 22.
As neither the presence of 21 nor 22 could be unambiguously proved in the extracts of the A. oryzae transformants due to their instability or quick conversion, the function of SpoG was further evaluated in vitro. The spoG gene was amplified from H. monticulosa MUCL 54604 cDNA and cloned into pET28a (Novagen), then transformed and expressed into Escherichia coli BL21.23 SpoG was purified by fast protein liquid chromatography (FPLC) as a soluble protein (see ESI Fig. S1.20–S1.22†). Sequence analysis indicated that SpoG requires α-ketoglutarate as a cofactor. Incubation of purified SpoG with intermediate 20, FeSO4, α-ketoglutarate, and ascorbate at 30 °C for 2 h led to the formation of three products, the main compound of which was identical with 22 (by mass, UV and retention time) from the heterologous expression experiments. In addition, 23 was formed as minor product alongside another compound with identical mass likely representing its proposed regioisomer 21 (Fig. 7B). Extended incubation times up to 16 h resulted in a complete conversion of 21 and 23 into 22 indicating that 21 and 23 are precursors of 22 and that SpoG catalyses two consecutive rounds of hydroxylation (Fig. 7C). These results show that SpoG catalyses hydroxylation at C-5 and C-6 of the saturated carbon chain, and that the pathway is conducted via21 and 23.24
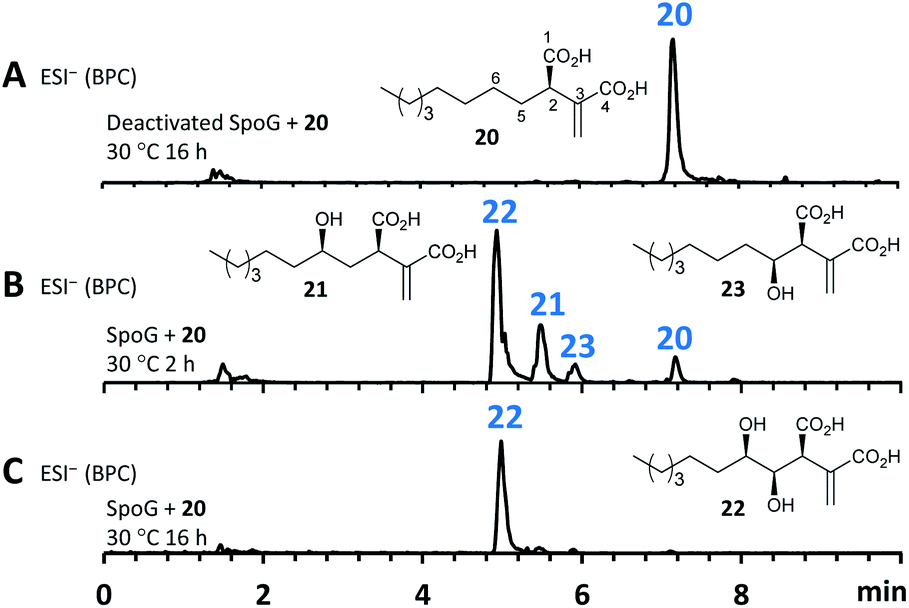 | ||
| Fig. 7 In vitro assay of SpoG using purified proteins. (A), ESI− trace of boiled SpoG (50 μm) incubated with 20 (2.5 mM), Tris buffer (50 mM, pH 7.5), ascorbate (4 mM), α-ketoglutarate (4 mM), and FeSO4 (0.2 mM) at 30 °C, 16 h; (B), ESI− trace of SpoG incubated with 20 under the same conditions for 2 h; (C), ESI− trace of SpoG incubated with 20 under the same conditions for 16 h. | ||
The presence of 21 and the lack of its putative shunts 15 and 9 in the in vitro setup indicates that 21 is stabilized in the buffer solution. In addition, the occurrence of 15 and 9 in relatively high titers in the A. oryzae transformants compared to the significant lower titers of 23, raises the question about regio-selectivity and -specificity of SpoG. Due to relative instability of 21 and 23 this question cannot be answered in this context. Compound 12, which occurred in the heterologous host (e.g. EXP 5), was not observed in the in vitro experiments, suggesting that A. oryzae can catalyse this transformation.
As previous reports demonstrated that lactonization can be pH-dependent,25in vitro generated 22 was acidified to pH 2 leading to partial conversion of 22 into 12 and minor quantities of 14 after 2 h of incubation (Fig. 8A). In addition to 12 and 14, very small amounts of 13 (derived from 1) could be found as well after increased incubation periods (Fig. 8B). This shows that lactone ring formation during sporothriolide biosynthesis can also occur spontaneously under acidic conditions, but is not enough to generate 1 in significant titers, indicating that further enzymes are required to form 1 in the natural producer.
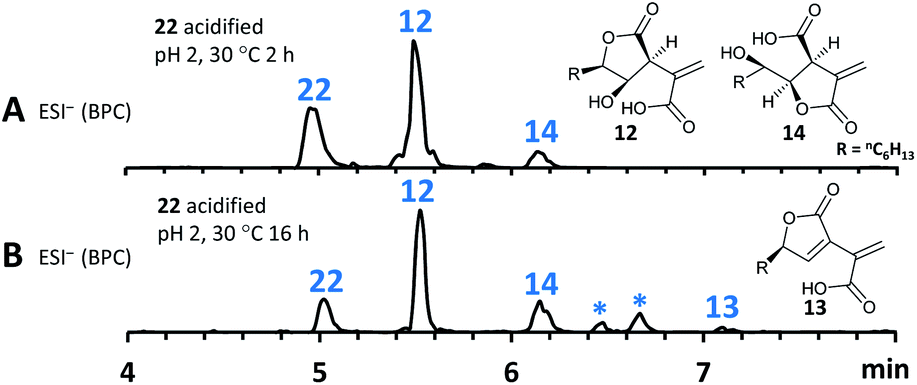 | ||
| Fig. 8 Lactonization under acidic conditions. (A), ESI− spectrum of 22 (obtained directly via incubation of 20 with SpoG) acidified with formic acid to pH 2, at 30 °C for 2 h; (B), as (A), 30 °C for 16 h. *Unknown degradation compounds. R = nC6H13. | ||
It is thought that ionised carboxylates which occur at higher pH values become less reactive due to being poor electrophiles,25 therefore explaining the observed in vitro results. As a side note, spontaneous lactonization between C-6 hydroxy functionality and the carboxyl group of the fatty acid derived chain seems to be favoured in this case.
The cofactor dependence of SpoG was also investigated showing that α-ketoglutarate is essential for turnover in vitro. Furthermore, monocarboxylic acid substrates such as trans-2-hexenoic acid and 2-methylhexanoic acid were tested with SpoG, but did not result in observable product formation (see ESI Fig. S1.20–S1.22†).
As previously indicated the heterologous expression of the full set of genes from the spo cluster led to the formation of high titers of 13, the supposed degradation product of 1. Thus, it seems likely that the final pathway steps are catalysed by one or both encoded lactonases (SpoH, SpoJ). Attempts to express SpoH and SpoJ and study them in vitro in analogy to SpoG were not successful, leading to insoluble and inactive proteins in E. coli and non-viable colonies in S. cerevisiae. Further experiments therefore involved heterologous expression in A. oryzae.
The genes spoH and spoJ were expressed together and individually with the remaining set of genes (Table 1, EXP 6–8). Individual expression of the lactonase genes showed 12 as predominant pathway product in both cases, whereas 1 or 13 could not be detected (Fig. 6C and D). Only when both lactonases were expressed together was the final pathway product 1 observed, which quickly converted into 13 when exposed to longer fermentation periods. In addition, small titers of 14 were observed at early fermentation time points, which disappeared at the end of the fermentation period, indicating that 14 may also be a pathway intermediate that is converted into 1.
Based on these studies a scheme for the biosynthesis of 1 can be proposed (Scheme 1). SpofasA, SpofasB, SpoE, SpoK and SpoL form octyl itaconic acid 20 as the first stable intermediate, which is converted by the dioxygenase SpoG into the C-5/C-6 hydroxylated product 22via the C-5 or C-6 hydroxylated intermediates 21 and 23. Either (or both) of 12 and 14 could be intermediates during the conversion of 22 to 1. However, our in vivo experiments were unable to resolve this question and lack of soluble protein obviated in vitro experiments. We propose that the lactonases SpoH and SpoJ form a bifunctional heterodimer to lactonize 22 into the final pathway product 1 as neither appears to catalyse the formation of 1 in isolation. The overall route is remarkable for the number of potential shunt pathways, and we have detected at least 10 possibilities for shunt formation, of which 8 compounds have been fully characterized here.
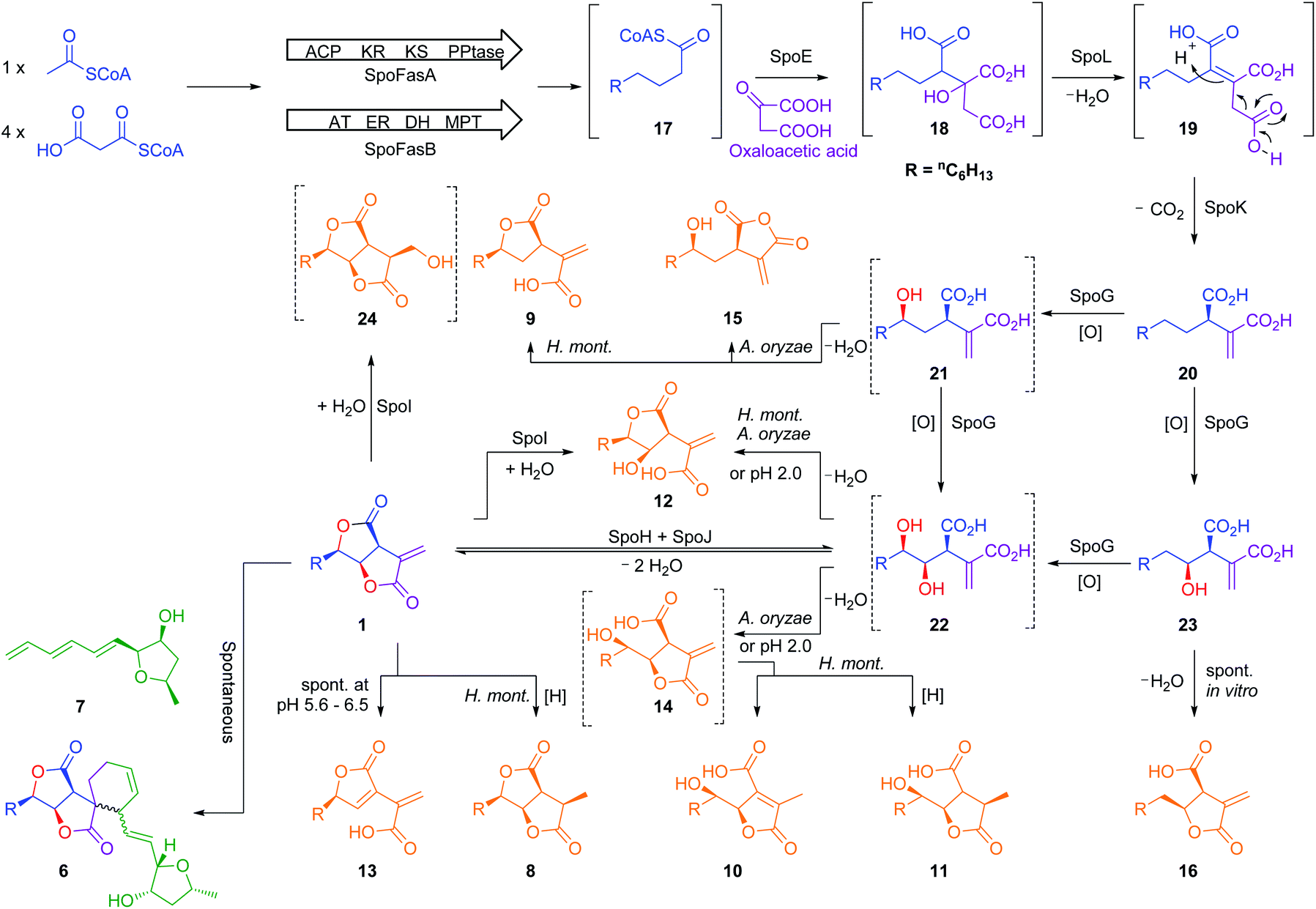 | ||
| Scheme 1 Proposed biosynthesis of sporothriolide 1 and sporochartines 6. Orange compounds result from shunt steps. Compounds in solid square brackets were not experimentally observed and those in dashed brackets were observed during experiments but structures could not be confirmed by NMR. Abbreviations: AT, acyl transferase; ER, enoyl reductase; MPT, malonyl palmitoyl transferase; ACP, acyl carrier protein; KR, ketoreductase; KS, ketosynthase; PPTase, phosphopantetheinyl transferase. | ||
As the biosynthesis of 1 can be fully explained by the activity of the investigated genes, the role of the unknown protein SpoI remained obscure. The function of spoI was especially intriguing as it showed high expression levels under both producing and non-producing conditions for 1 (see ESI Fig. S1.23–S1.26†). BLASTp17 analysis of SpoI showed it is a member of the cupin-domain-containing proteins. It is 35.7% identical to VirC which is involved in the fungal trichoxide biosynthetic pathway,26 although VirC is also of unknown function. Structural analysis using PHYRE-2 (ref. 18) also showed no relationship to known functionally-characterized enzymes.
To characterize SpoI, the gene was amplified from cDNA of H. monticulosa MUCL 54604, cloned into pET28a and overexpressed in E. coli. Purification of the protein in soluble form was achieved by FPLC. We first tested if SpoI could aid in the cycloaddition of sporothriolide 1 and trienylfuranol A 7 to afford sporochartines as we could not fully exclude the possibility of an enzyme catalysed reaction. Incubation of SpoI with 1 and 7 did not make the sporochartine adducts, but it resulted in the formation of small amounts of 12 and a new compound 24 which share the same molecular formula (HRMS m/z 255.1217 [M − H]−; calc. for C13H19O5, 255.1232). In addition, titers of 7 did not change throughout the experiment but 1 was consumed proving that 1 could be the substrate of SpoI (Fig. S1.26†).
The experiment was repeated with 1 as the sole substrate confirming that 1 is fully converted into 12 and 24 (Fig. 9). As the formation of 13 could be observed during the experiment it was not clear if 13 was used as substrate instead of 1. To test this, 13 was incubated with SpoI which did not lead to product formation demonstrating that 1 is indeed the substrate of SpoI. Isolation of 24 by upscaling the in vitro reaction proved unsuccessful, thus the structure could not be elucidated by NMR. As 24 and 12 share the same molecular formula C13H20O5, it was initially assumed the SpoI catalyses hydrolysis, which leads in the case of 12 to a ring opening of one of the lactones. However, the lack of 14, 10 and 11 allows only one possible structure for 24, where the exomethylene group is hydroxylated. As 24 was never observed in the wild type it is likely to be quickly degraded in vivo. The catalysed reaction of SpoI, may represent a resistance against the antifungal sporothriolide 1, protecting the producer organism from its toxic product when it accumulates in the cells.
 | ||
| Fig. 9 In vitro assay of SpoI using purified proteins. (A), ELSD chromatogram of boiled SpoI (50 μM) incubated with 1 (2.5 mM) in PBS buffer (pH 7.5) at 30 °C for 1 h; (B), ELSD chromatogram of SpoI (50 μM) incubated with 1 (2.5 mM) in PBS buffer (pH 7.5) at 30 °C for 1 h; (C), ELSD chromatogram of SpoI (50 μM) incubated with 13 (2.5 mM) in PBS buffer (pH 7.5) at 30 °C for 1 h. R = nC6H13. | ||
Finally, we investigated the distribution of fungal BGCs related to the spo pathway. Piliformic acid 2 is already known by labelling studies to be derived from acetate and an oxaloacetate derivative.3 It has been purified previously from the known producer Xylaria hypoxylon,27 the genome of which we have sequenced in a parallel project.10 Screening of the X. hypoxylon genome revealed a BGC with high homology to the spo BGC (see ESI Fig. S1.2†), but lacking spoG, spoH and spoJ, consistent with the lower level of oxygenation and cyclisation of 2 compared to 1.
Multi-gene BLAST28 analysis was performed using templates SpofasA (FAS α), SpofasB (FAS β), SpoE (citrate synthase), SpoF (transporter), SpoG (dioxygenase), SpoH (lactonase), SpoJ (lactonase), SpoK (decarboxylase) and SpoL (2-methyl citrate dehydratase) in a MultiGeneBLAST architecture search (see ESI S1.1.6 for details†). The results show that such FAS-based pathways are relatively common in fungi but have remained unrecognised to-date: modern bioinformatic tools such as antiSMASH15 do not yet recognize such BGCs, probably because of the high number of ‘primary-metabolism’ related steps. At least five homologous clusters encoding fungal FAS α and β components, and citrate synthase (spoE), dehydratase (spoL), decarboxylase (spoK), dioxygenase (spoG) and one or more lactonases (spoHJ) were detected, predominantly in Aspergillus species (see ESI Fig. S1.7†), and it is likely that similar pathways will appear elsewhere.
Discussion
The sporothriolide pathway (Scheme 1) shows several points of novelty. First, a dedicated fungal FAS system produces decanoate rather than the usual octadecanoate. This differs from other known pathways such as those involved in maleidride and squalestatin biosynthesis which begin with a dedicated PKS. FAS systems which selectively form short chains should find utility in the production of biofuels, for example, where C8 and C10 lipids are advantageous, and extensive efforts are underway to engineer short-chain synthases for this outcome.29 Fungal FASs appear to have already evolved the ability to selectively produce these valuable shorter compounds, such as decanoate in 1 and octanoate in 2 and 25, for example.The early steps of the spo pathway then follow the well-known primary metabolic steps from acetyl CoA to itaconic acid, but modified to accept a longer alkyl unit. The first four steps of the pathway thus closely mirror primary metabolic steps. The presence of the decarboxylase encoded by spoK differentiates the pathway from those proceeding towards the maleidrides6 where a different mode of decarboxylation appears to be coupled with multimerization catalysed by dedicated KI and PEBP enzymes,6,30 and from the squalestatins where no dehydration or decarboxylation reactions take place, but where much more extensive backbone oxygenation leads to more complex cyclisation modes.7
The oxygenase SpoG catalyses the hydroxylations required to form the bis-lactone, and this moves the pathway more conclusively towards secondary metabolism. These hydroxylated intermediates are clearly able to undergo many spontaneous cyclizations, but the presence of both lactonases SpoH and SpoJ directs the pathway to sporothriolide 1.
The presence and selectivity of the SpoG hydroxylase presumably controls the formation of related classes of metabolites in this family. For example in the case of the furofurandiones such as 1 double hydroxylation is required. Compounds such as oryzine A 25a (Fig. 10) requires only a single hydroxylation at C-5 performed by OryG (68% identity, 77% similarity),16 while oryzine B 25b may arise from double hydroxylation and 3,4-dehydration. Piliformic acid 2 requires no hydroxylation. Its 2,5 unsaturation most likely arises by isomerisation of 20, for example, consistent with the lack of a spoG homolog in its BGC. It also appears that the selectivity of the lactonase components (SpoHJ) may be able to control the formation of γ-lactones (e.g. 1) vs. δ-lactones (e.g.25). Further mix-and-match experiments will be required to verify these hypotheses.31
 | ||
| Fig. 10 Structure of oryzines isolated from A. oryzae. | ||
The identification of SpoI as a hydrolase involved in the putative self-resistance mechanism of the producer organism against 1 adds to the increasing knowledge about self-resistance genes in fungi32 and will enable the identification of similar enzymes in other biosynthetic pathways. Knowledge of the overall pathway may also help to elucidate the BGC of more esoteric compounds such as 3 where no biosynthetic information currently exists.
Finally, we showed that, unlike in many other fungal natural products,23,33 the DA reaction required to form the sporochartines 6a and 6b is spontaneous. Furthermore, no genes encoding proteins likely to be involved in the biosynthesis of trienylfuranol A 7, such as a PKS, or other oxidative proteins, were found within or near the spo gene cluster. Therefore, the biosynthesis of 7 remains cryptic for now, but experiments in this area will form part of our future investigations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Chinese Scholarship Council [Dong-Song Tian (201706670001)] and the German Research Foundation (DFG INST 187/686-1 and CO 1328/4-1). This work benefitted from the sharing of expertise within the DFG priority program “Taxon-Omics: New Approaches for Discovering and Naming Biodiversity” (SPP 1991). Prof. Marc Stadler (Helmholtz-Centre for Infection Research) and Dr Mark W Sumarah (London Research and Development Centre, Ottawa, Canada) are thanked for the gift of strains H. monticulosa MUCL 54604 and H. submonticulosa DAOMC 242471, respectively. Geraldine Le Goff is thanked for technical assistance. The bioinformatics support of the German Network for Bioinformatics Infrastructure (de.NBI) is gratefully acknowledged.Notes and references
- F. Surup, E. Kuhnert, E. Lehmann, S. Heitkämper, K. D. Hyde, J. Fournier and M. Stadler, Mycology, 2014, 5, 110–119 CrossRef CAS.
- K. Krohn, K. Ludewig, H.-J. Aust, S. Draeger and B. Schulz, J. Antibiot., 1994, 47, 113–118 CrossRef CAS.
- N. C. J. E. Chesters and D. O'Hagan, J. Chem. Soc., Perkin Trans. 1, 1997, 827–834 RSC.
- W. Weber, M. Semar, T. Anke, M. Bross and W. Steglich, Planta Med., 1992, 58, 56–59 CrossRef CAS.
- A. J. Szwalbe, K. Williams, D. E. O'Flynn, A. M. Bailey, N. P. Mulholland, J. L. Vincent, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Commun., 2015, 51, 17088–17091 RSC.
- K. Williams, A. J. Szwalbe, N. P. Mulholland, J. L. Vincent, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Angew. Chem. Int. Ed., 2016, 55, 6784–6788 CrossRef CAS.
- K. E. Lebe and R. J. Cox, Chem. Sci., 2019, 10, 1227–1231 RSC.
- C. Leman-Loubière, G. Le Goff, P. Retailleau, C. Debitus and J. Ouazzani, J. Nat. Prod., 2017, 80, 2850–2854 CrossRef.
- C. Leman-Loubière, G. Le Goff, C. Debitus and J. Ouazzani, Front. Mar. Sci., 2017, 4, 399 CrossRef.
- D. Wibberg, M. Stadler, C. Lambert, B. Bunk, C. Spröer, C. Rückert, J. Kalinowski, R. J. Cox and E. Kuhnert, Fungal Divers., 2020 DOI:10.1007/s13225-020-00447-5.
- L. L. Cao, Y. Y. Zhang, Y. J. Liu, T. T. Yang, J. L. Zhang, Z. G. Zhang, L. Shen, J. Y. Liu and Y. H. Ye, Pestic. Biochem. Physiol., 2016, 129, 7–13 CrossRef CAS.
- K. M. N. Burgess, A. Ibrahim, D. Sørensen and M. W. Sumarah, J. Antibiot., 2017, 70, 721–725 CrossRef CAS.
- C. Lambert, L. Wendt, A. I. Hladki, M. Stadler and E. B. Sir, Mycol. Prog., 2019, 18, 187–201 CrossRef.
- C. S. Jamieson, M. Ohashi, F. Liu, Y. Tang and K. N. Houk, Nat. Prod. Rep., 2019, 36, 698–713 RSC.
- K. Blin, S. Shaw, K. Steinke, R. Villebro, N. Ziemert, S. Y. Lee, M. H. Medema and T. Weber, Nucleic Acids Res., 2019, 47, W81–W87 CrossRef CAS.
- Z. Wasil, E. Kuhnert, T. Simpson and R. Cox, J. Fungi, 2018, 4, 96 CrossRef CAS.
- M. Johnson, I. Zaretskaya, Y. Raytselis, Y. Merezhuk, S. McGinnis and T. L. Madden, Nucleic Acids Res., 2008, 36, W5–W9 CrossRef CAS.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, Nat. Protoc., 2015, 10, 845–858 CrossRef CAS.
- F. Chen, P. Lukat, A. A. Iqbal, K. Saile, V. Kaever, J. van den Heuvel, W. Blankenfeldt, K. Büssow and F. Pessler, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 20644–20654 CrossRef CAS.
- M. L. Nielsen, L. Albertsen, G. Lettier, J. B. Nielsen and U. H. Mortensen, Fungal Genet. Biol., 2006, 43, 54–64 CrossRef CAS.
- S. Jenni, M. Leibundgut, D. Boehringer, C. Frick, B. Mikolásek and N. Ban, Science, 2007, 316, 254–261 CrossRef CAS.
- L. Liu, Y. Han, J. Xiao, L. Li, L. Guo, X. Jiang, L. Kong and Y. Che, J. Nat. Prod., 2016, 79, 2616–2623 CrossRef CAS.
- L. Kahlert, E. F. Bassiony, R. J. Cox and E. J. Skellam, Angew. Chem. Int. Ed., 2020, 59(14), 5816–5822 CrossRef CAS.
- J. Qi, D. Wan, H. Ma, Y. Liu, R. Gong, X. Qu, Y. Sun, Z. Deng and W. Chen, Cell Chem. Biol., 2016, 23, 935–944 CrossRef CAS.
- Z. Zhang, P. Gibson, S. B. Clark, G. Tian, P. L. Zanonato and L. Rao, J. Solution Chem., 2007, 36, 1187–1200 CrossRef CAS.
- L. Liu, M. C. Tang and Y. Tang, J. Am. Chem. Soc., 2020, 141, 19538–19541 CrossRef.
- J. R. Anderson, R. L. Edwards and A. J. S. Whalley, J. Chem. Soc., Perkin Trans. 1, 1985, 1481–1485 RSC.
- M. H. Medema, E. Takano and R. Breitling, Mol. Biol. Evol., 2013, 30, 1218–1223 CrossRef CAS.
- J. Gajewski, F. Buelens, S. Serdjukow, M. Janßen, N. Cortina, H. Grubmüller and M. Grininger, Nat. Chem. Biol., 2017, 13, 363–365 CrossRef CAS.
- R. Schor and R. Cox, Nat. Prod. Rep., 2018, 35, 230–256 RSC.
- C. Schotte, L. Li, D. Wibberg, J. Kalinowski and R. J. Cox, Angew. Chem. Int. Ed., 2020 DOI:10.1002/anie.202009914.
- N. P. Keller, Nat. Chem. Biol., 2015, 11, 671–677 CrossRef CAS.
- R. Schor, C. Schotte, D. Wibberg, J. Kalinowski and R. J. Cox, Nat. Commun., 2018, 9, 1963 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: including all experimental and spectroscopic details. See DOI: 10.1039/d0sc04886k |
| This journal is © The Royal Society of Chemistry 2020 |