
Chromophore amphiphile–polyelectrolyte hybrid hydrogels for photocatalytic hydrogen production†
Hiroaki
Sai
 ab,
Aykut
Erbas‡
b,
Adam
Dannenhoffer
b,
Dongxu
Huang
b,
Adam
Weingarten
c,
Erica
Siismets§
b,
Kyujin
Jang
b,
Karen
Qu
b,
Liam C.
Palmer
ac,
Monica
Olvera de la Cruz
bc and
Samuel I.
Stupp
*abcde
ab,
Aykut
Erbas‡
b,
Adam
Dannenhoffer
b,
Dongxu
Huang
b,
Adam
Weingarten
c,
Erica
Siismets§
b,
Kyujin
Jang
b,
Karen
Qu
b,
Liam C.
Palmer
ac,
Monica
Olvera de la Cruz
bc and
Samuel I.
Stupp
*abcde
aSimpson Querrey Institute, Northwestern University, Chicago, Illinois 60611, USA. E-mail: s-stupp@northwestern.edu
bDepartment of Materials Science and Engineering, 2220 Campus Drive, Evanston, IL 60208, USA
cDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
dDepartment of Medicine, Northwestern University, 251 East Huron St., Chicago, Illinois 60611, USA
eDepartment of Biomedical Engineering, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
First published on 5th December 2019
Abstract
Hybrid systems based on covalent polymers and supramolecular assemblies offer unique opportunities for functional materials based on the pathway-dependent dynamic structures of supramolecular assemblies and the mechanical stability of covalent polymers. We report here on the synthesis of functional hybrid hydrogels containing self-assembling chromophore amphiphiles and polyelectrolytes. Chromophore amphiphiles were introduced into non-aqueous solvent swollen polymer matrices and self-assembly of the chromophore amphiphiles into crystalline nanostructures was triggered in the confined environment of the covalent network upon solvent exchange for water. Opposite charges in the covalent polyelectrolyte and the chromophore amphiphiles and sterics entrap the supramolecular assemblies within the mechanically stable network. However, molecular components necessary for catalysis, byproducts from photocatalysis, and the hydrogen produced are able to diffuse in or out of the covalent network to create a reusable robust host for photocatalysis. By varying the monomer and crosslinker composition in the feed, we can tune the porosity of the network as well as the chemical environment in which supramolecular crystallization of the chromophore amphiphiles takes place. This allows optimization of the hydrogel mechanical properties, retention of the chromophore amphiphile assemblies, and the photocatalytic reaction efficiency. Coarse-grained molecular dynamics simulations revealed that the chromophore amphiphile assembly is guided by the polyelectrolyte network via ionic interactions. We also demonstrate successful photocatalytic hydrogen production from catalyst-laden hybrid hydrogels with the turnover frequency approaching that of the supramolecular hydrogel system, and also show that the hybrid hydrogels can be reused over multiple cycles as photosensitizers.
Introduction
Photocatalytic reactions to harness solar energy in the form of storable fuels is a major target in sustainable and clean energy production.1–4 Using soft materials in aqueous environments for such photocatalytic reactions is particularly interesting because of their similarity to biological photosynthetic systems and the versatility offered by the organic chemistry toolbox to tailor functions.5–11 In plants the photosynthetic process is carried out by a number of proteins in the chloroplasts, each responsible for photoinduced charge separation, charge transport and redox reactions yielding products that can be used for synthesis of fuels such as saccharides.12–14 Since the effective long range charge transport length scales in such aqueous environments is limited, these proteins are compartmentalized on the stacked lipid bilayer membranes.15,16 In order to mimic this compartmentalization inspired by artificial photosynthesis systems, spatial control of photoabsorbers and catalysts is highly desirable.Supramolecular assemblies of chromophore molecules that exhibit intermolecular charge transport have been highlighted by several research groups as promising materials for facile photocatalytic reactions.6,17–21 Use of supramolecular chemistry introduces multiple advantages such as flexibility in energy levels and modes of self-assembly by molecular design, potential pathway dependence structure control,22,23 ability to create nanoscale objects of high aspect ratio for preferred charge transport directions,24,25 and the possibility of self-repairing, self-healing features in synthetic systems.26,27 We previously reported on the self-assembly of chromophore amphiphile (CA) molecules into micrometer-sized ribbon-like nanostructures that form hydrogels and function as light harvesting assemblies for photocatalytic production of hydrogen.6 The perylene monoimide (PMI)-based-chromophores bearing hydrophilic carboxylate groups spontaneously assemble into two-dimensional crystalline lattices within the hydrogel network when electrostatic repulsion among carboxylate groups is screened by added salts. Crystallization of PMI assemblies is revealed by the appearance of Bragg peaks in wide-angle X-ray scattering patterns as well as changes in optical absorption indicating the existence of charge-transfer excitons.6,28,29 By tuning alkyl linker lengths between PMI cores and carboxylate groups,8 substitution at the 9-position of the PMI cores, which modulates molecular dipole moment,30 intermolecular packing sterics10 or electronic states,31 we accessed a number of variations in supramolecular morphology and optical absorption properties. Interestingly, attaching the bulky alkyl group 3-pentylamine at the 9-position of the perylene monoimide leads to a pathway-dependent polymorphism in the nature of the crystalline assemblies.32 Due to their tunable energy levels and high photochemical stability,33–35 PMI-based chromophore amphiphile assemblies hold promise as reusable substrates for photocatalysis. In our previous work the PMI assemblies in the hydrogel were crosslinked by adding the polyelectrolyte (PE) poly(diallyldimethylammonium chloride) (PDDA) with opposite charge to the negatively charged supramolecular assemblies prior to the addition of a sacrificial electron donor and a water soluble catalyst.6 In spite of PDDA crosslinking of the supramolecular hydrogels, the soft materials remain mechanically unstable and cannot be easily reused.
In this work we investigate possible pathways to encapsulate supramolecular photocatalytic hydrogels in a covalently crosslinked polyelectrolyte network. The objective has been to create mechanically robust systems that would allow photocatalysis to occur over multiple cycles by allowing substrates, catalysts, and products to flow freely in and out of the system. We were also motivated by the possibility of removing and reconstituting the supramolecular light harvesting component within these systems. For the function of a reusable photosensitizing material, it would be highly desirable to design a hybrid material in which supramolecular CA assemblies exist within a covalently crosslinked hydrogel. A covalent hydrogel as a hosting scaffold for CA photocatalytic assemblies also offer the possibility to chemically tune the mechanical and chemical properties as well as diffusivities for small molecules through crosslink densities and copolymerization of multiple monomers. Copolymerization would be useful in order to access properties such as the thermal, pH- or light-responsive behavior in the covalent network or ability to control its interactions with the supramolecular component. Specifically, strong PE hydrogels with backbones bearing opposite charge to that of CA assemblies would be amenable to facile incorporation of the chromophores in the covalent network.
We have incorporated PMI CAs in monomeric form into pre-synthesized PE networks followed by exchange from organic solvent to water in order to trigger their self-assembly within the covalent three-dimensional network. We have varied the monomer feed compositions for the PE hydrogels and employ experimental physicochemical characterization as well as coarse-grained molecular dynamics (MD) simulations to investigate the CA assembly process. Finally, we performed photocatalytic hydrogen production experiments using these hybrid hydrogels, focusing on the effect of the covalent network's chemical structure.
Experimental section
Materials
Unless otherwise noted, all chemicals were purchased in reagent grade or higher and used as received. The perylene monoimide (PMI) chromophore amphiphile (Fig. 1a, right) was synthesized according to the procedure described previously, except that the final purification by gel-permeation chromatography was excluded.6 The proton reduction catalyst, Na2[Mo3S13], was synthesized according to previously reported procedures.36,37 | ||
| Fig. 1 Synthesis of chromophore amphiphile–polyelectrolyte hybrid hydrogels. (a) Chemical structures of the monomers for PE hydrogels (shown in blue) and the chromophore amphiphile, PMI (shown in red). (b) Schematic representations and corresponding photographs of the materials at various processing steps. Large grids in the photographs measure 1 cm. | ||
Polyelectrolyte (PE) gel synthesis
PE hydrogels were synthesized via free radical polymerization, using sodium persulfate (Sigma-Aldrich, ≥99%) as the initiator and N,N′-methylenebisacrylamide (MBAA, Sigma-Aldrich, 99%) as the crosslinker. Table S1† shows the gels used for hybrid hydrogel synthesis. The 3-acrylamidopropyltrimethylammonium chloride (APTAC) monomer (Sigma-Aldrich, 75 wt% in aqueous solution) was diluted to 2 M in deionized (DI) water. In synthesizing copolymer hydrogels, 2 M aqueous solutions of acrylamide (Sigma-Aldrich, ≥99%, HPLC grade) and/or 2-acrylamido-2-methyl-1-propanesulfonic acid sodium salt (AMPS, Sigma-Aldrich, 50 wt% in aqueous solution) were mixed in varying ratios with the 2 M APTAC monomer solution. To this monomer solution, we added MBAA in the molar equivalence of 0.005–0.02 with respect to the monomers. The solution was then degassed by bubbling under a stream of argon for 10 min, and 0.005 molar equivalent sodium persulfate was added as a 10 wt% solution. The solution was quickly mixed, packed in 1 mL NormJect syringes with sealed needles as molds, and heated to 65 °C for 6 h.After the gels were cooled to room temperature, they were extruded from the molds and suspended in water baths for 48 h, with water exchange every 8 h. Once the hydrogels reached their equilibrium sizes, they were sectioned into approximately 2 mm-thick round disks and dried on a poly(tetrafluoroethylene) (PTFE) plate at 90 °C overnight to form dried gels. For ground gels, the dried gels were subject to manual grinding by mortar and pestle.
PMI/PE hybrid hydrogel synthesis
PMI were solubilized into water by the addition of one molar equivalence of 1 M sodium hydroxide aqueous solution. The PMI solution in water was then diluted to 11.6 mM, then freeze-dried. The dry powder was dissolved into 2,2,2-trifluoroethanol (TFE, Sigma-Aldrich, ≥99%) at 20 mg mL−1. The PE dried gels were placed at the bottom corner of 20 mL vials, and PMI solution in TFE was added at concentrations of 20 mg mL−1. Once the PMI solution was absorbed by the gel (overnight for monolithic dried gels and 3 min for ground gels) the gels containing were placed in baths of DI water. The DI water was exchanged at least three times to remove the organic solvent. For monolithic hydrogels the interval between exchanges is set to 8 h, while for the ground gels the exchange was performed every 3 min. A Falcon cell strainer (40 μm mesh size) was used to collect the ground hydrogel pieces while separating the liquid phase. Some of the hydrogels were then placed in 150 mM sodium chloride aqueous solution (NaClaq) baths. Thermal annealing of hydrogels was performed by heating closed vials containing the hydrogels in water or NaClaq to 95 °C for 30 min and cooling them down.Hybrid hydrogel characterization
Cryo-scanning electron microscopy was performed via high-pressure freezing with a Leica HPM100 high pressure freezer, followed by sample fracturing, etching and carbon coating on the Leica ACE100 sample cryo-preparation chamber and observation under Hitachi S-4800 microscope equipped with a cryogenic temperature stage. The etching was performed by raising the sample temperature to −105 °C in vacuo for 30 min. The coating thickness was set at 5 nm.Swelling ratio measurements were performed by measuring the weights of fully hydrated monolithic hydrogel and the corresponding dried gel on a pre-weighed aluminum dish. The drying was performed on a hotplate at 90 °C in air. Before PMI addition, the thickness of the monolithic samples was around 2–3 mm in hydrated states and the gels weighed a few to 10 milligrams in the dried state. In order to exclude free water from the weight measurement of the fully hydrated gel, the gels were placed on Kimwipe sheets before weighing. Triplicate measurements were performed to account for error in measurements.
UV-Vis absorption spectroscopy was performed on either a ThermoFisher NanoDrop spectrometer or a PerkinElmer Lambda Spec 1050 spectrometer. The hybrid hydrogels were sliced with a razor blade before being placed in a cuvette or on the measurement base. Confocal laser scanning microscopy (CLSM) of the hybrid hydrogels were performed on a Leica TCS SP5 microscope and on a Nikon A1R microscope. Samples were sliced with razor blades and sealed in Grace Bio-Labs silicone isolators with glass cover slips. The 633 nm laser was used for excitation, with emission window of 650 nm to 700 nm.
X-ray scattering patterns were obtained at the 5-ID-D beamline in the DND-CAT facility at Argonne National Laboratory, with the beam energy of 17 keV. Samples were sliced with a pair of razor blades placed with a 2 mm gap, and sealed in Grace Bio-Labs silicone isolators with Chemplex 7.5 μm Kapton windows before measurement. Air background, water background and parent hydrogel scattering patterns were obtained and used for background subtraction. The data analysis was performed with the Irena software package running on the Igor Pro software.42
Coarse-grain MD simulation
Coarse grained MD simulations were carried out using LAMMPS package at a constant effective pressure (P = 0), constant monomer number and a constant reduced temperature (T = 1.0).38 A detailed description is provided in the Methods section of the ESI.† Briefly, each simulation box contained 3 × 3 × 3 cubic hydrogel grids and a prescribed number of initially randomly distributed PMI molecules. Each monomer of the hydrogel matrix carries a positive charge (i.e., charge fraction is unity), whereas the head group for the PMI molecules bears an equal negative charge. Respective neutralizing counterions for the hydrogel and PMI molecules were added explicitly so that overall system is kept charge neutral. A bead-spring type of approach was used to model all molecular structures and counterions with the finite extensible nonlinear elastic bond potential and Lennard-Jones steric interactions. Electrostatic interactions were implemented through the Wolf scheme method with a cutoff distance of 6σ. The screening effect of the added salt was modeled by adding an exponential decay factor to the pair interaction potential between two charges. Periodic boundary conditions were applied in all three directions. The data production simulations were performed between 4 × 105 to 8 × 105 steps depending on the chain sizes and PMI concentrations, after initial relaxation steps. The simulation time step was 0.0025τ, where τ is the simulation time scale. For annealing procedures, the system temperature was increased to T = 4.0 (corresponding to the annealing temperature) at which PMI molecules form a nearly gaseous phase, and then decreased to T = 1.0 within 5 × 105 simulation steps. Snapshots and correlation functions were obtained via Visual Molecular Dynamics software package.Photocatalytic hydrogen production
For the control experiment with PMI/poly(diallyldimethylammonium chloride) supramolecular hydrogels, the samples for hydrogen production were produced according to a previously described procedure,5 except that 3 nmol of the molybdenum-sulfur catalyst Na2[Mo3S13] was used and the samples were placed in 2 dram clear vials sealed with PTFE/silicone septa. For the hybrid hydrogels, approximately 50–100 mg of the hybrid hydrogels were placed in 2 dram clear vials. To each vial, 176 μL of 17.8 μM Na2[Mo3S13] solution in water was added. The vials were sealed with PTFE/silicone septa, purged with argon and heated to 80 °C for 1 h. In a separate vial, 2 M ascorbic acid aqueous solution was balanced to pH 4 with 4 M sodium hydroxide aqueous solution unless otherwise noted. After the vials containing the hydrogels cooled to room temperature, 720 μL of the ascorbic acid solution was added to each vial. The vials were again purged with argon, and illuminated for 8 h with a Schott ACE 1 light source equipped with a broadband 21 V, 150 W EKE halogen bulb and fiber-optic goosenecks. After the illumination, the headspace gas was sampled from each vial for gas chromatography using a Shimadzu GC-2014 equipped with a 5 Å molecular-sieve column, argon carrier gas and a thermal conductivity detector. The H2 concentration in the sample headspace was determined by comparing the integrated peak area against an eight-point calibration curve created from injecting known volumes of 7% H2/93% N2 gas mixture. The turnover frequency (TOF) was determined by dividing the amount of H2 produced in moles by the amount of [Mo3S13]2− catalyst and by the illumination duration.Results and discussion
PMI/PE hybrid hydrogel synthesis
We synthesized the hybrid hydrogels consisting of PE network and PMI supramolecular assemblies via stepwise incorporation of PMI solution in 2,2,2-trifluoroethanol (TFE) into pre-synthesized 3-acrylamidopropyl-trimethylammonium chloride (APTAC) or APTAC-containing copolymer PE gels with acrylamide (AAm) or 2-acrylamido-2-methyl-1-propanesulfonic acid sodium salt (AMPS), followed by solvent exchange from organic solvents to water (Fig. 1). The choice of monomers aimed at investigating the effect of charge in the hydrogels as well as stability over a wide range of pH and in different solvents. The parent PE hydrogels were synthesized via conventional free-radical polymerization of the respective monomers in aqueous solution at 65 °C, using N,N-methylenebisacrylamide (MBAA) as the crosslinker. When fully hydrated, these parent hydrogels are optically transparent, and thus are suitable substrates to incorporate the photosensitizing chromophores assemblies (Fig. 1b, left). The stepwise incorporation process allowed the PMI molecules to penetrate the covalent network in monomeric form in TFE without any diffusion barrier, followed by induction of supramolecular self-assembly in the presence of the covalent PE network in water. PMIs were highly soluble in the fluorinated alcohols such as TFE and hexafluoroisopropanol up to 20 mg mL−1 and these solvents also had the ability to swell the APTAC-containing dried gels, thus allowing incorporation of the chromophores in the covalent network. Loading limits for the PE dried gels depend on the uptake capacity of the TFE solution. For example, the highest PMI loading in an APTAC gel with 2 mol% MBAA was thermogravimetrically determined to be 1.27 g PMI per gram of APTAC gel (0.3 mole fraction of PMI). Since AAm and AMPS homopolymers were both insoluble in TFE, the degree of swelling of the dried gel by TFE was correlated to the fraction of APTAC in the copolymer hydrogels (Fig. S1†). Other attempts to incorporate the PMI nanostructures into the PE network after the PMIs were unsuccessful, shedding light on the pathway-specific nature of this hybrid material formation (Fig. S2–S5, ESI discussion†).Structural characterization of the hybrid supramolecular-covalent networks
Due to the highly ionic nature of the hydrogel, conventional sample preparation techniques for SEM imaging such as freeze-drying and critical point drying proved unsuitable for observing network structures. As mentioned above, cryo-SEM of the high pressure-vitrified parent APTAC hydrogel showed randomly sized porosity, while the corresponding hybrid hydrogels showed ribbon-like structures forming across the covalent network (Fig. S4†). Both the parent hydrogels of varying compositions and the corresponding PMI/PE hybrid hydrogels were characterized with respect to composition, swelling ratio in water, and distribution of PMI molecules within the hydrogel. Fig. 2a shows the ratio of the weights of the hydrogels and the corresponding dried gels after being been fully swollen in DI water. The swelling ratio decreases with increasing PMI content within the hybrid gel. While the parent APTAC hydrogel with 2 mol% MBAA as the crosslinker swells 73-fold in DI water, the hybrid gel containing 30 mol% PMI in the parent hydrogel swells only 10-fold. In comparison, gels formed from a polymer containing 30 mol% negatively charged AMPS and 70 mol% positively charged APTAC swell 40-fold (Fig. S6†). This difference in swelling behavior suggests that the hydrogel swelling ratio is highly affected by the presence of PMI nanostructures within the PE network. This indicates that both ion condensation and hydrophobic effects from the PMI reduce the water uptake. The swelling equilibria of PE hydrogels have been extensively studied, and both empirical and theoretical scaling laws have been established correlating crosslinker density, hydrogel backbone charge density, ionic strength and the swelling ratio.39–43 Hydrogels synthesized using free-radical polymerization are known to contain structural and chemical heterogeneity due to the uncontrolled nature of polymerization kinetics, and therefore their swelling behavior is more complex than that predicted for theoretical models. Nevertheless, the following trends are clear: (1) a decrease in crosslinker concentration leads to an increase in swelling ratios, (2) an increase in content of the non-ionic AAm monomer leads to a decrease in swelling ratio in water, and (3) the ampholytic hydrogels consisting of APTAC and AMPS exhibit charge condensation and thus smaller swelling ratios as the APTAC/AMPS monomer ratios bearing opposite charge approach 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S6†). Finally, the AAm copolymer hydrogels are less prone to deswell upon solvent exchange to high ionic strength media44 and the ampholytic hydrogels can even swell through ion dissociation between opposite charges within the backbone.41 This relative insensitivity of swelling ratios to environmental ionic strength may lead to faster diffusion kinetics under conditions used for photocatalytic reactions.
:1 (Fig. S6†). Finally, the AAm copolymer hydrogels are less prone to deswell upon solvent exchange to high ionic strength media44 and the ampholytic hydrogels can even swell through ion dissociation between opposite charges within the backbone.41 This relative insensitivity of swelling ratios to environmental ionic strength may lead to faster diffusion kinetics under conditions used for photocatalytic reactions.
 | ||
| Fig. 2 Physicochemical properties of PMI/PE hybrid hydrogels. All hydrogels shown here are based on APTAC polyelectrolyte hydrogels with 2% crosslinker density. For other compositions, see ESI.† (a) Hydrogel swelling ratio as a function of PMI molar fraction in the hybrid gel; the swelling ratio is described as the weight of the hydrogels divided by the weight of the dried gels, while the PMI molar fraction is defined as the molar fraction of PMI in the hybrid dried gels. The red curve denotes an empirical power law fit to the data points, given as y = 1 + A(1 − 2x)B where A = 83 and B = 1.8. (b) UV-Vis absorption spectra of the PMI molecules in TFE (black), in water (red), and in the hybrid hydrogels in water (blue). (c–f) Confocal laser scanning microscopy images of hybrid gels at various stages of solvent processing: (c) in TFE, (d) in water, (e) in 150 mM NaClaq, and (f) after thermally annealing the hydrogel in 150 mM NaClaq. | ||
To understand how the PMI assemblies arrange within the PE hydrogel, we first used UV-Vis absorption spectroscopy to assess the extent of intermolecular excitonic coupling. Fig. 2b shows that while PMI molecules show a broad single peak at around 480–490 nm in fluorinated alcohols and in DI water, the PMI/APTAC hybrid hydrogels swollen in DI water reveal the two absorption peaks, one at 440 nm and one at 570 nm, which is consistent with our previous observations on PMI assemblies that acquire crystalline order and reveal absorption signatures arising from intermolecular excitonic coupling when electrostatically screened by ions.6,29 The large splitting of these two peaks correspond to the high degree of mixing between Frenkel excitation states and charge-transfer excitation states in the high dielectric aqueous environment, resulting in small exciton binding energies and thus efficient charge transfer from the site of exciton formation to reduction at the catalyst sites. The wavelength positions of this bimodal absorption are unchanged when the hybrid hydrogel is immersed in 150 mM sodium chloride aqueous solution (NaClaq) (Fig. S7†).
PMI molecules in TFE, as well as crystalline PMI assemblies in water, show a broad fluorescence centered around 650 nm, and thus can be imaged efficiently using confocal laser scanning microscopy (CLSM). Fig. 2c–f show the CLSM images of PMI/APTAC hybrid gels containing 2 mol% MBAA and ∼0.05 molar equivalence of PMI to APTAC at different stages of processing. Initially in TFE, the PMI signal appears uniformly distributed inside the organogels, with grain-like substructures (Fig. 2c). Once solvent exchange to water is performed, the hydrogel shows less fluorescence signals, with uniform distribution of the PMIs within the imaging resolution limit (Fig. 2d). Combined with the absorbance spectrum discussed above, we believe that the PMI molecules are locally assembled into ribbon-like small aggregates that have crystalline order. Treatment of the hybrid hydrogels with 150 mM NaClaq yields the formation of several micrometer length scale aggregates in the hybrid hydrogel, indicating the reorganization and growth of these chromophore amphiphiles into larger structures within the hydrogel (Fig. 2e). This change is sometimes coupled with macroscopically observed clouding of the hydrogel. When these hybrid hydrogels are thermally annealed in NaClaq at 95 °C, the aggregate size increases slightly (Fig. 2f). We note that these aggregates are comparable or larger than the mesh size of the parent hydrogels (see Fig. S5† for network size of the hydrogels and the ribbon size), thus supporting further the steric trapping mechanism discussed above.
To probe the crystallization of PMI assemblies in the presence of APTAC hydrogels, we carried out synchrotron X-ray scattering measurements. Fig. 3 shows the middle- to wide-angle (0.14 Å−1 < q < 4 Å−1) X-ray scattering profiles of the PMI/APTAC hydrogels containing 2 mol% MBAA and ∼0.05 molar equivalence of PMI to APTAC. The hybrid hydrogels placed under DI water exhibit three peaks at 0.74, 1.74 and 1.87 Å−1, consistent with previously reported Bragg reflections from ribbon-like two-dimensional crystalline PMI assemblies formed in water in the presence of salt.5 These peaks confirm that the APTAC polymer network provides sufficient ionic strength to induce crystallization within the entrapped PMI assemblies, estimated to be inversely proportional to the swelling ratios and on the order of 10 mM or higher in the absence of other ions. The presence of Bragg reflections is also consistent with the peak splitting observed in the absorption spectrum of the hybrid hydrogel. When this hydrogel was heated to 95 °C and cooled to room temperature, the Bragg peak intensity decreases slightly, thus indicating reduced crystallinity.
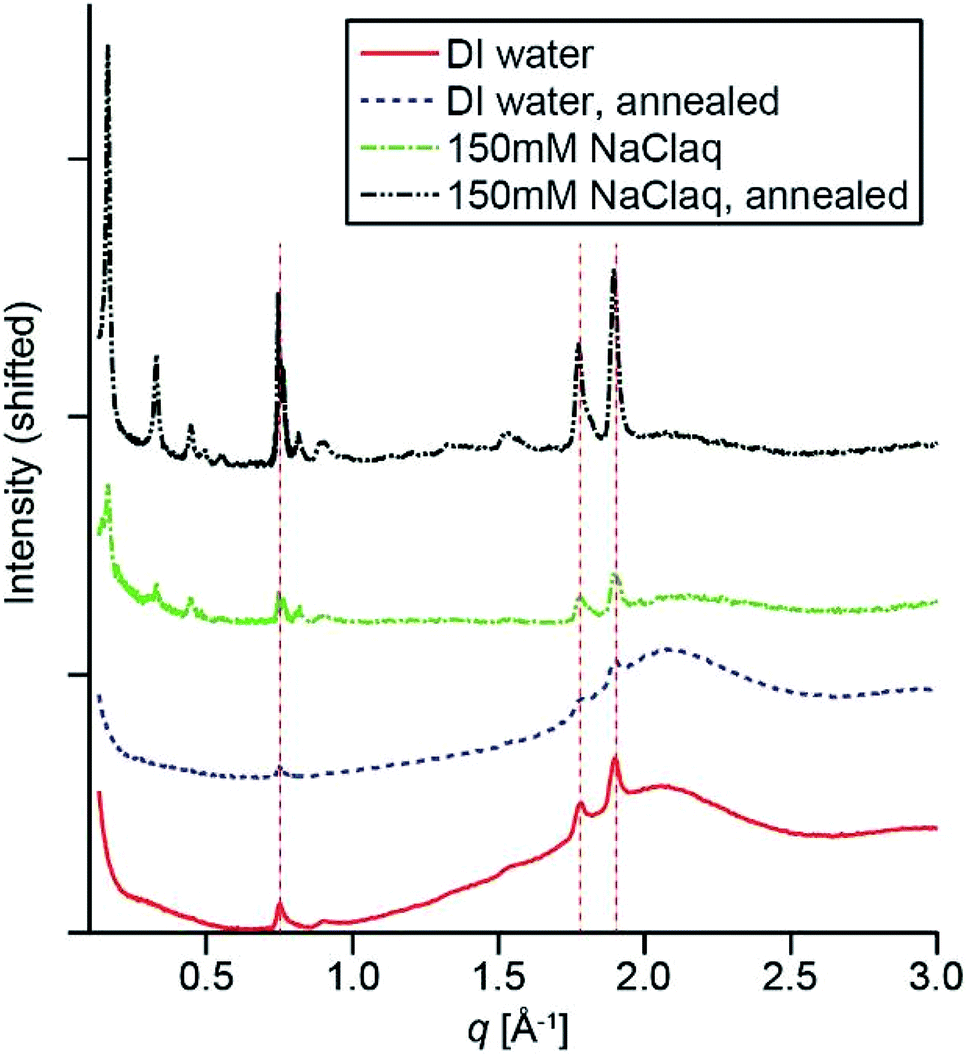 | ||
| Fig. 3 X-ray scattering patterns of PMI/APTAC hybrid hydrogels at various aqueous processing stages. Scattering intensities are in linear scale, and shifted vertically for clarity. The vertical dashed lines indicate the positions of the three Bragg peaks corresponding to the two-dimensional lattice of the PMI. | ||
When the hydrogel is placed in 150 mM aqueous NaCl solution, we observed a set of reflections appearing at 0.16, 0.33, 0.45, 0.50, 0.56 Å−1 in the middle-angle region, as well as a shoulder appearing next to the 0.74 Å−1 reflection. These peaks are indicative of the formation of three-dimensionally stacked crystallites, with a layer-to-layer distance of 37.7 Å, which is smaller than those observed for lyotropic ordering observed previously in PMI solutions at high concentration.5 Since the single layer thickness of these ribbons has been previously estimated to be ∼23 Å from atomic force microscopy,6 we hypothesize that these multilayered crystalline aggregates are expected to have hydrated gaps between the layers. When the hybrid hydrogels were dried under heat, this set of layered peaks shift to higher scattering angles, corresponding to a layer distance of 29.7 Å (Fig. S6†). Addition of water to the dried gel immediately changed the layer distance back to 37.7 Å, further supporting the hypothesis that water is incorporated between the layers in these aggregates. Upon heating this hybrid hydrogel to 95 °C and cooling, the Bragg peaks sharpen and their intensities increase, indicating higher PMI crystallinity consistent with the CLSM observation of larger ribbon-like structures (Fig. 2f). We observed similar assembly behavior for most of the copolymer hydrogels. Specifically, UV-Vis absorption spectra indicate the existence of intermolecular excitonic coupling upon solvent exchange to DI water, followed by an increase in crystallinity as observed in WAXS after solvent exchange to 150 mM NaClaq and thermal annealing (Fig. S8†).
Coarse-grain molecular dynamics simulation of the CA assembly in PEs
In order to shed light on the molecular details of self-assembly of PMI molecules within PE hydrogel networks, we performed coarse-grained molecular dynamics simulations. In our solvent-implicit simulations, we considered all the charges on the chains forming the gel network as well as those on the PMI molecules together with their neutralizing counterions. The idealized gel networks in these simulations have cubic topology provided by permanently fixed crosslinks.45 The molecular shape of the PMI molecules and related interaction parameters are coarse-grained from an atomistic model to account for hydrophobic and hydrophilic interactions (see ESI for simulation details†). We varied the degree of polymerization between two crosslinks as well as PMI concentration, and focused on their effects on structure in the hybrid hydrogels.We first constructed the coarse-grained PMI molecular model (Fig. 4a) and attempted to verify that this model represents supramolecular structures similar to the ribbon-like nanostructures observed experimentally in the absence of covalent polymer hydrogels.6 Simulating the self-assembly process of large molecular structures at low concentrations is computationally costly due to relatively long diffusion and healing times (i.e., on the orders of seconds), thus we instead monitored the stability of pre-assembled structures. PMI molecules were pre-assembled in a two-dimensional sheet packed in antiparallel conformation (i.e., tail to head) and thermalized. The PMI assemblies preserved the ribbon configurations over the course of our simulations over 100 ns in accordance with experimental observations (Fig. 4b), indeed proving that this coarse-grained model behaves consistently.
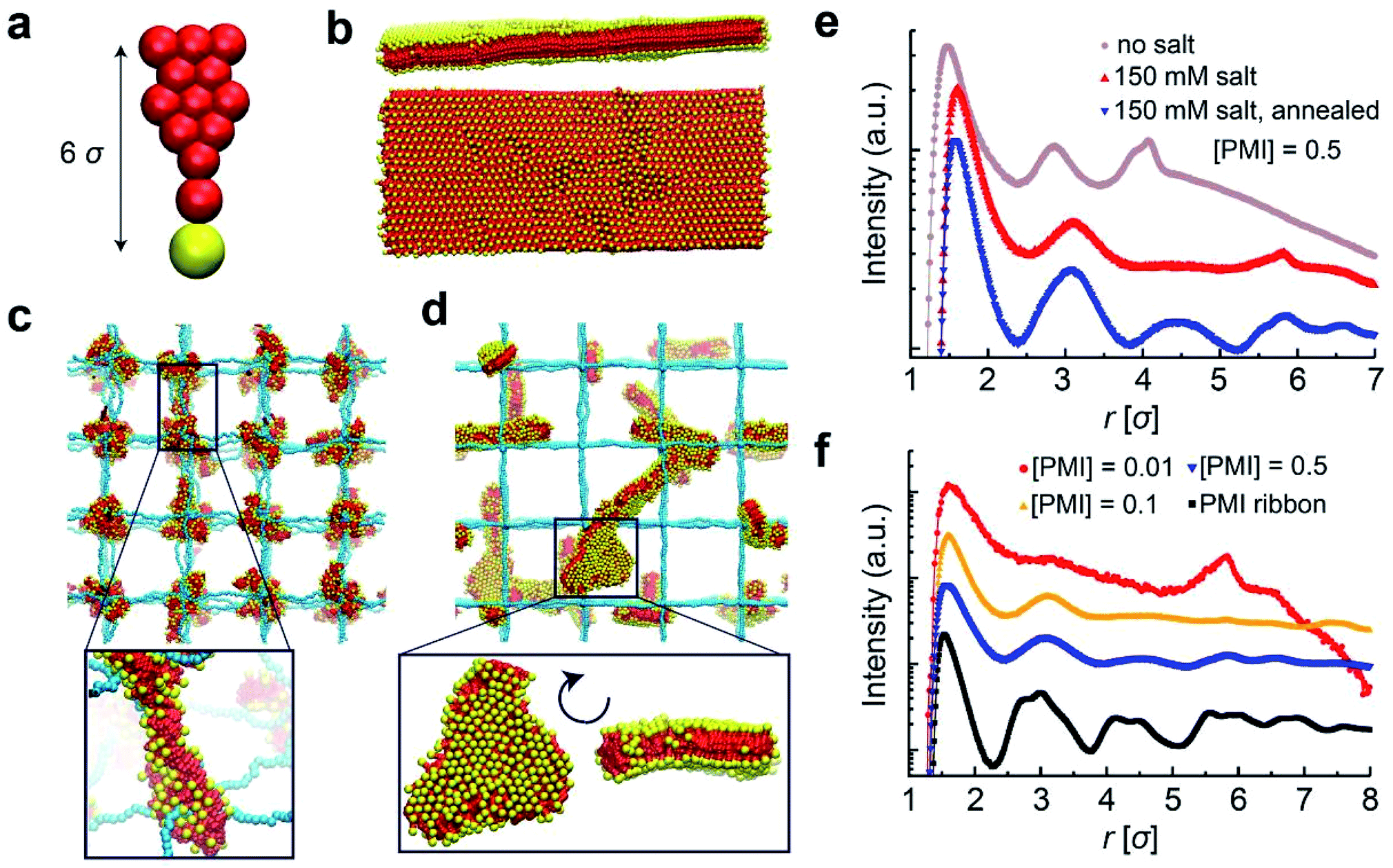 | ||
| Fig. 4 Coarse-grained MD simulations of PMI aggregation. (a) The coarse-grained PMI molecular model: all hydrophobic and charge neutral groups are rendered in red, whereas the charged group is represented by a yellow sphere; the counterions are not shown for clarity. (b) The side and top views of a simulated PMI ribbon with a thickness of 3.5σ in simulation units (1σ = 3.4 Å). The simulations are conducted with PMI molecules pre-assembled in an antiparallel packing configuration. (c) Hybrid hydrogel simulation snapshot for the degree of polymerization between crosslinks N = 30 and [PMI]/([PMI] + [APTAC]) = 0.01. An arbitrary node is shown in a zoomed in representation to highlight the nodal aggregate of chromophore amphiphiles near the covalent crosslinking points. (d) A zoomed out snapshot of the hybrid hydrogel simulation for [PMI]/([PMI] + [APTAC]) = 0.5. (e) Pair correlation functions (PCFs) between the charged head groups of PMI molecules in the ribbon configuration shown in (a) and hybrid hydrogels shown in (c) and (d). (f) PCFs between the hydrogel crosslinks and gel counterions, showing the PMI-driven depletion of the hydrogel counterions. | ||
Next, we incorporated PMI molecules at various concentrations into the PE hydrogel to monitor PMI self-assembly. In contrast to the gel-free PMI simulations, the PMI molecules were randomly distributed throughout the hydrogel at the beginning of each simulation. In the absence of added salt, the equilibrium configurations of the hybrid systems contain nodal aggregates accumulated at the crosslinks of the gel networks (Fig. 4c). While the electrostatic attraction between the oppositely charged hydrogel backbone and the head groups of the PMI molecules drives the aggregate formation, the preferred aggregation near the crosslinks interestingly highlights the higher local charge density near the nodal points of the network.46,47
To understand the effect of monovalent salt, we performed the same simulations with weaker electrostatic interactions between charged groups using a screened potential corresponding to an ionic strength of 150 mM NaClaq. Salt addition does not change the formation of the nodal aggregates in the simulation, but the PMI molecules are found to pack less tightly in the aggregates. Keeping the salt conditions at 150 mM, we then simulated the thermal annealing process in which we raised and then gradually lowered the temperature. The thermal annealing significantly changed the aggregate morphology inside the gels; the nodal aggregates disappear and ribbon-like structures form, in accordance with experimental observations (Fig. 4d). We believe the high electrostatic screening environment reduces the PMI–hydrogel interaction, thus rendering the PMI species more mobile and allowing them to probe larger volumes upon heating, leading to the growth of the self-assembled structures. Importantly, at our highest PMI concentration, some ribbon-like structures grow to dimensions larger than those of polymer chains connecting two nodes.
The effect of thermal annealing and salt on the formation of 2D aggregates in the gels also manifests itself in the pair correlation functions (PCFs). We calculated the PCFs for the center of masses of the PMI head groups (yellow beads in Fig. 4a) by using simulation trajectories. The PCFs in Fig. 4e demonstrates that hydrogel-incorporated PMIs in the salt-free environment have correlation distances that are smaller than those for PMIs in 150 mM NaClaq and that annealed hybrid systems show stronger correlation peaks at longer length scales r > 9 Å, or 3σ where σ is the simulation unit length. These correlation peaks and their ratios correspond to a 2D crystalline unit cell formed within PMI self-assemblies under charge-screening conditions.6 The PCF analysis confirms that the local crystallinity of PMI self-assembly is retained within the hydrogel network. The possibility that the annealing procedure leads to ribbon-like aggregates also depends on the PMI concentration incorporated in the hydrogels. Interestingly, for PMI concentrations around 1 mM, the simulations reveal an uneven distribution of the PMI aggregates among the hydrogel nodes, regardless of the salt condition or annealing procedure. We observe this behavior for various molecular weights of hydrogel chains. As the PMI concentration is increased, the size of aggregates near the nodes becomes larger and merges to form ribbon-like aggregates upon annealing (Fig. 4f).
These simulation results are consistent with experimental systems in terms of stabilization of the chromophore amphiphiles inside the covalent network and provide further molecular scale details on the PMI assembly process. The simulations indicate that in salt-free systems, PMI clusters start growing around hydrogel nodes. Upon annealing in salt solution, the nodal aggregates disassemble and reassemble into 2D crystalline structures throughout the 3D polymer network. Both simulations and experiment confirm that temperature annealing and screened electrostatic interactions are crucial to avoid kinetic traps to reach the crystalline structures. In the simulations in annealed systems with salt we observe at higher PMI concentrations large ribbon-like aggregates (Fig. 4d–f).
Photocatalytic hydrogen production
One of the key functions of the chromophore amphiphile assemblies is their ability to photosensitize and transfer electrons to appropriately matched catalysts.6 The resulting hybrid hydrogels of PMI/APTAC were tested for their photocatalytic proton reduction performance. Upon illumination of hybrid hydrogels loaded with a thiomolybdate catalyst, Na2[Mo3S13],30,32,37 in the ascorbic acid solution adjusted to pH 4, we observed H2 production as quantified by gas chromatography (see ESI and Fig. S9† for details on catalyst loading). We initially selected these reaction conditions based on previous photocatalysis studies from our group6,8,30–32 to protect the supramolecular and covalent components of the hybrid hydrogels from hydrolysis at extreme pH, and to prevent possible dissolution of the supramolecular assemblies at high pH. While photo- and electro-catalytic hydrogen production reactions are typically performed below pH 1 or in mixtures with organic solvents48,49 we found that neutralizing the ascorbic acid with base to pH 4 resulted in almost five-fold increase in hydrogen produced (see ESI, Fig. S10†), indicating that the moderately acidic pH is preferred for photocatalysis reactions in our case. We suspect that the combination of close energy level matching of the chromophore and catalyst in the fully aqueous system and the availability of protons contribute to the pH dependence.50,51Fig. 5 shows the effect of various parameters such as covalent crosslink density, PMI content and comonomer compositions on the photocatalytic hydrogen production performance. We observed a large variance in the photocatalytic hydrogen production values, which we believe arises from the inhomogeneous illumination of the macroscopic hydrogels as well as the variations in hydrogel shapes. Lowering the covalent crosslinker (MBAA) concentration from 2% to 0.5% in PMI/APTAC hydrogels resulted in increase of the turnover frequency (TOF) of the hydrogen production, defined as the moles of H2 molecules produced per moles of catalyst per hour (Fig. 5a). Interestingly, when more PMI was loaded on the same parent hydrogel, the TOF decreased (Fig. 5b). While incorporation of the non-charged AAm monomer in the parent PE hydrogels led to lower TOFs for hybrid hydrogels with MBAA content of 0.5 mol%, incorporating 10 mol% of the anionic AMPS led to an average turnover frequency of 107.4 h−1 (Fig. 5c), approaching that of the PMI/PDDA supramolecular hydrogels as shown in Fig. 5a. From these three sets of data we hypothesize that the degree of swelling in the 4 M ascorbic acid solution may be a dominating factor for photocatalytic hydrogen production kinetics. A reduction in covalent crosslink density and in the supramolecular network density are both contributing factors to increased swelling of the hybrid hydrogels and faster diffusion of dissolved species, which can accelerate the photocatalytic hydrogen production. The results indicate that structural parameters such as porosity create a trade-off between mechanical integrity and catalytic performance.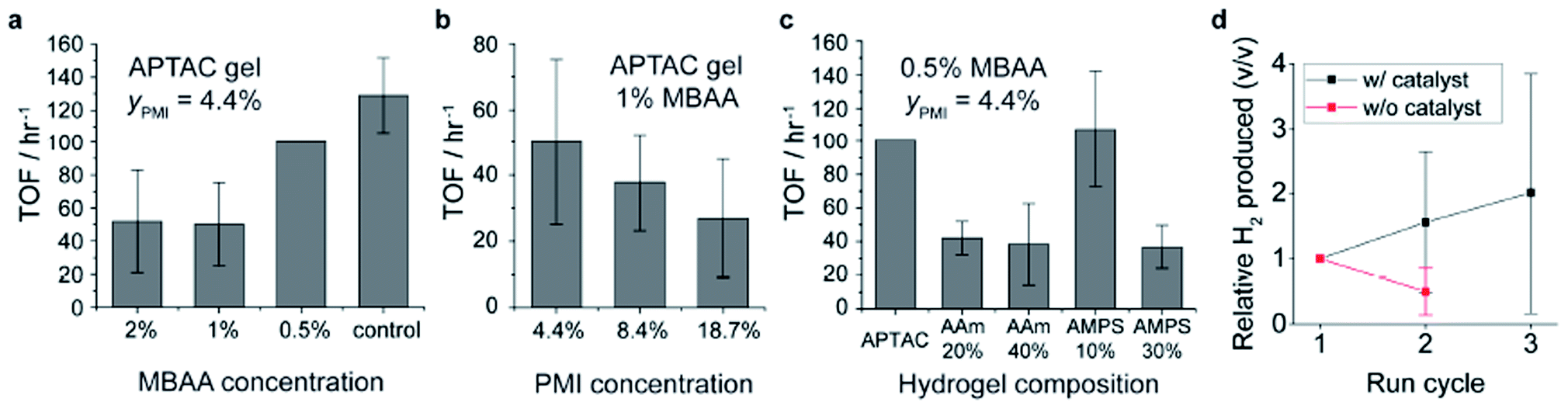 | ||
| Fig. 5 Photocatalytic hydrogen production performance for different hydrogel compositions, where the turnover frequency (TOF) is defined as (moles of hydrogen produced)/(moles of catalyst) per hour of illumination. (a) Effect of MBAA crosslinking agent concentration in the parent hydrogels in which the PE network is composed of APTAC monomers. The control experiment represents a physically crosslinked hydrogels of PMI with PDDA. (b) Effect of PMI molar concentration in the hybrid hydrogel from the same parent hydrogel (the 4.4% data point duplicates the 1% point in panel (a)). (c) Effect of AAm and AMPS comonomer molar concentration in the parent hydrogel (the 0% data point duplicates the second data point from the left in panel (a), and the APTAC data point duplicates the 0.5% data point in panel (a)). (d) Amount of hydrogen produced from subsequent rounds of illumination on the reused hybrid hydrogels normalized by the amount of hydrogen from the first round. Red points indicate test results in which no additional catalyst was incorporated; black line shows test results in which the catalyst was added before each test. | ||
An important feature of the hybrid hydrogels is the possibility to reuse them in multiple cycles of photocatalysis. After each cycle all that is required is to simply wash the gel in water to remove byproducts and replenish as necessary the catalyst and sacrificial electron donor. During this process, the entrapped supramolecular scaffold that serves as the light harvesting structure is retained and reused. To investigate the reusability of these hybrid hydrogels, we subjected the samples that were illuminated to DI water washing, and then carried out another round of photocatalytic H2 production (Fig. 5d). We note that the supramolecular hydrogels created from PMI and PDDA without the covalent hydrogel backbone cannot be handled without fracturing into fine suspensions, and thus are unsuitable for reuse. In contrast the hybrid hydrogels largely retained their shape and structure throughout the degassing, illumination and washing processes, likely aided by the deswelling of the hydrogel in the highly charge screened condition that relaxes chain stretching and renders the hydrogel less brittle.42 When the Na2[Mo3S13] catalyst was not added for the second round of tests, the hydrogels produced about half of the moles of H2 compared to the first round. This result indicates that the soluble, non-anchored thiomolybdate catalyst may be partially leaching out of the hybrid hydrogels or becoming inactive during the washing process. On the other hand, when the hydrogels were treated with another round of catalyst incorporation prior to the second round of the photocatalysis, the amount of H2 produced was 1.6 times higher than the original tests. Similarly, in the third round of tests the amount of H2 produced was 2.0 times relative to the original test. These results indicate that the hybrid hydrogels are not only reusable photosensitizers but can partially retain the catalysts in the hydrogels throughout the washing process. Future studies for sustained photocatalytic performance of these materials may include covalent linking of the catalyst clusters to PMIs52 and designing pore structures to further facilitate mass transport in the hydrogels while retaining the mechanical integrity and reusability.
Conclusions
We have demonstrated the possibility of entrapping supramolecular light harvesting assemblies for photocatalytic reactions within crosslinked polyelectrolytic hydrogels over very long time scales. This entrapment is possible by triggering self-assembly of chromophore amphiphile monomers through organic to water solvent exchange within the covalent network. The retention mechanism of the crystalline chromophore amphiphile assemblies in an aqueous environment results from both electrostatic interactions and physical entrapment in the mesh size of the network. Interestingly, coarse grained molecular dynamics simulations suggest that chromophore assemblies are nucleated at the nodes of the three-dimensional covalent network. Proof-of-principle experiments on photocatalysis of hydrogen production showed that optimized systems could be reused in multiple cycles of photocatalysis after refreshing the system for removal and addition of small molecule components. The trapping mechanism strategy of light harvesting assemblies would lend itself to compartmentalization of different chemical reactions in neighboring hydrogels with chromophore amphiphiles of different energy levels for multistep synthesis, as well as possibilities for using stimuli-responsive hydrogels as the covalent network to control the photocatalytic reactions.Author contributions
H. S., A. S. W., and S. I. S. conceived the experiment. H. S., A. J. D., A. S. W., E. S., K. J., and K. Q. synthesized the materials and performed measurements. A. E. and D. H. developed the coarse grain model and performed the simulations. L. C. P., M. O. d. l. C. and S. I. S. supervised the project. All authors participated in the writing and the revision of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was primarily supported by the Center for Bio-Inspired Energy Science, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES) under Award # DE-SC0000989. Molecular synthesis was supported by the National Science Foundation under NSF Award Number DMR-1121262. The authors thank the Sherman Fairchild Foundation for computational support. E. S. acknowledges the Materials Research Science and Engineering Center REU program, supported by the National Science Foundation under NSF Award Number DMR-1121262. The authors also thank Prof. Michael Wasielewski (Northwestern University) for access to the gas chromatography instrument, Zaida Alvarez-Pinto (Northwestern University) for assistance with optical imaging of the hydrogels, and Garrett Lau (Northwestern University) for assistance in acquiring thermogravimetric analysis data. This work made use of the following facilities at Northwestern University: the J. B. Cohen X-Ray Diffraction Facility, the EPIC facility (cryo-SEM), the Keck Biophysics facility (UV-Vis), the Biological Imaging Facility (CLSM), the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS) (X-ray scattering), and the IMSERC facility (TGA). The J. B. Cohen X-Ray Diffraction Facility is supported by the MRSEC program of the National Science Foundation (DMR-1121262) at the Materials Research Center of Northwestern University. The EPIC facility of the NUANCE Center has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the MRSEC program (NSF DMR-1121262) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN. The Keck Biophysics facility has received support from a Cancer Center Support Grant (NCI CA060553). DND-CAT is supported by Northwestern University, E. I. DuPont de Nemours & Co., and The Dow Chemical Company. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. IMSERC facility has received support from the NSF (CHE-1048773); Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the State of Illinois and International Institute for Nanotechnology (IIN).References
- A. Kay, I. Cesar and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 15714–15721 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS.
- S. Styring, Faraday Discuss., 2012, 155, 357–376 RSC.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695–698 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2008, 8, 76 CrossRef.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, D. J. Fairfield, A. R. Koltonow and S. I. Stupp, J. Am. Chem. Soc., 2015, 137, 15241–15246 CrossRef CAS.
- V. S. Vyas, V. W.-h. Lau and B. V. Lotsch, Chem. Mater., 2016, 28, 5191–5204 CrossRef CAS.
- R. V. Kazantsev, A. J. Dannenhoffer, A. S. Weingarten, B. T. Phelan, B. Harutyunyan, T. Aytun, A. Narayanan, D. J. Fairfield, J. Boekhoven, H. Sai, A. Senesi, P. I. O'Dogherty, L. C. Palmer, M. J. Bedzyk, M. R. Wasielewski and S. I. Stupp, J. Am. Chem. Soc., 2017, 139, 6120–6127 CrossRef CAS PubMed.
- J. Byun, K. Landfester and K. A. I. Zhang, Chem. Mater., 2019, 31, 3381–3387 CrossRef CAS.
- R. Hill and F. A. Y. Bendall, Nature, 1960, 186, 136 CrossRef CAS.
- P. Jordan, P. Fromme, H. T. Witt, O. Klukas, W. Saenger and N. Krauß, Nature, 2001, 411, 909 CrossRef CAS.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS.
- R. Danielsson, P.-Å. Albertsson, F. Mamedov and S. Styring, Biochim. Biophys. Acta, Bioenerg., 2004, 1608, 53–61 CrossRef CAS.
- A. V. Ruban and M. P. Johnson, Nat. Plants, 2015, 1, 15161 CrossRef CAS.
- P. G. Schouten, J. M. Warman, M. P. de Haas, M. Anne Fox and H.-L. Pan, Nature, 1991, 353, 736 CrossRef CAS.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- D. Liu, J. Wang, X. Bai, R. Zong and Y. Zhu, Adv. Mater., 2016, 28, 7284–7290 CrossRef CAS.
- A. Krieger, J. P. Fuenzalida Werner, G. Mariani and F. Gröhn, Macromolecules, 2017, 50, 3464–3475 CrossRef CAS.
- E. Cohen, H. Weissman, I. Pinkas, E. Shimoni, P. Rehak, P. Král and B. Rybtchinski, ACS Nano, 2018, 12, 317–326 CrossRef CAS.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492 CrossRef CAS.
- F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. Olvera de la Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469 CrossRef CAS.
- W. Zhang, W. Jin, T. Fukushima, A. Saeki, S. Seki and T. Aida, Science, 2011, 334, 340–343 CrossRef CAS.
- A. T. Haedler, K. Kreger, A. Issac, B. Wittmann, M. Kivala, N. Hammer, J. Köhler, H.-W. Schmidt and R. Hildner, Nature, 2015, 523, 196 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977 CrossRef CAS.
- M. Nakahata, S. Mori, Y. Takashima, H. Yamaguchi and A. Harada, Chem, 2016, 1, 766–775 CAS.
- B. Harutyunyan, A. Dannenhoffer, S. Kewalramani, T. Aytun, D. J. Fairfield, S. I. Stupp and M. J. Bedzyk, J. Phys. Chem. C, 2017, 121, 1047–1054 CrossRef CAS.
- N. J. Hestand, R. V. Kazantsev, A. S. Weingarten, L. C. Palmer, S. I. Stupp and F. C. Spano, J. Am. Chem. Soc., 2016, 138, 11762–11774 CrossRef CAS.
- A. S. Weingarten, A. J. Dannenhoffer, R. V. Kazantsev, H. Sai, D. Huang and S. I. Stupp, J. Am. Chem. Soc., 2018, 140, 4965–4968 CrossRef CAS PubMed.
- A. Dannenhoffer, H. Sai, D. Huang, B. Nagasing, B. Harutyunyan, D. J. Fairfield, T. Aytun, S. M. Chin, M. J. Bedzyk, M. Olvera de la Cruz and S. I. Stupp, Chem. Sci., 2019, 10, 5779–5786 RSC.
- R. V. Kazantsev, A. J. Dannenhoffer, T. Aytun, B. Harutyunyan, D. J. Fairfield, M. J. Bedzyk and S. I. Stupp, Chem, 2018, 4, 1596–1608 CAS.
- G. R. J. Müller, C. Meiners, V. Enkelmann, Y. Geerts and K. Müllen, J. Mater. Chem., 1998, 8, 61–64 RSC.
- J. A. A. W. Elemans, R. van Hameren, R. J. M. Nolte and A. E. Rowan, Adv. Mater., 2006, 18, 1251–1266 CrossRef CAS.
- A. Margineanu, J. Hotta, M. Van der Auweraer, M. Ameloot, A. Stefan, D. Beljonne, Y. Engelborghs, A. Herrmann, K. Müllen, F. C. De Schryver and J. Hofkens, Biophys. J., 2007, 93, 2877–2891 CrossRef CAS PubMed.
- A. Müller, V. Wittneben, E. Krickemeyer, H. Bögge and M. Lemke, Z. Anorg. Allg. Chem., 1991, 605, 175–188 CrossRef.
- V. P. Fedin, J. Czyzniewska, R. Prins and T. Weber, Appl. Catal., A, 2001, 213, 123–132 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- Z. Tong and X. Liu, Macromolecules, 1994, 27, 844–848 CrossRef CAS.
- X. Liu, Z. Tong and O. Hu, Macromolecules, 1995, 28, 3813–3817 CrossRef CAS.
- A. E. English, S. Mafé, J. A. Manzanares, X. Yu, A. Y. Grosberg and T. Tanaka, J. Chem. Phys., 1996, 104, 8713–8720 CrossRef CAS.
- M. Rubinstein, R. H. Colby, A. V. Dobrynin and J.-F. Joanny, Macromolecules, 1996, 29, 398–406 CrossRef CAS.
- D. Melekaslan and O. Okay, Polymer, 2000, 41, 5737–5747 CrossRef CAS.
- S. Durmaz and O. Okay, Polymer, 2000, 41, 3693–3704 CrossRef CAS.
- A. Erbas and M. Olvera de la Cruz, ACS Macro Lett., 2015, 4, 857–861 CrossRef CAS.
- H. H. Li, A. Erbas, J. Zwanikken and M. Olvera de la Cruz, Macromolecules, 2016, 49, 9239–9246 CrossRef CAS.
- T. Miyata, A. Endo, T. Ohmori, M. Nakaiwa, M. Kendo, K. Kurumada and M. Tanigaki, J. Chem. Eng. Jpn., 2002, 35, 640–648 CrossRef CAS.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248 CrossRef CAS PubMed.
- M.-L. Grutza, A. Rajagopal, C. Streb and P. Kurz, Sustainable Energy Fuels, 2018, 2, 1893–1904 RSC.
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321–1324 CrossRef CAS.
- M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2014, 136, 356–366 CrossRef CAS.
- Z. Ji, C. Trickett, X. Pei and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 13618–13622 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta08974h |
| ‡ Present affiliation: UNAM-National Nanotechnology Research Center and Institute of Materials Science & Nanotechnology, Bilkent University, Ankara 06800, Turkey. |
| § Present affiliation: School of Dentistry, University of Michigan, Ann Arbor, MI 48109, USA. |
| This journal is © The Royal Society of Chemistry 2020 |