
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Challenges and opportunities in the bottom-up mechanochemical synthesis of noble metal nanoparticles
Paulo F. M.
de Oliveira
 *ab,
Roberto M.
Torresi
a,
Franziska
Emmerling
*b and
Pedro H. C.
Camargo
*c
*ab,
Roberto M.
Torresi
a,
Franziska
Emmerling
*b and
Pedro H. C.
Camargo
*c
aDepartamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Av. Lineu Prestes 748, 05508000, São Paulo, Brazil. E-mail: paulofmo@usp.br
bBAM Federal Institute for Materials Research and Testing, Richard-Willstätter-Strasse 11, 12489 Berlin, Germany. E-mail: franziska.emmerling@bam.de
cDepartment of Chemistry, University of Helsinki, A. I. Virtasen aukio 1, Helsinki, Finland. E-mail: pedro.camargo@helsinki.fi
First published on 28th July 2020
Abstract
Mechanochemistry is a promising alternative to solution-based protocols across the chemical sciences, enabling different types of chemistries in solvent-free and environmentally benign conditions. The use of mechanical energy to promote physical and chemical transformations has reached a high level of refinement, allowing for the design of sophisticated molecules and nanostructured materials. Among them, the synthesis of noble metal nanoparticles deserves special attention due to their catalytic applications. In this review, we discuss the recent progress on the development of mechanochemical strategies for the controlled synthesis of noble metal nanostructures. We start by covering the fundamentals of different preparation routes, namely top-down and bottom-up approaches. Next, we focus on the key examples of the mechanochemical synthesis of non-supported and supported metal nanoparticles as well as hybrid nanomaterials containing noble metals. In these examples, in addition to the principles and synthesis mechanisms, their performances in catalysis are discussed. Finally, a perspective of the field is given, where we discuss the opportunities for future work and the challenges of mechanochemical synthesis to produce well-defined noble metal nanoparticles.
 Paulo F. M. de Oliveira | Paulo F. M. de Oliveira obtained his BS degree in Chemical Engineering from the University of São Paulo (USP), Lorena in 2012. His PhD at Centre RAPSODEE (2015), Mines-Albi, France, was dedicated to the investigation of mechanochemical organic reactions. He was a postdoc at University of Lille (2017) and studied phase transitions of molecular materials induced by mechanical stress. Currently, he holds a FAPESP postdoc fellowship and researches on nanoparticle mechanosynthesis at Institute of Chemistry USP São Paulo, for applications in catalysis and energy. His research interests include mechanochemistry, solid state phase transitions and mechanosynthesis of advanced materials. |
 Roberto M. Torresi | Roberto M. Torresi is full professor at the Institute of Chemistry of the University of São Paulo (São Paulo, Brazil). Graduated in chemistry at the National University of Córdoba (Argentina), where he obtained his PhD in 1986. After, he worked at Sorbonne University as postdoc. He served as visiting professor at Sorbonne University and Joseph Fourier University (France). His current lines of research include the study of nanostructures of redox materials and ionic liquids, aimed at electrochemical energy storage (mainly batteries and electrochemical capacitors). He is full member of the Brazilian Academy of Sciences and he is the author of over 195 peer-reviewed articles. |
 Franziska Emmerling | Franziska Emmerling is head of the Department Materials Chemistry at the Federal Institute for Materials Research and Testing (BAM) in Berlin. With an educational background in inorganic chemistry, she obtained her PhD at the Department of Chemistry at the Albert-Ludwigs University in Freiburg. Her field of research include in situ investigations of crystallization processes, nanoparticle formation, and mechanochemistry. |
 Pedro H. C. Camargo | Pedro H. C. Camargo received his BS and MS in Chemistry in 2003 and 2005, respectively, both from Federal University of Paraná in Brazil. In 2009, he obtained his PhD from Washington University in Saint Louis, where he worked in Prof. Younan Xia's group. In 2011, he started his independent career as an Assistant Professor at University of São Paulo, and relocated to a Professor position at the University of Helsinki in 2019. His research interests include the synthesis of nanomaterials for nanocatalysis and plasmonic photocatalysis. He serves as an Editor of the Journal of Materials Science, as an Associate Editor of the Journal of the Brazilian Chemical Society, and as an Editorial Board member for iScience. |
1. Introduction
The ancient interest in noble metals was based on their aesthetic properties such as their spectacular brightness. Gold (Au) and silver (Ag), for example, were employed in the composition of ornaments and jewelry.1,2 In the past century, extensive studies have emerged on the discovery and study of their optical, catalytic, electronic, and biomedical properties across the nanoscale, their clusters, and single atoms.3–8 These unique properties hold enormous potential for applications in several areas such as catalysis, sensing, energy conversion, photonics, and biomedicine.8–14The properties of metal nanoparticles (NPs) are strongly dependent on their physicochemical features, including size, shape, solid or hollow interiors, and composition. Moreover, in the case of bimetallic or multicomponent nanostructures, properties also depend on the elemental distribution within the particle (alloys or core–shell morphologies).15 By tuning and controlling these physical and chemical parameters, one can, therefore, alter and optimize the desired property for a target application. For instance, in the case of catalysis, it is expected that the surface area, size, surface composition, and the nature of exposed facets (shape) should strongly affect catalytic activities and selectivity (geometric and electronic effects).16–18 In this context, noble metal NPs have played a pivotal role in catalysis towards gas-phase hydrogenation/oxidation and carbon-activation, oxidations, coupling, reductions, and water splitting reactions, for example.5,19–27 When optical properties are considered, the shape, size, and composition in Ag and Au-based nanoparticles affect absorption and scattering efficiencies as well as the intensity and distribution of electric field enhancements as a result of the localized surface plasmon resonance (LSPR) excitation. These, for instance, have implications for surface-enhanced Raman scattering (SERS), plasmonic catalysis, and imaging applications.13,14,28–30 Therefore, it becomes clear the importance of controlling the synthesis of these materials (yielding controllable and well-defined physical and chemical features) not only to optimize catalytic performances but also to enable the unraveling of the structure–performance relationships.
Solution-based batch protocols comprising the reduction or decomposition of the salt precursor in the presence of capping and stabilizing agents have been achieving considerable success for the synthesis of size, shape, and composition-controlled noble metal NPs.4,15,20,27,31,32 Despite the versatility and success, several challenges remain. Solution-based syntheses are often carried out in diluted media, yielding suspensions with low nanoparticle concentrations. Hence, for many practical applications, it is necessary to scale up and to concentrate the suspension by precipitation and/or centrifugation. The scale-up can lead to problems regarding reproducibility and the necessary workup may also induce changes in or aggregation of the nanoparticles. There is also the need for purification to remove the excess of surfactants and unreacted precursors. Furthermore, the synthesis of NPs can be very sensitive to variations in reaction conditions and trace amounts of ions in the solution or solvent, which can lead to reproducibility issues.33 Another aspect is that solution-phase synthesis, such as the polyol34,35 and the Turkevich or Frens methods,36–39 generally requires high temperatures. Finally, the scale-up of solution-phase synthesis recipes under batch conditions remains limited, although recent progress has been achieved using flow conditions.40,41
Mechanochemical ball milling synthesis has gained prominence as efficient, eco-friendly, and versatile alternatives to replace some solvent-based protocols.42–45 In contrast to solution-based strategies, mechanochemical synthetic approaches are highly reproducible, avoid the use of solvents, and many of them can be easily scaled up.46,47 The earlier uses of mechanochemistry in the mineral and materials sciences48–51 have now been successfully expanded to areas including organic synthetic chemistry,52 crystal engineering and co-crystal formation,53,54 porous polymers construction,47,55,56 and studies of amorphization and polymorphism in molecular materials.57–60 Moreover, developments in mechanochemistry have facilitated the preparation of a wealth of advanced inorganic and hybrid organic–inorganic materials.61–63 Owing to its potential, IUPAC has recently highlighted mechanochemistry as one of the chemical technologies that will change the world.64 This has further encouraged the research community to push the field forward.
The synthesis of metal-based NPs by mechanochemical conditions has been explored for decades. The early focus in the field has been directed towards the preparation of metal alloys, metal oxides and sulfides, and the formation of composites.48,51,61,65 Milling has also been employed as a top-down approach to prepare metal-based nanoparticles by hard stress and comminution.61 More recently, the use of ball milling in the bottom-up synthesis of relatively monodispersed Ag66 and Au67 nanoparticles from the corresponding salt precursors has been described. Therefore, the recent advances in mechanochemistry have inspired this approach to emerge as a promising alternative for the synthesis of noble metal NPs.68,69 The simple reaction work-up (if needed), reproducibility, and process simplicity are very appealing in the context of Green Chemistry’.68,70
In this review, we cover the essential aspects of mechanochemical synthesis applied to noble metal NPs. Our goal is to provide some fundamentals and an update regarding the recent progress in this field. We start by presenting the fundamentals of the mechanosynthesis of nanoparticles and compares the two strategies for their preparation, named bottom-up (reduction of precursors, nucleation and growth) and top-down (comminution) (Section 2). This is followed by the discussion of the mechanochemical synthesis of non-supported noble metal NPs, with a focus on the bottom-up up approach (Section 3). Here, non-supported means the NPs that are not present at the surface of another solid matrix as supports. Thus, NPs are isolated and can be stabilized against aggregation by the presence of capping and/or stabilizing agents on their surfaces. We then focus on the mechanosynthesis of the supported noble metal NPs (where mechanochemical means were essential and employed in at least one step of the synthesis, Section 4). Here, supported NPs refer to those that are deposited over or within a solid matrix, such as oxides, porous materials, or biomass sources. In Sections 3 and 4, we present the advantages and limitations of the mechanochemical methodology in different examples. Particular attention is given to the mechanisms of noble metal NPs formation and the opportunities offered by the mechanochemical technique as a means to overcome the challenges of solution-phase protocols. We also highlight some examples of applications of noble metal NPs prepared by mechanochemistry. Finally, we present a perspective of the field where we discuss challenges and opportunities for future work on the mechanochemical synthesis of controllable and well-defined noble metal NPs (Section 5).
2. Mechanosynthesis of noble metal NPs – bottom-up and top-down approaches
Although several strategies have been described to prepare metal NPs, they can be classified into two main groups: bottom-up and top-down approaches. Different from most fluid phase techniques, the early mechanochemical methods for the synthesis of metal NPs were based on the milling and comminution of the bulk materials, regarded as a top-down method (Fig. 1). In these routes, the mechanical devices employed to produce NPs have included planetary, vibratory, and attritor mills. Each device presents a more prominent type of mechanical stress such as shear stress or shock-impact.61,71 As shown in Fig. 1 (upper region), starting from the bulk metal, the particles, and grains are reduced by the effect of mechanical action that breaks or dissociates the larger structures and induces severe plastic deformation as the milling time increases.61,69 After a milling period, NPs possessing grain boundary and intragranular defects with increased surface area (smaller sizes) are produced. Further milling can induce more deformation and stress accumulation. In some cases, sintered particles and amorphous phases can form upon continued milling, thereby decreasing the overall surface area.72–74 This mechanism is more likely to work in the case of brittle materials, although some reports have reported more malleable and ductile systems.75,76 Some advantages of this top-down route include its simplicity and the generation of relatively clean surfaces. Nevertheless, this approach might not be the most appropriate for the synthesis of controlled noble metal NPs, particularly when anisotropic morphologies and narrow size distribution are required. Comprehensive reviews on the top-down mechanochemical synthesis of nanoparticles focusing on the fundamentals of the technique can be found elsewhere.49,61,69,77–79 | ||
| Fig. 1 Mechanochemical synthesis of nanoparticles: top-down and bottom-up approaches. | ||
Regarding bottom-up strategies, recent methods for the synthesis of noble metal NPs from their salt precursors have involved manual grinding, alongside the use of ball milling devices. The success of this methodology has increased the scope of ball milling to the controlled synthesis of NPs.66–68Fig. 1 (bottom region) illustrates this method, where the NPs are produced by the reduction of precursor compounds generating the growth species (metal atoms). These atoms undergo nucleation and growth processes through different pathways (e.g. by atomic addition, coalescence, Ostwald ripening, oriented attachment) to generate the final NPs.80 Contrary to what was discussed in the top-down approach, in the bottom-up mechanochemical synthesis the increase in the milling time leads to an increase on the size of the NPs as a result of growth. Long milling periods can also further decrease the overall surface area even in the presence of capping agents due to the coalescence of smaller particles and welding (Fig. 1). In principle, bottom-up mechanochemical strategies present a huge potential for the control over the NPs physicochemical features by manipulating the nucleation and growth stages during the synthesis.15,31Table 1 summarizes a general overview of some fundamental features of the mechanochemical synthesis of noble metal NPs relative to solution phase strategies.
| Features | Mechanosynthesis | Solution-phase synthesis |
|---|---|---|
| Size control | Limited | Yes |
| Shape control | Very limited | Yes |
| Composition control | Limited | Yes |
| Scalability | Yes | Limited |
| Stability | Yes | Yes/no |
| Use of high temperature | No | Yes |
| Use of solvents | No | Yes |
| Purification steps | Yes or no | Yes |
Generally, the formation of metal NPs by bottom-up mechanosynthesis involves the use of three key components, which also apply to solution-phase synthesis: (i) the metal precursor, which it is the source of the growth species; (ii) a reducing agent or energy to drive a decomposition or reduction process to generate the growth species; and (iii) a capping agent, which it is used to stabilize the nanostructures against aggregation. The metal precursor is the container of the metal atoms. The precursors can be both inorganic stable salts (chlorides, bromides, and their complexes) or volatile organic compounds (acetylacetonates, acetates, etc.). The nature of the precursor, its stability its oxidation state and the precursor ligand can also affect the dynamic of the reaction and nanoparticle formation, yielding different NPs with distinct properties.81–83 More importantly, the redox potential of the precursor in a pair with the reducing agent will definitely affect the reaction kinetics, and thus, the final NPs' size and shape.84 Concerning the use of common reducing agents such as NaBH4, sodium citrate and ascorbic acid, they can have a more elaborated role than simply supplying electrons for the reduction of the metal ion. The physicochemical properties and the redox potential (face to a certain metal precursor) of the reductants can dictate the kinetics of reduction, nucleation, and growth of the crystalline nanoparticles in determined experimental conditions. At the end, this can also affect the final shape. Strong reducing agents tends to generate smaller nanoparticles and they are preferred for the generation of seeds. On the other hand, mild and weak reductants are generally employed in the evolution of anisotropic nanoparticles.84 The use of a capping agent is, finally, the last common parameter in both solution and mechanochemistry. The capping agent is important to prevent aggregation, coalescence, and overgrowth, keeping the nanosize. The capping agents (also called stabilizing agents) can stabilize the formed NPs by different mechanisms of surface interaction (coordination, electrostatic, chemisorption). At least in solution, the capping agent plays a pivotal role in the size and shape control.85,86 This happens due to the manipulation of the growth kinetics along with preferential interaction with some crystallographic directions.86 In mechanochemical systems it has to be present in a larger mass fraction compared to the metal load.87 Nonetheless, the use of common capping agents can be dismissed when the NPs are directly synthesized over a supporting material. It is noteworthy that these three parameters do not apply to top down mechanochemical syntheses methods that are based on comminution.
The role of capping and reducing agents can be sometimes played by the same chemical compound such as the case of sodium citrate and polyvinylpyrrolidone.66,88 The use of decomposable precursors (by heating or sonication) are also an alternative not to employ an additional reducing agent.89 The aggregation of the NPs can also be prevented, for example, by some byproduct formed in a larger volume fraction that hinders particle–particle contact, as in the case of multiphase systems.87 The kinetics of reduction, nucleation, and growth, which can, in turn, affect the size and shape of the produced NPs, will depend on the choice of these three main components, their concentrations, and their relative molar ratios. The influence of these parameters has been widely investigated in solution-based protocols, enabling the production of several noble metal NPs with controlled shapes, sizes, and compositions.10,15,84,90 Conversely, knowledge on the mechanochemical synthesis of noble metal NPs from the reduction of proper salt precursors remains limited and challenging. Here, important issues to consider are the type of milling device, the milling conditions, and the mass transfer in these systems, which are often diffusion-limited.
Temperature effects can also play a role in mechanochemical reactions. For instance, temperature increases during ball milling reactions originated from the comminution process and the mechanical action itself can occur. The heat generation may favor the target chemical reaction and/or induce precursor decomposition. Monitoring the overall temperature changes during milling organic compounds has shown that the temperature change does not surpass a few degrees.91 On the other hand, for purely inorganic systems such as in mechanical alloying92,93 and SiO2 (ref. 94 and 95) under vigorous milling conditions, the global temperatures can increase by tens to hundreds of degrees in vibratory/shaker mills94,95 and planetary mills,92–94 respectively. In the case of bottom-up mechanochemical synthesis of noble metal NPs, which is generally a hybrid system with inorganic (precursor) and organic compounds (capping and reducing agents, support), only a slight temperature increase (ΔT ∼ 2 °C) is reported.66 The exceptions include the case of volatile precursors milled with other thermal conductive inorganic materials (e.g., graphite), where decomposition occur, probably induced by the simultaneous effect of the mechanical input and temperature rising.89 Nonetheless, the overall temperature experienced by powder mixtures under continuous milling may not reflect the real temperature at the contact point between the milling ball and the reactants at the moment of the mechanical energy input (impact, friction, etc.).96 Therefore, a thermally induced reaction cannot be completely excluded from the mechanisms of NPs formation under milling conditions.
3. Synthesis of non-supported noble metal NPs
The synthesis of non-supported NPs is particularly interesting for the preparation of size- and shape-controlled NPs. These aspects are essential for fundamental and mechanistic understand of the NP formation. The synthesis of NPs detached from any solid matrix, generally requires the use of capping agents to stabilize them, preventing agglomeration or coalescence. Polymers such as PVP are the most used as capping agents in the bottom-up noble metal NPs mechanosynthesis. For the chemical reduction of the metal precursor, NaBH4 seems to be preferred. Table 2 summarizes the noble metal NP type, the reduction method and the capping agent used in the examples as well as some aspects of the mechanochemical method employed and the final material obtained.| System | Precursor and load | Reducing method | Stabilizing agent | Synthesis conditions | Conversion or yield | Products | Ref. |
|---|---|---|---|---|---|---|---|
| Monometallic | |||||||
| Au | KAuCl4, 20–40 mg | NaBH4 | PVP | Ball milling, agate set, 25 Hz, 15–120 min | — | Non-spherical NPs, 6.3 ± 2.1 to 27.9 ± 6.2 nm | 67 |
| HAuCl4 | Galvanic replacement | Aliphatic amines | Ball milling, stainless steel set, 30 Hz, 90 min | Quantitative conversion by XPS | Monodisperse round NPs, 1.3 ± 0.2 to 4.3 ± 1.2 nm | 97 | |
| AuCl, 16.4 mg | Sodium citrate | PVP | Ball milling, PTFE jar + ZrO2 balls + dispersion in water 30–120 min | Partial during milling | Nanotadpoles, head: 10–18 nm, tails: <22 nm | 98 | |
| Au(I)SC8H17, 25 mg | NaBH4 | Thiol molecules | Manual grinding, agate set, 3–5 min | Quantitative conversion by XRD | NPs with two family sizes, 2.0 ± 0.4 and 17.4 ± 5.7 nm | 99 | |
| Ag | AgNO3, 15–50 mg | PVP | PVP | Ball milling, agate set, 25 Hz, 15 min | Quantitative conversion checked with NaCl solution | Single crystals 3.5–4.5 nm, polycrystalline NPs > 6 nm | 66 |
| 15–50 mg | PEG | PEG | Ball milling, stainless steel set, 25 Hz, 15 min | Polydisperse NPs, 3.1 ± 1.4 to 22.8 ± 5.8 nm | 100 | ||
| 10–20 mg | Chitosan | Chitosan | Ball milling, stainless steel set, 25 Hz, 15 min | Aggregated flaked microparticles | 100 | ||
| 1 g | PVP | PVP | Manual grinding, agate mortar + aging (two steps of 24 h) | Only Ag0 phase by XRD | Ag@polypyrrole nanocomposites semi-spherical shape 35–44 nm | 101 | |
| 0.5 g | PEG-6000/8000 | PEG-6000/8000 | Ball milling, polyethylene jar + hardened steel balls, 200 rpm, 2–24 h | Only PEG and Ag0 phase by XRD | Polydisperse NPs, average crystalline size 10–12 nm | 102 | |
| 340 mg | NaH2PO2·H2O | PEG-400 | Manual grinding, agate set, 10 + 30 min | Quantitative conversion by XPS | 3D coral shaped Ag–Ag homojunction, individual particles: 35–50 nm | 103 | |
| Ag2O, 9.4 g | Carbon | — | Ball milling, hardened steel set, 250 rpm, up to 22 h | Quantitative conversion after 22 h by XRD | Polydisperse, 10–50 nm | 104 | |
| Ag2O, 2.0 g | Carbon | — | Ball milling, stainless steel set | — | 105 | ||
| Ag(I)SC8H17, 125 mg | NaBH4 | Thiol molecules | Manual grinding, agate set, 3–5 min | 78% yield (TGA) | Monodisperse NPs, 4.04 ± 0.45 nm | 99 | |
| AgNO3/Ag-thiolate, 47 mg | NaBH4 | Mercaptosuccinic acid (H2MSA) | Manual grinding, agate set, 10 min | — | Ag9 nanoclusters | 106 | |
| AgNO3/Ag-thiolate, 23 mg | NaBH4 | Thiolates of glutathione (GSH) or N-(2-mercaptopropionyl)glycine (MPGH) | Manual grinding, agate set, 10 min | — | Ag32 nanoclusters | 107 | |
| AgNO3/Ag-thiolate, 23 mg | NaBH4 | Phenylethylenethiolate (SCH2CH2Ph) | Manual grinding, agate set | 82% yield | Ag152 – 2 nm:, Ag92 core-Ag60(SCH2CH2Ph)60 shell | 108 | |
| Ag2C2O4, 2 g | Mechanochemical decomposition | PVP | Stainless steel mechanoreactor, Ar atmosphere | — | NPs, 5 nm mean size | 109 | |
| Pd | KPdCl4, 5 mg | PVP | PVP | Ball milling, agate set, 10–30 Hz, 10–30 min | Excellent conversion by UV-Vis | Different sizes e.g., 38.7 ± 0.6 nm at 20 Hz | 110 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||
| Multicomponent | |||||||
| Au–Ag | AgNO3, HAuCl4 | NaOH and chitosan, galvanic replacement | Chitosan | Manual grinding, agate set, 10–30 min | 50–60% conversion of Au3+ | AuAg alloy NPs of 3.0 ± 0.5 nm | 111 |
| AgnX@Ag (X = Cl−, Br−, CrO42−, PO43−) | Ag(I)acetate/AgnX | (NH2OH)2·H2SO4 | — | Manual grinding, 20 min | — | AgnX@Ag composites with 40 nm to 100–300 nm in size | 112 |
3.1. Monometallic NPs
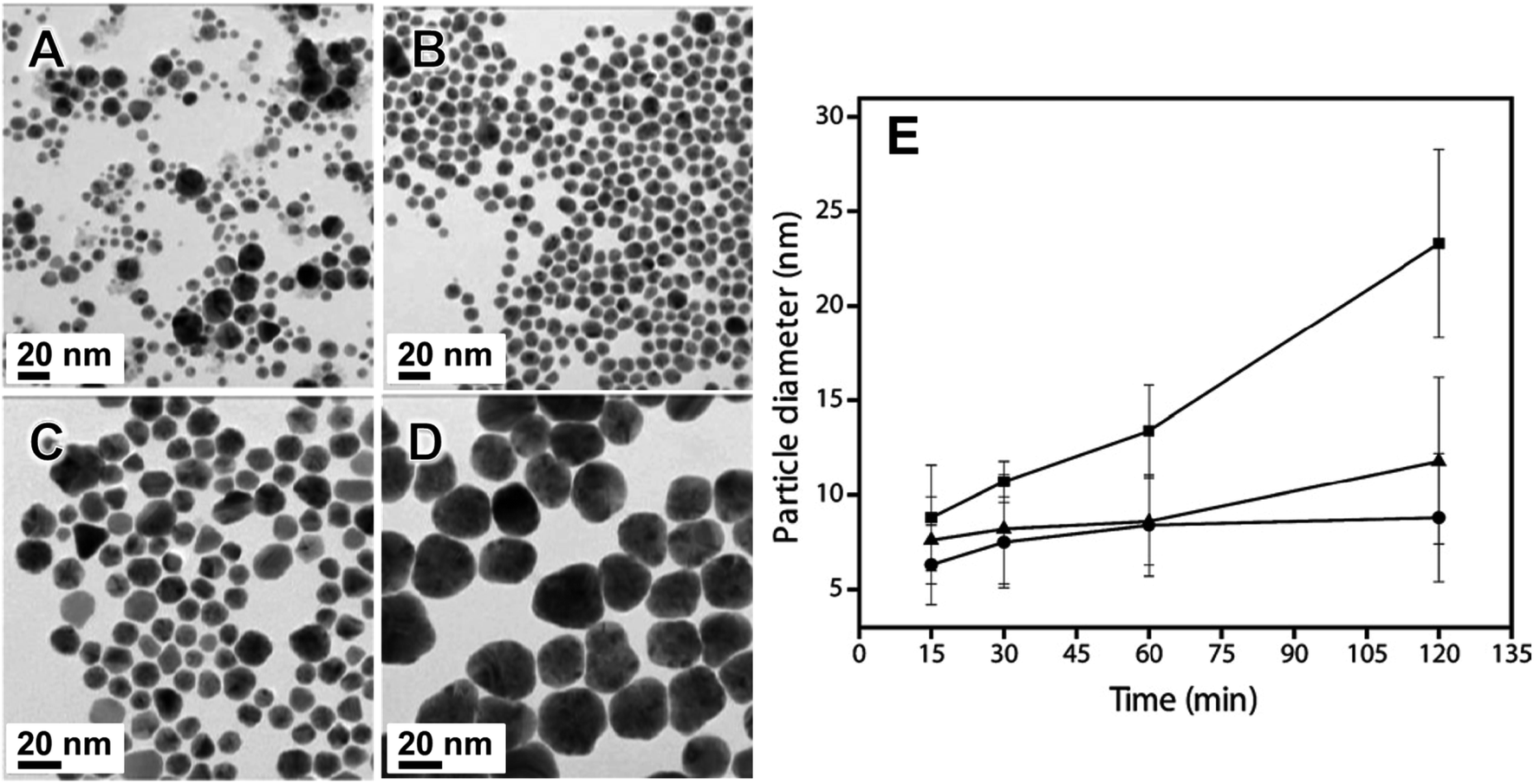 | ||
| Fig. 2 TEM images of Au nanoparticles synthesized in a vibratory ball mill (agate milling set) using KAuCl4 as Au precursor, PVP as capping agent, and NaBH4 as reducing agent as a function of milling time: (A) 15 min, (B) 30 min, (C) 60 min and (D) 120 min. Note that 30 min of milling time is optimal to obtain smaller sizes and relatively monodispersed nanoparticles. (E) Comparison of the average sizes as a function of time of the Au nanoparticles as a function of PVP average Mw and salt precursor concentration. (●) PVP (Mw 30 kg mol−1): KAuCl4 5:2; (▲) PVP (Mw 10 kg mol−1): KAuCl4 5:1; (■) PVP (Mw 10 kg mol−1): KAuCl4 5:2. Adapted from ref. 67 with permission of the Royal Society of Chemistry. | ||
Au NPs with sizes around 1 nm in diameter were obtained by milling the Au salt precursor with different amine ligands, as illustrated in Fig. 3.97 Other ligands were also studied, including N-heterocyclic and sulfur-based molecules. In this example, the use of amines with longer aliphatic chains enabled the synthesis of the ultra-small NPs with diameters in the range of 1.3 ± 0.2 to 4.3 ± 1.2 nm. The authors suggested that the longer chains enable the formation of an increased population of nuclei and/or better surface stabilization, thereby hindering the NPs' growth. Moreover, by increasing the milling time, the size of the produced nanoparticles could be increased.97 These results agree with the previous data concerning the effect of PVP chain length in the size control of Au NPs.67
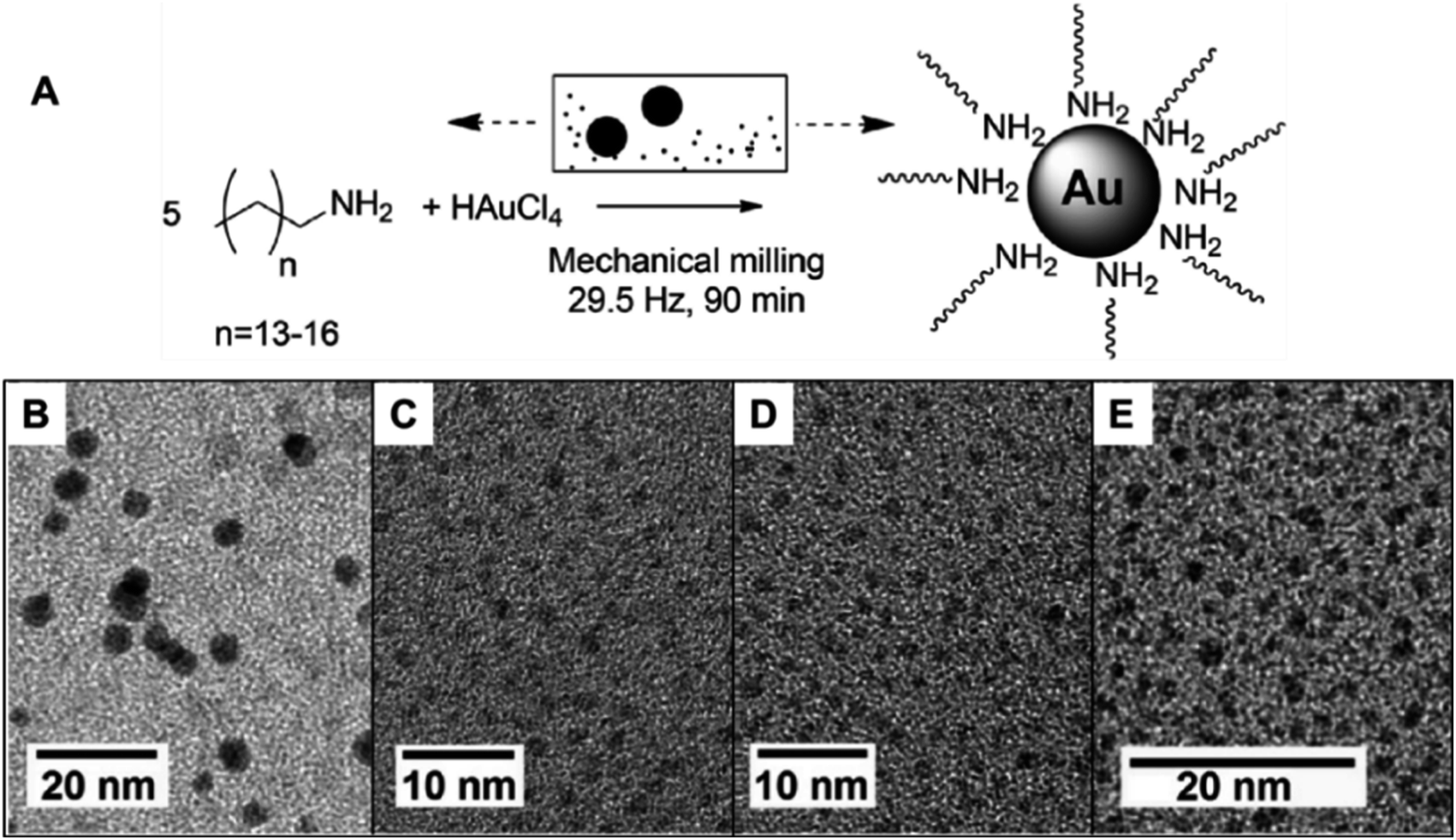 | ||
| Fig. 3 Mechanochemical synthesis of ultra-small Au NPs (1–4 nm) using amine ligands. The syntheses were carried out in a stainless-steel milling set that could be responsible for the reduction of gold salt through galvanic replacement. (A) Scheme of the synthetic procedure and (B–E) TEM images of the formed nanoparticles with different aliphatic amines: (B) pentadecylamine, (C) hexadecylamine, (D) heptadecylamine and (E) octadecylamine. Adapted from ref. 97 with permission of the Royal Society of Chemistry. | ||
During the preparation of the ultra-small NPs, it was noted that the use of a typical reducing agent was not required. This was due to the use of a stainless-steel milling set-up. When the mechanochemical synthesis was performed in this metal assembly, a galvanic replacement reaction took place between the metal salt precursor and the assembly. This galvanic process was responsible for the reduction of Au oxidized species. This could be an advantage to avoid or simplify the purification steps for the generated NPs. The size of the final Au NPs was determined using a combination of MALDI-TOF MS and TEM. According to MALDI-TOF MS, clusters were found to have sizes corresponding to Au59, A68, and Au76. UV-Vis also corroborates the small sizes as observed by the absence of LSPR of Au for particles smaller than 2 nm.97
Although control over size with limited monodispersity has been achieved via milling, control over shape remains limited and much more challenging. In this context, we recently described an approach combining mechanochemical and colloidal synthesis to allow for the formation of Au nanotadpoles, Fig. 4.98 This method allows utilizing the attractive features of both mechanochemical and colloidal syntheses, being comprised of two main stages. Firstly, mechanochemistry is employed to generate spherical NPs seeds (Fig. 4A). Then, a mixture containing these NPs seeds and the unreacted precursor undergoes a seeded growth stage in solution. This permits the separation between nucleation and growth in the second step. As a result, one can have better control over the size distribution and shape of the resulting nanostructures (which is often a limitation for shape control by mechanochemical synthesis). Moreover, the first mechanochemical step confers simplicity, scalability, and robustness to the synthesis (which is often challenging in colloidal methods). It follows that this mixed approach, therefore, allows combining the strengths of both the mechanochemical and colloidal synthetic strategies.98
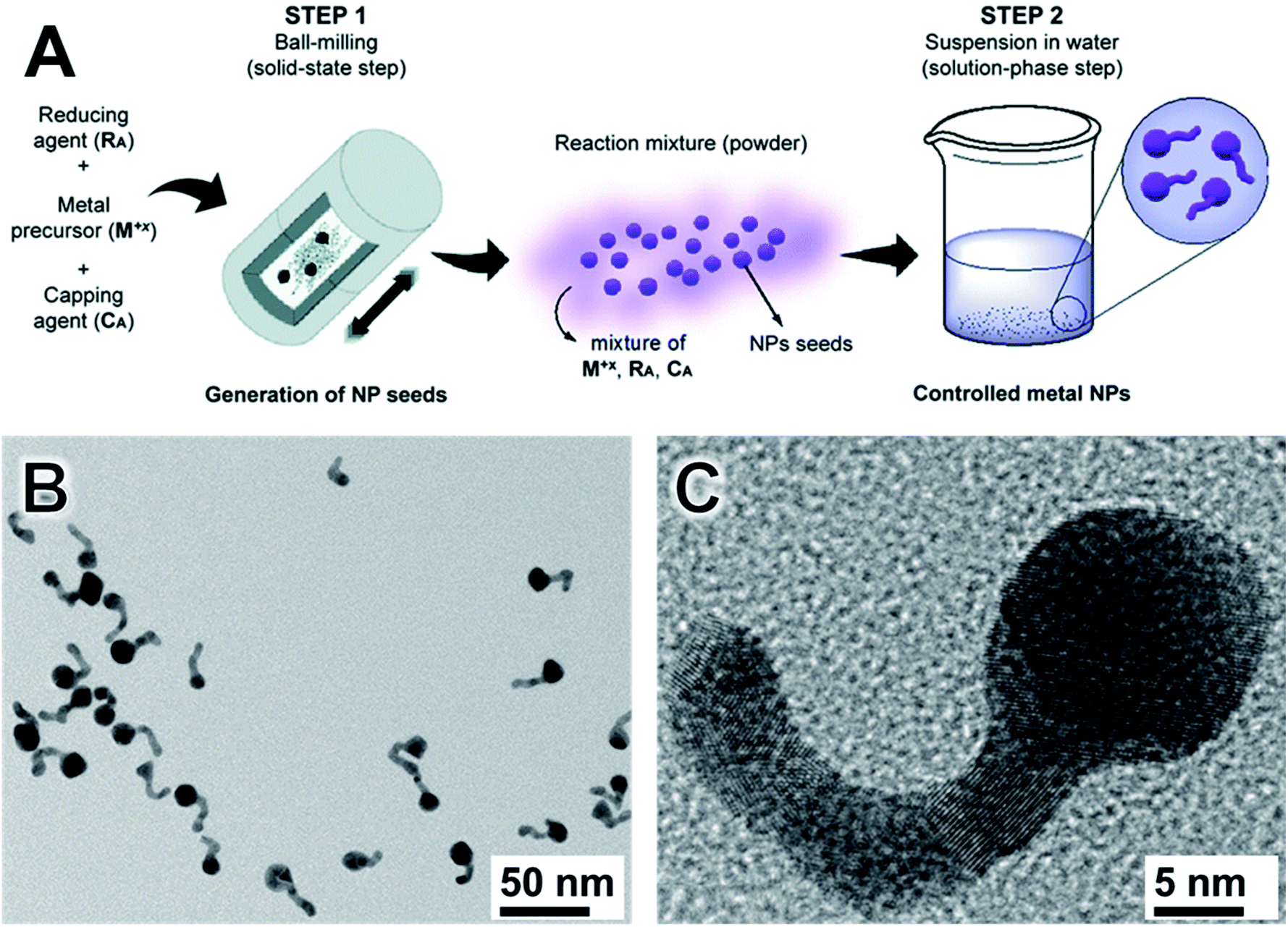 | ||
| Fig. 4 Two-step mechano-colloidal approach for the synthesis metal nanoparticles. (A) In the milling step, Au seeds are generated. When the reacting solid mixture is dispersed in water, the remaining salt precursor is reduced forming small nanoparticles that originate the tails by chaining and fusion. (B and C) TEM images of Au nanotadpoles synthesized by the methodology showing good size and shape control. Adapted from ref. 98 with permission of the Royal Society of Chemistry. | ||
By employing the synthesis of Au NPs as a proof-of-concept example, we demonstrate that this mechano-colloidal approach can lead to the synthesis of Au nanotadpoles (Fig. 4B and C) at room temperature by employing AuCl, sodium citrate, and polyvinylpyrrolidone (PVP) as starting materials. Here, the dispersion in water enabled the growth of the tails by progressive self-nucleation, linear agglomeration, and fusion of new particles. The final Au nanotadpoles displayed a nice dispersion and a defined morphology consisting of a round head of diameters ranging from 10–18 nm, and tails that were up to 22 nm in length and 5 nm in width. This procedure was performed in PTFE jar with zirconia balls to avoid any metal contamination. The reactions were studied by XRD, diffusive reflectance UV-Vis, and X-ray absorption spectroscopy in the XANES region.98
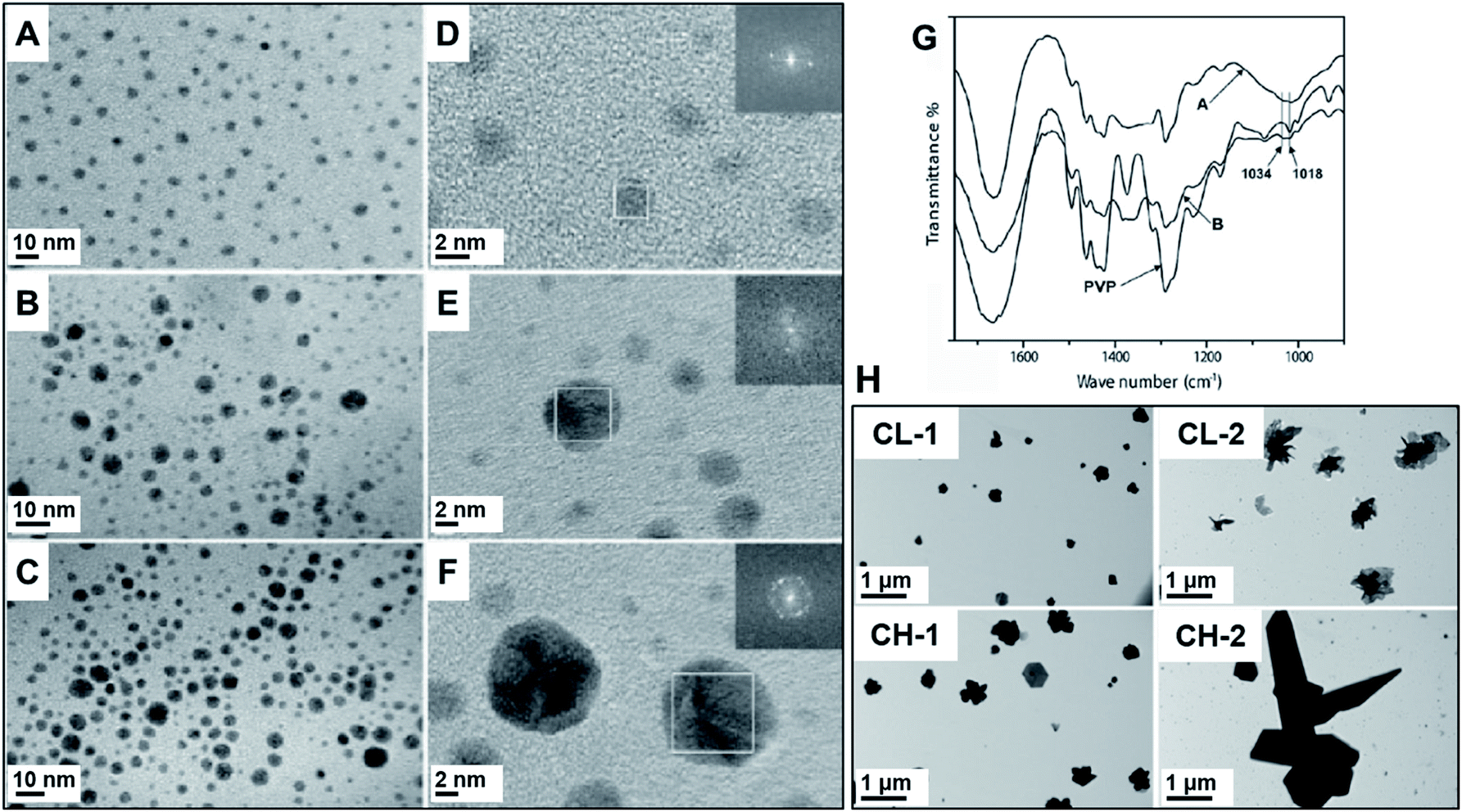 | ||
| Fig. 5 (A–F) PVP-capped Ag NPs. PVP was used as a capping and reducing agent. Ag NPs were synthesized in a vibratory mill (MM200, Retsch) using an agate milling set. Left: TEM images of samples prepared using different concentrations of AgNO3 (A/D, 15 mg, B/E, 30 mg, and C/F, 50 mg) for the same mass of PVP (100 mg). (G) FTIR spectra of the samples prepared using 15 mg of silver salt (sample A) and 30 mg (sample B) in comparison to pure PVP. There is an indication of participation of the pyrrolidyl nitrogen in the formation of the nanoparticles as can be seen by the shifts in the C–N vibrations at 1018 cm−1 for pure PVP to 1034 cm−1 when Ag nanoparticles are formed. Adapted from ref. 66 with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (H) TEM images of Ag NPs prepared by using chitosan, which also is responsible for the reduction of AgNO3 precursor. CL (chitosan low molecular weight) and CH (chitosan high molecular weight). The Ag NPs show unusual aggregated flake-shaped particles obtained along with some small nanocrystals. They were synthesized in a vibratory mill (MM200, Retsch) with a stainless-steel assembly. The syntheses were performed using 10 and 20 mg AgNO3 for CL-1/CH-1 and CL-2/CH-2, respectively. Adapted from ref. 100 with permission of the Society of Chemical Industry. | ||
The same group also studied different polymers in the ball milling synthesis of Ag NPs for biomedical applications as the anticancer agents.100 In addition to PVP, other biocompatible polymers including poly(ethylene glycol) (PEG) and chitosan were investigated. The mean size of the resulting Ag NPs ranged from 3.1 ± 1.4 to 23 ± 5.8 nm as a function of polymer composition and AgNO3:polymer mass ratio. The use of chitosan, for example, resulted in the formation of large aggregated flake-shape particles (Fig. 5H, CH, and CL samples). The formation of these anisotropic nanostructures was explained based on the reducing and low stabilization capability of chitosan, which contains different functional groups with lone-pair electrons capable of reducing Ag+. This presumably facilitates rapid reduction, thereby allowing particles to grow randomly as larger crystals, culminating in aggregated flake-like microparticles.100 Remarkably, both PVP66 and PEG-capped.100 nanoparticles were stable over one year period.
Very recently, PVP was also used as a simultaneous capping and reducing agent in the mechanochemical preparation of Ag@polypyrrole core–shell nanocomposites.101 The first step of the synthesis consists of the formation of Ag NPs by manual grinding AgNO3 and PVP. After an established time, the pyrrole monomer was introduced in different amounts and polymerized in the powder mixture. The obtained Ag NPs presented a semi-spherical shape with sizes from 35 to 44 nm and free from larger agglomerates. The core–shell morphology was formed through electrostatic interaction of the negatively charged carbonyl group of PVP and the polycationic polypyrrole. The core–shell material showed increased optical conductivity as a function of the pyrrole fraction due to the amplified polymerization of conducting polypyrrole.101
The effect of the molecular mass of polymeric stabilizers was again observed by using PEG-6000 and PEG-8000.102 Smaller spherical NPs around 10 nm in diameter were obtained by milling while employing the polymer with the longest polymeric chain. The steric stabilization was efficient even after 24 h of milling. The resulting Ag NPs exhibited very good antibacterial properties and good stability.102 The efficient use of low molecular mass polymers was also demonstrated in a different study.103 Low molecular mass PEG-400 was used for the synthesis of Ag NPs in the presence of NaH2PO2·H2O as a reductant.103 The reaction was conducted by manual grinding yielding a 3D coral shaped Ag–Ag homojunction architecture (Fig. 6A and B). This atypical assembly consists of quasi-spherical and irregular polyhedral nanoparticles (35–50 nm diameter) that aggregate through Ag–Ag homojunction (25–45 nm width) into 1D chains. The formation mechanism of theses shapes was proposed based on a template induction by the PEG linear chain molecule. The oxygen atoms of the PEG backbone provided the electrons to coordinate Ag+, which were reduced by the low valent phosphorous. Ag nucleated at these coordinating sites and grows along the polymer chain. Meanwhile, the free Ag+ species were also reduced and added to the growing NPs by atomic addition. The adjacent NPs joined each other along the polymer chain to form the homojunction by sharing the (110) crystalline plane. The NPs could further grow and aggregate via Ostwald ripening as shown in Fig. 6C. The resulting Ag NPs chains were tested as SERS substrates for the detection of rhodamine 6G showing an enhancement factor of 100 times stronger compared to the individual Ag NPs. This effect was attributed to the presence of the NPs junctions that enabled the generation of many electromagnetic hot spots.103 The Ag–Ag homojunction were stable under SERS tests and also under storage for more than two years.
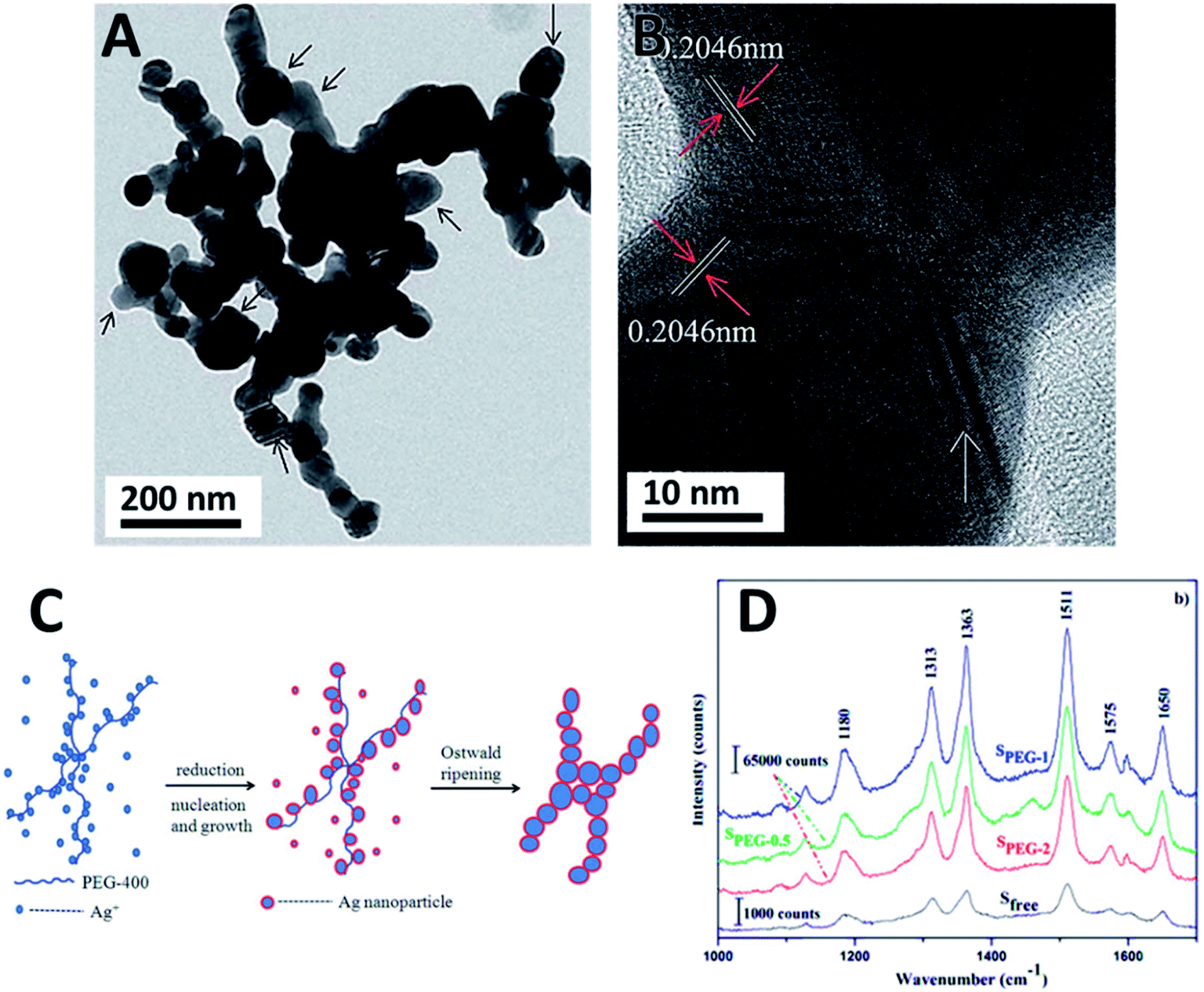 | ||
| Fig. 6 Ag NPs synthesized by manual grinding using PEG-400 as a surfactant. (A and B) TEM and HRTEM of Ag homojunction assemblies. (C) Proposed mechanism of formation of homojunction assemblies and (D) Raman spectra of rhodamine 6G in different substrates showing the SERS effect of the substrate over intensity: Sfree (AgNPs synthesized in the absence of PEG), SPEG-0.5, SPEG-1 and SPEG-2 (volume of PEG:AgNO3 mass ratio of 0.5, 1.0 and 2.0 respectively). Reproduced from ref. 103 with permission of the Royal Society of Chemistry. | ||
Other silver precursors have also been employed in the mechanochemical synthesis of Ag NPs.104,105 For instance, milling Ag2O with graphite afforded highly polydisperse NPs with particle size distribution ranging from 10 to 50 nm after 22 h of milling in a planetary mill using a steel milling jar.104 A mixture of Ag2O and Ag and unknown products according to XRD were obtained in a different study.105 More recently, monodispersed Ag NPs (4.04 ± 0.45 nm) were synthesized using a silver thiolate as a starting material.99 By manually grinding the metal precursor with NaBH4 in an agate mortar, the metal thiolates precursor were reduced to generate Ag and thiol stabilizing agents. The thiol molecules are responsible to control the particle growth and to stabilize the Ag NPs. This approach was also demonstrated for Au NPs, which presented two-family sizes: 2.0 ± 0.4 and 17.4 ± 5.7 nm. Interestingly, by grinding the metal-thiolate with octyl dithiocarbamic acid as sulfur source yielded the respective metal sulfites. This was mainly used for the preparation of semiconducting PbS, CdS, ZnS, and CuS NPs from Pb, Cd, Zn, and Cu thiolates, respectively.99
Other thiolates have been used in the synthesis of Ag clusters by simple manual grinding.107,108,120 The synthetic procedure afforded Ag9,120 Ag32 (ref. 107) and Ag152 (ref. 108) clusters by using mercaptosuccinic acid (H2MSA), glutathione, or N-(2-mercaptopropionyl)glycine and phenylethanethiol, respectively. The syntheses were performed by milling AgNO3 and the thiol-terminated molecules to form the Ag-thiolate precursor, which was followed by the addition of a considerable molar excess of NaBH4 as reductant. A higher nucleation rate, accompanied by the slower ion diffusion in the solid-state, was responsible for the formation of the small nanoclusters (in TEM seen as smaller as 1 nm). Besides, the absence of protic solvent contributed to better control over the nucleation and growth stages, thereby minimizing the formation of nanoclusters of different sizes when compared to solution-based approaches.107,120 TEM, XPS, NMR, MALDI MS, and ESI MS were used to characterize the Ag clusters.107,108,120 Ag9(H2MSA)7 clusters presented a remarkable stability for over two years in the solid state or in ethanol and water/solvent mixtures.120 This represents a great benefit of solid state mechanochemical synthesis as a unique alternative in the preparation of stable and ultrasmall nanostructures.
Other strategies have been used in the preparation of Ag NPs. An interesting one is the mechanochemical decomposition of silver oxalate (Ag2C2O4) to generate Ag NPs (Fig. 7A–C).109 The vibratory milling assembly is depicted in Fig. 7B. The Ag2C2O4 stayed settled at the bottom of the milling vessel that contained a PVP solution. As the milling ball hits the powder with a frequency of 0.2 Hz, Ag2C2O4 decomposes by releasing CO2 and forming metallic Ag. The nanoparticles transferred to water could be stabilized by PVP and presented the characteristic LSPR of Ag NPs as confirmed by UV-Vis spectroscopy. TEM images of the particles formed after 50 collisions showed nanoparticles with sizes ranging from 2 to 12 nm (average size around 5 nm) (Fig. 7C).109 This same strategy was used in the decomposition of pure or mixed silver and nickel oxalates.116 When the salts were processed separately or in a simple mixture, Ag and NiO nanoparticles in the range of 2–14 nm and 3–16 nm were formed respectively, as confirmed by TEM and XRD. On the contrary, when the starting salts were prepared by co-precipitation, as the fraction of Ag2C2O4 increased, the resulting mixtures upon decomposition indicated that Ag + NiO, Ag + Ni, and finally Ag–Ni alloy NPs are formed.116 In this and other examples, the kinetics of mechanochemical decomposition of Ag2C2O4 was used to study the powder mass trapped in each collision.121,122 These types of studies are of fundamental importance in the mechanistic comprehension of mechanochemical transformations and helps to differentiate from the thermal mediated events.
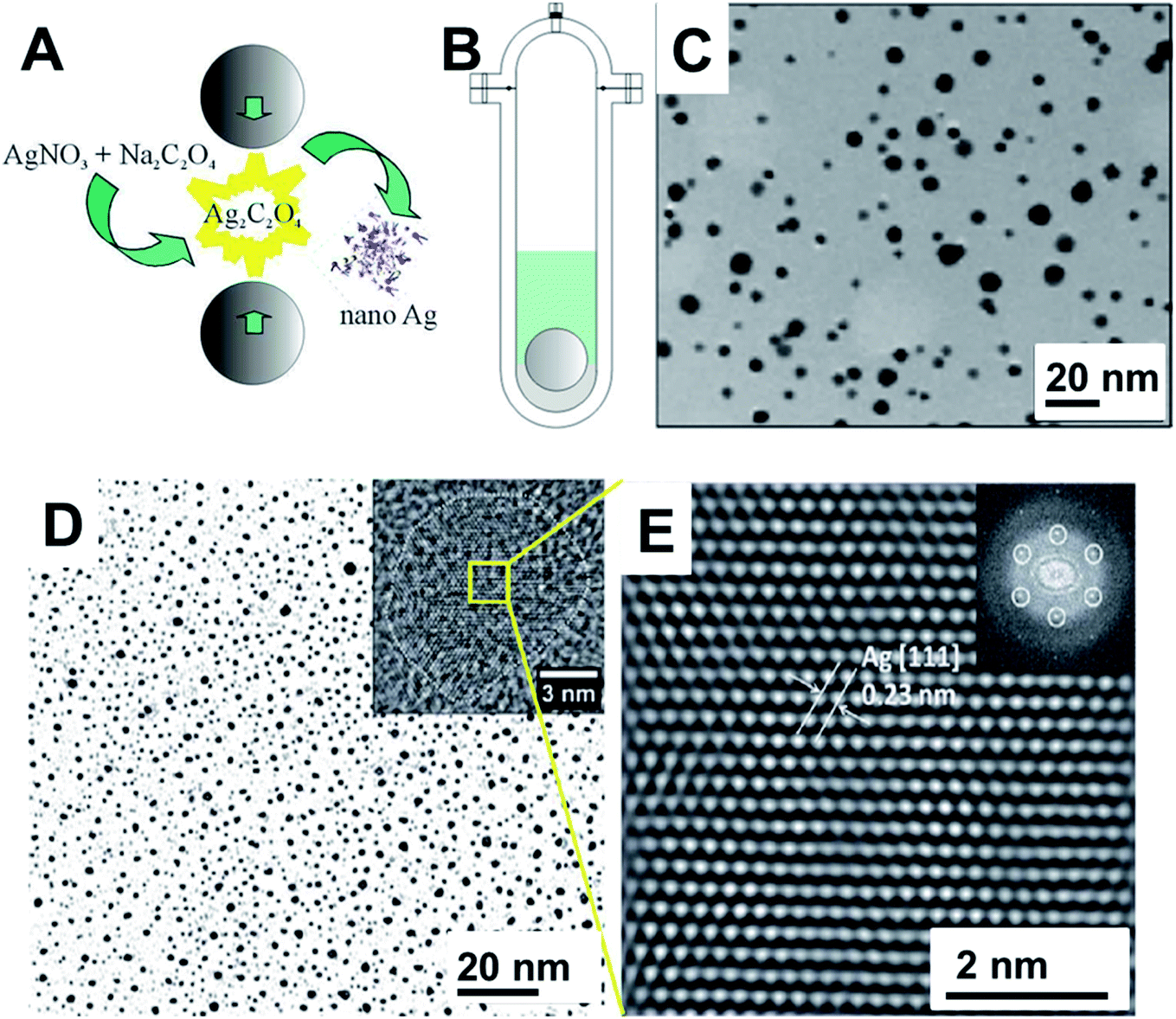 | ||
| Fig. 7 (A) Ag nanoparticles synthesis through the mechanochemical decomposition of Ag2C2O4. (B) Stainless still mechanoreactor used for the synthesis. It consists of a cylindrical reactor with a rounded bottom. A 20 g stainless steel ball was used for a total mass of 2 g of powder and 20 mL of PVP aqueous solution. As the reaction proceeds, the produced Ag NPs were transferred to water and stabilized by PVP. (C) TEM images of the resulting Ag NPs. Adapted and reproduced from ref. 109 with the permission of the American Chemical Society. (D and E) TEM images of Ag NPs prepared by cryo-milling. (D) TEM image of the Ag NPs after 7 hours of cryo-milling. (E) HRTEM image of the Ag NPs and the inset shows the FFT. Ag NPs were obtained from a top-down approach by cryo-milling Ag microparticles. The milling procedure was performed in a lab-adapted Analyzed 3/Pulverisette 0 (Fritsch) with fluxing Ar gas and liquid N2 inlet using a tungsten milling set. Adapted from ref. 118 with permission of the Royal Society of Chemistry. | ||
As an alternative to the above approaches, the use of cryomilling to produce ultrapure, non-coated Ag NPs, using a top-down methodology, has been described.117,118 In an adapted vibrating device that employed liquid N2 and fluxing with Ar gas to avoid Ag oxidation, Ag powder was milled using a tungsten carbide ball. This could lead to the formation of Ag NPs which remained stable for weeks when dispersed in polar solvents due to electrostatic stabilization. TEM images indicated an averaged particle size of 6 ± 2 nm (Fig. 7D) and high crystallinity (Fig. 7E).118 A similar procedure was used for the synthesis of Cu NPs.76
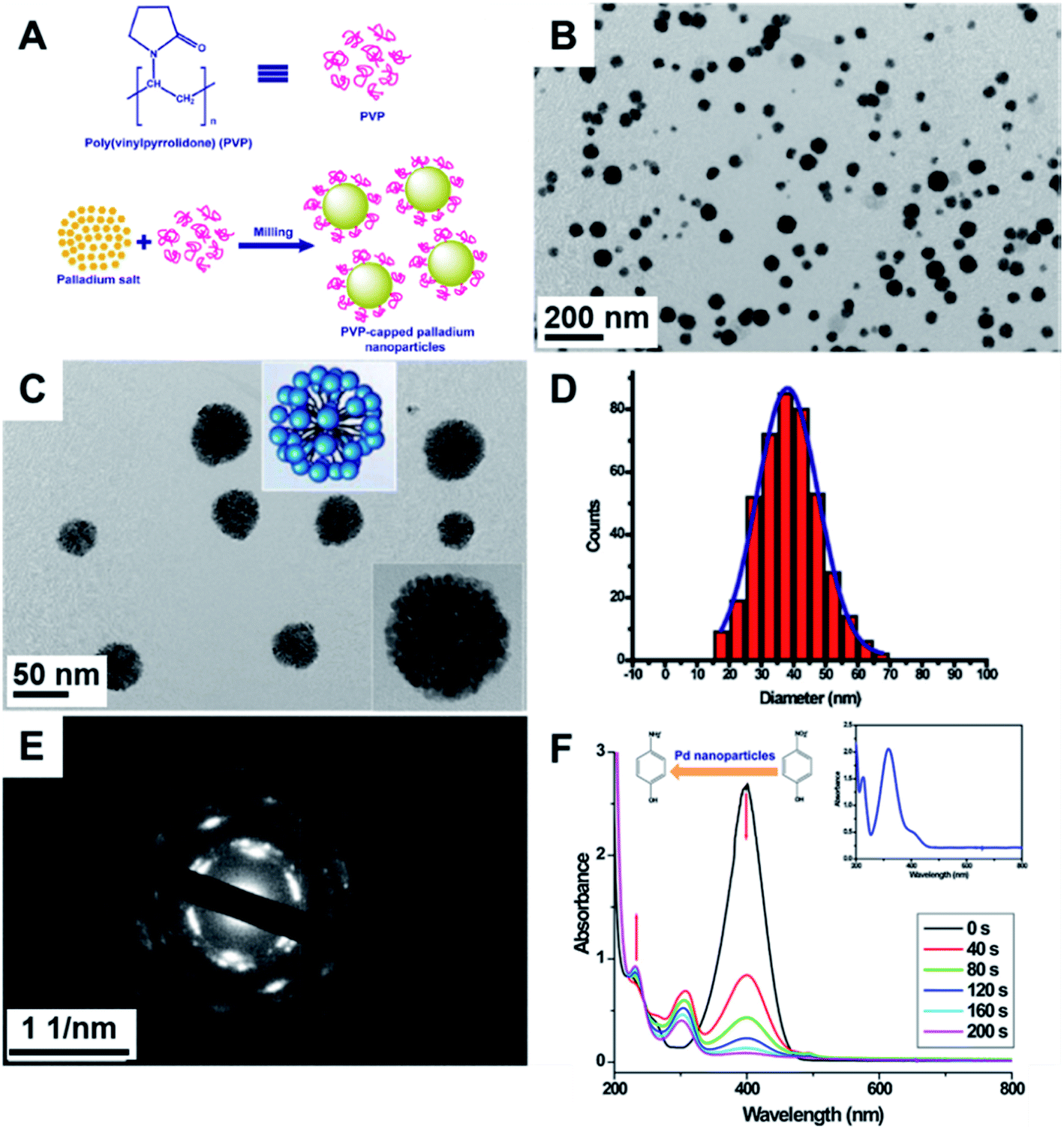 | ||
| Fig. 8 (A) Scheme for the synthesis of Pd NPs via agate milling for 10 min. The K2PdCl4:PVP mass ratio corresponded to 1:10. (B and C) TEM images of the Pd NPs presented a dendritic-like structure made up of smaller NPs. (D) Histogram of particle size distribution. (E) Electron diffraction showing the polycrystalline nature of the NPs, and (F) catalytic reduction of p-nitrophenol by NaBH4. The inset shows UV-Vis absorption spectra of p-nitrophenol. Reproduced from ref. 110 with permission of Elsevier. | ||
3.2. Multicomponent NPs
The preparation of multicomponent materials, such as alloys, is one of the earliest elaborated applications of mechanical milling.48 For such purposes, pure powders of the elements are milled in high-energy milling devices.48 Differently, the bottom-up mechanochemical approaches use the metal precursors and try to control their reduction, nucleation and growth. This intends to enable control over the composition and distribution of the components in the nanoparticle, such as the synthesis of alloys or core–shell NPs.5The control over NP composition – elemental distribution in multicomponent NPs – represents a powerful strategy to control performances. In this sense, manual grinding has been employed for the synthesis of a series of AgnX@Ag (X = Cl−, Br−, I− or CrO42−, PO43−) NPs from silver acetate.112 This protocol was simple and enabled good control of NP composition by changing the molar ratio of (NH2OH)2·H2SO4 to the anions. However, highly inhomogeneous particles were obtained: the Ag NPs in each of the AgnX@Ag composites displayed a broad distribution of shapes and sizes ranging from 40 nm to hundreds of nanometers. The prepared NPs were tested for their use in the photodegradation of methyl orange in aqueous solution, taking advantage of the LSPR effect in the NPs. The composites demonstrated high catalytic activity and stability during the performance tests under visible light irradiation. The type and charge of the anions affected the degradation rate of methyl orange.112
Chitosan-stabilized AuAg alloy NPs were prepared by manual grinding AgNO3 in an agate mortar in the presence of NaOH. This was followed by the addition of small amounts of a concentrated solution of AuCl4− during the grinding (Fig. 9A).111 The LSPR band of the NPs as a function of the composition is in agreement with the formation of bimetallic AgAu NPs (Fig. 9B). Specifically, a linear dependence (red-shift) between the Au content and the LSPR band position was detected.123 It is important to note that the presence of a single SPR peak indicates the formation of a spherical rather than a core–shell or hollow structures, in agreement with the TEM images (Fig. 9C). Increasing the amount of AuCl4− resulted in smaller NPs as indicated by TEM images (Fig. 9C).111 The formation of the AgAu NPs seemed to be a result of the galvanic replacement reaction, in which part of the Ag0 in the preformed Ag NPs was replaced by Au from AuCl4−. This led to the formation of very small Ag NPs that may further recombine to generate the final AgAu particles with the average mean size around 3 ± 0.5 nm. In turn, the etched Ag+ could be reduced again by chitosan chemical groups, generating new nuclei that grew into larger particles, with almost no Au, as supported by TEM and BF-STEM point EDS analysis. It is worth noting that the etching process is a surface driven, charge transfer reaction, where the diffusion of AuCl4− is hindered by the protective chitosan chains. This might be advantageous in the control of the final composition of the NPs. The AgAu bimetallic NPs composed by different Ag:Au molar ratio (Cat-1, 8:1; Cat-2, 3:1 and Cat-3, 2:1.3, respectively) were tested in the catalytic reduction of p-nitrophenol and other nitroarenes by NaBH4. The kinetics of reduction was followed by the absorbance of the substrate by UV-Vis spectroscopy. The studies at different temperatures enabled estimation of the activation energy from Arrhenius plot (Fig. 9D). The catalyst with lower relative Au content presented the best performance (Cat-1, Fig. 9D). The AuAg NPs also exhibited good recyclability and stability, which is of primary importance in catalysis.111
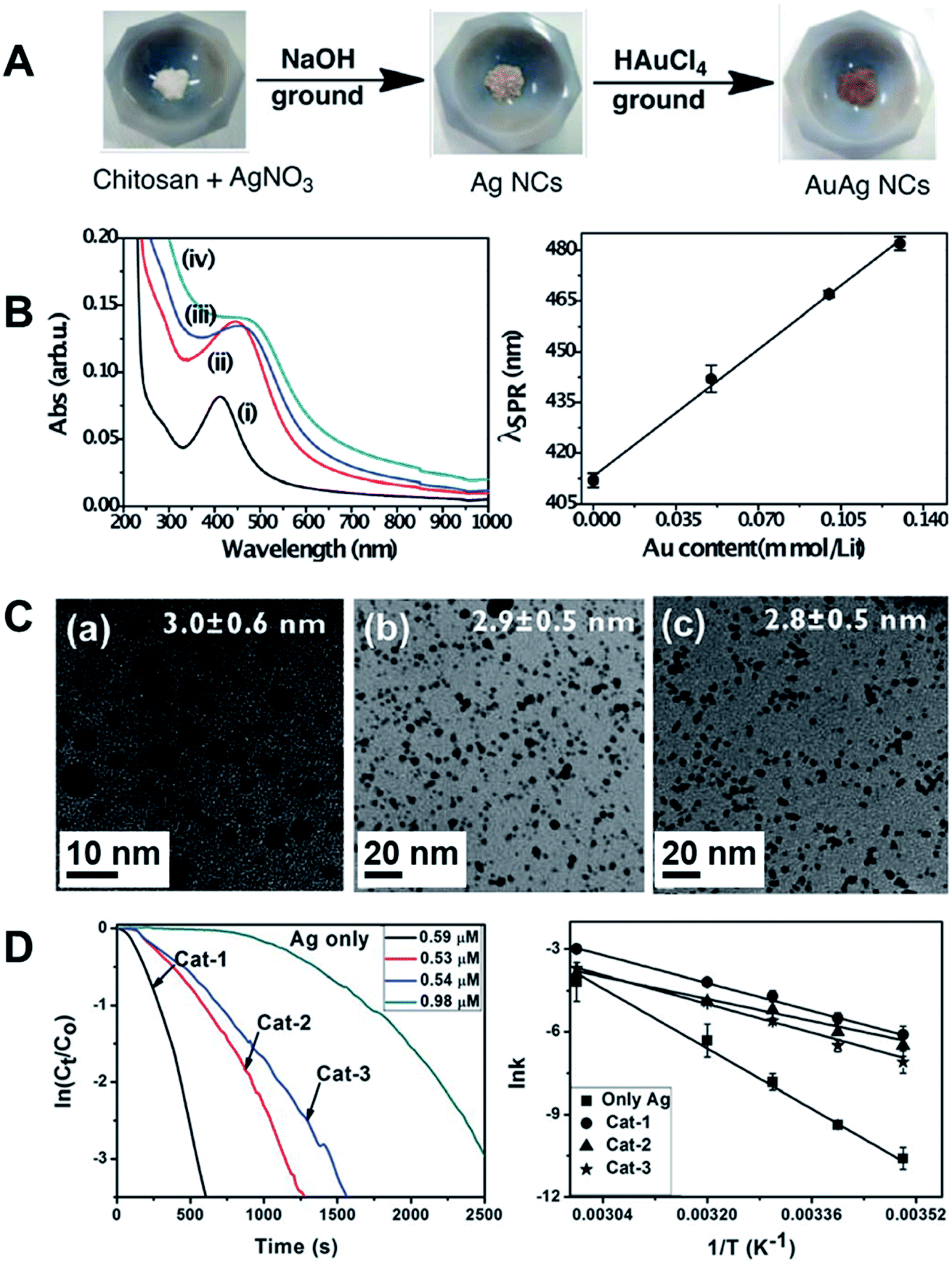 | ||
| Fig. 9 (A) Scheme for the synthesis of AuAg bimetallic alloy NPs by manual grinding using chitosan as a stabilizer; (B) UV-Vis extinction spectra of the nanoclusters composed of (i) pure Ag, and (ii–iv) AgAu bimetallic NCs (Cat-1, Cat-2 and Cat-3) with the following Ag:Au molar ratios: (ii) 8:1, Cat-1; (iii) 3:1, Cat-2 and (iv), 2:1.3, Cat-3. On the right, the linear correlation of the LSPR band position as a function of Au concentration is shown. (C) TEM images and mean particle size of the nanoclusters: (a) Cat-1, (b) Cat-2 and (c) Cat-3. (D) The NPs were tested in the catalytic reduction of p-nitrophenol by NaBH4. Left: Logarithmic concentration of p-nitrophenol as a function of time for different catalysts. Right: Arrhenius plots for reactions catalyzed by Ag and the AuAg NPs. Reproduced from ref. 111 with permission of the Royal Society of Chemistry. | ||
It is noteworthy that there is only a limited number of multicomponent NPs prepared mechanochemically from the reduction of metal precursors, which makes it difficult a rational analysis of the systems. Despite that, the results seem promising. For example, for AgnX@Ag (X = Cl−, Br−, I− or CrO42−, PO43−) NPs, the composition of Ag0 could be controlled by using appropriate amounts of the reductant during the synthesis. The same was not directly possible for AuAg nanoalloys, in which the Au content varied from the corresponding precursor concentration added in the synthesis. Nonetheless, this relatively simple approach was able to yield nanoparticles with final sizes smaller than 4 nm, which is attractive for catalytic applications.
3.3. Overview and comparison among metal NPs
Overall, the bottom-up mechanochemical synthesis of non-supported noble metal NPs share some common aspects regardless of the metal. Except top-down approaches,118 in all cases, organic ligands and polymers have been used to stabilize the systems. PVP is a well-known surfactant in colloidal chemistry and it has been also successfully used in solid state mechanochemistry for Au, Ag and Pd NPs synthesis as discussed above. However, its use seems to be more effective for NPs bigger than 5 nm. On the other hand, the preparation of ultrasmall nanoparticles and clusters has been achieved by using amine97 and thiolate-based molecules as stabilizing agents.107,108,120 This is a great advantage for application purposes that demands higher surfaces, such as catalysis. In terms of the synthetic protocols, both ball milling and manual grinding have been successfully employed. While ball milling allows a better reproducibility and gram-scale synthesis,109 manual grinding has enabled the preparation of monodispersed Ag nanoclusters when associated with organic precursors.107,108,120 Interestingly, Ag particles can take unexpected arrangements by using chitosan100 and low molecular mass PEG,103 while for Au this was only detected when associated with solution so far.98 Another important aspect is the use of the milling assembly also as reactant for the synthesis of Au NPs, enabling galvanic replacement and the reduction of the metal precursor without any additional capping agents.97 Regarding the multicomponent systems, a better control of composition is expected by using automated milling devices and compatible reducing agents.There are still several issues that still need to be addressed in the mechanochemical synthesis of noble-metal NPs when we compare with the current state of development of solution-phase synthesis. Specifically, it is known that: (i) the reactivity of solid reductants is different as compared to the solution phase (it remains poorly understood how different reducing agents affect the kinetics formation and shape of the resulting NPs);98 (ii) diffusion processes in the liquid and solid phases are changed; (iii) the interaction of surfactants or stabilizers with the growth species in the mechanochemical synthesis should also be altered relative to the solution phase, and (iv) the nature of the metal precursors is different. While in the solution phase synthesis of metal NPs precursors can dissociate or form dimers in solution before reduction or decomposition to generate the growth species, these processes do not occur in mechanochemistry and can lead to significant changes in reactivity, reduction kinetics, and morphology/size of generated nuclei.
Due to these challenges, the types of noble metal NPs that can be produced via mechanochemical synthesis remains limited to only a few systems, spherical morphologies, and in terms of control over size, monodispersity, and composition (bimetallic NPs, core–shell, and alloys). Also, most of the mechanochemical synthesis of metal NPs have been focused or restricted to systems for catalytic applications, although some examples of sensing via surface-enhanced Raman scattering have also been described. Therefore, it becomes clear that it is essential to shedding light on the mechanisms of NPs formation and therefore enable the development of methods for the tightly controlled synthesis of noble metal NPs by mechanochemical routes to increase the scope of materials, NPs shapes, and sizes, and applications.
4. Synthesis of supported noble metal NPs
One of the greatest advantages of nanocatalysts is the ease in recovery and reuse. Ideally, the surface of the catalysts must be fully available to reach higher activities. Ensuring surface availability is can be an issue in nanosized materials that are traditionally synthesized in the presence of stabilizing capping agents. Often, high temperatures or several washing steps can be employed to remove these surface agents. However, these treatments can result in the modification of the nanostructures. One alternative is to synthesize the active material already on a solid support that can stabilize the NPs, thereby eliminating the use of additional stabilizers and impregnation steps. In this context, the mechanochemical synthesis of supported NPs has emerged as an attractive strategy. Table 3 displays the selected examples of supported NPs synthesized entirely or partially mechanochemically over a range of supporting materials (bio-, SiO2−, carbon-based materials and porous coordination polymers).| System | Precursor | Reducing method | Support | Synthesis conditions | Final products | Conversion or yield | Stability comments | Ref. |
|---|---|---|---|---|---|---|---|---|
| Au | AuCl3 | NaBH4 | Cyclodextrins | Ball milling, zirconia set, 30 Hz, 5 min | 1.54 ± 0.4 nm (for β-cyclodextrin), (5 wt% AuCl3 → 3.25 wt% Au) | Complete conversion according to XPS | TEM shows morphological changes after 3 cycles of catalytic tests | 124 |
| HAuCl4 | NaBH4 | SiO2-Schiff base | Wet impregnation + manual grinding, agate set, 0.5–3 h | NPs ∼ 1 nm, nanoclusters and single atoms, 0.07–1.48 wt% Au | Complete conversion according to XPS | — | 125 | |
| HAuCl4·xH2O | Reducing groups in lignin backbone | Lignin | Ball milling, stainless steel set, 29.5 Hz, 90 min | AuNPs in lignin, 14.8 ± 6.8 nm | Complete conversion according to XPS | — | 126 | |
| Au(III)acetate | Decomposition | MWCNTs | Ball milling, zirconia set, 10 min | AuNPs (∼3 nm) over MWCNTs | — | — | 89 | |
| HAuCl4·3H2O | Milling + laser sintering | Porous glass | Ball milling, zirconia set (30 Hz, 60 min) + laser sintering | AuNPs of 30 nm dispersed in the glass matrix ∼ 1 wt% Au | — | Stable during catalytic test | 127 | |
| Au(III)dimethyl(acetylacetonate) | H2 | Fluorinated Cd-based MOF | Hand grinding, agate set (30 min) + heat treatment in H2/He atmosphere | Highly dispersed Au clusters (Au2, Au3 based on EXAFS), ∼1 wt% Au | — | Stable in air for six months | 128 | |
| ZIF-8 | Hand grinding, agate set (35 min) + heat treatment in H2/He atmosphere | AuNPs of 3.4 ± 1.4 nm (1 wt% Au) and 4.2 ± 2.6 nm (5 wt% Au load) | — | Stable during catalytic test | 129 | |||
| MOF-5 | Hand grinding, agate set (20 min) + heat treatment in H2/N2 atmosphere | AuNPs of 3.3 ± 2.3 nm, 2 wt% of Au load | — | — | 130 | |||
| CPL-2 | Uniform cluster size (2.2 ± 0.3 nm), 2 wt% of Au load | — | 130 and 131 | |||||
| Al-MIL-53 | Au clusters of 1.5 ± 0.7 nm, 2 wt% of Au load | — | 130 and 131 | |||||
| Ag | AgNO3 | — | Eggshell membrane | Ball milling, tungsten carbide set, 500 rpm, 30 min | 3.9 ± 1.1 nm | Complete conversion by XRD | — | 132 |
| — | Wet milling, steel set, 5 h | 10–15 nm, 9.72 wt% Ag (XPS) | Complete conversion by XRD | — | 133 | |||
| Carboxylic acid groups | Wet milling, steel set, 2 h | 5–20 nm | Complete conversion by XRD | — | 134 | |||
| Reducing groups in Origanum vulgare L. plant | Origanum vulgare L. plant | Ball milling, tungsten carbide set, 500 rpm, 45 min | Bimodal distribution, 2.9 ± 1.3 and 34.4 ± 16.1 nm | Complete conversion by XRD | — | 132 | ||
| Reducing groups in lignin backbone | Lignin | Ball milling, stainless steel set, 30 Hz, 90 min | AgNPs (1–30 nm) on the surface of lignin. A better distribution within the matrix is possible by addition of polyacrylamide | XPS and XRD supports Ag0 only | Stable | 135 | ||
| Ag(I)acetate | Decomposition | MWCNTs | Ball milling, zirconia set, 5–30 min | Mostly Ag single crystals, mean size 2.5 nm with narrow distribution | 40–60% (10 min milling) | — | 89 | |
| AgNO3 | Mechanochemical decomposition + thermal | Al-SBA-15 | Ball milling, stainless steel set (350 rpm, 10 min) + thermal treatment in air | AgNPs (mean size 5 nm) inside and outside the channels of Al-SBA-15, 1.9 wt% Ag (ICP-AES) | XPS indicates only Ag0 | Stable under catalytic tests | 136 | |
| Pd | Pd(II)acetate | Reducing groups in lignin backbone | Lignin | Ball milling, stainless steel set, 29.5 Hz, 90 min | PdNP throughout lignin, 2.8 ± 0.7 nm | Pd0 + PdO (XPS) | — | 126 |
| Pd(II)acetylacetonate | PdNP throughout lignin, 3.6 ± 0.9 nm | Pd0 + PdO (XPS) | — | 126 | ||||
| PdCl2 | PdNPs outside lignin matrix, 3.2 ± 1.6 nm | Pd0 + PdO (XPS) | — | 126 | ||||
| Pd(II) acetate | Decomposition | MWCNTs | Ball milling, zirconia set, 10 min | PdNPs (∼3 nm) homogenously distributed over MWCNT surface | — | — | 89 | |
| Pd(II) acetate | Mechanochemical decomposition + H2 | Al-SBA-15, Al-MCF | Ball milling (5 min, 350 rpm) + thermal treatment in N2/H2 atmosphere | PdNPs onto the support's surface and in the pores, 0.2–2.1 wt% Pd | — | High stability of recyclability | 137 | |
| PdCl2 | H2 | ZIF-8 | Multiple ball milling treatments (2 h + 0.5 h, 40 Hz) + thermal treatment in H2 | Pd@ZIF-8. PdNPs of ∼4.5 nm, 1 wt% Pd | Pd0 by XPS | Stable under catalytic tests | 138 | |
| Pt | Pt(II)acetylacetonate | Decomposition | MWCNTs | Ball milling, zirconia set, 10 min | AuNPs (∼3 nm) over MWCNTs | — | — | 89 |
| PtCl2 | H2 | ZIF-8 | Multiple ball milling treatments + thermal treatment in H2 | Pt@ZIF-8. PdNPs not determined | — | — | 138 | |
| AuPd | Au(III)acetate, Pd(II) acetate | Decomposition | TiO2 | Manual grinding (1–20 min) + thermal treatment in He | — | AuNPs > 20 nm, AuPd alloys 2–5 nm (2.4 wt% Au, 2.5 wt% Pd) | — | 139 |
4.1. NPs supported on bio-based materials
Saccharide additives have been used in the synthesis of Au NPs employing NaBH4 as a reducing agent in a vibratory ball mill with a zirconia milling assembly.124 The hydration degree of the additives affected the kinetics of NPs formation, showing that water played an important role for NaBH4 dissociation. The best result in terms of NPs size and dispersion (1.54 ± 0.4 nm) within the solid matrix was achieved with native β-cyclodextrin (β-CD), as showed in the TEM images (Fig. 10A). The saccharides played the role of both stabilizing support for Au NPs and as a selective supramolecular structure for the subsequent applications. The prepared materials were used as nanocatalysts without further treatments towards the reduction of nitrobenzene derivatives by NaBH4 (Fig. 10B). Here, the catalytic investigations were also carried out under mechanochemical conditions. The conversion of the nitrobenzenes was chemically selective towards the amino derivatives regardless of the cyclodextrin additive. Host–guest supramolecular interactions strongly affected the catalytic performances. The dynamics of exchange of the substrate and product within the CDs cavities determined the conversion for 15 min of milling (Fig. 10C). Besides, this dynamic was also influenced by the nature and the relative position of the halogen in the phenyl ring. para-Isomers fit well within the CD cavity. This led to stronger interactions which slowed the product–substrate exchange, poisoning the site and resulting in lower conversions. On the other hand, meta and ortho-substituted halogenonitrobenzenes, which have a poor fit and consequently can quickly leave the reaction site, favor the conversion (Fig. 10D). Despite some morphological changes in the AuNPs seen by TEM after the tests, the catalysts still displayed a good performance after recycling.124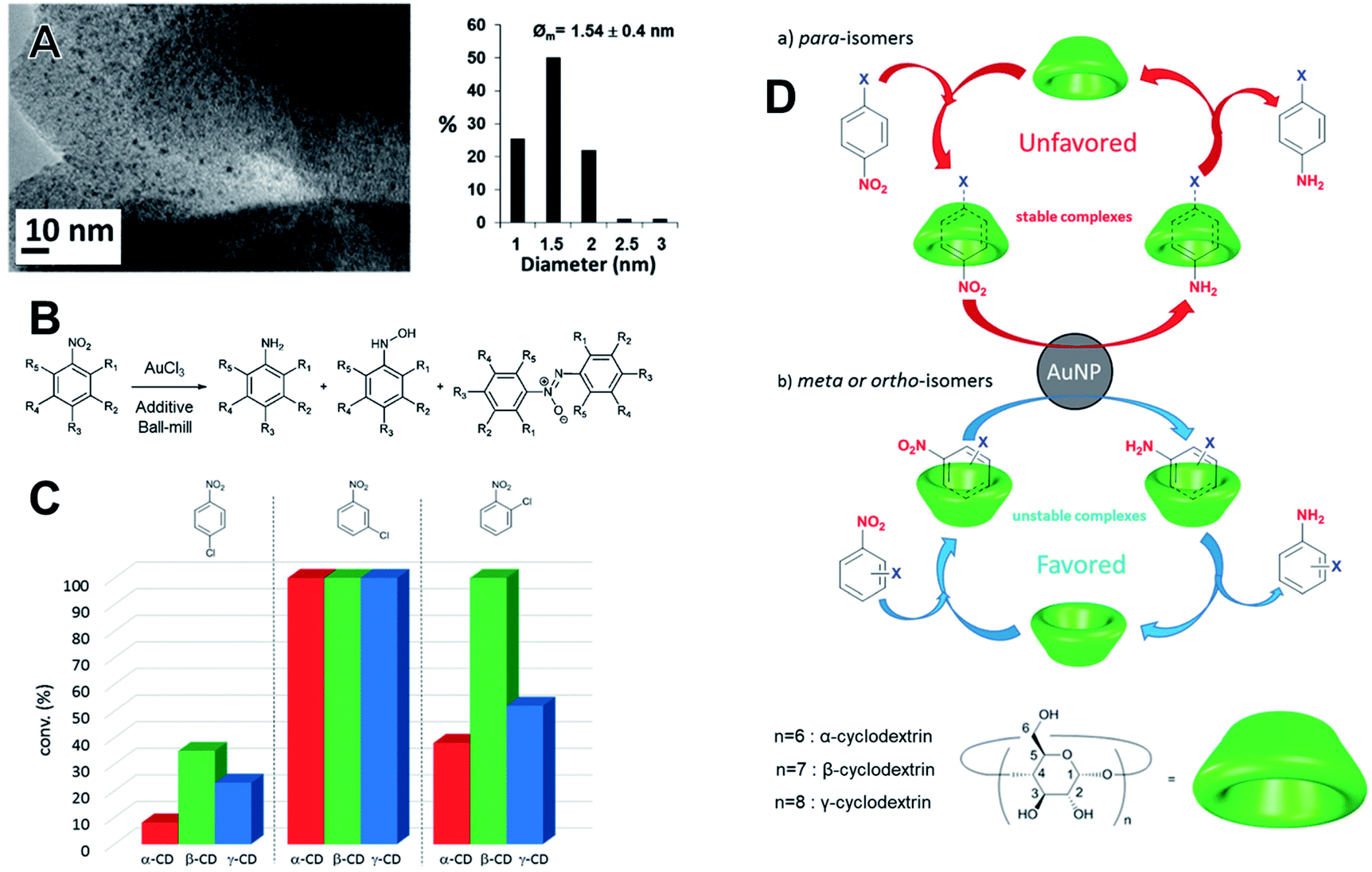 | ||
| Fig. 10 (A) TEM and histogram of size distributions of Au NPs stabilized by saccharide β-cyclodextrin. The Au NPs were prepared by direct reduction of AuCl4− precursor over the saccharide using NaBH4 as the reductant in a vibratory mill (MM400, Retsch). (B) The as-prepared materials were employed as nanocatalysts in the reduction of substituted nitrobenzene derivatives. The reactions were conducted in the same milling device in the sequence of AuCl4− reduction over the cyclodextrins. (C and D) The conversion could also be tuned as a function of halogenonitrobenzene derivative as well by the dynamic of exchange in the host–guest complex. Adapted and reproduced from ref. 124 with permission of the Royal Society of Chemistry. | ||
The use of biomass as support for nanoparticles represents a very attractive approach. For instance, lignin,126,135 eggshell,132–134 and plant biomass132 have been reported as solid support for noble metal NPs in mechanochemical synthesis. At the same time, the reducing groups present in these materials were able to convert Ag+ to Ag0. For example, eggshell membrane and Origanum vulgare L. plant were employed in the synthesis of Ag NPs for application as antibacterial agents.132 The NPs were synthesized in a planetary mill using a tungsten carbide assembly. The characterization of the materials by UV-Vis spectroscopy, TEM, and XRD confirmed the formation of nanosized Ag. While using eggshell membranes led to the formation of smaller Ag NPs (3.9 ± 1.1 nm in size, Fig. 11A and C), the use of plant biomass led to a bimodal particle size distribution around 2.9 ± 1.3 and 34.4 ± 16.1 nm (Fig. 11B and D). This indicates the enhanced capacity of the former to act as a stabilizing matrix. The agglomerates observed in the case of the plant matrix can also be a result of the reducing kinetics as well as of a more heterogeneous distribution of the reducing sites present in this material. The antibacterial activity of the NPs was tested against different bacterial cultures (Fig. 11E). The results indicated preferential activity of Ag NPs supported in the Origanum vulgare L. plant, even if the NPs were bigger. The authors attributed this effect to the Ag NPs being exposed on the surface (the case of the agglomerates) making them more available to act as antibacterial agent.132 The use of eggshell in the reduction of silver nitrate to yield Ag NPs/eggshell composites was also the subject of other reports that further demonstrated the antibacterial activity of the materials prepared under ball milling conditions.133,134
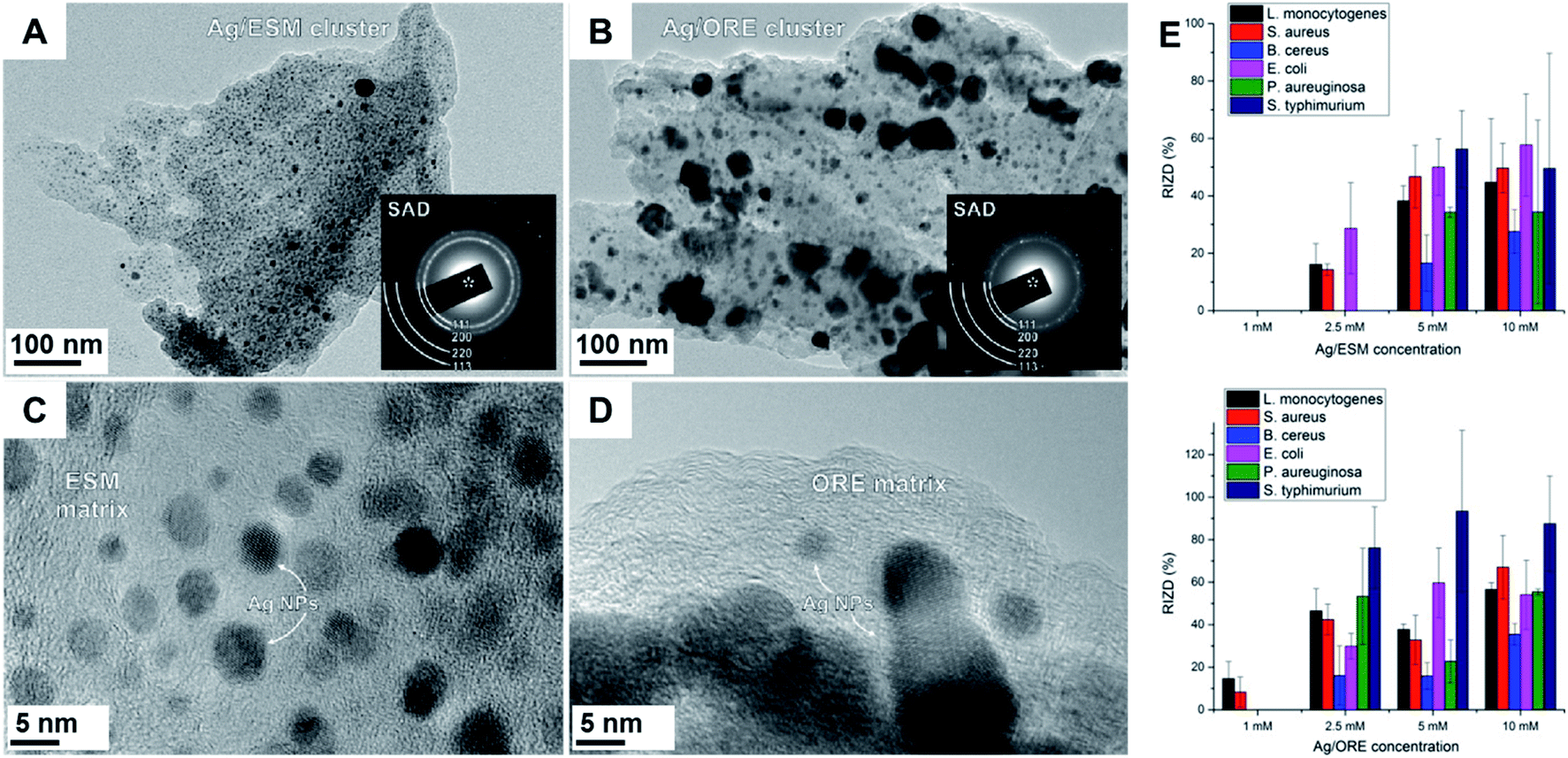 | ||
| Fig. 11 Ag NPs synthesis using biomaterials as reducing agent and support. TEM images of Ag/eggshell membrane (A and C) and Ag/Origanum vulgare L. plant (B and D) composites. Selected area electron diffraction (SAD) patterns, shown in the insets, reveal the crystallinity of Ag NPs in both samples. Milling procedures carried out in a planetary mill with a tungsten milling set. (E) Antibacterial activity of the Ag/ESM (top) and Ag/ORE (bottom) samples against different species. RIZD (%) represents the percentage of relative inhibition zone diameter. Reproduced from ref. 132 with the permission of Elsevier. | ||
Lignin has attracted considerable attention as solid support for the immobilization of NPs.126,135 In this sense, the mechanochemical synthesis of noble metal NPs with lignin are also studied (Fig. 12A). For example, by using AgNO3 as a salt precursor, antibacterial filters based on Ag NPs stabilized in the lignin matrix under ball milling conditions were prepared.135 The final Ag NPs displayed high polydispersity with sizes in the range of 1 to 30 nm, but the composites were stable. The reducing groups present in the lignin polymeric chain were responsible for the formation of Ag NPs. No significant effect was observed by changing the lignin employed in the synthesis (kraft or thermal-treated). Moreover, the Ag NPs produced by milling with lignin were primarily observed to form on the surface of the matrix or were otherwise completely detached, as illustrated by the TEM images. When polyacrylamide was used in addition to lignin in the milling procedure, the Ag NPs were mostly encased within the organic matrix.135 This effect was also observed in another study where, by changing the nature of the salt precursor, Pd NPs could be better distributed within or in the outer parts of the lignin matrix.126 TEM images of Pd NPs prepared in a vibratory ball mill showed that by using Pd(II)acetate or Pd(II) acetylacetonate, the particles in sizes of 2.8 ± 0.7 and 3.6 ± 0.9 nm respectively, tended to be encased within the biopolymer. This encasement is a result of the chemical affinity of the metal precursor to the organic nature of lignin. In contrast, the use of PdCl2 produced NPs of a mean size of 3.2 ± 1.6 nm located predominantly on the surfaces of lignin (Fig. 12B–D).126 The same tendency was observed for Ru NPs synthesized using an inorganic salt precursor or a metal–organic complex. X-ray photoemission spectroscopy (XPS) showed that Pd and Pd(II) species were present, while only Ru(II) was detected. FTIR-ATR clearly showed that the lignin was oxidized after the milling procedure, indicating its participation in the reduction of the precursors. In this same study, Au and Re NPs were prepared also prepared using lignin. In the case of Re, XPS indicated the formation of ReO (6.2 ± 1.5 nm in size), resulting in an uncommon snail eye-like shape (Fig. 12E).126
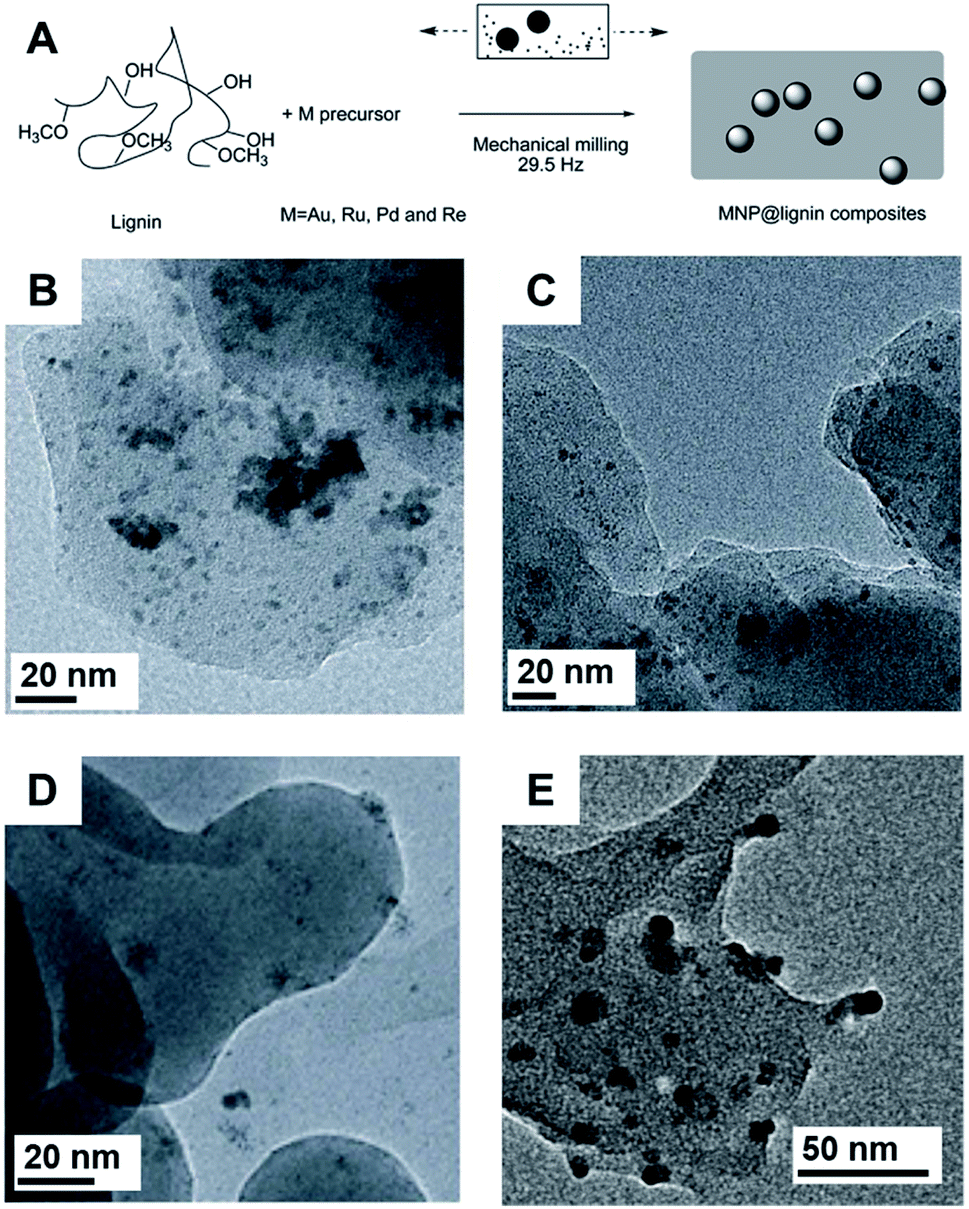 | ||
| Fig. 12 (A) Scheme for the mechanochemical synthesis of NPs@lignin composites with Au, Pd, Ru, and Re. A similar procedure was used for Ag NPs. The phenol terminations of lignin were responsible for the reduction of the salt precursors and it also plays the role of stabilizing the matrix. (B–D) TEM images of Pd NPs-kraft lignin synthesized using different Pd salt precursors. (B) Pd(II) acetate; (C) Pd(II) acetylacetonate and (D) PdCl2. The use of organic precursors favors the stabilization of the NPs within the lignin matrix. (E) TEM image of Re NPs-lignin composite obtained by milling of Re(CO)5Br and lignin. Reproduced from ref. 126 with permission of the Royal Society of Chemistry. | ||
4.2. NPs supported on carbon-based materials
Carbon-based materials, such as multi-walled carbon nanotubes (MWCNTs), have also been employed as supports. One strategy to obtain smaller NPs immobilized on the surface of MWCNTs is presented in Fig. 13 (top).89 The decomposition of organic metal salts upon high energy milling in the presence of thermally conductive substrates resulted in the formation of Ag, Pd, Pt and Au NPs around 3 nm in size (Fig. 13A–D, respectively), regardless of the metal loading (1–10 wt%) or the MWCNT diameter. Long milling periods are required for further decomposition of the precursors. Unfortunately, this prolonged milling time partially destroyed the carbon nanotubes. As such, a compromise had to be found between milling time and product yields to maximize the quality of the final materials. This was achieved by using 10 min milling. Prolonged milling treatments in high metal loadings (20 wt%) also resulted in particle sintering and hence the formation of bigger NPs. Moreover, the MWCNT-facilitated mechanosynthesis of noble metal NPs demonstrated the need for thermally conductive material (MWCNTs, SWCNTs, expanded graphite, of expanded graphite (EG), hexagonal boron nitride) as substrates to generate the metallic NPs. The use of insulating supports only led to low metal NP yields. This study might be an illustration of the importance of heat transfer in mechanochemical systems, where a temperature increase induced by the milling treatment can be efficiently used to drive the transformation. Although the product outcomes from this mechanochemical procedure were equivalent to the purely thermal treatment (i.e. metallic NPs, acetic acid), the particle sizes obtained by purely milling were much smaller, presumably due to the mechanical mixing.89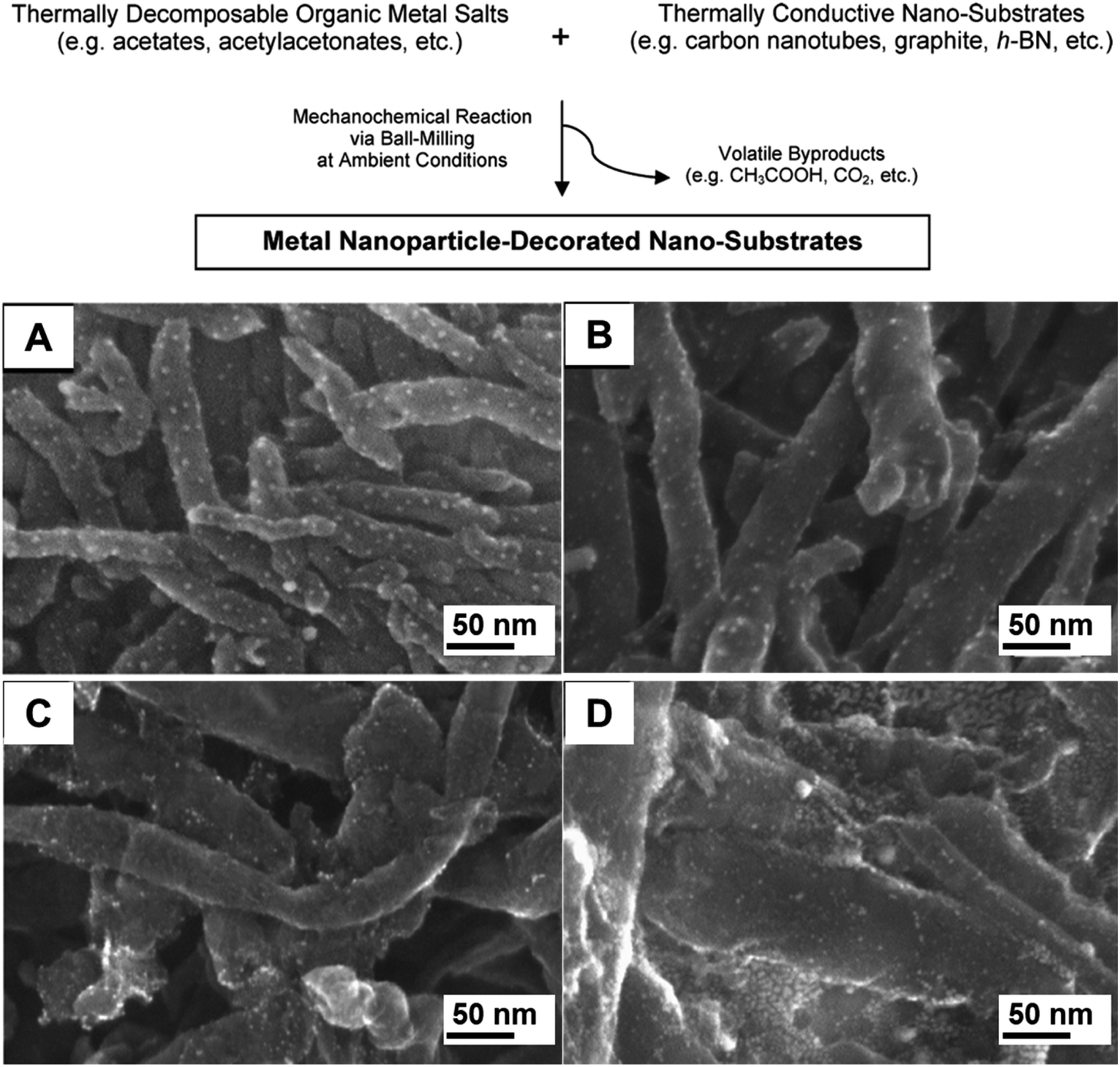 | ||
| Fig. 13 Scheme for the mechanochemical synthesis of noble metal NPs on carbon nanotubes (A) and SEM images of the NMNPs-supported MWCNTs: (A) Ag (from silver acetate), (B) Pd (from PdII acetate), (C) Pt (Pt acetylacetonate) and (D) Au NPs (from AuIII acetate). The syntheses were performed in a high energy shaker mill (SPEX CertiPrep 8000D) using a zirconia milling set. Adapted from ref. 89 with permission of the American Chemical Society. | ||
The preparation of noble metal NPs supported on MWCNTs has also been reported by a two-step process. In this preparative strategy, the precursors (the carbon materials and the metal acetates) are first milled followed by a thermal treatment step.140 After thermal treatment, NPs grew because of the continued reaction with the remaining salt after the ball milling step. Further particle growth occurred by aggregation and Ostwald ripening processes. The average particle size of 28 nm was obtained for Ag NPs for a milling treatment of 10 min. In the case of Ag/MWCNTs, the electron beam during HRTEM imaging reduced the remaining Ag(I)acetate and showed that the Ag NPs were formed away from MWCNT surfaces.140 In another study, Pd/MWCNTs, and Pd/SWCNTs hybrids were prepared by ball milling using Pd(II)acetate, which was partially reduced during the mechanochemical procedure, resulting in low Pd NPs size dispersion of 3.4 ± 1.3 nm in size.141 The product obtained after subsequent heating afforded bigger and poorly dispersed NPs in the carbon matrix (9.4 ± 3.6 nm), according to TEM. The materials were further characterized by XRD and XPS, which demonstrated the partial reduction of the salt during milling. They exhibited excellent catalytic activity in a series of Suzuki cross-coupling reactions, with the material prepared by ball milling presented superior performances even at low concentrations.141
4.3. NPs supported in SiO2-based materials
Organic volatile metal precursors have been used to generate small NPs by thermal decomposition, free from halogenates. These halogenates can be detrimental for catalytic activities.128–131,137,139,142 Simple manual grinding of Au or Pd acetates along with solid supports such as TiO2 and SiO2 was used in the preparation of catalysts for alcohol oxidation and formation of hydrogen peroxide. After grinding, the mixtures were heat-treated at 350 °C to generate the final materials. The best catalytic activity, superior to the solution-prepared counterparts, was obtained when Au and Pd acetates were mixed, generating sub-5 nm AuPd NPs alloys.139Supported Pd NPs (<2 nm) were prepared by milling together Pd(II) acetate and ordered mesoporous SBA-15 silica (Al-SBA-15) or mesostructured cellular foam (Al-MCF) (Fig. 14A).137 After thermal treatment in the reductive H2/N2 atmosphere at 723 K, the material was used in the hydrogenation of p-nitrophenol using NaBH4 in solution. Materials prepared with milling-dispersed NPs presented higher per-metal-content activity as compared to that prepared by wet impregnation (Fig. 14B).137 In a different study, an analogous protocol was used for the synthesis of Ag NPs with a mean size of 5 nm supported in similar porous materials. The catalysts were evaluated in the synthesis of imidazolones via cycloisomerizations of propargylic ureas143 and the dearomatization of indoles.136 In both cases the material showed to be recyclable and stable under catalytic tests.
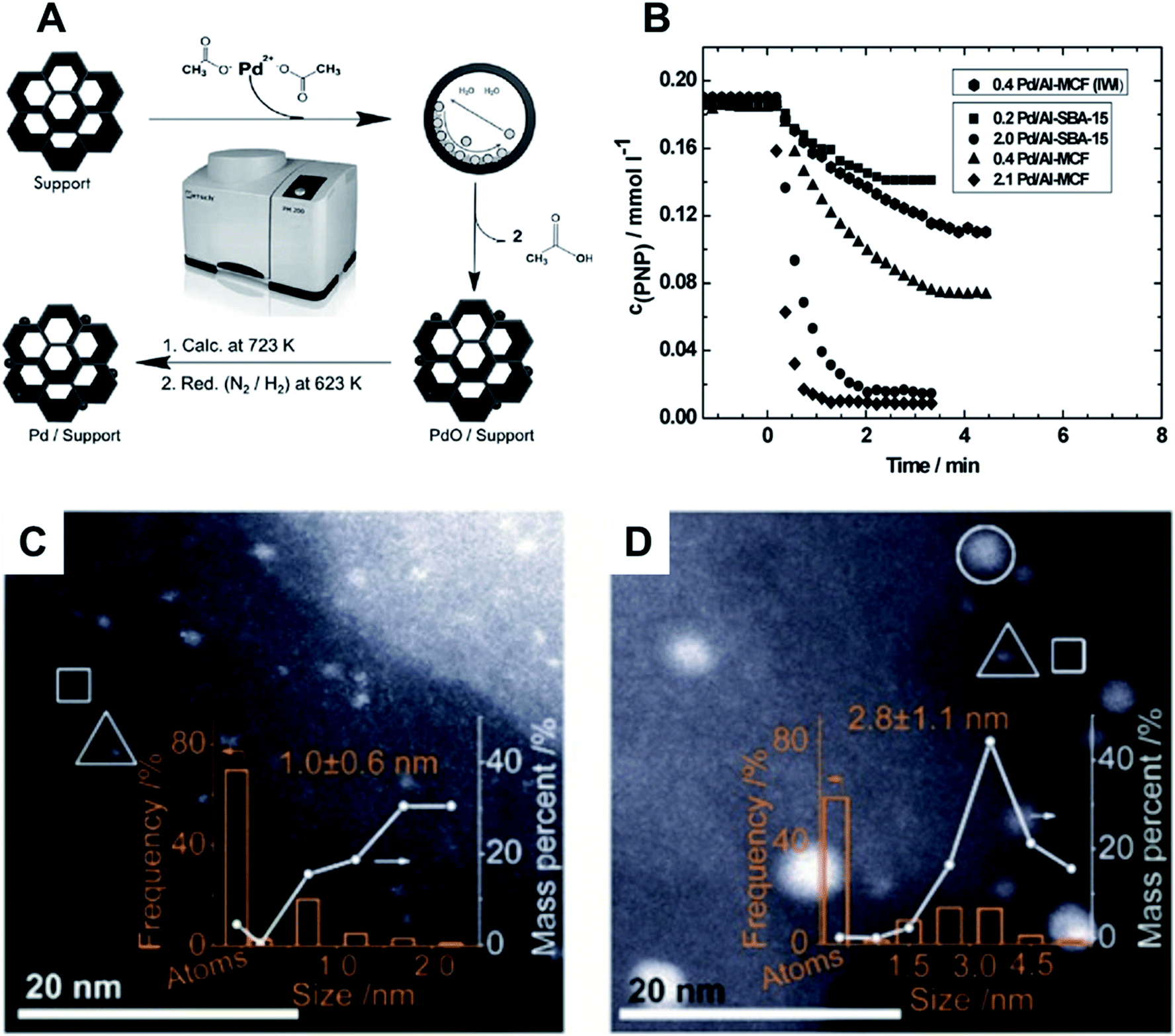 | ||
| Fig. 14 (A) Scheme for the mechanochemical synthesis of Pd NPs supported on ordered nanoporous materials (Al-SBA-15 and Al-MCF) via ball milling. The approach employs a subsequent reduction in H2 atmosphere before catalytic tests towards the reduction of p-nitrophenol. (B) Catalytic studies showing the decrease of p-nitrophenol concentration upon catalytic reduction by NaBH4 for the different nanocatalysts. Adapted from ref. 137 with permission of the Royal Society of Chemistry. (C and D) Aberration-corrected HAADF-STEM images of Au/SiO2-Schiff: (A) manual grinding mechanochemical method and (B) solution-phase synthesis. The NPs, clusters, and isolated Au atoms are indicated by white circles, triangles, and squares, respectively. Reproduced from ref. 125 with permission of SIOC, CAS, Shanghai, & Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In another example using silica, Au/SiO2-Schiff nanocatalysts were prepared in two steps.125 Wet impregnation was used to disperse the salt precursor in the solid matrix. After drying, the powders were treated by manual grinding in the presence of NaBH4. The resulting material consisted mainly of Au NPs of 1 nm in size. Au clusters and single atoms could also be formed and stabilized as determined by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. 14C, white triangles, and squares). The material prepared by solution-phase synthesis consisted of Au NPs that were bigger and were polydisperse (Fig. 14D). This demonstrates the advantages of the grinding process in the control of size and distribution with the solid support. The resulting sizes of the Au NPs were influenced by the relative humidity as the hydride formation and diffusion is facilitated by water.125 As mentioned above, the influence of water was also noted in the case of Au NPs synthesized with CD in different hydration degrees.124 The final Au/SiO2-Schiff catalysts were tested towards the reduction of CO2.125
Au NPs supported on a glass surface were prepared by milling the salt precursor with porous glass. The milled mixture was subsequently dispersed in ethanol and deposited onto glass slides for drying. Finally, the dispersed material was laser sintered, during which most of the salt was reduced, producing AuNPs of 30 nm according to SEM and UV-Vis. The final material was used in the plasmon-driven photocatalytic degradation of ethanol in a bypass-flow reactor and remained stable.127
4.4. NPs supported in porous coordination polymers
Porous coordination polymers such as metal–organic frameworks (MOFs), were used as support of Au NPs by manual grinding with an organogold complex.128–131,142 Among several advantages, these materials provide the highest known surface areas. Furthermore, the association of MOFs and NPs presents synergistic effects with enhanced performance.144 In the synthesis of these hybrid materials, dimethyl(acetylacetonate)Au(III) was used as metal precursor due to its volatility.128–131,142 The metal precursor was manually ground with the MOF and then thermally treated in a reducing atmosphere of H2/N2. In the synthesis performed using a Cd-based fluorinated MOF and metal loading of Au 1 wt%, TEM studies were not able to identify any NPs. The lack of NPs indicated that the Au(0) was probably present as small clusters. The cluster formation was further supported by EXAFS which showed Au⋯Au distances in agreement to Au2 and Au3 clusters. The final composites were found to be stable in air for over six months, demonstrating the efficiency of the methodology in preparing ultrafine, stable NPs.128Au NPs@ZIF-8 stable catalysts were also prepared by manual grinding + H2 reduction approach, leading to AuNPs sizes of 3.4 ± 1.4 nm and 4.2 ± 2.6 nm for 1 and 5 wt% of Au load, respectively. The final materials were tested in the CO oxidation, demonstrating good activities (Fig. 15).129 Other examples of MOF-Au NPs prepared by grinding are known, including MOF-5, CPL-2, and Al-MIL-53 porous polymer. Each of these systems was tested for the N-alkylation of primary amines130 and in the oxidation of alcohols.131 Curiously, the size of the Au NPs (from 1.6 ± 1.0 to 3.3 ± 2.3 nm) were influenced by the type of porous polymer used.130
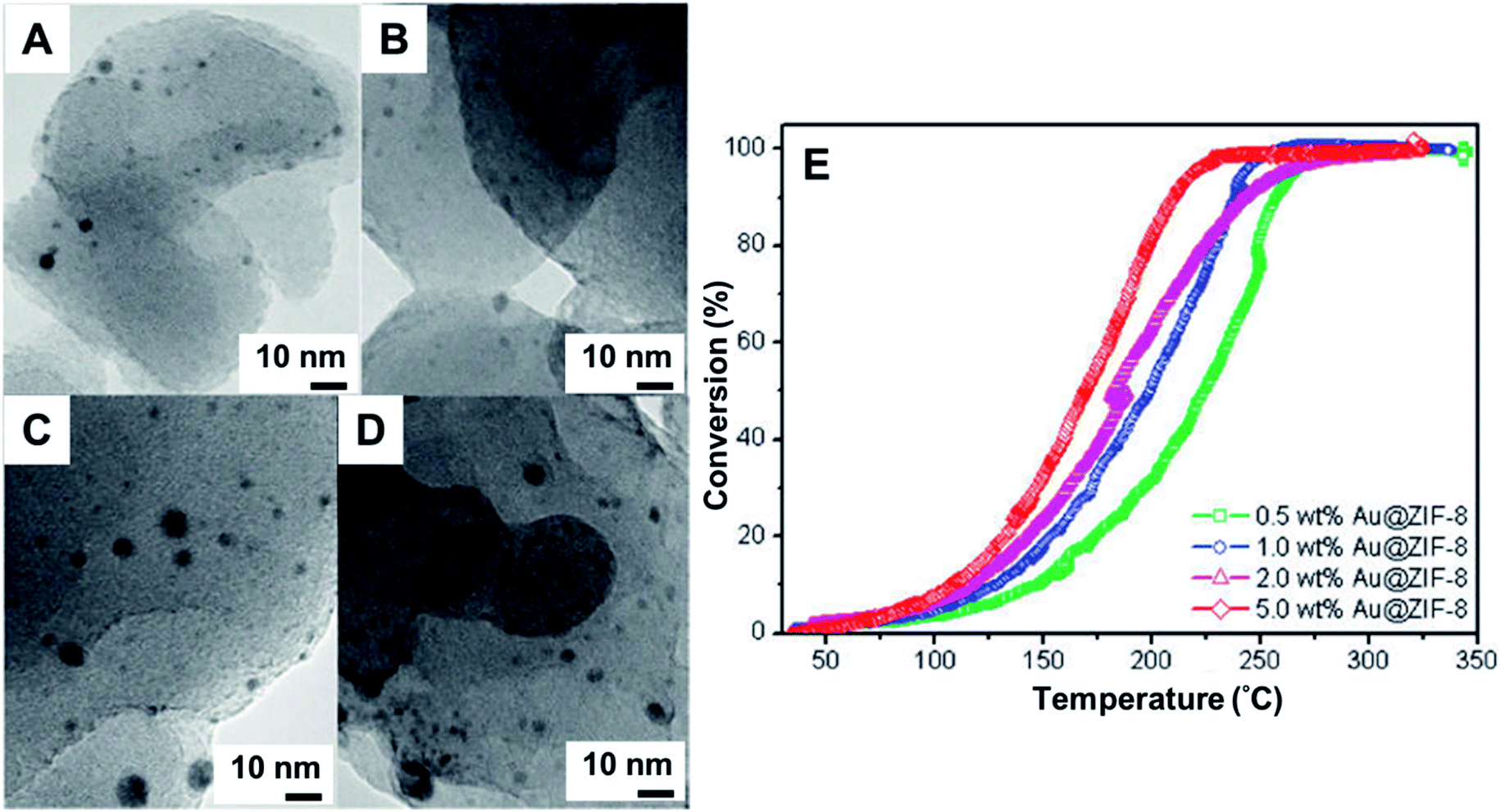 | ||
| Fig. 15 (A–D) TEM images of Au NPs supported on ZIF-8. Manual grinding in agate was used to incorporate the volatile organogold complex within the ZIF-8 matrix. The material was heated at 230 °C in a gas mixture of 10 vol% H2 in He for 2.5 h to yield Au@ZIF-8. The materials were used as nanocatalysts in the CO oxidation. (A and B) Au loading was 1 wt% and (C and D) Au loading wt%, before (A and C) and after (B and D) catalytic reaction. (E) Conversion% as a function of temperature s for CO oxidation over Au@ZIF-8 catalysts with different Au loadings. Adapted from ref. 129 with permission of the American Chemical Society. | ||
Very recently, ball milling was used to facilitate the synthesis of stable metal NPs@MOFs composites.138 The synthetic procedure is depicted in Fig. 16A for the case of Pd@ZIF-8. The procedure involves the impregnation of the support, here ZnO, with the noble metal salt by ball milling. Subsequently, the solid mixture was treated at 150 °C under H2 flow to reduce Pd2+ to metallic Pd. In a second milling step, the Pd/ZnO was milled under ILAG (ion- and liquid assisted grinding) conditions along with the organic linker, 2-methylimidazole (Hmim). This second milling step resulted in the formation of the final Pd@ZIF-8. As can be seen in the HRTEM image (Fig. 16B), the material contains well-dispersed, crystalline Pd NPs with a mean size of around 4.5 nm. The versatility of this approach was demonstrated by preparing other ZIF-8 systems such as Pt@ZIF-8 and Ru@ZIF-8, as well as for other coordination polymers resulting in Pd@[Zn2(ta)2(dabco)], Au@HKUST and Pd@[Y(BTC)(H2O)]. Pd@ZIF-8 was tested in hydrogenation reactions, showing alkyne size selectivity due to the pore size of ZIF-8. Comparable size selectivity was not observed for the non-encapsulated Pd (Fig. 16C). The activity of the reused catalyst towards 1-hexene hydrogenation only shows a slight decrease after five cycles (Fig. 16D).138
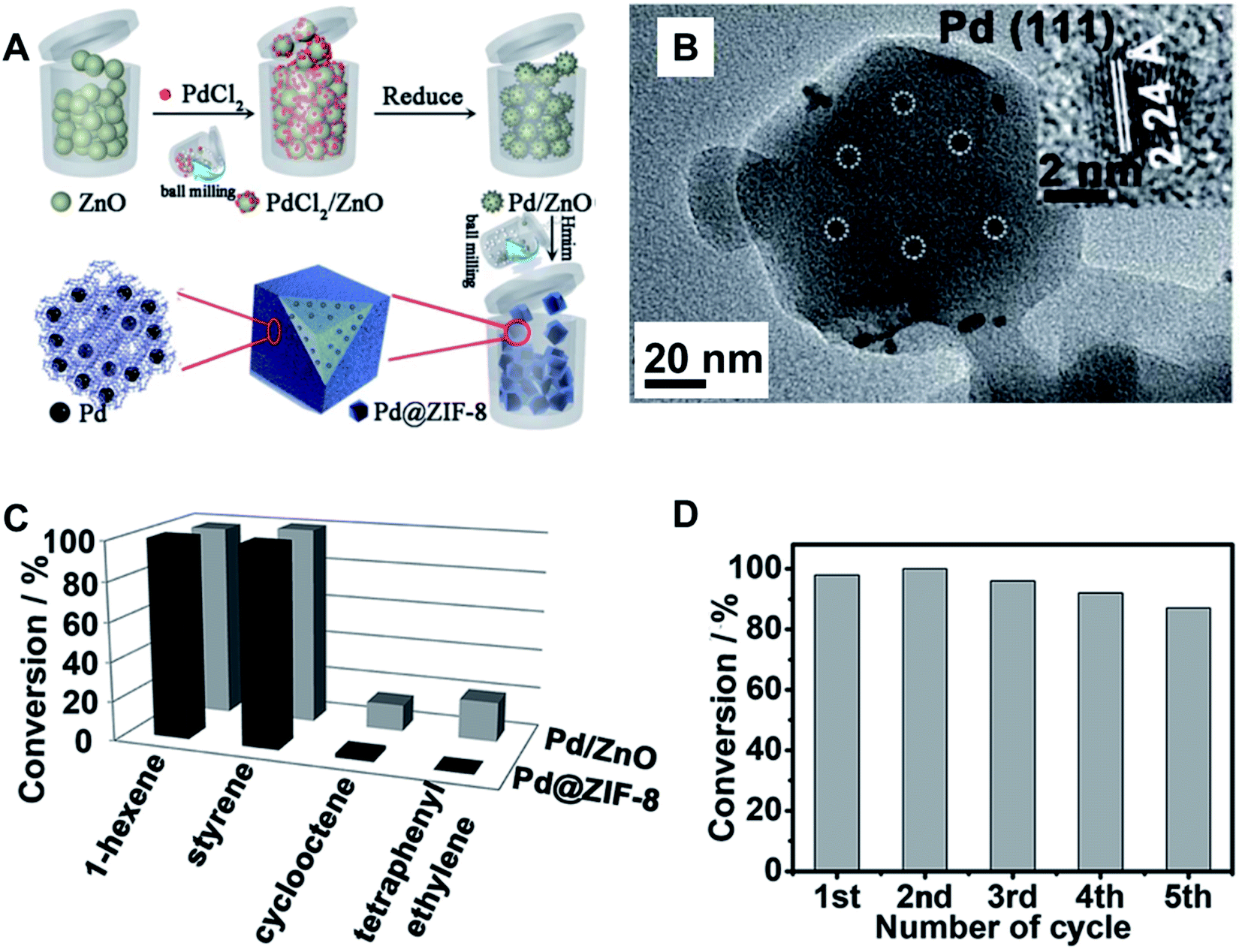 | ||
| Fig. 16 Mechanochemically-assisted synthesis of metal NPs@MOFs. (A) Synthetic protocol for preparation of Pd@ZIF-8 through mechanical mixing of PdCl2/ZnO, thermal treatment in H2 flow to produce Pd/ZnO, and finally, ball milling with the organic linker 2-methylimidazole (Hmim) to form Pd@ZIF-8. (B) HRTEM of Pd@ZIF-8 showing the Pd NPs size and dispersion; (C) conversion of alkynes upon hydrogenation using of Pd@ZIF-8 as catalysts and (D) conversion of 1-hexene with reused catalyst. Adapted from ref. 138 with permission of the Royal Society of Chemistry. | ||
The mechanosynthesis of noble-metal NPs directly over the support (one-pot approach) has many advances for practical applications. For example, non-supported NPs require high-speed centrifugation or precipitation for isolation and washing. In this case, the separation from the reaction media is greatly facilitated in the case of the supported NPs. More importantly, this enables, at least in principle, to prepare in one-pot supported NPs with their surface free from surfactants or stabilizers, which is attractive for applications in catalysis. This can represent a direct advantage over solution-phase NPs for catalytic applications, that may require additional steps for removing surface ligands or capping agents before their catalytic applications. This is because surface ligands and stabilizing agents can block active sites, leading to detrimental effects in catalysis. If the mechanosynthesis method to produce the target supported NPs also involves the use of stabilizers, these steps for their removal before catalytic applications may also be required or employed to optimize catalytic activity. However, as described in this section, several supported NPs obtained by mechanochemistry displayed interesting catalytic activities that are parallel to supported NPs produced by solution-phase syntheses.
Moreover, the presence of the support can influence the size of the NPs, providing heterogeneous nucleation sites during the mechanochemical synthesis. This can help in controlling the final size and monodispersity (affect nucleation and growth stages) and avoid NPs aggregation during the synthesis due to metal support interactions (interesting in the context of catalysis). Also, this can avoid nonclassical growth events based on aggregation and oriented attachment, for example. Thus, this strategy becomes attractive for the synthesis of supported ultrasmall NPs, small clusters, and single-atoms, which have emerged as the ultimate frontier in the field of nanocatalysis.
As discussed in this review, challenges on the mechanochemical synthesis of supported NPs remain on the control over size and size distribution. It is still required greater knowledge and tuning over the interaction between the support and the precursor and possible degradation of the support itself by prolonged milling times. On the other hand, the mechanochemical synthesis of NPs over a supporting material is considerably more capable to yield ultrasmall NPs or even small clusters and single atoms. Overall, the approach to follow in the synthesis of NPs by mechanochemical means must be application- or fundamental-driven.
5. Summary and outlook
Mechanochemistry has been promoted as a promising tool to perform environmentally friendly transformations. It holds the potential to circumvent the need for high temperatures and solvents and purification steps in many syntheses relative to traditional solid- and solution-phase methods.145,146 The mechanochemical synthesis of noble metal NPs has shown substantial progress in recent years, as discussed in this review, and its practical guidelines are summarized in Table 4. Here, the primary focus was placed on supported and non-supported NPs by a bottom-up approach, in which NPs are grown from atomic building blocks rather than by reducing the size of larger particles. In traditional colloidal, solution-based synthetic approaches, bottom-up strategies based on the chemical reduction or decomposition of the metal precursor provides better control over the kinetics of NPs nucleation and growth, enabling the tailoring over NPs shapes and sizes and versatility in terms of synthesis conditions that can be optimized. Borrowing these concepts to mechanochemistry, the role of mechanical energy – either by automated ball milling of manual grinding – in the bottom-up growth of NPs is likely two-fold. First, the mechanical treatment facilitates the mixing and renewal of particle surfaces. This is required to ensure continued reaction between solid bodies. Second, the energy imparted by the mechanical treatment is believed to trigger the chemical reaction necessary for reduction of the metal precursors and generation of the corresponding growth species. In the example of supported NPs, a milling step can also be employed to finely and homogenously disperse the metal precursor over the surface of the support before reduction/decomposition, nucleation, and growth processes take place. This can facilitate the synthesis of surfactant-free materials, which are attractive for catalytic applications.| Parameter | Effect |
|---|---|
| Milling/grinding time | Long milling duration tends to increase the particle size, yet small NPs are possible at high conversions of the metal precursor for protected NPs |
| A compromise in between milling time and conversion of the precursor is necessary in the case of surfactant free NPs | |
| Milling/grinding bodies | Nonmetallic milling assembly (jar + balls, or mortar and pestle) are preferred to avoid contamination (ZrO2, agate, PTFE, etc.) |
| The use of metallic milling set can be beneficial if a galvanic replacement can take place, dismissing the use of common reducing agents | |
| Capping agent | For synthetic polymers (e.g., PVP), a high molecular mass provides a better stabilization and afford smaller NPs |
| Capping agents can be dismissed in the case for supported NPs | |
| Reducing agent | Not yet investigated in detail. Similar to solution, strong reducing agents such as NaBH4 or H2 (after dispersion of the precursor and support by grinding) tends to generate smaller NPs. Protecting agents such as PVP or PEG, and supporting materials (e.g. Lignin) can also play the role of reductants |
| Metal salt precursor | Organic volatile precursors tend to yield smaller NPs |
| The chemical nature/affinity of the salt determines the NP distribution face to the supporting material, when employed |
The stability of the materials prepared mechanochemically deserves emphasis. The noble metal NPs prepared and stored in the solid state can present extraordinary stability over time (e.g., Ag9 nanoclusters are stable for over two years).120 The fact that they are produced directly in the solid state prevents fast diffusion, agglomeration and overgrowth by coalescence or Ostwald repining for example. Thus, the original properties of the materials can be preserved. This is an important factor when it comes to scalability of the systems and for industrial applications purposes.
Unfortunately, it remains poorly understood how different reducing agents affect the kinetics formation and shape of the resulting NPs.98 Several issues still need to be addressed in the mechanochemical synthesis of metal NPs relative to solution-phase strategies. These are related to the understanding the reactivity of solid reductants, diffusion processes in the solid phase, interaction of surfactants or stabilizers with the growth species, and the true nature of the metal precursors. The knowledge in terms of milling conditions, capping agents, salt precursors, etc., has just started to be rationalized as discussed in the examples presented herein. Up to date, the mechanochemical synthesis is still restricted to a few systems and it does not allow to reach the same level of sophistication and versatility relative to solution-phase strategies. It is clear that most shapes produced from the mechanochemical synthesis of noble-metal NPs have been restricted to spherical morphology, and samples have shown only limited size monodispersity and compositional control (for multicomponent nanoparticles). This is probably due to the difficulties in separating the events that lead to the generation of growth species, nucleation, growth, and non-classical growth in the solid phase as opposed to colloidal synthesis. This can also be related to the high heterogeneity of the media. Even in efficient milling process it is difficult to achieve a homogeneous local mixture due to reduced mass transfer and diffusion limitations in comparison to solvated media. This makes it more challenging to selectively maneuver the rate of growth along with certain crystallographic directions and induce symmetry breaking. Similarly, it remains challenging the synthesis of bimetallic NPs with well-defined elemental distributions and surface morphologies (conformal core–shell coating, branched morphologies, alloys, etc.). In this context, the development of in situ techniques to study and unravel each step in the mechanochemical synthesis is required to provide insights on how these processes happen and how to manipulate the several processes occurring in the solid mixture under mechanochemical conditions.
For multicomponent NPs, the control of composition and the element distribution are essential. In the bottom-up mechanochemical synthesis of noble metal NPs, such control is, however, particularly challenging. Probably diffusion limitations in the solid state and different reduction rates for each metal prevent the formation of more homogenous materials directly under milling or grinding. Further studies in order to determine the appropriate milling and reducing conditions for the formation of alloys, core–shell, etc., is an important direction for the field. On the other hand, even with these shortcomings, the use of mechanochemical means has produced exceptional noble metal materials for catalytic applications.111,139 This makes the bottom-up mechanochemical synthesis still a great candidate for the preparation of multicomponent materials in a robust and scalable manner.
The fundamentals of the mechanochemical bottom-up synthesis of noble-metal NPs with controlled physicochemical features are in the early stages of development. In particular, controlling the physicochemical features of such mechanochemically prepared NPs remains an exceptional challenge. It is worth mentioning that multi-phase solid reactions do not occur in the whole bulk of the sample. Rather, these reactions occur only in the regions where a small fraction of contact between phases takes place.50 Therefore, continuous mechanical action aims to enhance these contact events through mass transport or comminution. Fluid phases can also be formed upon mechanical action. At a deeper level, mechanical input enhances diffusion and generates strain and defects. Moreover, mechanical action induces pulses of pressure and temperature at the site of impact. The mechanical perturbation of inorganic solids (e.g. metal salt precursors) might result in the redistribution of electrons, holes, and structural defects.50 Consequently, the reactivity of the solid changes. However, which of these mentioned effects (mixing-diffusion and/or electronic perturbation) of mechanochemical energy will play a pivotal role in the synthesis of NPs remains elusive.
An important perspective of the field is related to the detailed understanding of the nucleation and growth stages happening in the solid phase. For instance, elucidating the mechanisms of nucleation and growth are of vital importance if one aims to truly control the physical and chemical features of the produced NPs. Although insights can be borrowed from solution-based systems, there are critical differences in solid-state approaches. As such, the continued and dedicated investigation into solid-state NPs growth mechanisms is fundamental. This includes in situ studies147,148 into the high heterogeneity of the reacting media, diffusion limitations, and even the effective temperatures experienced during milling.149,150 For instance, it is crucial to identify homogenous, if possible, vs. heterogeneous nucleation events and the symmetry reduction to generate anisotropic nanoparticles. In situ monitoring during milling147,148 might help to unravel some of the unknown variables, particularly some kinetic aspects.
We believe that the knowledge gained on the mechanochemical synthesis of noble metal NPs can serve as a foundation to enable the controlled synthesis of bimetallic systems with controlled morphologies (core–shell, alloys, dimers, and core-satellite, for example). Another future direction is to extend controlled mechanochemical synthesis to other metals, oxides and sulfides nanoparticles.151–153 Although most of the examples discussed herein focused on applications in catalysis, it is clear that the development of the synthetic strategies will benefit and allow the mechanochemical synthesis for applications in other fields, such as sensing, biomedicine, plasmonics, and others. In this context, future directions should concentrate on moving beyond trial and error (observation driven) strategies towards a more rational, design-driven approach based on mechanistic understanding. This would be of invaluable help to expand the field for more diverse systems, including oxides and non-noble metal materials. It can be envisioned that the emerging areas of machine learning and artificial intelligence applied to materials sciences can contribute to enabling the community to move in this direction from data acquired from in situ studies. We believe that mechanochemistry presents an enormous potential to greatly impact the synthesis of noble metal NPs in the upcoming decades.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Adam Michalchuk for the careful reading of the review. P. F. M. O. thanks the Sao Paulo Research Foundation FAPESP for the fellowships (FAPESP Grant 2017/15456-0 and 2019/01619-0). R. M. T. thanks the CNPq, CAPES, and FAPESP (2015/26308-7) for the financial support. P. H. C. C. thanks the University of Helsinki and the Jane and Aatos Erkko Foundation for the financial support.References
- W. Niu and X. Lu, in Metallic Nanostructures, ed. Y. Xiong and X. Lu, Springer International Publishing, Cham, 2015, pp. 1–47 Search PubMed.
- R. Vajtai, in Springer Handbook of Nanomaterials, ed. R. Vajtai, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 1–36 Search PubMed.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- Y. Liu, X.-M. Lin, Y. Sun and T. Rajh, J. Am. Chem. Soc., 2013, 135, 3764–3767 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- A. G. M. da Silva, T. S. Rodrigues, S. J. Haigh and P. H. C. Camargo, Chem. Commun., 2017, 53, 7135–7148 RSC.
- T. S. Rodrigues, A. G. M. da Silva and P. H. C. Camargo, J. Mater. Chem. A, 2019, 7, 5857–5874 RSC.
- M. Azharuddin, G. H. Zhu, D. Das, E. Ozgur, L. Uzun, A. P. F. Turner and H. K. Patra, Chem. Commun., 2019, 55, 6964–6996 RSC.
- J. Patarroyo, N. G. Bastús, A. Genç, J. Sancho-Parramon, V. Puntes and J. Arbiol, Nanophotonics, 2017, 6, 193–213 Search PubMed.
- J. Langer, S. M. Novikov and L. M. Liz-Marzán, Nanotechnology, 2015, 26, 322001 CrossRef PubMed.
- K. L. Göeken, V. Subramaniam and R. Gill, Phys. Chem. Chem. Phys., 2015, 17, 422–427 RSC.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- C. T. Campbell, S. C. Parker and D. E. Starr, Sci. 80, 2002, 298, 811–814 CrossRef CAS PubMed.
- C. Tabor, R. Narayanan and M. A. El-Sayed, in Model Systems in Catalysis: Single Crystals to Supported Enzyme Mimics, 2010, pp. 395–414 Search PubMed.
- A. Zecchina, E. Groppo and S. Bordiga, Chem.–Eur. J., 2007, 13, 2440–2460 CrossRef CAS PubMed.
- M.-L. Cui, Y.-S. Chen, Q.-F. Xie, D.-P. Yang and M.-Y. Han, Coord. Chem. Rev., 2019, 387, 450–462 CrossRef CAS.
- Z. Fan and H. Zhang, Chem. Soc. Rev., 2016, 45, 63–82 RSC.
- H. Liu, F. Nosheen and X. Wang, Chem. Soc. Rev., 2015, 44, 3056–3078 RSC.
- X. Liu, J. Iocozzia, Y. Wang, X. Cui, Y. Chen, S. Zhao, Z. Li and Z. Lin, Energy Environ. Sci., 2017, 10, 402–434 RSC.
- I. S. Park, M. S. Kwon, K. Y. Kang, J. S. Lee and J. Park, Adv. Synth. Catal., 2007, 349, 2039–2047 CrossRef CAS.
- C. A. Stowell and B. A. Korgel, Nano Lett., 2005, 5, 1203–1207 CrossRef CAS PubMed.
- S. Xie, X. Y. Liu and Y. Xia, Nano Res., 2015, 8, 82–96 CrossRef CAS.
- Y. Yan, J. S. Du, K. D. Gilroy, D. Yang, Y. Xia and H. Zhang, Adv. Mater., 2017, 29, 1605997 CrossRef PubMed.
- S. Zhou, M. Zhao, T.-H. Yang and Y. Xia, Mater. Today, 2019, 22, 108–131 CrossRef CAS.
- G. Baffou and R. Quidant, Chem. Soc. Rev., 2014, 43, 3898 RSC.
- C. L. Nehl and J. H. Hafner, J. Mater. Chem., 2008, 18, 2415 RSC.
- X. Zhang, Y. L. Chen, R.-S. Liu and D. P. Tsai, Rep. Prog. Phys., 2013, 76, 04401 CrossRef PubMed.
- Y. Xia, X. Xia and H.-C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310–325 CrossRef CAS.
- L. Scarabelli, A. Sánchez-Iglesias, J. Pérez-Juste and L. M. Liz-Marzán, J. Phys. Chem. Lett., 2015, 6, 4270–4279 CrossRef CAS PubMed.
- F. Fievet, J. P. Lagier and M. Figlarz, MRS Bull., 1989, 14, 29–34 CrossRef CAS.
- F. Fiévet, S. Ammar-Merah, R. Brayner, F. Chau, M. Giraud, F. Mammeri, J. Peron, J.-Y. Piquemal, L. Sicard and G. Viau, Chem. Soc. Rev., 2018, 47, 5187–5233 RSC.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
- M. Wuithschick, A. Birnbaum, S. Witte, M. Sztucki, U. Vainio, N. Pinna, K. Rademann, F. Emmerling, R. Kraehnert and J. Polte, ACS Nano, 2015, 9, 7052–7071 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- G. Frens, Nature, Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- S. Tao, M. Yang, H. Chen and G. Chen, ACS Sustainable Chem. Eng., 2018, 6, 8719–8726 CrossRef CAS.
- H. Huang, H. du Toit, M. O. Besenhard, S. Ben-Jaber, P. Dobson, I. Parkin and A. Gavriilidis, Chem. Eng. Sci., 2018, 189, 422–430 CrossRef CAS.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- C. Bolm and J. G. Hernández, Angew. Chem., Int. Ed., 2019, 58, 3285–3299 CrossRef CAS PubMed.
- J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed.
- D. E. Crawford, C. K. G. Miskimmin, A. B. Albadarin, G. Walker and S. L. James, Green Chem., 2017, 19, 1507–1518 RSC.
- D. Crawford, J. Casaban, R. Haydon, N. Giri, T. McNally and S. L. James, Chem. Sci., 2015, 6, 1645–1649 RSC.
- C. Suryanarayana, Prog. Mater. Sci., 2001, 46, 1–184 CrossRef CAS.
- V. V Boldyrev, Russ. Chem. Rev., 2006, 75, 177–189 CrossRef.
- E. Boldyreva, Chem. Soc. Rev., 2013, 42, 7719–7738 RSC.
- P. Baláž, Mechanochemistry in Nanoscience and Minerals Engineering, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008 Search PubMed.
- G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668 RSC.
- D. Braga, L. Maini and F. Grepioni, Chem. Soc. Rev., 2013, 42, 7638 RSC.
- A. Delori, T. Friščić and W. Jones, CrystEngComm, 2012, 14, 2350 RSC.
- C. Mottillo and T. Friščić, Molecules, 2017, 22, 144 CrossRef PubMed.
- T. Friščić, in Metal-Organic Frameworks: Design and Application, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010, pp. 267–299 Search PubMed.
- M. Descamps and J. F. Willart, Adv. Drug Delivery Rev., 2016, 100, 51–66 CrossRef CAS PubMed.
- M. Descamps, J. F. Willart, E. Dudognon and V. Caron, J. Pharm. Sci., 2007, 96, 1398–1407 CrossRef CAS PubMed.
- M. Descamps, J. F. Willart, E. Dudognon, R. Lefort, S. Desprez and V. Caron, in Mater. Res. Soc. Symp. Proc., 2007, vol. 979, pp. 116–131 Search PubMed.
- P. F. M. Oliveira, J.-F. Willart, J. Siepmann, F. Siepmann and M. Descamps, Cryst. Growth Des., 2018, 18, 1748–1757 CrossRef CAS.
- P. Baláž, M. Achimovičová, M. Baláž, P. Billik, Z. Cherkezova-Zheleva, J. M. Criado, F. Delogu, E. Dutková, E. Gaffet, F. J. Gotor, R. Kumar, I. Mitov, T. Rojac, M. Senna, A. Streletskii and K. Wieczorek-Ciurowa, Chem. Soc. Rev., 2013, 42, 7571 RSC.
- K. Ralphs, C. Hardacre and S. L. James, Chem. Soc. Rev., 2013, 42, 7701–7718 RSC.
- C. Xu, S. De, A. M. Balu, M. Ojeda and R. Luque, Chem. Commun., 2015, 51, 6698–6713 RSC.
- F. Gomollón-bel, Chem. Int., 2019, 12–17 Search PubMed.
- C. Suryanarayana and N. Al-Aqeeli, Prog. Mater. Sci., 2013, 58, 383–502 CrossRef CAS.
- D. Debnath, C. Kim, S. H. Kim and K. E. Geckeler, Macromol. Rapid Commun., 2010, 31, 549–553 CrossRef CAS PubMed.
- D. Debnath, S. H. Kim and K. E. Geckeler, J. Mater. Chem., 2009, 19, 8810 RSC.
- A. Moores, Curr. Opin. Green Sustain. Chem., 2018, 12, 33–37 CrossRef.
- P. Baláž, in Mechanochemistry in Nanoscience and Minerals Engineering, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, pp. 1–102 Search PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- P. Baláž, in Mechanochemistry in Nanoscience and Minerals Engineering, Springer Berlin Heidelberg, Berlin, Heidelberg, 1st edn, 2008, pp. 103–132 Search PubMed.
- H. J. Fecht, Nature, 1992, 356, 133–135 CrossRef CAS.
- H.-J. Fecht, Nanostruct. Mater., 1995, 6, 33–42 CrossRef CAS.
- A. N. Streletskii, D. G. Permenov, B. B. Bokhonov, I. V. Kolbanev, A. V. Leonov, I. V. Berestetskaya and K. A. Streletzky, J. Alloys Compd., 2009, 483, 313–316 CrossRef CAS.
- B. W. McMahon, J. P. L. Perez, J. Yu, J. A. Boatz and S. L. Anderson, ACS Appl. Mater. Interfaces, 2014, 6, 19579–19591 CrossRef CAS PubMed.
- K. Barai, C. S. S. Tiwary, P. P. P. Chattopadhyay and K. Chattopadhyay, Mater. Sci. Eng., A, 2012, 558, 52–58 CrossRef CAS.
- T. Prasad Yadav, R. Manohar Yadav and D. Pratap Singh, Nanosci. Nanotechnol., 2012, 2, 22–48 CrossRef.
- N. F. Uvarov and V. V Boldyrev, Russ. Chem. Rev., 2001, 70, 265–284 CrossRef CAS.
- E. Gaffet and G. Le Caër, in Encyclopedia of nanoscience and nanotechnology, American Scientific Publishers Stevenson Ranch, Calif, USA, 2004, vol. 5, pp. 91–129 Search PubMed.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- A. P. LaGrow, K. R. Knudsen, N. M. AlYami, D. H. Anjum and O. M. Bakr, Chem. Mater., 2015, 27, 4134–4141 CrossRef CAS.
- A. Alshammari, A. Köckritz, V. N. Kalevaru and A. Martin, in Studies in Surface Science and Catalysis, Elsevier Masson SAS, 2010, vol. 175, pp. 409–412 Search PubMed.
- V. Sashuk and K. Rogaczewski, J. Nanopart. Res., 2016, 18, 261 CrossRef.
- T. S. Rodrigues, M. Zhao, T. Yang, K. D. Gilroy, A. G. M. da Silva, P. H. C. Camargo and Y. Xia, Chem.–Eur. J., 2018, 24, 16944–16963 CrossRef CAS PubMed.
- L. M. Rossi, J. L. Fiorio, M. A. S. Garcia and C. P. Ferraz, Dalton Trans., 2018, 47, 5889–5915 RSC.
- T. Yang, Y. Shi, A. Janssen and Y. Xia, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.201911135.
- T. Tsuzuki and P. G. McCormick, J. Mater. Sci., 2004, 39, 5143–5146 CrossRef CAS.
- J. Polte, M. Herder, R. Erler, S. Rolf, A. Fischer, C. Würth, A. F. Thünemann, R. Kraehnert and F. Emmerling, Nanoscale, 2010, 2, 2463 RSC.
- Y. Lin, K. A. Watson, S. Ghose, J. G. Smith, T. V. Williams, R. E. Crooks, W. Cao and J. W. Connell, J. Phys. Chem. C, 2009, 113, 14858–14862 CrossRef CAS.
- P. H. C. Camargo, T. S. Rodrigues, A. G. M. da Silva and J. Wang, in Metallic Nanostructures, eds. Y. Xiong and X. Lu, Springer International Publishing, Cham, 2015, pp. 49–74 Search PubMed.
- H. Kulla, S. Haferkamp, I. Akhmetova, M. Röllig, C. Maierhofer, K. Rademann and F. Emmerling, Angew. Chem., Int. Ed., 2018, 57, 5930–5933 CrossRef CAS PubMed.
- S. Q. Xi, J. G. Zhou and X. T. Wang, Powder Metall., 2007, 50, 367–373 CrossRef CAS.
- Y.-S. Kwon, K. B. Gerasimov and S.-K. Yoon, J. Alloys Compd., 2002, 346, 276–281 CrossRef CAS.
- L. Takacs and J. S. McHenry, J. Mater. Sci., 2006, 41, 5246–5249 CrossRef CAS.
- R. Schmidt, H. Martin Scholze and A. Stolle, Int. J. Ind. Chem., 2016, 7, 181–186 CrossRef.
- F. K. h. Urakaev, in High-Energy Ball Milling - Mechanochemical processing of nanopowders, ed. M. Sopicka-Lizer, Woodhead Publishing, 2010, pp. 9–44 Search PubMed.
- M. J. Rak, N. K. Saadé, T. Friščić and A. Moores, Green Chem., 2014, 16, 86–89 RSC.
- P. F. M. de Oliveira, J. Quiroz, D. C. de Oliveira and P. H. C. Camargo, Chem. Commun., 2019, 55, 14267–14270 RSC.
- A. Bera, B. Busupalli and B. L. V. Prasad, ACS Sustainable Chem. Eng., 2018, 6, 12006–12016 CrossRef CAS.
- D. Debnath, Y. Lee and K. E. Geckeler, Polym. Int., 2017, 66, 512–520 CrossRef CAS.
- S. S. M. Ali, W. H. Eisa and A. Abouelsayed, Composites, Part B, 2019, 176, 107289 CrossRef CAS.
- T. Jayaramudu, G. M. Raghavendra, K. Varaprasad, G. V. S. Reddy, A. B. Reddy, K. Sudhakar and E. R. Sadiku, J. Appl. Polym. Sci., 2016, 133, 43027 CrossRef.
- B. Lu, F. Zhan, G. Gong, Y. Cao, Q. Zhen and P. Hu, RSC Adv., 2016, 6, 74662–74669 RSC.
- G. R. R. Khayati and K. Janghorban, Adv. Powder Technol., 2012, 23, 393–397 CrossRef CAS.
- H. Szwarc and M. Gasgnier, J. Solid State Chem., 1998, 136, 51–55 CrossRef CAS.
- T. U. B. Rao, B. Nataraju and T. Pradeep, J. Am. Chem. Soc., 2010, 132, 16304–16307 CrossRef CAS PubMed.
- T. Udayabhaskararao, M. S. Bootharaju and T. Pradeep, Nanoscale, 2013, 5, 9404 RSC.
- I. Chakraborty, A. Govindarajan, J. Erusappan, A. Ghosh, T. Pradeep, B. Yoon, R. L. Whetten and U. Landman, Nano Lett., 2012, 12, 5861–5866 CrossRef CAS PubMed.
- F. Delogu, Langmuir, 2012, 28, 10898–10904 CrossRef CAS PubMed.
- T. Premkumar and K. E. Geckeler, Colloids Surf., A, 2014, 456, 49–54 CrossRef CAS.
- A. Murugadoss, N. Kai and H. Sakurai, Nanoscale, 2012, 4, 1280 RSC.
- P. Hu, Y. Cao, D. Jia, Q. Li and R. Liu, Sci. Rep., 2015, 4, 4153 CrossRef PubMed.
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS.
- X. Yang, M. Yang, B. Pang, M. Vara and Y. Xia, Chem. Rev., 2015, 115, 10410–10488 CrossRef CAS PubMed.
- L. Qin, G. Zeng, C. Lai, D. Huang, P. Xu, C. Zhang, M. Cheng, X. Liu, S. Liu, B. Li and H. Yi, Coord. Chem. Rev., 2018, 359, 1–31 CrossRef CAS.
- F. Delogu, Mater. Chem. Phys., 2014, 147, 629–635 CrossRef CAS.
- N. Kumar and K. Biswas, Rev. Sci. Instrum., 2015, 86, 083903 CrossRef.
- N. Kumar, K. Biswas and R. K. Gupta, RSC Adv., 2016, 6, 111380–111388 RSC.
- B. J. Wiley, S. H. Im, Z.-Y. Li, J. McLellan, A. Siekkinen and Y. Xia, J. Phys. Chem. B, 2006, 110, 15666–15675 CrossRef CAS PubMed.
- T. U. B. Rao, B. Nataraju and T. Pradeep, J. Am. Chem. Soc., 2010, 132, 16304–16307 CrossRef CAS PubMed.
- F. Delogu, Mater. Chem. Phys., 2012, 137, 297–302 CrossRef CAS.
- G. Ligios, A. M. Bertetto and F. Delogu, J. Alloys Compd., 2013, 554, 426–431 CrossRef CAS.
- J. L. Wang, R. A. Ando and P. H. C. Camargo, ACS Catal., 2014, 4, 3815–3819 CrossRef CAS.
- S. Menuel, B. Léger, A. Addad, E. Monflier and F. Hapiot, Green Chem., 2016, 18, 5500–5509 RSC.
- Q. Liu, X. Wang, Y. Ren, X. Yang, Z. Wu, X. Liu, L. Li, S. Miao, Y. Su, Y. Li, C. Liang and Y. Huang, Chin. J. Chem., 2018, 36, 329–332 CrossRef CAS.
- M. J. Rak, T. Friščić and A. Moores, Faraday Discuss., 2014, 170, 155–167 RSC.
- H. L. K. S. Stolle, F. Garwe, R. Müller, T. Krech, B. Oberleiter, T. Rainer, W. Fritzsche and A. Stolle, RSC Adv., 2018, 8, 30289–30297 RSC.
- H. L. Jiang, Q. P. Lin, T. Akita, B. Liu, H. Ohashi, H. Oji, T. Honma, T. Takei, M. Haruta and Q. Xu, Chem.–Eur. J., 2011, 17, 78–81 CrossRef CAS PubMed.
- H. Jiang, B. Liu, T. Akita, M. Haruta, H. Sakurai and Q. Xu, J. Am. Chem. Soc., 2009, 131, 11302–11303 CrossRef CAS PubMed.
- T. Ishida, N. Kawakita, T. Akita and M. Haruta, Gold Bull., 2009, 42, 267–274 CrossRef CAS.
- T. Ishida, M. Nagaoka, T. Akita and M. Haruta, Chem.–Eur. J., 2008, 14, 8456–8460 CrossRef CAS PubMed.
- M. Baláž, N. Daneu, Ľ. Balážová, E. Dutková, Ľ. Tkáčiková, J. Briančin, M. Vargová, M. Balážová, A. Zorkovská and P. Baláž, Adv. Powder Technol., 2017, 28, 3307–3312 CrossRef.
- B. J. Tiimob, G. Mwinyelle, W. Abdela, T. Samuel, S. Jeelani and V. K. Rangari, J. Agric. Food Chem., 2017, 65, 1967–1976 CrossRef CAS PubMed.
- V. Apalangya, V. Rangari, B. Tiimob, S. Jeelani and T. Samuel, Appl. Surf. Sci., 2014, 295, 108–114 CrossRef CAS.
- M. J. Rak, T. Friščić and A. Moores, RSC Adv., 2016, 6, 58365–58370 RSC.
- F. Schröder, U. K. Sharma, M. Mertens, F. Devred, D. P. Debecker, R. Luque and E. V. Van der Eycken, ACS Catal., 2016, 6, 8156–8161 CrossRef.
- M. Al-Naji, A. M. Balu, A. Roibu, M. Goepel, W.-D. Einicke, R. Luque and R. Gläser, Catal. Sci. Technol., 2015, 5, 2085–2091 RSC.
- X. Li, Z. Zhang, W. Xiao, S. Deng, C. Chen and N. Zhang, J. Mater. Chem. A, 2019, 7, 14504–14509 RSC.
- S. A. Kondrat, G. Shaw, S. J. Freakley, Q. He, J. Hampton, J. K. Edwards, P. J. Miedziak, T. E. Davies, A. F. Carley, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Chem. Sci., 2012, 3, 2965 RSC.
- Y. Lin, K. A. Watson, M. J. Fallbach, S. Ghose, J. G. Smith, D. M. Delozier, W. Cao, R. E. Crooks and J. W. Connell, ACS Nano, 2009, 3, 871–884 CrossRef CAS PubMed.
- A. R. Siamaki, Y. Lin, K. Woodberry, J. W. Connell and B. F. Gupton, J. Mater. Chem. A, 2013, 1, 12909 RSC.
- T. Ishida, N. Kawakita, T. Akita and M. Haruta, Deposition of gold clusters onto porous coordination polymers by solid grinding, Elsevier Masson SAS, 2010, vol. 175 Search PubMed.
- A. Zuliani, P. Ranjan, R. Luque and E. V Van der Eycken, ACS Sustainable Chem. Eng., 2019, 7, 5568–5575 CrossRef CAS.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- H. Chen, W. Lin, Z. Zhang, K. Jie, D. R. Mullins, X. Sang, S.-Z. Yang, C. J. Jafta, C. A. Bridges, X. Hu, R. R. Unocic, J. Fu, P. Zhang and S. Dai, ACS Mater. Lett., 2019, 1, 83–88 CrossRef CAS.
- Y. X. Shi, K. Xu, J. K. Clegg, R. Ganguly, H. Hirao, T. Friščić and F. García, Angew. Chem., Int. Ed., 2016, 55, 12736–12740 CrossRef CAS PubMed.
- L. Batzdorf, F. Fischer, M. Wilke, K.-J. Wenzel and F. Emmerling, Angew. Chem., Int. Ed., 2015, 54, 1799–1802 CrossRef CAS.
- T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. a J. Kimber, V. Honkimäki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66–73 CrossRef PubMed.
- G. Martin, Phys. Rev. B, 1984, 30, 1424–1436 CrossRef CAS.
- J. F. Willart and M. Descamps, Mol. Pharm., 2008, 5, 905–920 CrossRef CAS.
- M. Y. Malca, H. Bao, T. Bastaille, N. K. Saadé, J. M. Kinsella, T. Friščić and A. Moores, Chem. Mater., 2017, 29, 7766–7773 CrossRef CAS.
- Z. Shalabayev, M. Baláž, N. Daneu, E. Dutková, Z. Bujňáková, M. Kaňuchová, Z. Danková, Ĺ. Balážová, F. Urakaev, Ĺ. Tkáčiková and M. Burkitbayev, ACS Sustainable Chem. Eng., 2019, 7, 12897–12909 CrossRef CAS.
- V. Šepelák, A. Düvel, M. Wilkening, K.-D. Becker and P. Heitjans, Chem. Soc. Rev., 2013, 42, 7507 RSC.
| This journal is © The Royal Society of Chemistry 2020 |