
A vanadium–nickel oxynitride layer for enhanced electrocatalytic nitrogen fixation in neutral media†
Bin
Chang
a,
Lequan
Deng
a,
Shouzhi
Wang
a,
Dong
Shi
a,
Zizheng
Ai
a,
Hehe
Jiang
a,
Yongliang
Shao
 a,
Lei
Zhang
a,
Jianxing
Shen
b,
Yongzhong
Wu
*a and
Xiaopeng
Hao
*a
a,
Lei
Zhang
a,
Jianxing
Shen
b,
Yongzhong
Wu
*a and
Xiaopeng
Hao
*a
aState Key Laboratory of Crystal Materials, Shandong University, Jinan, 250100, China. E-mail: xphao@sdu.edu.cn; wuyz@sdu.edu.cn
bDepartment of Materials Science and Engineering, Qilu University of Technology, Jinan, 250353, China
First published on 3rd December 2019
Abstract
The application of transition-metal oxides (TMOs) in electrocatalytic nitrogen fixation still is hindered by their sluggish reaction kinetics and weak stabilization. In this work, a vanadium–nickel oxynitride (VNiON) layer is designed and synthesized on the corresponding oxide nanosheets to solve the above crucial issues. The first-principles kinetics analyses theoretically prove that the delocalized electron environment of VNiON enhanced π backdonation, which is conducive to nitrogen absorption and activation. Experimentally, both the ammonia production rate (∼6.78 μg h−1 cmcat.−2) and faradaic efficiency (∼5.57%) of VNiON are enhanced by 2-fold relative to those of its corresponding oxide under neutral conditions. Meanwhile, the stability of oxide is enormously improved by introducing a VNiON layer. The mechanism of improving the nitrogen fixation performance of oxides is investigated. This work provides a novel strategy of constructing oxides with advantageous structures for extensive electrochemical applications.
Artificial nitrogen fixation provides an effective route to synthesize the vital nitrogen building block ammonia (NH3), which plays a key role in the manufacture of fertilizers, explosives, plastics, pharmaceuticals and so on.1–4 However, due to the strong triple-bond (N
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, bond energy of 940.95 kJ mol−1), it is difficult to mildly reduce N2 to NH3.5–7 Tremendous efforts have been devoted to ammonia synthesis including biosynthesis, plasma-induced strategies, photocatalysis and electrocatalysis.8–11 Among all the appealing routes, the electrocatalytic nitrogen reduction reaction (NRR) has been emerging as an environmentally and sustainably benign process for NH3 production.12–16 More importantly, the NRR system can be easily powered by renewable electricity (e.g. solar and wind).17,18 Even though various materials exhibit NRR activity in aqueous systems, the chemical inertness of N2 (the sluggish reaction kinetics) and the competition with the hydrogen evolution reaction (HER) hinder the NRR performance.19–25
N, bond energy of 940.95 kJ mol−1), it is difficult to mildly reduce N2 to NH3.5–7 Tremendous efforts have been devoted to ammonia synthesis including biosynthesis, plasma-induced strategies, photocatalysis and electrocatalysis.8–11 Among all the appealing routes, the electrocatalytic nitrogen reduction reaction (NRR) has been emerging as an environmentally and sustainably benign process for NH3 production.12–16 More importantly, the NRR system can be easily powered by renewable electricity (e.g. solar and wind).17,18 Even though various materials exhibit NRR activity in aqueous systems, the chemical inertness of N2 (the sluggish reaction kinetics) and the competition with the hydrogen evolution reaction (HER) hinder the NRR performance.19–25
Heretofore, transition-metal oxides (TMOs) were generally acknowledged as NRR active materials owing to their inappropriate hydrogen adsorption energy.26–31 However, the electron-rich surface of pure TMOs limits the N2 adsorption and their poor conductivity increases energy consumption. These issues severely hinder the NRR application of TMOs. In view of this, the research emphasis shifts to other transition-metal-based materials.32–35 Recent theoretical calculations and experiments both suggest that transition-metal nitrides (TMNs) are promising NRR electrocatalysts via a Mars–van Krevelen mechanism under ambient conditions.36–38 TMNs possess excellent conductivity and stronger chemical stability characteristics than the corresponding TMOs, but TMNs are prone to cause HER side reactions. All of the above issues may be settled by use of a novel composite through combining the advantages of TMOs and TMNs.
In our previous work, transition-metal oxynitride (TMON) layers have been proven to exhibit improved electrochemical performances.39 When nitride anions with −3 charge replace the oxygen anions with −2 charge, the TMONs will possess additional metal cation valence states,40 which is conducive to absorbing the lone-pair electrons of nitrogen. Meanwhile, the atomic/ionic radiuses of single nitrogen and oxygen elements are approximate. Considering Pauling's second rule, TMON layers can effectively reduce the superposition lattice distortion effect and weaken the negative influences of structural stability.39,41 Therefore, a TMON layer on its corresponding TMO was designed to simultaneously improve NRR activity and stability. In addition to the HER inert of TMOs and the excellent conductivity and structural stability of TMNs, the synthesized TMON layer also possesses outstanding nitrogen adsorption ability.
Herein, a novel vanadium–nickel oxynitride (VNiON) layer covered on the corresponding oxide nanosheets is designed and synthesized to enhance the electrochemical NRR under ambient conditions (0.05 M Na2SO4). The ammonia formation rate (6.78 μg h−1 cmcat.−2) and faradaic efficiency (FE) (5.57%) of VNiON are twice that of vanadium–nickel oxide (VNiO) and quadruple of nitride (VNiN). Moreover, the NRR stability of VNiON is superior to that of VNiO. Both theoretical and experimental results show that oxides with advantageous structures (oxynitride layer) have advanced electrochemical applications.
First-principles DFT calculations were first performed to verify the feasibility of the NRR experiments of oxynitride layers. Vanadium–nickel based materials were investigated to fabricate metal oxynitride layers (Fig. S1, ESI†). After the N atoms partially replaced the O atoms, the electron environment clearly delocalized (Fig. 1a). This delocalized environment enhances the π backdonation from the metal to the π* orbitals of N2, which enhances N2 absorption, strengthens the metal (M)–N2 interaction and reduces the tendency for N2 to dissociate.42,43 The introduction of more nitrogen elements not only changed the basic structure and electron distribution of the material, but also made it easier to adsorb reactive hydrogen or H2O, which subsequently promotes HER side reactions (Fig. 1a). Furthermore, in the plots of the partial density of states (PDOS), the effective density of states is slightly delocalized when partial O–M–O bondings are replaced by O–M–N bondings (Fig. 1b and S2–S5, ESI†). The introduced N 2p states exhibit a broad DOS peak at the Fermi surface and some new electronic state energy near the Fermi level (cyan shaded area in Fig. 1b), which directly enhances the electronic conductivity of VNiON materials.39,44 This result is also consistent with previous results.39 The nitrogen contents in the structure are further increased. As shown in Fig. 1b, the DOS of VNiN(O) displays slightly increased charge density around the Fermi level with respect to that of VNiON, which can provide more charge carriers. And the DOS distribution of the VNiN(O) structure is more delocalized near the Fermi surface, which is beneficial for improving the conductivity. However, good conductivity does not necessarily mean excellent catalytic activity. The good conductivity and the outstanding H* adsorption ability of nitrides will directly promote the occurrence of HER side reactions.45–47 As shown in Fig. 1c, the N2 adsorption ability of VNiON is superior to that of VNiO. Considering the reaction mechanism of electrocatalytic hydrogen production and nitrogen fixation, the adsorption energy of N2, H and H2O on VNiON was further investigated according to the as-built models. VNiON had a much smaller ΔG value of H adsorption (ΔGH) than N2 adsorption (ΔGN2). However, ΔGN2 was smaller than ΔGH2O. The above phenomenon indicates that the VNiON material has great potential application in nitrogen fixation in neutral solution.
 | ||
| Fig. 1 (a) Charge density differences of the pristine NiO, VNiO, VNiON and VNiN(O). The yellow and blue isosurfaces denote electron gain and loss, respectively. The white, purple, red and blue spheres represent nickel, vanadium, oxygen and nitrogen, respectively. (b) DFT calculated density of states (DOS) of pristine NiO, VNiO, VNiON and VNiN(O). The Fermi levels were set to zero. (c) The adsorption energy of different reaction groups on VNiO and VNiON. | ||
Based on the above theoretical basis, VNiON was successfully designed and the fabrication process is illustrated in Fig. 2a. The VNiO nanosheets with the VNiON layer on carbon fibres were obtained after NH3 annealing. The morphology characterization results demonstrate that the two-dimensional (2D) nanosheet structure is well preserved, which confirms the good structural stability (Fig. 2b, c and S6, ESI†). The grain size shrinkage during calcination induced the granulation inside nanosheets.39 The corresponding high-resolution TEM (HRTEM) image clearly illustrates that the nanosheets are covered by an amorphous shell (approximately 9 nm) (Fig. 2d). The inside of the shell preserves the highly crystalline nature of NiO with clearly identified lattice fringe spaces of 0.241 nm and 0.209 nm corresponding to the (101) and (012) planes of hexagonal NiO, respectively. The corresponding EDS mapping images show the uniform distribution of V, Ni, O and N (Fig. 2e, S7 and Table S1, ESI†). Characteristic XRD peaks of VNiON are close to those in the corresponding oxide and nitride samples (Fig. 2f). Furthermore, the M–O–N peaks located at ∼820 cm−1 appear between the peaks of M–O and M–N in FTIR spectra (Fig. S8, ESI†). The above elemental compositions and structure analysis provide direct evidence for the successful formation of metal oxynitride (VNiON). Meanwhile, Brunauer–Emmett–Teller (BET) gas absorptiometry measurements were conducted to examine the porous nature of VNiO, VNiN and VNiON samples. Nitrogen adsorption–desorption isotherms and pore size distribution of the structures are shown in Fig. S8.† The specific surface areas are 75.4, 63.8 and 56.4 m2 g−1, corresponding to VNiON, VNiN and VNiO, respectively. The isotherm of VNiON shows a slightly narrow range of H3-type hysteresis loops in the range P/P0 = 0.45–1.0, which indicates that the mesopore has a wide pore diameter distribution (2–30 nm). This is consistent with the pore size distribution. The granular 2D morphology of VNiON nanosheets also presents a high specific surface area that enables the catalyst to contact with more nitrogen in the electrolyte (Fig. S9, ESI†).
 | ||
| Fig. 2 Schematic illustration of the formation process of VNiON (a). SEM images of VNiON under high and low magnifications (inset image) (b). TEM (c), high resolution TEM (d) and EDS elemental mappings (e) of VNiON. XRD patterns of VNiON and control samples (f). | ||
The surface chemical composition and bonding configuration of VNiON have been further investigated. The survey XPS spectrum of VNiON reveals that the material is composed of V, Ni, O and N,39 which is consistent with the EDS results (Fig. S10, ESI†). Compared to the control oxide and nitride, two additional peaks, located at ∼523.5 eV and ∼515.5 eV in the V 2p region, correspond to VNiON oxynitride layers (Fig. 3a). Meanwhile, Ni–O–N peaks also exist in the Ni 2p high-resolution XPS spectra (Fig. 3b). The absence of M–O–N peaks in nitrides may be caused by the minimal content of the corresponding oxynitrides after adequate nitridation. Furthermore, several additional peaks at ∼529.7 eV, ∼531.6 eV and ∼395.5 eV, ∼397.2 eV corresponding to Ni–O–N and V–O–N bondings appear in O 1s and N 1s spectra of VNiON (Fig. 3c and d). These observations effectively confirm the successful synthesis of VNiON oxynitride layers. The replacement of oxygen anions in O–M–O bonding with nitride anions in M–O–N bonding has induced additional metal cation valence states,39,48 which promote the lone-pair electrons of nitrogen absorption, and these effects, in turn, further enhance NRR ability.
 | ||
| Fig. 3 XPS spectra of VNiO, VNiN and VNiON samples: V 2p (a), Ni 2p (b), O 1s (c) and N 1s (d). | ||
To shed light on the superiority of oxynitrides, the electrochemical NRR performances were investigated in a 0.05 M Na2SO4 solution using an ultraviolet spectrophotometer. Linear-sweep voltammetric (LSV) curves were first explored in both Ar and N2 saturated electrolytes. As shown in Fig. S11,† when the potential is more negative than −0.4 V vs. RHE, the LSV curve in N2 saturated electrolyte delivers lower potential and higher current density, indicative of a N2 reduction event. The chronoamperometry curves of VNiON at various potentials are illustrated in Fig. 4a, and it shows that the current density remains stable for 3 hours. The corresponding highest ammonia yield is 6.79 μg h−1 cm−2 at −0.40 V versus RHE with a FE of 3.04% (Fig. 4b and S12, ESI†). The highest FE (5.57%) is achieved at a potential of −0.20 V. Meanwhile, the same experiments were also explored under acidic and alkaline conditions (Fig. S13 and S14, ESI†). Compared to the neutral NRR, both ammonia yields and FE of the NRR in acidic and alkaline media obviously decreased (Fig. S15, ESI†). Since the catalysts employed in this work contain nitrogen species which may cause false positive results, a series of control experiments were explored to exclude possible influences from any contaminants. A trace amount of ammonia production under an Ar atmosphere indicates that only N2 provides the nitrogen source to NH3 (Fig. S16a, ESI†). The comparison of VNiON and bare CC further provides an accurate assessment that excludes residual influences (Fig. S16b, ESI†). The control experiments rule out the influence of the nitrogen element in the materials. This NRR activity of VNiON is superior to that of the corresponding oxide and nitride and previous reports (Fig. 4c, S17–S19 and Table S2, ESI†). Both the ammonia production rate and the FE are approximately twice that of VNiO and quadruple of VNiN. As illustrated, the highest NH3 yield of 3.25 μg h−1 cm−2 for VNiO is achieved at −0.45 V versus RHE (Fig. S20, ESI†). The highest NH3 yield of 2.64 μg h−1 cm−2 for VNiN is achieved at −0.30 V. The highest FEs of VNiO (3.29%) and VNiN (2.59%) are both achieved at a potential of −0.20 V. VNiN exhibits larger current and less output of ammonia (Fig. S20, ESI†). The main reason is the inhibition of the NRR by the appropriate hydrogen adsorption energy of nitrides. This is also consistent with the LSV results. The corresponding electrochemical impedance spectroscopy (EIS) results illustrate that VNiON shows a smaller semicircle than VNiO and VNiN in the Nyquist plot, indicating the lower charge transfer resistance and superior charge transport kinetics of the former (Fig. S21, ESI†). The electrochemically effective surface area (EESA) for each material was further estimated from the electrochemical double-layer capacitance (Cdl) (Fig. S22, ESI†). The VNiON material exhibits the largest Cdl of 88.5 mF cm−2 among all the control samples. The result of electrode–electrolyte contact area is consistent with that of intrinsic surface area tested by BET. Considering the great influence of the V/Ni ratio on the catalytic performance, several control samples with different V/Ni ratios were further synthesized and named VNiON-1/2/3/4/5 (Fig. S23 and Table S3, ESI†). The similar morphology of the control samples eliminates the influence of different morphology on the NRR activity. Due to the competitive adsorption of nitrogen and hydrogen species, both ammonia yield rates and FE significantly decreased with the increase of the Ni content (Fig. S24, ESI†). Although the FE can be increased up to 9.64% by increasing the V content, the ammonia yields obviously decreased to one third of the optimal sample (Fig. S25, ESI†). Electrochemical impedance spectroscopy and electrochemically effective surface area illustrate the superior charge transport kinetics and abundant electrochemically active sites of VNiON. Furthermore, the NH3 yield rate and FE remain stable after 20 cycles of chronoamperometric runs (60 hours) (Fig. 4d), which is indicative of a better NRR stability of VNiON than VNiO and VNiN (Fig. S26, ESI†). After the first three cycles, nearly no current density loss is observed during the long-term electrolysis process (Fig. 4e). Even for 24 h per cycle, the current density remains stable during the 24 h long-term electrolysis process and the stabilized current density verifies the electrochemical stability (Fig. S27, ESI†). Furthermore, the XRD patterns and the morphology of VNiON nanosheets are nearly unchanged after the durability test (Fig. 4e, inset and S28, ESI†). As illustrated in the corresponding HRTEM image, the oxynitride layer is completely maintained after the NRR process. The presence of M–O–N peaks in XPS spectra further verifies the structural stability of VNiON oxynitride layers, which is consistent with the TEM results (Fig. S29 and S30, ESI†). The proposed NRR pathway on VNiON proceeds via a Mars–van Krevelen mechanism.36 An ammonia molecule is formed by extracting a surface N atom with adsorbed hydrogen atoms and leaves behind a nitrogen vacancy, and the active site is regenerated by activating N2 and thus healing the vacancy. Hence, the electrocatalytic NRR durability of the oxynitride layer is effectively realized. The above results indicate the outstanding structural stability of VNiON.
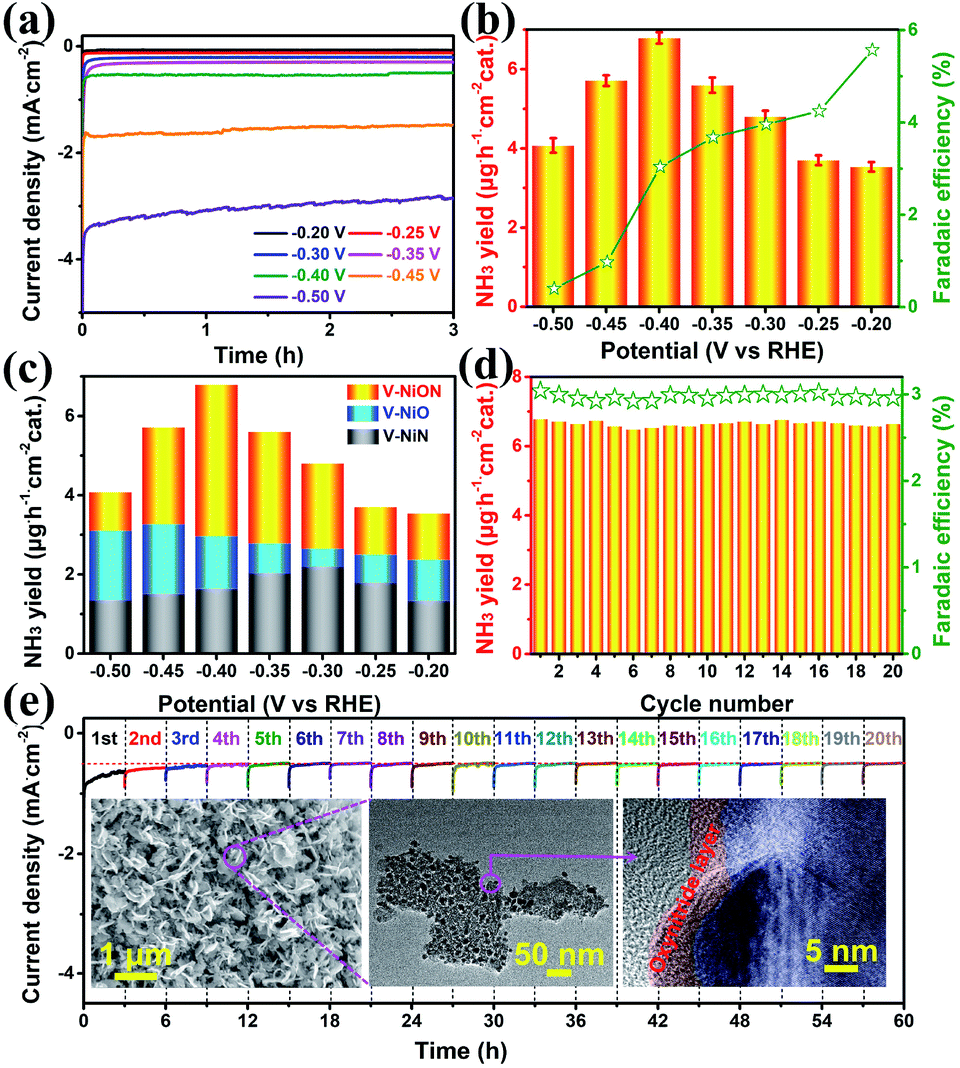 | ||
| Fig. 4 Chronoamperometry curves of VNiON at various potentials for 3 hours in N2-saturated 0.05 M Na2SO4 (a). NH3 yields and FE of VNiON for the NRR at various potentials (b). Comparison of NH3 yields of VNiO, VNiN and VNiON (c). NH3 yields and FE of VNiON at a potential of −0.40 V vs. RHE during 20-times recycling tests (d). Recycling chronoamperometry curves of VNiON at a potential of −0.40 V for 60 hours (e). The insets show the SEM, TEM and HRTEM images of VNiON after durability tests. | ||
In summary, the delocalized electron environment and plentiful active sites of VNiON enhance the ability of nitrogen absorption and activation. Such a catalyst exhibits excellent neutral NRR activity with an ammonia yield of ∼6.78 μg h−1 cmcat.−2 and FE of ∼5.57% and continuous NRR durability for 60 hours. The enhanced activity and ability indicate that the semiconductor oxides with advantageous structures (oxynitride layer) possess enormous potential for improving their corresponding applications in energy conversion and storage.
Experimental
Density functional theory (DFT) calculations
All calculations were carried out by using the projector augmented wave method in the framework of density functional theory (DFT),49 as implemented in the Vienna Ab initio Simulation Package (VASP). The generalized gradient approximation (GGA) and Perdew–Burke–Ernzerhof (PBE) exchange functional were used.50 Structural relaxation calculations were performed by using the spin-polarized GGA method.51 The convergence criteria of energy and force calculations were set to 10−5 eV per atom and 0.01 eV Å−1, respectively. The VNiON model was constructed from the 4 × 4 × 1 supercell of pristine NiO by replacing two Ni atoms and eight O atoms with two V atoms and eight N atoms, respectively. The plane-wave energy cutoff was set to 500 eV, and the Monkhorst–Pack method with the 2 × 2 × 3 meshes was employed for the Brillouin zone sampling of the 4 × 4 × 1 supercell of the pristine NiO and VNiON.51 Considering the strong Coulomb repulsion interactions of transition metals, the spin-polarized GGA + U method with a value of Ueff = 6.0 and 3.1 eV was applied for Ni 3d and V 3d states, respectively.52Synthesis of bimetallic oxynitride (VNiON)
The synthesis procedure for VNiON is as follows: 2.4 mmol Ni(NO3)2·6H2O, 0.8 mmol NH4VO3 and 5 mmol urea were first dissolved in 30 mL deionized water. The aqueous solution was transferred into a 35 mL Teflon-lined stainless steel autoclave. A 1 cm × 2 cm piece of clean carbon cloth (CC) treated with air plasma was immersed in the precursor solution in the autoclave. The autoclave was sealed and maintained at 120 °C for 12 h under self-generated pressure and then cooled down to room temperature naturally. Bimetallic hydroxides grown on the CC were washed with deionized water and ethanol and dried at 80 °C in air. The as-prepared hydroxide samples were further thermally annealed at 400 °C under an NH3 atmosphere for 2 h at a heating rate of 2 °C min−1, and both the heating and cooling processes were under argon atmosphere protection. For comparison, the VNiO sample was obtained by annealing at 400 °C under an argon atmosphere for 2 h, and the VNiN sample was obtained by annealing at 400 °C under an ammonia (NH3) atmosphere for 4 h. The control NiON sample was produced by a similar synthesis process of VNiON without NH4VO3.Physicochemical characterization
Powder X-ray diffraction (XRD) patterns of the materials were obtained on a diffractometer (Bruker D8) using a Cu Kα radiation source (λ = 0.15418 nm) with a 2θ scan from 10° to 90° with a step size of 0.04. An X-ray photoelectron spectrometer with a monochromatic Al Kα source (hv 1/4 1486.6 eV) and a charge neutralizer was used. All the binding energies were calibrated to the C 1s peak at 284.6 eV of the surface adventitious carbon. Scanning electron microscopy (SEM) images were collected using a Hitachi S-4800 microscope equipped with an energy-dispersive X-ray analyser (EDS, Horiba EMAX Energy EX-350). High-resolution TEM (HRTEM) images were obtained using a Philips Tecnai 20U-Twin microscope at an acceleration voltage of 200 kV. The solution of samples was achieved after 20 min of ultrasonic pretreatment. The TEM samples were prepared by dropping the primed solution onto a copper grid with a polyvinyl formal support film and dried in air. FT-IR spectra were obtained using a Bruker Tensor 27 infrared spectrometer.Electrochemical measurements
The electrocatalytic NRR tests were performed by using a two-compartment H-type like electrolytic cell, which was separated using a Nafion 117 membrane (DuPont). The Nafion membrane was pretreated by boiling it in H2O2 (5%) at 80 °C for 1 h and deionized water for another 1 h, sequentially. The electrochemical experiments were conducted with an electrochemical workstation (CHI 760C) by using a three-electrode configuration (working electrode is the as-synthesized material@CC, the counter electrode is the Pt plate, and the reference electrode is Ag/AgCl/saturated KCl). Before NRR tests, the cathode electrolyte was purged with high purity nitrogen (99.999%, 40 mL min−1) for 0.5 hour and then the flow rate was adjusted to 15 mL min−1 and maintained stable during the constant potential test for 3 hours. The ammonia formation rate presented in the manuscript is the average data for the reaction of 3 hours. In this work, all potentials were converted to the reversible hydrogen electrode (RHE) potential using the equation ERHE = EAg/AgCl + 0.0591 × pH + 0.194, resulting in a shift of +0.6077 V versus RHE (0.05 M Na2SO4, pH ≈ 7.1). Polarization curves were obtained using linear sweep voltammetry (LSV) with a scan rate of 2 mV s−1 at 25 °C in an aqueous solution (0.05 M Na2SO4) with constant N2 (g) or Ar (g) continually purging for 30 min prior to the measurements. The polarization curves were the steady-state ones after several cycles. Electrochemical impedance spectroscopy (EIS) was carried out in potentiostatic mode from 105 to 0.01 Hz. The long-term stability test was carried out using chronoamperometry measurements.Statement of contributions
B. Chang synthesized the material and performed most of the electrochemical tests; L. Q. Deng was involved in the complementary electrocatalytic tests for revision; B. Chang carried out the theoretical DFT calculation and analysed the DFT results with Y. Z. Wu and X. P. Hao; S. Z. Wang and D. Shi were involved in the synthesis of materials; Z. Z. Ai and H. H. Jiang were involved in the electrochemical studies; Y. L. Shao, L. Zhang and J. X. Shen assisted with the SEM, TEM and EDX measurements; Y. Z. Wu and X. P. Hao designed the study; B. Chang, Y. Z. Wu and X. P. Hao prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Contract No. 51872162 and 11890700), and the Major Basic Program of the Natural Science Foundation of Shandong Province (Contract ZR2017ZB0317 and ZR2018MEM013).References
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, 873–879 CAS.
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 146–157 CrossRef PubMed.
- C. Tang and S. Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- B. H. R. Suryanto, H. L. Du, D. B. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- S. D. Minteer, P. Christopher and S. Linic, ACS Energy Lett., 2019, 4, 163–166 CrossRef CAS.
- K. Ithisuphalap, H. G. Zhang, L. Guo, Q. G. Yang, H. P. Yang and G. Wu, Small Methods, 2018, 1800352 Search PubMed.
- M. Ali, F. L. Zhou, K. Chen, C. Kotzur, C. L. Xiao, L. Bourgeois, X. Y. Zhang and D. R. MacFarlane, Nat. Commun., 2016, 7, 11335–11341 CrossRef CAS PubMed.
- Y. Ashida, K. Arashiba, K. Nakajima and Y. Nishibayashi, Nature, 2019, 568, 536–540 CrossRef CAS PubMed.
- R. Hawtof, S. Ghosh, E. Guarr, C. Y. Xu, R. M. Sankaran and J. N. Renner, Sci. Adv., 2019, 5, 5778 CrossRef PubMed.
- Q. C. Wang, Y. P. Lei, D. S. Wang and Y. D. Li, Energy Environ. Sci., 2019, 12, 1730–1750 RSC.
- S. L. Zhao, X. Y. Lu, L. Z. Wang, J. L. Gale and R. Amal, Adv. Mater., 2019, 31, 1805367 CrossRef PubMed.
- X. Yan, D. L. Liu, H. H. Cao, F. Hou, J. Liang and S. X. Dou, Small Methods, 2019, 1800501 CrossRef.
- W. B. Qiu, X. Y. Xie, J. D. Qiu, W. H. Fang, R. P. Liang, X. Ren, X. Q. Ji, G. W. Cui, A. M. Asiri, G. L. Cui, B. Tang and X. P. Sun, Nat. Commun., 2018, 9, 3485–3491 CrossRef PubMed.
- J. C. Liu, X. L. Ma, Y. Li, Y. G. Wang, H. Xiao and J. Li, Nat. Commun., 2018, 9, 1610–1618 CrossRef PubMed.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- S. K. Ritter, ACS Cent. Sci., 2017, 3, 512–514 CrossRef CAS PubMed.
- X. Ren, J. X. Zhao, Q. Wei, Y. J. Ma, H. R. Guo, Q. Liu, Y. Wang, G. W. Cui, A. M. Asiri, B. H. Li, B. Tang and X. P. Sun, ACS Cent. Sci., 2019, 5, 116–121 CrossRef CAS PubMed.
- Y. Yao, H. J. Wang, X. Z. Yuan, H. Li and M. H. Shao, ACS Energy Lett., 2019, 4, 1336–1341 CrossRef CAS.
- L. L. Zhang, L. X. Ding, G. F. Chen, X. F. Yang and H. H. Wang, Angew. Chem., Int. Ed., 2019, 58, 2612–2616 CrossRef CAS PubMed.
- X. M. Yu, P. Han, Z. X. Wei, L. S. Huang, Z. X. Gu, S. J. Peng, J. M. Ma and G. F. Zheng, Joule, 2018, 2, 1–13 CrossRef.
- L. Q. Li, C. Tang, B. Q. Xia, H. Y. Jin, Y. Zheng and S. Z. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS.
- C. Y. Hu, X. Chen, J. B. Jin, Y. Han, S. M. Chen, H. X. Ju, J. Cai, Y. R. Qiu, C. Gao, C. M. Wang, Z. M. Qi, R. Long, L. Song, Z. Liu and Y. J. Xiong, J. Am. Chem. Soc., 2019, 141, 7807–7814 CrossRef CAS PubMed.
- T. W. Wu, X. J. Zhu, Z. Xing, S. Y. Mou, C. B. Li, Y. X. Qiao, Q. Liu, Y. L. Luo, X. F. Shi, Y. N. Zhang and X. P. Sun, Angew. Chem., Int. Ed., 2019, 58, 1–6 CrossRef.
- N. Cao, Z. Chen, K. T. Zang, J. Xu, J. Zhong, J. Luo, X. Xu and G. F. Zheng, Nat. Commun., 2019, 10, 2877–2888 CrossRef PubMed.
- G. Zhang, Q. H. Jia, K. Zhang, Y. Chen, Z. H. Lie, H. J. Liu, J. H. Li and J. H. Qu, Nano Energy, 2019, 59, 10–16 CrossRef CAS.
- J. R. Han, Z. C. Liu, Y. J. Ma, G. W. Cui, F. Y. Xie, F. X. Wang, Y. P. Wu, S. Y. Gao, Y. H. Xu and X. P. Sun, Nano Energy, 2018, 52, 264–270 CrossRef CAS.
- X. F. Wu, L. Xia, Y. Wang, W. B. Lu, Q. Liu, X. F. Shi and X. P. Sun, Small, 2018, 14, 1803111 CrossRef PubMed.
- T. W. Wu, W. H. Kong, Y. Zhang, Z. Xing, J. X. Zhao, T. Wang, X. F. Shi, Y. L. Luo and X. P. Sun, Small Methods, 2019, 1900356 CrossRef.
- R. Zhang, H. R. Guo, L. Yang, Y. Wang, Z. G. Niu, H. Huang, H. Y. Chen, L. Xia, T. S. Li, X. F. Shi, X. P. Sun, B. H. Li and Q. Liu, ChemElectroChem, 2019, 6, 1014–1018 CrossRef CAS.
- W. Ye, M. Arif, X. Y. Fang, M. A. Mushtaq, X. B. Chen and D. P. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 28809–28817 CrossRef CAS PubMed.
- Y. F. Fang, Z. C. Liu, J. R. Han, Z. Y. Jin, Y. Q. Han, F. X. Wang, Y. S. Niu, Y. P. Wu and Y. H. Xu, Adv. Energy Mater., 2019, 1803406 CrossRef.
- Y. X. Guo, Z. Y. Yao, B. J. J. Timmer, X. Sheng, L. Z. Fan, Y. Y. Li, F. G. Zhang and L. C. Sun, Nano Energy, 2019, 62, 282–288 CrossRef CAS.
- R. Zhang, J. R. Han, B. Z. Zheng, X. F. Shi, A. M. Asiri and X. P. Sun, Inorg. Chem. Front., 2019, 6, 391–395 RSC.
- X. Yang, J. Nash, J. Anibal, M. Dunwell, S. Kattel, E. Stavitski, K. Attenkofer, J. G. Chen, Y. S. Yan and B. J. Xu, J. Am. Chem. Soc., 2018, 140, 13387–13391 CrossRef CAS PubMed.
- H. Y. Jin, L. Q. Li, X. Liu, C. Tang, W. J. Xu, S. M. Chen, L. Song, Y. Zheng and S. Z. Qiao, Adv. Mater., 2019, 1902709 CrossRef PubMed.
- C. He, Z. Y. Wu, L. Zhao, M. Ming, Y. Zhang, Y. P. Yi and J. S. Hu, ACS Catal., 2019, 9, 7311–7317 CrossRef CAS.
- S. Z. Wang, L. L. Li, Y. L. Shao, L. Zhang, Y. L. Li, Y. Z. Wu and X. P. Hao, Adv. Mater., 2019, 31, 1806088 CrossRef PubMed.
- H. Tan, Z. H. Liu, D. L. Chao, P. Hao, D. D. Jia, Y. H. Sang, H. Liu and H. J. Fan, Adv. Energy Mater., 2018, 8, 1800685 CrossRef.
- L. Pauling, J. Am. Chem. Soc., 1929, 51, 1010–1026 CrossRef CAS.
- C. X. Guo, J. R. Ran, A. Vasileff and S. Z. Qiao, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- M. A. Légaré, G. B. Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed.
- H. Y. Liang, H. J. Xi, S. Q. Liu, X. M. Zhang and H. Q. Liu, Nanoscale, 2019, 11, 18183–18190 RSC.
- K. Xu, P. Z. Chen, X. L. Li, Y. Tong, H. Ding, X. J. Wu, W. S. Chu, Z. M. Peng, C. Z. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef CAS PubMed.
- B. Chang, J. Yang, Y. L. Shao, L. Zhang, W. L. Fan, B. B. Huang, Y. Z. Wu and X. P. Hao, ChemSusChem, 2018, 11, 3198–3207 CrossRef CAS PubMed.
- A. P. Wu, Y. Xie, H. Ma, C. G. Tian, Y. Gu, H. J. Yan, X. M. Zhang, G. Y. Yang and H. G. Fu, Nano Energy, 2018, 44, 353–363 CrossRef CAS.
- A. Fuertes, Mater. Horiz., 2015, 2, 453–461 RSC.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133–1138 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- Y. Gao, X. Wang, J. Ma, Z. Wang and L. Chen, Chem. Mater., 2015, 27, 3456–3461 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The DFT calculation details; the detailed characterization on the materials; additional electrochemical results and stability test results. See DOI: 10.1039/c9ta11378a |
| This journal is © The Royal Society of Chemistry 2020 |